Abstract
Tolerance to water deficits was evolutionarily relevant to the conquest of land by primitive plants. In this context, epigenetic events may have played important roles in the establishment of drought stress responses. We decided to inspect epigenetic marks in the plant organ that is crucial in the sensing of drought stress: the root. Using tomato as a crop model plant, we detected the methylated epialleles of Asr2, a protein-coding gene widespread in the plant kingdom and thought to alleviate restricted water availability. We found 3 contexts (CG, CNG, and CNN) of methylated cytosines in the regulatory region of Solanum lycopersicum Asr2 but only one context (CG) in the gene body. To test the hypothesis of a link between epigenetics marks and the adaptation of plants to drought, we explored the cytosine methylation status of Asr2 in the root resulting from water-deficit stress conditions. We found that a brief exposure to simulated drought conditions caused the removal of methyl marks in the regulatory region at 77 of the 142 CNN sites. In addition, the study of histone modifications around this model gene in the roots revealed that the distal regulatory region was rich in H3K27me3 but that its abundance did not change as a consequence of stress. Additionally, under normal conditions, both the regulatory and coding regions contained the typically repressive H3K9me2 mark, which was lost after 30 min of water deprivation. As analogously conjectured for the paralogous gene Asr1, rapidly acquired new Asr2 epialleles in somatic cells due to desiccation might be stable enough and heritable through the germ line across generations, thereby efficiently contributing to constitutive, adaptive gene expression during the evolution of desiccation-tolerant populations or species.
Keywords: :
Introduction
Epigenetic research has generated a fair amount of information on certain plant models, such as Arabidopsis,Citation1,Citation2 rice,Citation3 and maize.Citation4 Eukaryotic DNA methylation is one of the most studied epigenetic processes as it results in a direct and heritable covalent modification triggered by external stimuli. While methylation in animal genomes occurs mostly in regulatory regions, methylation in Arabidopsis is also found in transcribed sequences at not only canonical CG sites but also CNG (N denotes A, C, or T) and CNN (asymmetric) sites. The latter sites are preferentially methylated in repetitive elements and transposons.Citation5,Citation6
Mutant analyses in Arabidopsis ascertained that methylation in each context arises from specialized enzymatic machineries, such as the following examples: (1) the enzyme MET1, orthologous to mammalian DNMT1, which maintains CG methylation;Citation7 (2) the enzyme CMT3,Citation6 the structure of which was recently elucidated by X-ray diffraction crystallography,Citation8 which, together with the histone methyltransferase KYP, is responsible for transferring the methyl group in the CNG context;Citation9 and (3) the methyltransferase DRM2,Citation10 guided by short sequence-specific RNAs, which appears to catalyze de novo methylation in all contexts, including CNN.Citation11 In any case, intragenic DNA methylation mechanisms are essential as they regulate gene expression and plant development,Citation12 but there are still additional molecular players to be explored in more depth, such as those that prevent genes from undergoing ectopic deposition of methylation.Citation13
We are particularly interested in investigating the epigenetics of plant species with larger and more complex genomes than Arabidopsis, specifically with respect to the alterations elicited by abiotic stress. Among the different types of environmental stresses amenable to study, we selected water shortage because primitive plants have coped with this stress since they colonized land habitats approximately 400 MYA.Citation14 Our studies centered on the tomato plant (Solanum lycopersicum), an edible plant crop (http://mips.helmholtz-muenchen.de/plant/tomato/index.jsp) of great economic importance with a genomeCitation15 that is almost 10 times larger than that of Arabidopsis and with very few epigenetics surveys.Citation16-Citation18 Using this model system, we previously investigated the cytosine methylation status in the leaf of Asr1,Citation17 a non-transposon, protein-coding, desiccation stress-inducible gene of the LEA superfamilyCitation19 that is conserved in the plant kingdom but lacks an orthologous counterpart in Arabidopsis. The ancestral (300 MYA) ASR gene familyCitation20 has been extensively studied by us and other groups at the DNA,Citation20 RNA,Citation21 and proteinCitation22-Citation24 levels and in terms of physiological function.Citation25
We now report a methylation study on the paralogous gene Asr2, which has been a target of positive selection during the evolution of the Solanum genus in arid environments.Citation20 Moreover, this model gene displays intraspecific nucleotide variation, evidence of non-neutral evolution in different populations ascribed to different rainfall regimes.Citation26 In addition to the two latter relevant reasons related to the adaptation to threatening environmental demands, we selected this gene because it has a very simple organization, consisting of exon 1 and exon 2 with 159 and 186 nt, respectively, separated by a short intron. In addition, its sequence upstream of the transcription start site, also analyzed in this work, has been functionally tested to efficiently drive the transcription of a reporter gene in transgenic plants.Citation27 We also chose the root as the source of genomic DNA for these epigenetic studies for two reasons: (1) it is the organ, through its hairs, involved in the primary sensing of drought stress;Citation28 and (2) it is the organ in which Asr2 expression is the highest upon water stress.Citation21
In summary, our group’s main interest was the link between epigenetics and stress in plants,Citation29-Citation31 particularly water-deficit stressCitation32 in species with large genomes. For the above-mentioned reasons, Asr2 is a suitable and attractive model gene for this purpose. As stress-induced physiological responses in Arabidopsis are thought to depend on altered DNA methylationCitation33 and histone modifications,Citation34 we tested this hypothesis experimentally in a crop species by examining the gains and losses of cytosine methylation and histone marks in the roots as a consequence of imposing water stress conditions on tomato plants.
Results
Indicator of drought conditions
We first assessed that drought conditions really occurred. To this end, we applied a simple procedure consisting of weighing thoroughly wiped roots before and after the stress treatments (Fig. S1). The results indicated that the weight decreased, strongly suggesting the loss of internal water as a consequence of the stress.
Overall non-CG methylation in the Asr2 promoter/enhancer region and gene body
We wanted to gain insight into the methylation events in cytosine contexts other than CG. To this end, we used the bisulfite conversion procedure,Citation35 taking care to address methodological concerns, namely parallel bisulfite control reactions on non-methylated or in vitro-methylated plasmid DNA (see Materials and Methods). In addition, to understand the molecular mechanisms underlying the adaptation of plants to abiotic stress, we analyzed root DNA methylation after imposing water-shortage stress on whole tomato plants through root drying.
For that purpose, we performed an inspection of the methylated sites at the functionally studied regulatory region of tomato Asr2,Citation27 spanning 968 nt upstream of the transcription start site (), in the root. As all the observed epialleles showed different methylation patterns at CNN sites, we did not need to address the concerns of Henderson et al.Citation36 or the model of Peng and EckerCitation37 to eliminate sibling clones. After grouping the results according to each methylation type, we concluded that there were substantial levels of the three types of methylation (CG, CNG, and CNN) in that region under non-water stress conditions (). After stress, the overall CNN methylation showed a slight but significant (P < 0.05) decrease (; Fig. S2).
Figure 1. Overall methylation levels in the Asr2 regulatory region and gene body. Data were grouped by methylation context in the regulatory region, which is located upstream of the transcription start site (A), and coding region (B) under both normal conditions and after 30 min of water stress. (C) Schematic representation of the entire gene, aligned to (Aand B), showing the annealing positions of the primers relative to the +1 site designed for the post-bisulfite PCR analysis. The amplicon sizes and number of cytosine residues in each context existing in the amplicons analyzed for (Aand B) are also shown. The error bars indicate the standard error (SEM). The slight decrease in the overall CNN methylation in the regulatory region was significant (*P < 0.05). The increased values in the CNG (***P < 0.001) and CNN (**P < 0.01) methylation in the coding region were also significant. The bisulfite treatments were performed as indicated in Materials and Methods. The primers for post-bisulfite PCR are listed Table S1. GenBank accession numbers for Asr2: L20756, CU468249, and X74907.
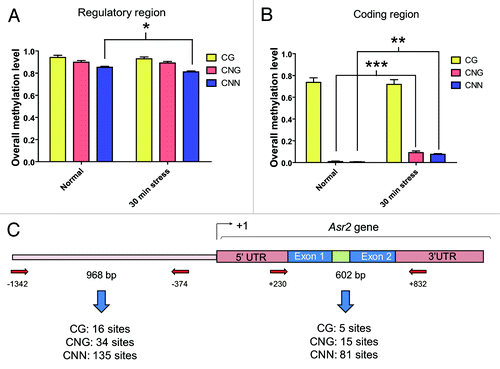
In parallel, we analyzed the cytosine methylation in the gene body of Asr2, consisting of 2 exons and one intron and spanning 603 nt, in the root (). The raw data (Fig. S3) were grouped by methylation type, leading to the conclusion that a considerable proportion (near 80%) of the clones showed methylation at the few existing CG sites () under both environmental conditions. In contrast, very few of the numerous CNG and CNN sites were methylated under either normal or non-water stress conditions (). It is worth mentioning that after the water stress, 2 distinct clones appeared to be strongly methylated in all contexts (Fig. S3B), leading to an overall increase in CNG and CNN methylation when analyzed comprehensively.
Detailed picture of CNN demethylation in the Asr2 promoter/enhancer upon stress
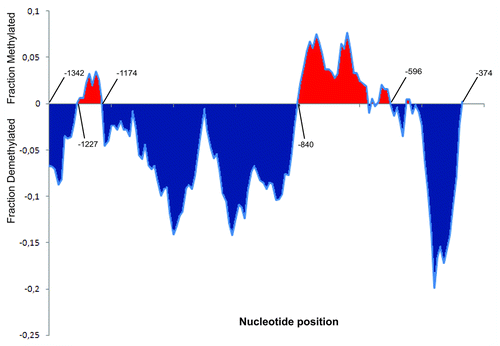
We next decided to carry out a close-up inspection of the upstream regulatory region by performing calculations of the mean (methylation level values) in a recursive fashion, that is, depending on a previously calculated value. This computational tool is known as a “moving average” (http://lorien.ncl.ac.uk/ming/filter/filmav.htm). In our case, this procedure consisted of a moving window of 10 contiguous cytosines (see Materials and Methods for an explanation). This approach allowed the observation of a slight increase due to stress in cytosine methylation distant (−1227/−1174) to the gene body and more evidently in the −840 to −596 interval. The sequence between these 2 regions and the proximal regulatory region showed a marked trend toward demethylation (). In particular, we found that the simulated drought conditions caused the removal of methyl marks at 77 of the 142 asymmetric (CNN) sites present in the upstream regulatory region. When focusing on other regions of Asr2, no striking tendencies were revealed.
Figure 2. Detailed inspection of the stress-induced changes in asymmetric methylation (CNN) within the Asr2 regulatory region. The moving average statistical tool (http://lorien.ncl.ac.uk/ming/filter/filmav.htm) was employed. The X-axis represents 10-cytosine (5 left + 5 right) windows for each position corresponding to the 142 cytosine residues (CNN) existing in this region. Y-axis units represent the moving average of the differences between the methylation level for each cytosine under stress and the methylation level for the same cytosine under no stress. Some position numbers relative to the +1 transcription start site are indicated.
Histone marks in Asr2
As gene expression is also influenced by post-translational histone modifications,Citation34 we decided to explore H3K27me3, H3K9me2, and H3K4me3, abundant signatures of gene repression (H3K27me3, H3K9me2), and activation (H3K4me3) in Arabidopsis.Citation38 H3K27me3 and H3K4me3 marks in the upstream region closest to the gene (−316/−207 bp) were barely detected (). However, in the upstream distal region (−987/−820 bp), the H3K27me3 repressive mark was detected (P < 0.05, P < 0.01), but it did not change as a result of stress (). Surprisingly, these histone methylation marks within the Asr2 coding region were not appreciably perceived (). In contrast, the usually repressive H3K9me2 mark was clearly evident in all regions analyzed (), decreasing after the water shortage.
Figure 3. Histone marks on the Asr2 regulatory and coding regions. The H3K4me3 (A and B), H3K27me3 (C and D), and H3K9me2 (E and F) levels in the regulatory (A, C, and E) and coding (B, D, and F) regions under both normal conditions and after 30 min of water stress are expressed as % input. ChIP was performed as described in Materials and Methods. (G) Schematic representation of the entire gene, aligned to (A–F), showing the annealing positions of the primers designed for qPCR and the sizes of the analyzed amplicons. The error bars indicate the standard error (SEM). Statistically significant (*P < 0.05, **P < 0.01, ***P < 0.001) histone marks are highlighted. Statistical significance between no-antibody (noise signal) and real ChiP signal is shown except for the evident H3K9me2 mark (E and F) under normal conditions (P < 0.001) for the sake of simplicity, not to mask the pronounced effect of stress on demethylation in both gene regions. The qPCR primers are listed in Table S1.
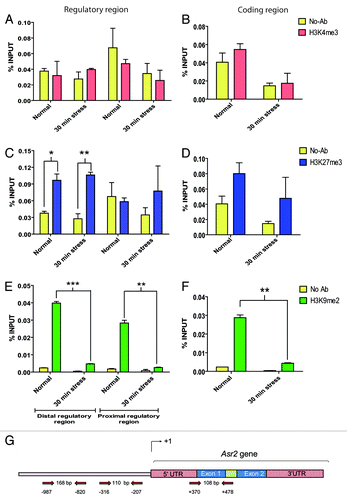
Given that some negative results were obtained, we assessed the performance of the antibodies on the constitutively expressed actin gene and the transposable element ToRTL, which were selected as control loci. Although we detected only H3K9me2 mark on the body of actin, which was reduced after stress (P < 0.001, Fig. S4C), immunoprecipitation with the same antibodies effectively captured all the studied methylation marks in the chromatin wrapping the transposon; however, these marks were lost as a result of water stress (P < 0.05, P < 0.01) (Fig. S4). These results confirmed the good quality of the antibodies specific to the inspected histone marks.
Asr2 expression in roots upon water stress
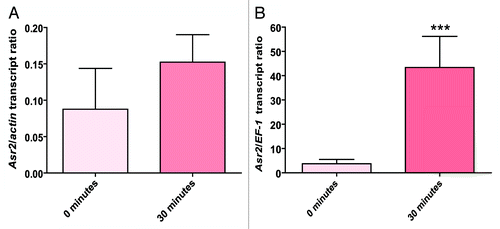
To identify an eventual correlation between any type of methylation (CG, CNG, and CNN) and expression of our model gene, we performed qRT-PCR for both the normal and stress conditions (). After normalization against the housekeeping EF-1 mRNA, the results showed a slight increase in the Asr2 mRNA levels at as early as 10 min of water stress (P < 0.05, data not shown) and an even higher expression at 30 min of stress, the time point at which we performed the methylation analysis (P < 0.001, ). Longer times resulted in the occurrence of massive cell death (Fig. S5), which would have made an epigenetic evaluation impossible.
Figure 4.Asr2 expression as a consequence of water stress. The root mRNA steady-state levels were measured by qRT-PCR as described in Materials and Methods. The data were normalized to actin (A) or EF-1 (B) mRNA at each stress time before comparing the effects of the different stress treatments. Normalized against EF-1, the difference in the expression levels of Asr2 was statistically significant at 30 min (***P < 0.001). The qRT-PCR primers are listed in Table S1. The error bars indicate the standard error (SEM).
Discussion
In plants, the classical CG methylation context not only is linked to the regulatory region of the gene, as in mammals, but also is found in the coding region of the gene.Citation39,Citation40 Another particularity of plant species is that this methylation context is not necessarily associated with gene silencing.Citation31 Moreover, these marks are often present even in the body of housekeeping genes, such as actin,Citation41 and appear to provide a conserved pattern in different organs of the same species. For example, the few methylated CG sites in the Asr2 gene body we found in the roots strikingly matched those found in the same gene in fruit by the recent genome-wide methylome project performed by Giovannoni’s group.Citation18 However, we observed no stress-provoked higher CG methylation, in contrast to our previous report on another model gene of the same family in tomato leaves.Citation17
Regarding non-CG methylation, its existence in tandem repeats has been demonstrated by Jacobsen’s groupCitation42 along with its conservation across duplicated regions of the genome.Citation43 In this work on the drought-responsive gene Asr2 in roots, in contrast to our previous report on leaf Asr1, no asymmetric CNN methylation in the transcribed sequence was found in the absence of stress. However, we uncovered extensive methylation at these nonconventional acceptor sites in the upstream regulatory region. Moreover, we noticed that this epigenetic mark in combination with CG and CNG methylation correlated with poor expression, consistent with previous evidence.Citation44 We also showed that upon stress, many of these CNN sites were demethylated, similar to what occurs with those in the gene body of leaf Asr1.Citation17 Analogously, a null DRM2 mutant was reported to block non-CG methylation, which allowed for full desilencing of the FWA gene, resulting in a late-flowering phenotype.Citation45 Along the same lines, evidence from Matzke’s lab showed that the combination of CG and non-CG methylation in at least some coding regions was associated with gene silencing in synergid cells.Citation46
It is worth noting at this point that the various clones with dissimilar patterns displayed in the Kismeth schemes may have arisen from different cell types present together in the root samples under examination, with each having a distinct epigenetic behavior, as conjectured by Peng and Ecker (2012).Citation37
With respect to the chromatin structure, a link between cytosine methylation and histone methylation is currently known to exist.Citation47,Citation48 A clear example of this relationship is given by KYP and VIM1, which bind to methylated cytosines and act as histone methyltransferases.Citation49 Other protein complexes, however, lack methyltransferase activity but bind to methylated cytosines and cooperate with the DNA methylation machinery, such as in the case of SUVH9 and DRM2, the methyltransferase devoted, but not exclusively, to the asymmetric context.Citation45 It is also worth mentioning that any conclusions regarding the assignment of different plant cytosine methyltransferases to particular substrate sites have been derived solely through reverse genetics by analyzing mutantsCitation6,Citation10,Citation50 and not from in vitro experiments with purified enzymes. However, there are recent advances on the structure of the maize crystallized CMT3 protein, highlighting its domains involved in binding to nucleosomes and catalysis on CNG sites.Citation8 In this context of chromatin modifications, we found no significant stress-induced variations in the levels of either H3K4me3 or H3K27me3 marks (associated with the opposite effects on transcription in ArabidopsisCitation38) in our model gene. Because methylated H3K27 is independent of and potentially mutually exclusive with DNA methylation,Citation51 this mark is not very informative for understanding the relationship among chromatin modifications, DNA methylation and gene regulation at this locus. Therefore, we also explored the repressive mark H3K9me2, which turned out to correlate with CNN methylation, both pointing in the same direction and, in turn, inversely associated with Asr2 gene expression.
The data presented herein provide a new example of a not very frequent genomic location for CNN methylation in plants, namely, a non-repetitive, non-transposon gene. However, there are certainly other examples, e.g., Baek et al. (2010) in Arabidopsis,Citation52 Zhang et al. (2011) in sorghum,Citation53 and Li et al. (2012) in rice young panicles.Citation54 Certainly, more biochemical studies will help elaborate new models to understand the in vivo maintenance of CNN methylation during DNA replication, which is difficult to envisage, as there are no local cytosine residues to be methylated in the nascent complementary strand.
As high throughput technologies for uncovering specific methylomes are becoming more and more accessible, comparisons of genome-wide cytosine methylation in all contexts or of average methylation levels can be easily made among different plant sources. For example, in rice, the average methylation levels in the 3 contexts are approximately 44%, 24%, and 5%, respectively,Citation55 revealing that the rice genome has a much higher level of DNA methylation than Arabidopsis.Citation56 Strikingly, in tomato, the percentages of mC in the CG, CNG, and CNN contexts are even higher: approximately 85%, 56%, and 8%, respectively, for leaves and oscillating from 74–79%, 52–54%, and 13–14%, respectively, for fruits at different maturation stages.Citation18 A noteworthy bonus of the present work was that it was conducted on the root, a plant organ almost neglected at the epigenetic level, particularly with respect to methylation, except for a few works in ArabidopsisCitation37,Citation52 and sorghum.Citation53
With respect to the appealing connection between plant epigenetics and stress, our findings in the tomato plant were consistent with the hypothesis highlighted by the Kovalchuk groupCitation30 in Arabidopsis and experimentally supported in rice,Citation55 in which at least some stress-induced phenotypes depend on altered DNA methylation. In this regard, the change in asymmetric epigenetic marks we observed upon environmental drought conditions may represent an alternative mechanism for adaptation in plants, not only in Arabidopsis but also in species with larger and more complex genomes. The rapid emergence of these newly acquired epialleles in roots, coupled with the unique ability of plants to produce germ line cells late during development, may allow for the inheritance of these marks across (not necessarily stressed) generations,Citation57-Citation60 thus efficiently contributing to the constitutive, adaptive gene expression during evolution of drought tolerance in crop species. We have no evidence of such a conjecture, and this issue might be resolved by future research.
Materials and Methods
Plant material
Commercial tomato (Solanum lycopersicum) seeds were bleached by sinking in a 20 g/l sodium hypochlorite solution for 30 min. After the treatment, the seeds were placed on dampened blotting paper and left in the dark for 72 h. Plantlets were placed in a growth chamber at 23 °C with a photoperiod of 12 h light/12 h dark for 5 d followed by transplantation to pots. The plants were then returned to the growth chamber and watered twice weekly until the experiments were performed.
Water stress conditions
Three-week-old plants were taken from the pots, and their roots were carefully cleaned. The roots were cut off, wiped, weighed, and frozen in liquid nitrogen (non-stressed plants) or placed onto blotting paper for different times up to 30 min, weighed, and immediately frozen (stressed plants). The decrease in weight (Fig. S1) was indicative of water loss. For each condition, the roots of 8 plants were pooled together to achieve the amount of DNA necessary for the subsequent bisulfite treatment.
DNA extraction
Peralta and Spooner’s protocolCitation61 was followed with some modifications. This procedure included the use of CTAB as a detergent instead of SDS, which is appropriate for tomato due to its high content of sugars and polyphenols. The DNA quality was assessed by spectrophotometry using the A260/A280 ratio. Only samples with an A260/A280 ratio between 1.7 and 2.0 were used.
Bisulfite conversion procedure
We used the protocol described by Clark et al.Citation35 with some modifications. The DNA was digested with Dra I (5′-TTTAAA-3′) at 37 °C overnight to obtain DNA fragments of approximately 2,000 bp in average length, which were purified by extraction with phenol:chloroform (1:1). Total genomic DNA (1 μg) was then treated with bisulfite. The conversion step was performed for 16 h at 55 °C. The treated DNA was then purified using the commercial Wizard DNA Clean-Up System kit (Promega).
Post-bisulfite PCR
Both the regulatory region of Asr2, which was defined as comprising 1,500 bp upstream of the +1 transcription start siteCitation23 on chromosome 4, and the gene body of Asr2 were separately amplified. The primers were designed using the Beacons Designer software (http://www.premierbiosoft.com/molecular_beacons/index.html) to have the highest possible content of T residues, especially at the 3′ ends, to favor the selective amplification of bisulfite-converted molecules.Citation62
For the amplification reaction of the regulatory region (968-bp amplicon), 5 μl of bisulfite-treated product was amplified by Taq DNA Polymerase (Invitrogen) in an MJ Research PTC-100 (MJ Research Inc.) according to the following program: 40 cycles of denaturation (94 °C, 30 s), annealing (61 °C, 30 s), and elongation (72 °C, 1.30 min). This PCR was performed using the primers listed in Table S1.
For the body region (602-bp amplicon), we used the same conditions, but the program was the following: 40 cycles of denaturation (94 °C, 30 s), annealing (58 °C, 45 s), and elongation (72 °C, 45 s). For this purpose, we used the primers listed in Table S1.
In all PCR reactions, we used 0.625 U of Taq DNA polymerase, 6 μM MgCl2, 0.2 μM dNTPs, and 0.2 μM of each primer in a final volume of 50 μl.
Validation of bisulfite conversion efficiency
Full conversion was assessed as described in Gonzalez et al.;Citation17 essentially, p-Bluescript SK (+) plasmid DNA (Stratagene) was in vitro methylated with mHaeIII methyltransferase (NEB) and incubated with bisulfite in parallel with the genomic DNA samples, followed by PCR with specific primers. The bisulfite treatment of the plasmid destroyed two pre-existing Bfa I restriction sites and created a new BfaI site, as expected. In addition, we ruled out a possible unintentional overestimation of cytosine methylation due to an eventual inefficient bisulfite conversion by using primers specifically designed to amplify the converted template but incapable of annealing to the natural sequence.Citation62
Subcloning and sequencing
Subcloning was performed with the pGEM-T “easy vector” (Promega). Plasmid minipreps were processed from randomly picked insert-positive colonies using the GeneJET Plasmid Miniprep Kit (Fermentas). Sanger sequencing was performed with SP6 and T7 universal primers. For the regulatory region, 13 clones were sequenced in the case of plants under normal conditions and 16 for plants after 30 min of drought. For the coding region, 22 clones each were sequenced in both cases.
Methylation data analysis
To analyze the methylation data, we used the Kismeth softwareCitation63 (http://katahdin.mssm.edu/kismeth/revpage.pl), which allows visualization of the proportion of clones (epialleles) showing methylation at every particular site. Once the methylation level data for each site were gathered, GraphPad software (www.GraphPad.com) was used for the statistical analysis. The data were grouped according to methylation type (CpG, CpNpG, and CpNpN). Statistical significance was determined using the Mann–Whitney test at the 95% significance level. For a detailed analysis of the CNN methylation level on the regulatory region (), we employed the moving average statistical tool (http://lorien.ncl.ac.uk/ming/filter/filmav.htm), which calculates the mean in a recursive fashion, that is, depending on a previously calculated value. In our case, this method involved taking a window of a fixed size of 10 contiguous cytosines, calculating the average methylation value for those 10 cytosines, and then displacing the window one position (one cytosine) at a time for the successive calculations of the mean.
Chromatin immunoprecipitation (ChIP) specific for histone modifications
We followed the protocol of Ricardi et al.Citation64 but used Dynabeads protein A (Invitrogen) instead of agarose beads. We used 2 μg of DNA for the input and 8 μg for all the incubations with the antibody. To ensure that those amounts remained constant, the volumes were variable according to the DNA initial concentrations. Anti-H3K4me3, H3K9me2, and H3K27me3 antibodies were purchased from Abcam.
For the qPCR, we used 0.625 U of Maxima Hot Start Taq (Fermentas), 3 mM magnesium chloride, 2 mM dNTP mixture (Fermentas), 0.2 μM of each primer (IDT Inc.), and 2 μl of the template in a final volume of 25 μl. As the fluorophore, we used Sybr Green® (Roche). The reactions were conducted in a Stratagene Mx3000P equipment under the following cycling conditions: 2 min of denaturation at 94 °C followed by 40 cycles of 30 s of denaturation at 94 °C, 30 s of annealing at 67 °C, and 30 s of elongation at 72 °C. The melting curve was performed between 65 °C and 95 °C, with readings every 0.5 °C. The primers are listed in Table S1.
Expression analysis (RNA extraction, reverse transcription, and qRT-PCR)
Total RNA was extracted with Trizol (Invitrogen) from 100-mg mortar-ground roots in liquid nitrogen followed by incubation with 12.5 U of DNAseI (Invitrogen). The reverse transcription was achieved using 2 μl RNA, 50 U MMLV-RT (Promega), and oligo-dT (50 pmoles) in a 25-μl final volume for 1 h at 42 °C. To prevent RNA degradation, 10 U RNaseOUT (Invitrogen) were added. The qRT-PCR was performed under the same conditions indicated for the ChIP experiments, using the primers listed in Table S1.
The data obtained for Asr2 mRNA were normalized to actin or EF-1 mRNA at each stress time before comparing the effects of the different stress treatments.
Determination of cell viability under water stress conditions by staining
We followed the protocol described by Tamás,Citation65 with some modifications, based on the use of Evans Blue, a dye capable of penetrating only dead cells that is subsequently extracted by dimethylformamide. In detail, the plants were stressed for different lengths of time. The roots were then cut into fragments of approximately 5 mm with a scalpel and weighed before the test. Subsequently, a solution of 0.25% Evans Blue was added with gently shaking for 15 min, followed by three washes with mQ water for 10 min each with stirring. The root pieces were then incubated for 1 h with dimethylformamide with continuous stirring and then centrifuged at 21,500 × g for 5 min. Finally, the supernatant was removed. The absorbance at 595 nm was measured using a microplate reader (Biorad) and normalized to the starting mass of the roots.
| Abbreviations: | ||
| Asr1 | = | abscisic acid stress and ripening 1 |
| Asr2 | = | abscisic acid stress and ripening 2 |
| bp | = | base pairs |
| DNA | = | deoxyribonucleic acid |
| RNA | = | ribonucleic acid |
| MET1 | = | methyltransferase 1 |
| DNMT1 | = | DNA methyltransferase 1 |
| CMT3 | = | chromomethylase 3 |
| H3K4me3 | = | tri-methylated lysine 4 of histone 3 |
| H3K9me2 | = | di-methylated lysine 9 of histone 3 |
| H3K27me3 | = | tri-methylated lysine 27 of histone 3 |
| KYP, kryptonite | = | histone 3 lysine 9 methyltransferase |
| DRM2 | = | domains rearranged methyltransferase 2 |
| LEA | = | late embryogenesis abundant |
| MYA | = | million years ago |
| PCR | = | polymerase chain reaction |
| qRT-PCR | = | quantitative real time – polymerase chain reaction |
| FWA | = | flowering wageningen |
| VIM1 | = | variant in methylation 1 |
| SUVH9 | = | SU (Var) 3-9 homolog 9 |
| CTAB | = | cetyl trimethyl ammonium bromide |
| SDS | = | sodium dodecyl sulfate |
| ToRTL | = | tomato copia-type retrotransposon |
| EF-1 | = | elongation factor 1 |
| nt | = | nucleotide(s) |
| mC | = | methylcytosine |
Additional material
Download Zip (95.5 KB)Acknowledgments
This work was supported by grants from Universidad de Buenos Aires (UBA), Agencia Nacional de Promoción Científica y Tecnológica (ANPCyT), and Consejo Nacional de Investigaciones Científicas y Tecnológicas (CONICET), Argentina.
RMG performed all the experimental work, generated the data, and extensively revised the manuscript together with MMR and NDI. MMR set up the experimental conditions for ChIP. NDI introduced the theoretical frame, coordinated the project, and drafted the manuscript. All 3 authors read and approved the final manuscript.
MMR and RMG hold post-doctorate fellowships from Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET), Argentina. NDI is an Independent Researcher at CONICET.
Disclosure of Potential Conflicts of Interest
The authors declare that they have no competing interests.
Supplemental Materials
Supplemental materials may be found here:
http://www.landesbioscience.com/journals/epigenetics/article/25524
References
- Reinders J, Paszkowski J. Unlocking the Arabidopsis epigenome. Epigenetics 2009; 4:557 - 63; http://dx.doi.org/10.4161/epi.4.8.10347; PMID: 19934651
- Zhang M, Kimatu JN, Xu K, Liu B. DNA cytosine methylation in plant development. J Genet Genomics 2010; 37:1 - 12; http://dx.doi.org/10.1016/S1673-8527(09)60020-5; PMID: 20171573
- Akimoto K, Katakami H, Kim HJ, Ogawa E, Sano CM, Wada Y, et al. Epigenetic inheritance in rice plants. Ann Bot 2007; 100:205 - 17; http://dx.doi.org/10.1093/aob/mcm110; PMID: 17576658
- Wang X, Elling AA, Li X, Li N, Peng Z, He G, et al. Genome-wide and organ-specific landscapes of epigenetic modifications and their relationships to mRNA and small RNA transcriptomes in maize. Plant Cell 2009; 21:1053 - 69; http://dx.doi.org/10.1105/tpc.109.065714; PMID: 19376930
- Chen M, Lv S, Meng Y. Epigenetic performers in plants. Dev Growth Differ 2010; 52:555 - 66; http://dx.doi.org/10.1111/j.1440-169X.2010.01192.x; PMID: 20646028
- Meyer P. DNA methylation systems and targets in plants. FEBS Lett 2011; 585:2008 - 15; http://dx.doi.org/10.1016/j.febslet.2010.08.017; PMID: 20727353
- Finnegan EJ, Peacock WJ, Dennis ES. Reduced DNA methylation in Arabidopsis thaliana results in abnormal plant development. Proc Natl Acad Sci U S A 1996; 93:8449 - 54; http://dx.doi.org/10.1073/pnas.93.16.8449; PMID: 8710891
- Du J, Zhong X, Bernatavichute YV, Stroud H, Feng S, Caro E, et al. Dual binding of chromomethylase domains to H3K9me2-containing nucleosomes directs DNA methylation in plants. Cell 2012; 151:167 - 80; http://dx.doi.org/10.1016/j.cell.2012.07.034; PMID: 23021223
- Jackson JP, Lindroth AM, Cao X, Jacobsen SE. Control of CpNpG DNA methylation by the KRYPTONITE histone H3 methyltransferase. Nature 2002; 416:556 - 60; http://dx.doi.org/10.1038/nature731; PMID: 11898023
- Cao X, Jacobsen SE. Role of the Arabidopsis DRM methyltransferases in de novo DNA methylation and gene silencing. Curr Biol 2002; 12:1138 - 44; http://dx.doi.org/10.1016/S0960-9822(02)00925-9; PMID: 12121623
- Zhang X. The epigenetic landscape of plants. Science 2008; 320:489 - 92; http://dx.doi.org/10.1126/science.1153996; PMID: 18436779
- Feng S, Jacobsen SE, Reik W. Epigenetic reprogramming in plant and animal development. Science 2010; 330:622 - 7; http://dx.doi.org/10.1126/science.1190614; PMID: 21030646
- Miura A, Nakamura M, Inagaki S, Kobayashi A, Saze H, Kakutani T. An Arabidopsis jmjC domain protein protects transcribed genes from DNA methylation at CHG sites. EMBO J 2009; 28:1078 - 86; http://dx.doi.org/10.1038/emboj.2009.59; PMID: 19262562
- Qiu YL, Palmer JD. Phylogeny of early land plants: insights from genes and genomes. Trends Plant Sci 1999; 4:26 - 30; http://dx.doi.org/10.1016/S1360-1385(98)01361-2; PMID: 10234267
- Tomato Genome Consortium. The tomato genome sequence provides insights into fleshy fruit evolution. Nature 2012; 485:635 - 41; http://dx.doi.org/10.1038/nature11119; PMID: 22660326
- Teyssier E, Bernacchia G, Maury S, How Kit A, Stammitti-Bert L, Rolin D, et al. Tissue dependent variations of DNA methylation and endoreduplication levels during tomato fruit development and ripening. Planta 2008; 228:391 - 9; http://dx.doi.org/10.1007/s00425-008-0743-z; PMID: 18488247
- González RM, Ricardi MM, Iusem ND. Atypical epigenetic mark in an atypical location: cytosine methylation at asymmetric (CNN) sites within the body of a non-repetitive tomato gene. BMC Plant Biol 2011; 11:94; http://dx.doi.org/10.1186/1471-2229-11-94; PMID: 21599976
- Zhong S, Fei Z, Chen YR, Zheng Y, Huang M, Vrebalov J, et al. Single-base resolution methylomes of tomato fruit development reveal epigenome modifications associated with ripening. Nat Biotechnol 2013; 31:154 - 9; http://dx.doi.org/10.1038/nbt.2462; PMID: 23354102
- Battaglia M, Olvera-Carrillo Y, Garciarrubio A, Campos F, Covarrubias AA. The enigmatic LEA proteins and other hydrophilins. Plant Physiol 2008; 148:6 - 24; http://dx.doi.org/10.1104/pp.108.120725; PMID: 18772351
- Frankel N, Carrari F, Hasson E, Iusem ND. Evolutionary history of the Asr gene family. Gene 2006; 378:74 - 83; http://dx.doi.org/10.1016/j.gene.2006.05.010; PMID: 16822623
- Maskin L, Gudesblat GE, Moreno JE, Carrari FO, Frankel NS, Sambade A, et al. Differential expression of the members of the Asr gene family in tomato (Lycopersicon esculentum). Plant Sci 2001; 161:739 - 46; http://dx.doi.org/10.1016/S0168-9452(01)00464-2
- Konrad Z, Bar-Zvi D. Synergism between the chaperone-like activity of the stress regulated ASR1 protein and the osmolyte glycine-betaine. Planta 2008; 227:1213 - 9; http://dx.doi.org/10.1007/s00425-008-0693-5; PMID: 18270732
- Maskin L, Frankel N, Gudesblat G, Demergasso MJ, Pietrasanta LI, Iusem ND. Dimerization and DNA-binding of ASR1, a small hydrophilic protein abundant in plant tissues suffering from water loss. Biochem Biophys Res Commun 2007; 352:831 - 5; http://dx.doi.org/10.1016/j.bbrc.2006.11.115; PMID: 17157822
- Ricardi MM, Guaimas FF, González RM, Burrieza HP, López-Fernández MP, Jares-Erijman EA, et al. Nuclear import and dimerization of tomato ASR1, a water stress-inducible protein exclusive to plants. PLoS One 2012; 7:e41008; http://dx.doi.org/10.1371/journal.pone.0041008; PMID: 22899993
- Moretti MB, Maskin L, Gudesblat G, García SC, Iusem ND. ASR1, a stress-induced tomato protein, protects yeast from osmotic stress. Physiol Plant 2006; 127:111 - 8; http://dx.doi.org/10.1111/j.1399-3054.2006.00664.x
- Giombini MI, Frankel N, Iusem ND, Hasson E. Nucleotide polymorphism in the drought responsive gene Asr2 in wild populations of tomato. Genetica 2009; 136:13 - 25; http://dx.doi.org/10.1007/s10709-008-9295-1; PMID: 18636230
- Rossi M, Carrari F, Cabrera-Ponce JL, Vázquez-Rovere C, Herrera-Estrella L, Gudesblat G, et al. Analysis of an abscisic acid (ABA)-responsive gene promoter belonging to the Asr gene family from tomato in homologous and heterologous systems. Mol Gen Genet 1998; 258:1 - 8; http://dx.doi.org/10.1007/s004380050700; PMID: 9613566
- Jones VA, Dolan L. The evolution of root hairs and rhizoids. Ann Bot 2012; 110:205 - 12; http://dx.doi.org/10.1093/aob/mcs136; PMID: 22730024
- Finnegan EJ. Epialleles - a source of random variation in times of stress. Curr Opin Plant Biol 2002; 5:101 - 6; http://dx.doi.org/10.1016/S1369-5266(02)00233-9; PMID: 11856603
- Boyko A, Kovalchuk I. Epigenetic control of plant stress response. Environ Mol Mutagen 2008; 49:61 - 72; http://dx.doi.org/10.1002/em.20347; PMID: 17948278
- Chinnusamy V, Zhu JK. Epigenetic regulation of stress responses in plants. Curr Opin Plant Biol 2009; 12:133 - 9; http://dx.doi.org/10.1016/j.pbi.2008.12.006; PMID: 19179104
- Caramelo JJ, Iusem ND. When cells lose water: Lessons from biophysics and molecular biology. Prog Biophys Mol Biol 2009; 99:1 - 6; http://dx.doi.org/10.1016/j.pbiomolbio.2008.10.001; PMID: 18977383
- Boyko A, Blevins T, Yao Y, Golubov A, Bilichak A, Ilnytskyy Y, et al. Transgenerational adaptation of Arabidopsis to stress requires DNA methylation and the function of Dicer-like proteins. PLoS One 2010; 5:e9514; http://dx.doi.org/10.1371/journal.pone.0009514; PMID: 20209086
- Kouzarides T. Chromatin modifications and their function. Cell 2007; 128:693 - 705; http://dx.doi.org/10.1016/j.cell.2007.02.005; PMID: 17320507
- Clark SJ, Statham A, Stirzaker C, Molloy PL, Frommer M. DNA methylation: bisulphite modification and analysis. Nat Protoc 2006; 1:2353 - 64; http://dx.doi.org/10.1038/nprot.2006.324; PMID: 17406479
- Henderson IR, Chan SR, Cao X, Johnson L, Jacobsen SE. Accurate sodium bisulfite sequencing in plants. Epigenetics 2010; 5:47 - 9; http://dx.doi.org/10.4161/epi.5.1.10560; PMID: 20081358
- Peng Q, Ecker JR. Detection of allele-specific methylation through a generalized heterogeneous epigenome model. Bioinformatics 2012; 28:i163 - 71; http://dx.doi.org/10.1093/bioinformatics/bts231; PMID: 22689757
- Feng S, Jacobsen SE. Epigenetic modifications in plants: an evolutionary perspective. Curr Opin Plant Biol 2011; 14:179 - 86; http://dx.doi.org/10.1016/j.pbi.2010.12.002; PMID: 21233005
- Lister R, O’Malley RC, Tonti-Filippini J, Gregory BD, Berry CC, Millar AH, et al. Highly integrated single-base resolution maps of the epigenome in Arabidopsis. Cell 2008; 133:523 - 36; http://dx.doi.org/10.1016/j.cell.2008.03.029; PMID: 18423832
- Shibuya K, Fukushima S, Takatsuji H. RNA-directed DNA methylation induces transcriptional activation in plants. Proc Natl Acad Sci U S A 2009; 106:1660 - 5; http://dx.doi.org/10.1073/pnas.0809294106; PMID: 19164525
- Zhang X, Yazaki J, Sundaresan A, Cokus S, Chan SW, Chen H, et al. Genome-wide high-resolution mapping and functional analysis of DNA methylation in Arabidopsis. Cell 2006; 126:1189 - 201; http://dx.doi.org/10.1016/j.cell.2006.08.003; PMID: 16949657
- Henderson IR, Jacobsen SE. Tandem repeats upstream of the Arabidopsis endogene SDC recruit non-CG DNA methylation and initiate siRNA spreading. Genes Dev 2008; 22:1597 - 606; http://dx.doi.org/10.1101/gad.1667808; PMID: 18559476
- Widman N, Jacobsen SE, Pellegrini M. Determining the conservation of DNA methylation in Arabidopsis. Epigenetics 2009; 4:119 - 24; http://dx.doi.org/10.4161/epi.4.2.8214; PMID: 19384058
- Diéguez MJ, Bellotto M, Afsar K, Mittelsten Scheid O, Paszkowski J. Methylation of cytosines in nonconventional methylation acceptor sites can contribute to reduced gene expression. Mol Gen Genet 1997; 253:581 - 8; http://dx.doi.org/10.1007/s004380050360; PMID: 9065691
- Greenberg MV, Ausin I, Chan SW, Cokus SJ, Cuperus JT, Feng S, et al. Identification of genes required for de novo DNA methylation in Arabidopsis. Epigenetics 2011; 6:344 - 54; http://dx.doi.org/10.4161/epi.6.3.14242; PMID: 21150311
- You W, Tyczewska A, Spencer M, Daxinger L, Schmid MW, Grossniklaus U, et al. Atypical DNA methylation of genes encoding cysteine-rich peptides in Arabidopsis thaliana. BMC Plant Biol 2012; 12:51; http://dx.doi.org/10.1186/1471-2229-12-51; PMID: 22512782
- Johnson LM, Bostick M, Zhang X, Kraft E, Henderson I, Callis J, et al. The SRA methyl-cytosine-binding domain links DNA and histone methylation. Curr Biol 2007; 17:379 - 84; http://dx.doi.org/10.1016/j.cub.2007.01.009; PMID: 17239600
- Johnson LM, Law JA, Khattar A, Henderson IR, Jacobsen SE. SRA-domain proteins required for DRM2-mediated de novo DNA methylation. PLoS Genet 2008; 4:e1000280; http://dx.doi.org/10.1371/journal.pgen.1000280; PMID: 19043555
- Woo HR, Pontes O, Pikaard CS, Richards EJ. VIM1, a methylcytosine-binding protein required for centromeric heterochromatinization. Genes Dev 2007; 21:267 - 77; http://dx.doi.org/10.1101/gad.1512007; PMID: 17242155
- Lindroth AM, Cao X, Jackson JP, Zilberman D, McCallum CM, Henikoff S, et al. Requirement of CHROMOMETHYLASE3 for maintenance of CpXpG methylation. Science 2001; 292:2077 - 80; http://dx.doi.org/10.1126/science.1059745; PMID: 11349138
- Saze H, Tsugane K, Kanno T, Nishimura T. DNA methylation in plants: relationship to small RNAs and histone modifications, and functions in transposon inactivation. Plant Cell Physiol 2012; 53:766 - 84; http://dx.doi.org/10.1093/pcp/pcs008; PMID: 22302712
- Baek D, Jiang J, Chung JS, Wang B, Chen J, Xin Z, et al. Regulated AtHKT1 gene expression by a distal enhancer element and DNA methylation in the promoter plays an important role in salt tolerance. Plant Cell Physiol 2011; 52:149 - 61; http://dx.doi.org/10.1093/pcp/pcq182; PMID: 21097475
- Zhang M, Xu C, von Wettstein D, Liu B. Tissue-specific differences in cytosine methylation and their association with differential gene expression in sorghum. Plant Physiol 2011; 156:1955 - 66; http://dx.doi.org/10.1104/pp.111.176842; PMID: 21632971
- Li X, Zhu J, Hu F, Ge S, Ye M, Xiang H, et al. Single-base resolution maps of cultivated and wild rice methylomes and regulatory roles of DNA methylation in plant gene expression. BMC Genomics 2012; 13:300; http://dx.doi.org/10.1186/1471-2164-13-300; PMID: 22747568
- Wang WS, Pan YJ, Zhao XQ, Dwivedi D, Zhu LH, Ali J, et al. Drought-induced site-specific DNA methylation and its association with drought tolerance in rice (Oryza sativa L.). J Exp Bot 2011; 62:1951 - 60; http://dx.doi.org/10.1093/jxb/erq391; PMID: 21193578
- Stroud H, Greenberg MV, Feng S, Bernatavichute YV, Jacobsen SE. Comprehensive analysis of silencing mutants reveals complex regulation of the Arabidopsis methylome. Cell 2013; 152:352 - 64; http://dx.doi.org/10.1016/j.cell.2012.10.054; PMID: 23313553
- Henderson IR, Jacobsen SE. Epigenetic inheritance in plants. Nature 2007; 447:418 - 24; http://dx.doi.org/10.1038/nature05917; PMID: 17522675
- Gehring M, Henikoff S. DNA methylation dynamics in plant genomes. Biochim Biophys Acta 2007; 1769:276 - 86; http://dx.doi.org/10.1016/j.bbaexp.2007.01.009; PMID: 17341434
- Hunter B, Hollister JD, Bomblies K. Epigenetic inheritance: what news for evolution?. Curr Biol 2012; 22:R54 - 6; http://dx.doi.org/10.1016/j.cub.2011.11.054; PMID: 22280908
- Lang-Mladek C, Popova O, Kiok K, Berlinger M, Rakic B, Aufsatz W, et al. Transgenerational inheritance and resetting of stress-induced loss of epigenetic gene silencing in Arabidopsis. Mol Plant 2010; 3:594 - 602; http://dx.doi.org/10.1093/mp/ssq014; PMID: 20410255
- Peralta IE, Spooner DM. Granule-bound starch synthase (GBSSI) gene phylogeny of wild tomatoes (Solanum L. section Lycopersicon [Mill.] Wettst. subsection Lycopersicon). Am J Bot 2001; 88:1888 - 902; http://dx.doi.org/10.2307/3558365; PMID: 21669622
- Wojdacz TK, Hansen LL, Dobrovic A. A new approach to primer design for the control of PCR bias in methylation studies. BMC Res Notes 2008; 1:54; http://dx.doi.org/10.1186/1756-0500-1-54; PMID: 18710507
- Gruntman E, Qi Y, Slotkin RK, Roeder T, Martienssen RA, Sachidanandam R. Kismeth: analyzer of plant methylation states through bisulfite sequencing. BMC Bioinformatics 2008; 9:371; http://dx.doi.org/10.1186/1471-2105-9-371; PMID: 18786255
- Ricardi MM, González RM, Iusem ND. Protocol: fine-tuning of a Chromatin Immunoprecipitation (ChIP) protocol in tomato. Plant Methods 2010; 6:11; http://dx.doi.org/10.1186/1746-4811-6-11; PMID: 20380723
- Tamás L, Simonovicova M, Huttova J, Mistrik I. Aluminium stimulated hydrogen peroxide production of germinating barley seeds. Environ Exp Bot 2004; 51:281 - 8; http://dx.doi.org/10.1016/j.envexpbot.2003.11.007