Abstract
MyoD1 is a key regulator that orchestrates skeletal muscle differentiation through the regulation of gene expression. Although many studies have focused on its role in transcriptional control at gene promoters, less is known regarding the role of MyoD1 in the assembly of active enhancers. Here, we discuss novel data that point to the ability of MyoD1 to mediate the assembly of active enhancers that augment the transcription of genes essential for muscle development and lineage specification. Based on genome-wide studies of epigenetic marks that typify active enhancers, we recently identified the compendium of distal regulatory elements that dictate transcriptional programs during myogenesis. Superimposition of MyoD1 binding sites upon the locations of muscle enhancers revealed its unequivocal binding to a core region of nearly a third of condition-specific muscle enhancers. Further studies exploring deposition of enhancer-related epigenetic marks in myoblasts lacking MyoD1 demonstrate the dependence of muscle enhancer assembly on the presence of MyoD1. We propose a model wherein MyoD1 mediates recruitment of Set7, H3K4me1, H3K27ac, p300, and RNAP II to MyoD1-bound enhancers to establish condition-specific activation of muscle genes. Moreover, muscle enhancers are modulated through coordinated binding of transcription factors, including c-Jun, Jdp2, Meis, and Runx1, which are recruited to muscle enhancers in a MyoD1-dependent manner. Thus, MyoD1 and enhancer-associated transcription factors function coordinately to assemble and regulate enhancers, thereby augmenting expression of muscle-related genes.
Lineage specification demands precise and tightly coordinated patterns of gene expression that dictate cell fate choices. In metazoans, cell type specificity of gene expression patterns is largely regulated by distal regulatory elements, called enhancers. Similar to promoters, enhancers harbor arrays of sequences that are recognized by multiple transcription factors (TFs), but unlike promoters, enhancers can activate transcription independent of their location, distance, or orientation with respect to the transcription start site (TSS) of each gene (reviewed in refs. Citation1–Citation4). Seminal studies have revealed that chromatin states at promoters are highly correlated across different cell types, whereas histone modifications at enhancers are frequently cell type-specific and tightly associated with gene expression patterns.Citation5,Citation6 This fundamental observation suggests that enhancers can be regarded as a major determinant of cell type-specific gene expression in mammalian genomes. Indeed, evidence is emerging that due to their potent effect on gene transcription, enhancers are capable of influencing a variety of fundamental cellular phenomena, including stem cell multipotencyCitation7,Citation8 and nuclear and chromosomal architecture.Citation9,Citation10
Along these lines, aberrant enhancer function was recently demonstrated to have important implications for human disease and development. It has been estimated that the total coverage of human enhancers amounts to nearly 10% of the human genome, representing a 5-fold greater coverage compared with protein-coding sequences.Citation2 Although most well established disease-related mutations are within protein-coding sequences, it is logical to assume that mutations within enhancers could similarly result in human disease. These mutations could impair recruitment of TFs to enhancers, potentially disrupting tissue- and lineage-specific gene expression and, consequently, development. Indeed, sequencing analyses and epigenetic comparisons have implicated mutations in non-coding distal enhancers as a pathological basis of multiple diseases and cancer.Citation11-Citation15 In addition, single-nucleotide polymorphisms (SNPs) can confer genetic variation within enhancers as a result of altered TF binding to enhancer sites. For example, recent studies have demonstrated that a single nucleotide alteration in the enhancer of the sodium channel gene, SCN10A, functionally disrupts TBX3/TBX5 binding and leads to reduced cardiac activity of this enhancer in vivo.Citation16 In another report, allelic variation in a common non-coding SNP located in the distal enhancer of the pigment gene, OCA2, was shown to disrupt its regulatory function and promoter interactions, resulting in allelic gene expression differences that influence pigmentation.Citation17
Given the essential function of enhancers, it is important to comprehensively deduce the locations of these elements in each cell type. It is now clear that deposition of monomethylated H3K4 (H3K4me1), coupled with robust acetylation of histone H3K27 (H3K27ac), constitutes a predominant chromatin “signature” for transcriptional enhancers associated with actively transcribed genes.Citation18,Citation19 Recruitment of enzymes that facilitate this unique set of histone modifications, such as histone acetyltransferases, p300 and CREB binding protein (CBP),Citation20 which acetylate H3K27, and methyltransferases Set7,Citation13 MLL3 and MLL4,Citation14 which monomethylate H3K4, are also associated with active enhancers. Transcriptional enhancers were further shown to recruit RNA polymerase II (RNAP II), and such recruitment is associated with transcription of small RNAs and large non-coding RNAs.Citation20,Citation21 These observations, catalyzed by the advent of massively parallel sequencing, have accelerated enhancer identification and analysis and allowed enhancer mapping in numerous cell types, including forebrain,Citation22 cortical neurons,Citation20 bone marrow macrophages,Citation23 heart,Citation24,Citation25 and adipocytes.Citation26 Tissue-specific TF binding represents an additional attribute of active enhancers, and it is logical to postulate that such TFs facilitate recruitment of histone-modifying enzymes.Citation8 Motif searches on genomic regions identified by ChIP-seq analysis as potential enhancers reveal the recruitment of cell type-specific TFs that were previously implicated as essential regulators of lineage-specific differentiation. This list of factors includes Foxa1 and Foxa2 in liver cells,Citation8,Citation27 PU.1 in pro-B lymphocytes,Citation23 Rfx1 in neural progenitors,Citation8 and STAT1, 4 and 6 proteins in T cells,Citation28 among others. Thus, the recent discovery of the role of these TFs positions them as critical master regulators of enhancer activity.
Based on the combination of chromatin marks observed in a given state, distal enhancers can be separated into several classes: “active” enhancers marked by H3K4me1 and H3K27ac, “inactive” (or “poised”) enhancers associated with H3K4me1 but not H3K27ac, and “latent” enhancers, which, in terminally differentiated cells, are not bound by TFs and lack the histone marks characteristic of enhancers but are capable of rapidly acquiring both features in response to specific stimuli.Citation8,Citation29-Citation31 Thus, prior to their differentiation into pro-B lymphocytes, liver, or neuronal cells, ES cells contain poised enhancers that are converted to their active state upon differentiation.Citation8 Complementary evidence showed that in addition to H3K4me1 (and the absence of H3K27ac), poised enhancers in ES or undifferentiated cells are associated with the repressive modification H3K27me3.Citation30,Citation32 Interestingly, epigenetic studies in pluripotent ES cells have shown that during the earliest stages of development, poised enhancers are bound by pioneer factors (also known as “place-holders”) that act to maintain developmental enhancers in a poised/inactive state.Citation2,Citation3,Citation33 Upon differentiation, and depending upon the lineage that is adopted, the presence of such factors at enhancers decays, concomitant with the appearance of a master regulator and chromatin marks signifying an active enhancer. Thus, upon differentiation of ES cells into endoderm, FoxD3 is replaced by the activating factor, FoxA1Citation27; in addition, upon differentiation to neurons, Sox2 is replaced by Sox3 and Sox11.Citation34 Although the biology of placeholders needs further exploration, one critical aspect is their timely replacement by enhancer activating factors and the acquisition of active enhancer signature. It is possible that defects in this transition could be deleterious to differentiation, as they are likely to lock cells in a proliferative state, which could promote cancer.
MyoD1 as a Central Regulator of Active Muscle Enhancers
MyoD1 is a key master regulator of skeletal muscle differentiation,Citation35 and its role in myogenic differentiationCitation36 and trans-differentiation of fibroblastsCitation37 and other cell typesCitation38 has been well established. Indeed, enforced expression of MyoD1 alone is sufficient to trans-differentiate fibroblasts and other cell types to muscle.Citation36-Citation38 Upon induction of differentiation, MyoD1 interacts with E-proteins to form DNA-binding heterodimers that synergistically activate promoter-specific transcription of myogenic genesCitation39 in a temporally defined manner.Citation40 The MyoD1:E-protein heterodimer complex exhibits preference for an E-box element with the VCASCTGT consensus sequence (where V stands for A, C or G, and S stands for C or G), which was found to be enriched within promoter and enhancer regions associated with muscle-related genes.Citation41-Citation44 In addition, in vivoCitation45 and in vitroCitation46,Citation47 reporter assays established that MyoD1 activates transcription of genes containing multiple E-box motifs. Although the mechanisms by which MyoD1 contributes to muscle differentiation have primarily focused on promoter regulation, classical enhancer-reporter assays revealed a dependence on MyoD1 for enhancer activity as well. Enhancer regions critical for the myogenic program and enriched with E-box elements that recruit MyoD1 have been identified distal to the myogenin,Citation48,Citation49 MyoD1,Citation50 myosin chain 1,Citation51-Citation53 Ckm,Citation54,Citation55 Myf5 and Mrf4,Citation56,Citation57 γ-sarcoglycan,Citation58 Id1Citation59 and Adamts5Citation60 genes.
To identify active enhancers in skeletal muscle genome-wide and to elucidate the molecular mechanisms that regulate their activity during differentiation, we recently performed ChIP-seq analysis (high-throughput sequencing of DNA enriched by chromatin immunoprecipitation) on four of the well-established enhancer marks (H3K4Me1, H3K27ac, p300 and RNAP II) before and after myogenic differentiation.Citation61 Our analysis revealed that the total number of muscle enhancers increased throughout differentiation from approximately 4,000 in myoblasts to as many as 1.5-times that number in myotubes. Of these, ~3,000 were exclusively active before differentiation, and ~5,000 were only active after differentiation (whereas the remaining enhancers were constitutively marked in both conditions). Interestingly, this finding is in line with the previously known increase in active enhancer assembly in the transition from blastula to gastrula stagesCitation31 and from embryonic stem cells (ESC) to neuronal progenitor cells (NPC),Citation8 and thus, it may reflect a general feature of enhancers related to differentiation. Our compendium of distal enhancers recovered many previously identified enhancer regions, and overall there was a strong correlation between the activity of condition-specific enhancers and the transcription level of their adjacent genes, indicating that these enhancers augment transcription of thousands of protein-coding genes. Consistent with other tissue-specific elements whose evolution occurs at higher rates,Citation62,Citation63 we have shown that muscle-specific enhancers are strongly- but not ultra-conserved.Citation61
Furthermore, by linking condition-specific enhancers to their nearest associated condition-specific genes, we showed that the median enhancer-promoter distance for myotubes was significantly shorter (by more than 13 kb) than the corresponding distance in myoblasts, suggesting that reduction in the distances between enhancers and their active, linked promoters may also be a characteristic of muscle differentiation. Future studies will determine whether this reduction could be related to differentiation-dependent, architectural changes in chromatin. In addition, approximately 10% of our condition-specific enhancers were associated with non-coding transcripts,Citation64 and ~60% of these enhancers displayed significant levels of RNAP II recruitment. These findings are in line with previous reports that suggest a cis-regulatory role for transcripts emanating from enhancers.Citation20,Citation21
In-silico analysis indicated that nearly half of myoblast-specific and approximately 80% of myotube-specific enhancers exhibited predicted MyoD1 binding sites.Citation61 However, overlap of our enhancer data sets with experimentally determined MyoD1-binding eventsCitation44 revealed that approximately 30% of condition-specific enhancers were bound with MyoD1, suggesting that additional epigenetic cues, such as binding of sequence-specific factors and/or chromatin architecture, dictated by histone modifications, play an important role in limiting MyoD1 recruitment to muscle enhancers. The strong trans-activation potential of MyoD1 at muscle enhancers is likely augmented through its ability to interact with multiple transcriptional regulatory factors. Motif enrichment analysis of ChIP-seq data sets indicated that sequence-specific TFs can be recruited in a spatially constrained manner around MyoD1-binding sites. Importantly, several of these identified TFs, such as Jdp2,Citation65 Meis,Citation66 c-Jun,Citation67 and Runx1,Citation68 are well known regulators of myogenesis, and in the absence of MyoD1, the recruitment of these factors, as well as RNAP II, was dramatically reduced at MyoD1-bound enhancers (). These findings suggest that MyoD1 plays an essential role in enhancer assembly by recruiting TFs that have established roles in muscle differentiation. Indeed, genome-wide analysis of c-Jun binding sites in myoblasts indicated that MyoD1 and c-Jun co-localize within a narrow window on 54% of muscle enhancers.Citation61 Further, suppression of c-Jun expression in myoblasts led to strong reductions in the levels of H3K4me1 and H3K27ac at selected myoblast enhancers bound by c-Jun but not at other enhancers that were not bound by this TF.Citation61 These studies, which strongly suggest that MyoD1 and c-Jun coordinately regulate enhancer assembly, are consistent with biochemical studies showing that these proteins interact in vitro.Citation67 It will be interesting in the future to determine whether mutations and SNPs that map to our compendium of enhancers—particularly those that map to MyoD1, c-Jun, and other factor binding sites—could underlie human skeletal muscle disease.
Figure 1. Model for the coordinate assembly of active MyoD1 enhancers in muscle. (A) MyoD1 co-binds to enhancers in conjunction with a putative pioneer factor (“placeholder”) that maintains them in a poised/inactive state. (B) Eviction (or inactivation) of enhancer-bound placeholder allows the recruitment of other transcription factors that positively regulate enhancer activity, leading to acquisition of a transcriptionally active state, characterized by deposition of H3K4me1 and H3K27ac and often in non-coding transcription. See text for further details.
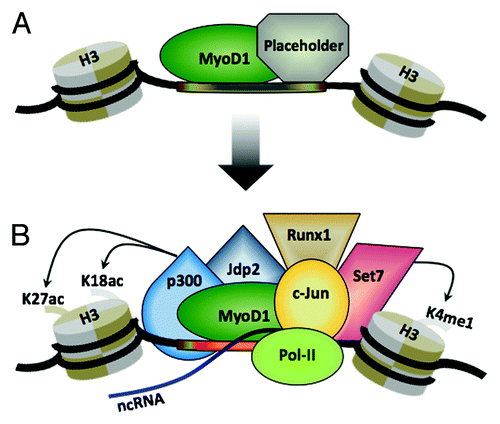
Several studies have indicated that once tethered to its binding sites at promoters, MyoD1 recruits the acetyltransferases, p300 and PCAF, leading to acetylation of histones H3 and H4Citation44,Citation69-Citation73 and MyoD,Citation47,Citation74 respectively. Our analysis indicated that the overwhelming majority of condition-specific enhancers bound by MyoD1 were co-occupied by p300. Statistically, the fraction of MyoD1-bound enhancers that recruit p300 was significantly higher than the fraction of MyoD1 enhancers that lack p300, suggesting that MyoD1 might have a strong impact on p300 recruitment to active muscle enhancers. Indeed, MyoD1-null myoblasts exhibited sharply diminished levels of p300 and H3K27ac at each of the MyoD1-bound enhancers that we tested.Citation61 qChIP experiments revealed that Set7 was recruited to muscle enhancers in a MyoD1-dependent manner, since recruitment of Set7 and deposition of H3K4me1 were significantly diminished at MyoD1-bound enhancers in MyoD1−/− myoblasts, as compared with their wild-type counterparts.Citation61 These results are consistent with another study that demonstrated direct MyoD1-Set7 interactions on the MCK enhancer and delineated the importance of Set7 for promoting myoblast differentiation via regulation of H3K4me1 deposition.Citation75 Furthermore, we detected MyoD1-independent recruitment of Set7 to enhancers, suggesting that this methyltransferase may be recruited by TFs other than MyoD1. Despite these observations, one important question—the functional role of mono-methylation of H3K4 in enhancer regulation—will need to be addressed to complete our understanding of MyoD1 function.
Another intriguing question pertains to the role of placeholders as regulators of the myogenic program. It is interesting to speculate that myogenic muscle precursor cells (myoblasts) found within the developing limb bud and regenerating satellite cells might show widespread binding of such putative factors to their enhancers (). In each of these cases, MyoD1 is expressed at high levels, yet these populations remain undifferentiated until appropriate environmental conditions are satisfied. Attempts to identify a specific modification that can convert MyoD1 from an inactive to an active form have not succeeded.Citation76 Of note, our studies have shown that among MyoD1-bound enhancers that become exclusively active in myotubes, the majority of them (~72%), are already bound by MyoD1 in myoblasts, prior to their assembly into active enhancers.Citation61 These results suggest that in many cases, binding of MyoD1 by itself is not sufficient to promote the transition to an active enhancer, and it is plausible that an additional trigger is required to facilitate the removal or acquisition of a specific factor that co-occupies the enhancer with MyoD1. It is possible that co-repressors (or other placeholders) can suppress the action of MyoD1 bound to muscle enhancers before it becomes active.Citation33,Citation77,Citation78 Future investigations into how placeholders restrain the transcriptional activity of MyoD1 (and possibly other myogenic regulatory factors, MRFs) will greatly advance our understanding of mechanisms through which the myogenic program is induced.
Signaling to Enhancers: Conversion to an Active or Inactive State
Recent studies have delineated the signaling pathways that govern specification of mesodermal precursor cells into myoblasts and subsequently into differentiated myotubes (reviewed in ref. Citation79). Such specification of these precursor cells, located in the dorsal region of the somites (dermomyotome), into myoblasts is mediated by a variety of molecular signals emanating from surrounding tissues. A combination of stimulatory signals, including Wnts, Sonic hedgehog (Shh), and Noggin, and suppressive signals, such as BMP4, ultimately dictate expression patterns of the primary MRFs, MyoD1 and Myf5, and therefore propel commitment to the myogenic fate. In the lateral part of the dermomyotome, myoblasts that preferentially express MyoD1 migrate to form the myotome, which eventually forms the skeletal musculature. Increased expression of the secondary MRFs—myogenin and MRF4—stimulates the next step in myogenesis, as skeletal myoblasts fuse and eventually form bundles of multinucleated myofibers.Citation79
Several studies suggest that extracellular signals could ultimately be converted into epigenetic modifications that directly affect transcription. For example, one of the major pathways associated with muscle differentiation is the p38 MAP kinase signaling cascade. At the onset of differentiation, p38 is activated and phosphorylates E47 at Ser140, leading to its dimerization with MyoD1 and to subsequent binding to E-box motifs of different muscle promoters.Citation80 In addition, phosphorylation of the SWI/SNF component, BAF60, by p38α/β stimulates the recruitment of this multi-subunit chromatin-remodeling complex to regulatory regions of muscle-specific genes.Citation71 Further, forced inhibition of p38α/β blocks the engagement between MyoD1 and BRG1 and BRM, two ATPase subunits of SWI/SNF.Citation71 Another aspect of p38 activity is its ability to directly phosphorylate the transactivation domains of MEF2A and MEF2C, which, through interactions with MyoD1, results in stimulation of their transcriptional activity.Citation81-Citation84 By contrast, p38γ is known to phosphorylate MyoD1, resulting in its enhanced promoter occupancy but reduced transcriptional activity.Citation85 Another signaling pathway that is known to mediate intracellular events in response to external growth factors, such as IGF1, is PI3K/AKT signaling. This pathway, known to function in parallel with the p38 cascade during early myogenic differentiation, is critically involved in activation of muscle differentiation,Citation86 muscle cell survival,Citation87 and regeneration.Citation88,Citation89 Consistent with this notion, suppression of each of these pathways was shown to alter the patterns of specific chromatin modifiers on the Mck enhancer.Citation90
Given the fact that MyoD1 plays a prominent role at enhancers, and since several of these signaling pathways impinge on MyoD1 activity, it is reasonable to postulate that these cascades may indeed regulate MyoD1 activity at enhancers, converting them to an activated or inactivated state. Thus, although the mechanisms through which these signaling pathways participate in regulation of enhancer activity remain largely unknown, future studies will determine the extent to which these signaling pathways play a role at enhancers, before and during muscle differentiation.
Conclusion
In summary, we propose that while MyoD1 binds to a large number of promoters that augment expression of genes essential for specifying the muscle lineage, a key role for this factor appears to be its ability to bind to enhancers. Our model establishes a role for MyoD1 in mediating the co-recruitment of several chromatin modifying enzymes, among them Set7 and p300, which subsequently deposit the histone marks that typify active enhancers (). Importantly, several TFs, including c-Jun, are recruited to active enhancers bound by MyoD1, suggesting that interactions between MyoD1 and other TFs are necessary for assembly of active enhancers. Interestingly, unlike MyoD1, whose expression is restricted to skeletal muscle, c-Jun is expressed in multiple tissue types. Thus, it is interesting to speculate that in other tissues, alternative bHLH-type transcriptional activators could collaborate with c-Jun to regulate enhancer activity through a similar mechanism.
Acknowledgments
We thank members of the Dynlacht laboratory, in particular J. Cheng and C. Bowman, for helpful discussions. Work in the Dynlacht laboratory was supported by NIH grant 2R01 GM067132.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Ong CT, Corces VG. Enhancer function: new insights into the regulation of tissue-specific gene expression. Nat Rev Genet 2011; 12:283 - 93; http://dx.doi.org/10.1038/nrg2957; PMID: 21358745
- Bulger M, Groudine M. Functional and mechanistic diversity of distal transcription enhancers. Cell 2011; 144:327 - 39; http://dx.doi.org/10.1016/j.cell.2011.01.024; PMID: 21295696
- Spitz F, Furlong EE. Transcription factors: from enhancer binding to developmental control. Nat Rev Genet 2012; 13:613 - 26; http://dx.doi.org/10.1038/nrg3207; PMID: 22868264
- Calo E, Wysocka J. Modification of enhancer chromatin: what, how, and why?. Mol Cell 2013; 49:825 - 37; http://dx.doi.org/10.1016/j.molcel.2013.01.038; PMID: 23473601
- Heintzman ND, Hon GC, Hawkins RD, Kheradpour P, Stark A, Harp LF, et al. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature 2009; 459:108 - 12; http://dx.doi.org/10.1038/nature07829; PMID: 19295514
- Visel A, Blow MJ, Li Z, Zhang T, Akiyama JA, Holt A, et al. ChIP-seq accurately predicts tissue-specific activity of enhancers. Nature 2009; 457:854 - 8; http://dx.doi.org/10.1038/nature07730; PMID: 19212405
- Cui K, Zang C, Roh TY, Schones DE, Childs RW, Peng W, et al. Chromatin signatures in multipotent human hematopoietic stem cells indicate the fate of bivalent genes during differentiation. Cell Stem Cell 2009; 4:80 - 93; http://dx.doi.org/10.1016/j.stem.2008.11.011; PMID: 19128795
- Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, Steine EJ, et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc Natl Acad Sci U S A 2010; 107:21931 - 6; http://dx.doi.org/10.1073/pnas.1016071107; PMID: 21106759
- Blackwood EM, Kadonaga JT. Going the distance: a current view of enhancer action. Science 1998; 281:60 - 3; http://dx.doi.org/10.1126/science.281.5373.60; PMID: 9679020
- Chepelev I, Wei G, Wangsa D, Tang Q, Zhao K. Characterization of genome-wide enhancer-promoter interactions reveals co-expression of interacting genes and modes of higher order chromatin organization. Cell Res 2012; 22:490 - 503; http://dx.doi.org/10.1038/cr.2012.15; PMID: 22270183
- Akhtar-Zaidi B, Cowper-Sal-lari R, Corradin O, Saiakhova A, Bartels CF, Balasubramanian D, et al. Epigenomic enhancer profiling defines a signature of colon cancer. Science 2012; 336:736 - 9; http://dx.doi.org/10.1126/science.1217277; PMID: 22499810
- Emison ES, McCallion AS, Kashuk CS, Bush RT, Grice E, Lin S, et al. A common sex-dependent mutation in a RET enhancer underlies Hirschsprung disease risk. Nature 2005; 434:857 - 63; http://dx.doi.org/10.1038/nature03467; PMID: 15829955
- Kleinjan DA, van Heyningen V. Long-range control of gene expression: emerging mechanisms and disruption in disease. Am J Hum Genet 2005; 76:8 - 32; http://dx.doi.org/10.1086/426833; PMID: 15549674
- Visel A, Rubin EM, Pennacchio LA. Genomic views of distant-acting enhancers. Nature 2009; 461:199 - 205; http://dx.doi.org/10.1038/nature08451; PMID: 19741700
- Noonan JP, McCallion AS. Genomics of long-range regulatory elements. Annu Rev Genomics Hum Genet 2010; 11:1 - 23; http://dx.doi.org/10.1146/annurev-genom-082509-141651; PMID: 20438361
- van den Boogaard M, Wong LY, Tessadori F, Bakker ML, Dreizehnter LK, Wakker V, et al. Genetic variation in T-box binding element functionally affects SCN5A/SCN10A enhancer. J Clin Invest 2012; 122:2519 - 30; http://dx.doi.org/10.1172/JCI62613; PMID: 22706305
- Visser M, Kayser M, Palstra RJ. HERC2 rs12913832 modulates human pigmentation by attenuating chromatin-loop formation between a long-range enhancer and the OCA2 promoter. Genome Res 2012; 22:446 - 55; http://dx.doi.org/10.1101/gr.128652.111; PMID: 22234890
- Heintzman ND, Stuart RK, Hon G, Fu Y, Ching CW, Hawkins RD, et al. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat Genet 2007; 39:311 - 8; http://dx.doi.org/10.1038/ng1966; PMID: 17277777
- Zentner GE, Tesar PJ, Scacheri PC. Epigenetic signatures distinguish multiple classes of enhancers with distinct cellular functions. Genome Res 2011; 21:1273 - 83; http://dx.doi.org/10.1101/gr.122382.111; PMID: 21632746
- Kim TK, Hemberg M, Gray JM, Costa AM, Bear DM, Wu J, et al. Widespread transcription at neuronal activity-regulated enhancers. Nature 2010; 465:182 - 7; http://dx.doi.org/10.1038/nature09033; PMID: 20393465
- De Santa F, Barozzi I, Mietton F, Ghisletti S, Polletti S, Tusi BK, et al. A large fraction of extragenic RNA pol II transcription sites overlap enhancers. PLoS Biol 2010; 8:e1000384; http://dx.doi.org/10.1371/journal.pbio.1000384; PMID: 20485488
- Visel A, Taher L, Girgis H, May D, Golonzhka O, Hoch RV, et al. A high-resolution enhancer atlas of the developing telencephalon. Cell 2013; 152:895 - 908; http://dx.doi.org/10.1016/j.cell.2012.12.041; PMID: 23375746
- Ghisletti S, Barozzi I, Mietton F, Polletti S, De Santa F, Venturini E, et al. Identification and characterization of enhancers controlling the inflammatory gene expression program in macrophages. Immunity 2010; 32:317 - 28; http://dx.doi.org/10.1016/j.immuni.2010.02.008; PMID: 20206554
- May D, Blow MJ, Kaplan T, McCulley DJ, Jensen BC, Akiyama JA, et al. Large-scale discovery of enhancers from human heart tissue. Nat Genet 2012; 44:89 - 93; http://dx.doi.org/10.1038/ng.1006; PMID: 22138689
- Narlikar L, Sakabe NJ, Blanski AA, Arimura FE, Westlund JM, Nobrega MA, et al. Genome-wide discovery of human heart enhancers. Genome Res 2010; 20:381 - 92; http://dx.doi.org/10.1101/gr.098657.109; PMID: 20075146
- Mikkelsen TS, Xu Z, Zhang X, Wang L, Gimble JM, Lander ES, et al. Comparative epigenomic analysis of murine and human adipogenesis. Cell 2010; 143:156 - 69; http://dx.doi.org/10.1016/j.cell.2010.09.006; PMID: 20887899
- Xu J, Watts JA, Pope SD, Gadue P, Kamps M, Plath K, et al. Transcriptional competence and the active marking of tissue-specific enhancers by defined transcription factors in embryonic and induced pluripotent stem cells. Genes Dev 2009; 23:2824 - 38; http://dx.doi.org/10.1101/gad.1861209; PMID: 20008934
- Vahedi G, Takahashi H, Nakayamada S, Sun HW, Sartorelli V, Kanno Y, et al. STATs shape the active enhancer landscape of T cell populations. Cell 2012; 151:981 - 93; http://dx.doi.org/10.1016/j.cell.2012.09.044; PMID: 23178119
- Ostuni R, Piccolo V, Barozzi I, Polletti S, Termanini A, Bonifacio S, et al. Latent enhancers activated by stimulation in differentiated cells. Cell 2013; 152:157 - 71; http://dx.doi.org/10.1016/j.cell.2012.12.018; PMID: 23332752
- Rada-Iglesias A, Bajpai R, Swigut T, Brugmann SA, Flynn RA, Wysocka J. A unique chromatin signature uncovers early developmental enhancers in humans. Nature 2011; 470:279 - 83; http://dx.doi.org/10.1038/nature09692; PMID: 21160473
- Bogdanovic O, Fernandez-Miñán A, Tena JJ, de la Calle-Mustienes E, Hidalgo C, van Kruysbergen I, et al. Dynamics of enhancer chromatin signatures mark the transition from pluripotency to cell specification during embryogenesis. Genome Res 2012; 22:2043 - 53; http://dx.doi.org/10.1101/gr.134833.111; PMID: 22593555
- Asp P, Blum R, Vethantham V, Parisi F, Micsinai M, Cheng J, et al. Genome-wide remodeling of the epigenetic landscape during myogenic differentiation. Proc Natl Acad Sci U S A 2011; 108:E149 - 58; http://dx.doi.org/10.1073/pnas.1102223108; PMID: 21551099
- Zaret KS, Carroll JS. Pioneer transcription factors: establishing competence for gene expression. Genes Dev 2011; 25:2227 - 41; http://dx.doi.org/10.1101/gad.176826.111; PMID: 22056668
- Bergsland M, Ramsköld D, Zaouter C, Klum S, Sandberg R, Muhr J. Sequentially acting Sox transcription factors in neural lineage development. Genes Dev 2011; 25:2453 - 64; http://dx.doi.org/10.1101/gad.176008.111; PMID: 22085726
- Tapscott SJ. The circuitry of a master switch: Myod and the regulation of skeletal muscle gene transcription. Development 2005; 132:2685 - 95; http://dx.doi.org/10.1242/dev.01874; PMID: 15930108
- Rudnicki MA, Schnegelsberg PN, Stead RH, Braun T, Arnold HH, Jaenisch R. MyoD or Myf-5 is required for the formation of skeletal muscle. Cell 1993; 75:1351 - 9; http://dx.doi.org/10.1016/0092-8674(93)90621-V; PMID: 8269513
- Davis RL, Weintraub H, Lassar AB. Expression of a single transfected cDNA converts fibroblasts to myoblasts. Cell 1987; 51:987 - 1000; http://dx.doi.org/10.1016/0092-8674(87)90585-X; PMID: 3690668
- Weintraub H, Tapscott SJ, Davis RL, Thayer MJ, Adam MA, Lassar AB, et al. Activation of muscle-specific genes in pigment, nerve, fat, liver, and fibroblast cell lines by forced expression of MyoD. Proc Natl Acad Sci U S A 1989; 86:5434 - 8; http://dx.doi.org/10.1073/pnas.86.14.5434; PMID: 2748593
- Lassar AB, Davis RL, Wright WE, Kadesch T, Murre C, Voronova A, et al. Functional activity of myogenic HLH proteins requires hetero-oligomerization with E12/E47-like proteins in vivo. Cell 1991; 66:305 - 15; http://dx.doi.org/10.1016/0092-8674(91)90620-E; PMID: 1649701
- Bergstrom DA, Penn BH, Strand A, Perry RL, Rudnicki MA, Tapscott SJ. Promoter-specific regulation of MyoD binding and signal transduction cooperate to pattern gene expression. Mol Cell 2002; 9:587 - 600; http://dx.doi.org/10.1016/S1097-2765(02)00481-1; PMID: 11931766
- Murre C, McCaw PS, Vaessin H, Caudy M, Jan LY, Jan YN, et al. Interactions between heterologous helix-loop-helix proteins generate complexes that bind specifically to a common DNA sequence. Cell 1989; 58:537 - 44; http://dx.doi.org/10.1016/0092-8674(89)90434-0; PMID: 2503252
- Blackwell TK, Weintraub H. Differences and similarities in DNA-binding preferences of MyoD and E2A protein complexes revealed by binding site selection. Science 1990; 250:1104 - 10; http://dx.doi.org/10.1126/science.2174572; PMID: 2174572
- Blais A, Tsikitis M, Acosta-Alvear D, Sharan R, Kluger Y, Dynlacht BD. An initial blueprint for myogenic differentiation. Genes Dev 2005; 19:553 - 69; http://dx.doi.org/10.1101/gad.1281105; PMID: 15706034
- Cao Y, Yao Z, Sarkar D, Lawrence M, Sanchez GJ, Parker MH, et al. Genome-wide MyoD binding in skeletal muscle cells: a potential for broad cellular reprogramming. Dev Cell 2010; 18:662 - 74; http://dx.doi.org/10.1016/j.devcel.2010.02.014; PMID: 20412780
- Davis RL, Weintraub H. Acquisition of myogenic specificity by replacement of three amino acid residues from MyoD into E12. Science 1992; 256:1027 - 30; http://dx.doi.org/10.1126/science.1317057; PMID: 1317057
- Bengal E, Flores O, Rangarajan PN, Chen A, Weintraub H, Verma IM. Positive control mutations in the MyoD basic region fail to show cooperative DNA binding and transcriptional activation in vitro. Proc Natl Acad Sci U S A 1994; 91:6221 - 5; http://dx.doi.org/10.1073/pnas.91.13.6221; PMID: 8016142
- Dilworth FJ, Seaver KJ, Fishburn AL, Htet SL, Tapscott SJ. In vitro transcription system delineates the distinct roles of the coactivators pCAF and p300 during MyoD/E47-dependent transactivation. Proc Natl Acad Sci U S A 2004; 101:11593 - 8; http://dx.doi.org/10.1073/pnas.0404192101; PMID: 15289617
- Yee SP, Rigby PW. The regulation of myogenin gene expression during the embryonic development of the mouse. Genes Dev 1993; 7:7A 1277 - 89; http://dx.doi.org/10.1101/gad.7.7a.1277; PMID: 8391506
- Cheng TC, Wallace MC, Merlie JP, Olson EN. Separable regulatory elements governing myogenin transcription in mouse embryogenesis. Science 1993; 261:215 - 8; http://dx.doi.org/10.1126/science.8392225; PMID: 8392225
- Goldhamer DJ, Brunk BP, Faerman A, King A, Shani M, Emerson CP Jr.. Embryonic activation of the myoD gene is regulated by a highly conserved distal control element. Development 1995; 121:637 - 49; PMID: 7720572
- Donoghue M, Ernst H, Wentworth B, Nadal-Ginard B, Rosenthal N. A muscle-specific enhancer is located at the 3′ end of the myosin light-chain 1/3 gene locus. Genes Dev 1988; 2:12B 1779 - 90; http://dx.doi.org/10.1101/gad.2.12b.1779; PMID: 3240859
- Rosenthal N, Berglund EB, Wentworth BM, Donoghue M, Winter B, Bober E, et al. A highly conserved enhancer downstream of the human MLC1/3 locus is a target for multiple myogenic determination factors. Nucleic Acids Res 1990; 18:6239 - 46; http://dx.doi.org/10.1093/nar/18.21.6239; PMID: 2243772
- Wentworth BM, Donoghue M, Engert JC, Berglund EB, Rosenthal N. Paired MyoD-binding sites regulate myosin light chain gene expression. Proc Natl Acad Sci U S A 1991; 88:1242 - 6; http://dx.doi.org/10.1073/pnas.88.4.1242; PMID: 1847512
- Johnson JE, Wold BJ, Hauschka SD. Muscle creatine kinase sequence elements regulating skeletal and cardiac muscle expression in transgenic mice. Mol Cell Biol 1989; 9:3393 - 9; PMID: 2796990
- Horlick RA, Benfield PA. The upstream muscle-specific enhancer of the rat muscle creatine kinase gene is composed of multiple elements. Mol Cell Biol 1989; 9:2396 - 413; PMID: 2761536
- Carvajal JJ, Keith A, Rigby PW. Global transcriptional regulation of the locus encoding the skeletal muscle determination genes Mrf4 and Myf5. Genes Dev 2008; 22:265 - 76; http://dx.doi.org/10.1101/gad.442408; PMID: 18198342
- Chang TH, Primig M, Hadchouel J, Tajbakhsh S, Rocancourt D, Fernandez A, et al. An enhancer directs differential expression of the linked Mrf4 and Myf5 myogenic regulatory genes in the mouse. Dev Biol 2004; 269:595 - 608; http://dx.doi.org/10.1016/j.ydbio.2004.02.013; PMID: 15110722
- Wakabayashi-Takai E, Noguchi S, Ozawa E. Identification of myogenesis-dependent transcriptional enhancers in promoter region of mouse gamma-sarcoglycan gene. Eur J Biochem 2001; 268:948 - 57; http://dx.doi.org/10.1046/j.1432-1327.2001.01954.x; PMID: 11179961
- Tournay O, Benezra R. Transcription of the dominant-negative helix-loop-helix protein Id1 is regulated by a protein complex containing the immediate-early response gene Egr-1. Mol Cell Biol 1996; 16:2418 - 30; PMID: 8628310
- Barthel KK, Liu X. A transcriptional enhancer from the coding region of ADAMTS5. PLoS One 2008; 3:e2184; http://dx.doi.org/10.1371/journal.pone.0002184; PMID: 18478108
- Blum R, Vethantham V, Bowman C, Rudnicki M, Dynlacht BD. Genome-wide identification of enhancers in skeletal muscle: the role of MyoD1. Genes Dev 2012; 26:2763 - 79; http://dx.doi.org/10.1101/gad.200113.112; PMID: 23249738
- Ponting CP. The functional repertoires of metazoan genomes. Nat Rev Genet 2008; 9:689 - 98; http://dx.doi.org/10.1038/nrg2413; PMID: 18663365
- Prabhakar S, Poulin F, Shoukry M, Afzal V, Rubin EM, Couronne O, et al. Close sequence comparisons are sufficient to identify human cis-regulatory elements. Genome Res 2006; 16:855 - 63; http://dx.doi.org/10.1101/gr.4717506; PMID: 16769978
- Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 2010; 28:511 - 5; http://dx.doi.org/10.1038/nbt.1621; PMID: 20436464
- Ostrovsky O, Bengal E, Aronheim A. Induction of terminal differentiation by the c-Jun dimerization protein JDP2 in C2 myoblasts and rhabdomyosarcoma cells. J Biol Chem 2002; 277:40043 - 54; http://dx.doi.org/10.1074/jbc.M205494200; PMID: 12171923
- Knoepfler PS, Bergstrom DA, Uetsuki T, Dac-Korytko I, Sun YH, Wright WE, et al. A conserved motif N-terminal to the DNA-binding domains of myogenic bHLH transcription factors mediates cooperative DNA binding with pbx-Meis1/Prep1. Nucleic Acids Res 1999; 27:3752 - 61; http://dx.doi.org/10.1093/nar/27.18.3752; PMID: 10471746
- Bengal E, Ransone L, Scharfmann R, Dwarki VJ, Tapscott SJ, Weintraub H, et al. Functional antagonism between c-Jun and MyoD proteins: a direct physical association. Cell 1992; 68:507 - 19; http://dx.doi.org/10.1016/0092-8674(92)90187-H; PMID: 1310896
- Wang X, Blagden C, Fan J, Nowak SJ, Taniuchi I, Littman DR, et al. Runx1 prevents wasting, myofibrillar disorganization, and autophagy of skeletal muscle. Genes Dev 2005; 19:1715 - 22; http://dx.doi.org/10.1101/gad.1318305; PMID: 16024660
- Puri PL, Sartorelli V, Yang XJ, Hamamori Y, Ogryzko VV, Howard BH, et al. Differential roles of p300 and PCAF acetyltransferases in muscle differentiation. Mol Cell 1997; 1:35 - 45; http://dx.doi.org/10.1016/S1097-2765(00)80005-2; PMID: 9659901
- Rampalli S, Li L, Mak E, Ge K, Brand M, Tapscott SJ, et al. p38 MAPK signaling regulates recruitment of Ash2L-containing methyltransferase complexes to specific genes during differentiation. Nat Struct Mol Biol 2007; 14:1150 - 6; http://dx.doi.org/10.1038/nsmb1316; PMID: 18026121
- Simone C, Forcales SV, Hill DA, Imbalzano AN, Latella L, Puri PL. p38 pathway targets SWI-SNF chromatin-remodeling complex to muscle-specific loci. Nat Genet 2004; 36:738 - 43; http://dx.doi.org/10.1038/ng1378; PMID: 15208625
- Cao Y, Kumar RM, Penn BH, Berkes CA, Kooperberg C, Boyer LA, et al. Global and gene-specific analyses show distinct roles for Myod and Myog at a common set of promoters. EMBO J 2006; 25:502 - 11; http://dx.doi.org/10.1038/sj.emboj.7600958; PMID: 16437161
- Yuan W, Condorelli G, Caruso M, Felsani A, Giordano A. Human p300 protein is a coactivator for the transcription factor MyoD. J Biol Chem 1996; 271:9009 - 13; http://dx.doi.org/10.1074/jbc.271.15.9009; PMID: 8621548
- Sartorelli V, Puri PL, Hamamori Y, Ogryzko V, Chung G, Nakatani Y, et al. Acetylation of MyoD directed by PCAF is necessary for the execution of the muscle program. Mol Cell 1999; 4:725 - 34; http://dx.doi.org/10.1016/S1097-2765(00)80383-4; PMID: 10619020
- Tao Y, Neppl RL, Huang ZP, Chen J, Tang RH, Cao R, et al. The histone methyltransferase Set7/9 promotes myoblast differentiation and myofibril assembly. J Cell Biol 2011; 194:551 - 65; http://dx.doi.org/10.1083/jcb.201010090; PMID: 21859860
- Ludolph DC, Konieczny SF. Transcription factor families: muscling in on the myogenic program. FASEB J 1995; 9:1595 - 604; PMID: 8529839
- Wang JC, Waltner-Law M, Yamada K, Osawa H, Stifani S, Granner DK. Transducin-like enhancer of split proteins, the human homologs of Drosophila groucho, interact with hepatic nuclear factor 3beta. J Biol Chem 2000; 275:18418 - 23; http://dx.doi.org/10.1074/jbc.M910211199; PMID: 10748198
- Sekiya T, Zaret KS. Repression by Groucho/TLE/Grg proteins: genomic site recruitment generates compacted chromatin in vitro and impairs activator binding in vivo. Mol Cell 2007; 28:291 - 303; http://dx.doi.org/10.1016/j.molcel.2007.10.002; PMID: 17964267
- Chargé SB, Rudnicki MA. Cellular and molecular regulation of muscle regeneration. Physiol Rev 2004; 84:209 - 38; http://dx.doi.org/10.1152/physrev.00019.2003; PMID: 14715915
- Lluís F, Ballestar E, Suelves M, Esteller M, Muñoz-Cánoves P. E47 phosphorylation by p38 MAPK promotes MyoD/E47 association and muscle-specific gene transcription. EMBO J 2005; 24:974 - 84; http://dx.doi.org/10.1038/sj.emboj.7600528; PMID: 15719023
- Ornatsky OI, Cox DM, Tangirala P, Andreucci JJ, Quinn ZA, Wrana JL, et al. Post-translational control of the MEF2A transcriptional regulatory protein. Nucleic Acids Res 1999; 27:2646 - 54; http://dx.doi.org/10.1093/nar/27.13.2646; PMID: 10373581
- Zhao M, New L, Kravchenko VV, Kato Y, Gram H, di Padova F, et al. Regulation of the MEF2 family of transcription factors by p38. Mol Cell Biol 1999; 19:21 - 30; PMID: 9858528
- Puri PL, Wu Z, Zhang P, Wood LD, Bhakta KS, Han J, et al. Induction of terminal differentiation by constitutive activation of p38 MAP kinase in human rhabdomyosarcoma cells. Genes Dev 2000; 14:574 - 84; PMID: 10716945
- Black BL, Olson EN. Transcriptional control of muscle development by myocyte enhancer factor-2 (MEF2) proteins. Annu Rev Cell Dev Biol 1998; 14:167 - 96; http://dx.doi.org/10.1146/annurev.cellbio.14.1.167; PMID: 9891782
- Gillespie MA, Le Grand F, Scimè A, Kuang S, von Maltzahn J, Seale V, et al. p38-gamma-dependent gene silencing restricts entry into the myogenic differentiation program. J Cell Biol 2009; 187:991 - 1005; http://dx.doi.org/10.1083/jcb.200907037; PMID: 20026657
- Wu Z, Woodring PJ, Bhakta KS, Tamura K, Wen F, Feramisco JR, et al. p38 and extracellular signal-regulated kinases regulate the myogenic program at multiple steps. Mol Cell Biol 2000; 20:3951 - 64; http://dx.doi.org/10.1128/MCB.20.11.3951-3964.2000; PMID: 10805738
- Lawlor MA, Rotwein P. Insulin-like growth factor-mediated muscle cell survival: central roles for Akt and cyclin-dependent kinase inhibitor p21. Mol Cell Biol 2000; 20:8983 - 95; http://dx.doi.org/10.1128/MCB.20.23.8983-8995.2000; PMID: 11073997
- Musarò A, McCullagh K, Paul A, Houghton L, Dobrowolny G, Molinaro M, et al. Localized Igf-1 transgene expression sustains hypertrophy and regeneration in senescent skeletal muscle. Nat Genet 2001; 27:195 - 200; http://dx.doi.org/10.1038/84839; PMID: 11175789
- Barton ER, Morris L, Musaro A, Rosenthal N, Sweeney HL. Muscle-specific expression of insulin-like growth factor I counters muscle decline in mdx mice. J Cell Biol 2002; 157:137 - 48; http://dx.doi.org/10.1083/jcb.200108071; PMID: 11927606
- Serra C, Palacios D, Mozzetta C, Forcales SV, Morantte I, Ripani M, et al. Functional interdependence at the chromatin level between the MKK6/p38 and IGF1/PI3K/AKT pathways during muscle differentiation. Mol Cell 2007; 28:200 - 13; http://dx.doi.org/10.1016/j.molcel.2007.08.021; PMID: 17964260