Abstract
Reduced representation bisulfite sequencing (RRBS) has been used to profile DNA methylation patterns in mammalian genomes such as human, mouse and rat. The methylome of the zebrafish, an important animal model, has not yet been characterized at base-pair resolution using RRBS. Therefore, we evaluated the technique of RRBS in this model organism by generating four single-nucleotide resolution DNA methylomes of adult zebrafish brain. We performed several simulations to show the distribution of fragments and enrichment of CpGs in different in silico reduced representation genomes of zebrafish. Four RRBS brain libraries generated 98 million sequenced reads and had higher frequencies of multiple mapping than equivalent human RRBS libraries. The zebrafish methylome indicates there is higher global DNA methylation in the zebrafish genome compared with its equivalent human methylome. This observation was confirmed by RRBS of zebrafish liver. High coverage CpG dinucleotides are enriched in CpG island shores more than in the CpG island core. We found that 45% of the mapped CpGs reside in gene bodies, and 7% in gene promoters. This analysis provides a roadmap for generating reproducible base-pair level methylomes for zebrafish using RRBS and our results provide the first evidence that RRBS is a suitable technique for global methylation analysis in zebrafish.
Introduction
DNA methylation provides a stable mechanism of epigenetic regulation of gene expression that is prevalent in all vertebrates.Citation1,Citation2 DNA methylation has a crucial role in gene silencing, tissue differentiation, genomic imprinting, X chromosome inactivation, phenotypic plasticity, and disease susceptibility.Citation3–Citation7 In addition, aberrant DNA methylation is a associated with several human diseases and has a well-established role in tumorigenesis.Citation8
Zebrafish (Danio rerio) is a vertebrate model organism that is used for investigating organogenesis, embryogenesis, and for modeling several human diseases such as cancer, including tumors induced by environmental carcinogens.Citation9–Citation16 Comparative gene homology studies and genome-scale epigenetic profiling in zebrafish reveal that the DNA methylation landscape of the zebrafish genome closely approximates mammalian genomes (e.g., human and mice).Citation17,Citation18 Therefore, zebrafish represent an attractive model for investigating vertebrate-specific DNA methylation and the evolution of methylation signatures in vertebrates. Moreover, since zebrafish do not require either imprinting of genes or sex chromosomes for viability, as is the case in mammals, they provide a simplified system for exploring the roles of DNA methylation in vertebrate development.Citation18 Further, as a result of their fast generation time, their numerous progeny and the ease with which they can be observed during early development, zebrafish promise to be an excellent model for studying trans-generational epigenetic inheritance and reprogramming.
Recently, whole genome bisulfite sequencing of zebrafish has been reported.Citation19,Citation20 These studies have described the distribution of DNA methylation, especially in gametes and during early development. The other methylation studies on zebrafish have used methylated DNA immunoprecipitation (MeDIP) on promoter arrays or sequencing,Citation21–Citation24 and a major limitation of this antibody-based method is that it does not allow investigation of CpG sites at base-pair resolution. Further, the enrichment of the genome depends on the CpG density; therefore, MeDIP preferentially isolates methylated CpG-rich regions, potentially failing to detect methylation at the CpG-poor regions of the genome.Citation25 Reduced representation bisulfite sequencing (RRBS) is an effective alternative approach to whole genome methylation sequencing that generates multiple base-pair resolution methylomes at a reduced cost.Citation26–Citation29 However, the RRBS technique has been primarily used for human and mouse genomes, and its effectiveness has not yet been demonstrated in zebrafish. Here, we evaluate the technique of RRBS in DNA from zebrafish brain. Brain is probably the most complex vertebrate organ, and epigenetic events have roles in memory formation and learning,Citation30,Citation31 brain development,Citation32 early life stress,Citation30 neurodegeneration,Citation33 neurological and neuropsychiatric disorders,Citation34,Citation35 and establishment of neuronal identity.Citation36
To our knowledge, this study provides the first single-nucleotide resolution DNA methylation map of the zebrafish brain. We compare previously described RRBS methylome for human, mouse and rat with zebrafish RRBS wherever applicable to highlight the technical and biological differences, such as DNA sequence alignment, CpG density and enrichment, CpG island features and overall methylation profile. As an additional control, we performed RRBS analysis on zebrafish liver to corroborate the findings from zebrafish brain RRBS. Furthermore, we provide an overview of the distribution of methylation and mapped CpG dinucleotides in the zebrafish brain as captured by the reduced representation method.
Results
The zebrafish reduced representation (RR) genome
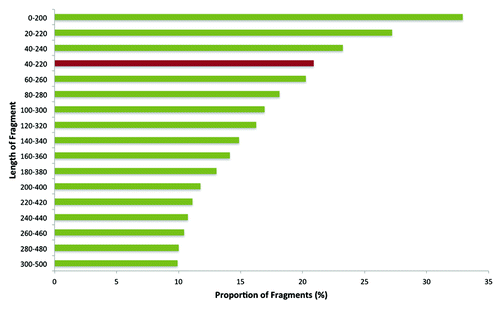
In silico digestion of the zebrafish genome (Zv9) with MspI enzyme resulted in 1,265,636 fragments with a total length of 1,407,827,316 bp. We first performed a simulation of the distribution of MspI fragments: by taking a 200 bp rectangular window and a 20 bp gradient, the proportion of MspI fragments contained in each bin was determined (). The simulation showed that the reduced representation (RR) genome of 40–220 bp contained 20.9% of the total MspI fragments. Selection of fragments shorter than 40 bp included an additional 12.5% fragments. However, we selected 40–220 bp fragments to generate the RRBS library for two reasons. First, very short fragments are likely to map to multiple locations in the genome. Second, selection of 40–220 bp fragments provided the opportunity for a frame-by-frame comparison of zebrafish RRBS with previously published human and mouse RRBS data on the same size selection.Citation37
Figure 1. Distribution of MspI digested fragments in zebrafish genome. The red bar represents the size selection performed for the current study, i.e., 40–220 bp.
CpG enrichment in the zebrafish RR genome
For zebrafish, the total size of the in silico RR genome was 31 Mb comprising 264,598 MspI fragments of 40–220 bp lengths. It contained 1.43 million CpG sites representing 5.3% of the genomic total, indicating a 2.4-fold enrichment of CpG sites in the RR genome (). By comparison, the human RR genome has a size of 74 Mb consisting of 647,626 MspI fragments. The human RR genome is more than double the size of the zebrafish RR genome, and both are proportional to the size of the whole genome of these organisms. However, the human RR genome, which represents 2.3% of the whole genome, contained 4.1 million CpG sites (13.5% of the genomic total), corresponding to a 5.7-fold enrichment of CpG sites. In mice, the RR genome represents 1.4% of the genome and contains 7.0% of the CpG sites in the genome, corresponding to a 5-fold enrichment. These comparisons demonstrate that CpG dinucleotides are more enriched in the human and mice MspI-generated RR genome of 40–220 bp than that of zebrafish.
Table 1. Comparison of human and zebrafish RR genome*
RRBS and alignment
We obtained an average of 1.2 Gb of sequence from each of the four zebrafish RRBS libraries. For each sample, 24.5 million sequenced reads of 49 bp in length were supplied. For all four samples, more than 70% of the reads mapped to the reference genome of Zv9 (). However, the percentage of reads that mapped to multiple locations of the genome was higher (range: 43.6% to 45.5%) than the percentage of the reads that uniquely mapped (range: 27% to 32.7%). The percentage of multiple mapping in zebrafish was 5-fold higher than in our previous Bismark alignment of the human RRBS library, where only 7.7% of the reads (75 bp length) showed multiple mapping against the whole human genome (GRCh37 build).Citation37
Table 2. Details of output and mapping of zebrafish RRBS libraries
High coverage CpG dinucleotides (CpG10)
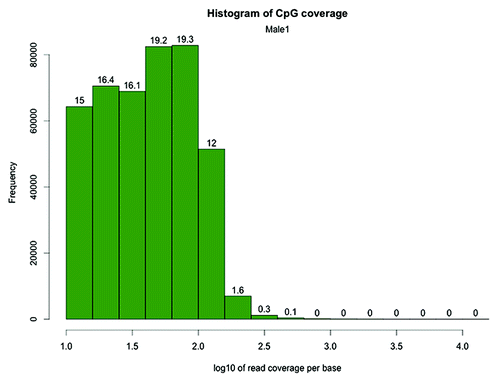
After alignment, we filtered the CpG dinucleotides based on coverage. Only CpG sites covered by 10 or more reads (CpG10) were retained for further analysis. For four of our samples, 429,088, 404,563, 303,757 and 405,903 CpG10 were obtained, with a mean coverage ranging from to 55 to 77 (). The distribution of sequenced read coverage of CpG10 in the Male1 sample is shown in as an example (see also Fig. S3–S5 for coverage histograms of other samples). The distribution demonstrates that although the filtered CpGs received high mean coverage, the libraries did not suffer from bias due to excessive amplification of a subset of fragments, should a spurious amplification of fragments exist, an extra peak would be visible on the right hand side of the histogram.
Table 3. Number and coverage of CpG10 in zebrafish RRBS methylome after alignment*
Figure 2. CpG site coverage histogram of the zebrafish Male1 RRBS library. The x-axis shows log 10 values corresponding to the number of reads per CpG. The numbers on the bars denote the percentage of CpG sites contained in the respective bins. Log10Citation10 = 1, Log10Citation31 = 1.5, Log10100 = 2, Log10177 = 2.25, Log10317 = 2.5, Log1010000 = 4
Global CpG methylation profile of the zebrafish RR genome
The global CpG methylation ranged from 69.6–75.0% () in male and female zebrafish RRBS libraries. The distribution of methylation at each CpG site revealed heavy methylation (> 95% methylation) of 40.7 to 42.8% of CpG10 (). By contrast, 10.9% to 12.2% of CpG10 were completely unmethylated bases (< 5% methylation). Between 15.8% and 16.6% of the CpG10 showed intermediate methylation (> 20% and < 80%) (; Fig. S6–S8).
Figure 3. CpG methylation distribution in zebrafish brain. The X-axis shows percent methylation for each CpG. The numbers on the bars denote the percentage of CpGs contained in the respective bins.
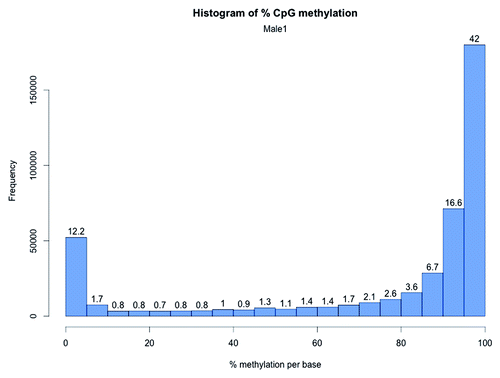
In mammalian genomes, the RRBS protocol has been shown to enrich for CG rich regions (CpG islands)Citation26 and, as CpG islands remain largely unmethylated in mammalian genomes, the percent methylation of CpGs in RRBS libraries is expected to be lower than the average methylation of the genome. For example, RRBS on rat dorsal root ganglia (analyzed CpG sites = 2.8 million) showed more than half of the CpG sites captured by RRBS were hypomethylated (0–10% methylation) and a fifth of the sites demonstrated hypermethylation (90–100% methylation).Citation38 However, for zebrafish brain RRBS, the trend was opposite, i.e., > 50% of the CpG10 were hypermethylated (> 90% methylation) and less than 15% of the CpG sites were hypomethylated (0% -10% methylation). Meissner and colleagues performed RRBS for the first time in mouse embryonic stem cells (analyzed CpG sites = 543,678 with coverage of ≥ 10) and reported a similar methylation pattern to rat RRBS, i.e., > 40% analyzed CpG sites showed hypomethylation.Citation26 In the same study, the median percentage CpG methylation for mice brain was shown to be 10 (analyzed CpG sites = 906,010 with a median coverage of 14). As a result of much higher prevalence of hypermethylated CpG10, zebrafish brain RRBS showed higher median methylation ranging from 92.4 to 93.3 ().
Table 4. Per quartile methylation distribution of CpG10 in zebrafish RRBS samples
Although RRBS and whole genome bisulfite sequencing (WGBS) methylation are not directly comparable (as RRBS covers 5–15% of the CpG sites in the genome enriching for CpG rich regions, whereas WGBS includes 80–100% of the CpG sites including those in repetitive elements), we extracted WGBS data for human and mouse brain and compared their CpG methylation with that from zebrafish RRBS. The results suggested that the proportion of methylated (80–100% methylation) CpG sites is higher in zebrafish RRBS brain methylome compared with the WGBS methylome of human and mice. Further, the percentage of unmethylated (0–20% methylation) bases was higher in zebrafish RRBS samples as well, reflecting the relative enrichment of RRBS for CpG features compared with WGBS (Table S1).
To test whether the striking difference in global CpG methylation in zebrafish RRBS compared with the other mammalian genomes is due to the tissue under investigation (i.e., brain) we performed RRBS of adult zebrafish liver. The global CpG methylation for liver was 68.8% and the median methylation of the CpG10 was 90.9 (number of CpG10 = 219947 with a mean coverage of 21.4) similar to that observed for zebrafish brain samples (). The distribution of methylation at each CpG site revealed that 63.4% of the CpG10 in zebrafish liver have a methylation percentage ranging from 80–100%; whereas, for 23.8% of the CpG10, the methylation percentage ranged from 0–20% (), with 12.8% CpG10 showing intermediate methylation. Overall, the comparison of the CpG10 methylation distribution between brain and liver () indicates the zebrafish methylome, as captured by RRBS, is different than the mammalian genome. In fact, our results are concordant with recent WGBS studies on zebrafish sperm, oocytes, mid-blastula embryo, and muscle (). In all these tissues, WGBS analysis showed a high proportion of methylated CpGs (80–100%) compared with unmethylated (0–20%).Citation19,Citation20
Figure 4. CpG methylation distribution in zebrafish liver. The X-axis shows percent methylation for each CpG site. The numbers on the bars denote the percentage of CpGs contained in the respective bins.
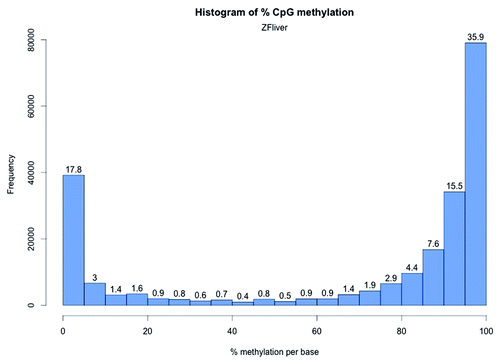
Table 5. Percentage distribution of methylated and unmethylated CpG zebrafish RRBS samples and comparison with zebrafish WGBS*
Non-CpG methylation in the zebrafish genome
For all samples, both Bismark and methylKit results suggested successful bisulfite conversion of the genome. We found that the percentage of non-CpG methylation was low in adult zebrafish brain, ranging from 1.7% to 2.9% (). Previously, heavy non-CpG methylation in zebrafish was reportedCitation39; however, other bisulfite sequencing experiments have demonstrated very low levels of non-CpG methylation.Citation22,Citation40,Citation41 RRBS studies on rat brain suggested less than 1% non-CpG methylation.Citation38 On the contrary, whole genome methylation analysis of mice brain suggested 35% of DNA methylation could occur in a non-CpG contextCitation42 (CHH and CHG). Our results, however, show that non-CpG methylation in adult zebrafish brain is minimal. Further analysis in zebrafish liver revealed 3.7% non-CpG methylation, supporting the idea that globally non-CpG methylation in zebrafish genome is negligible and are concordant with recent WGBS studies on zebrafish.Citation19,Citation20
Reproducibility of zebrafish RRBS methylomes
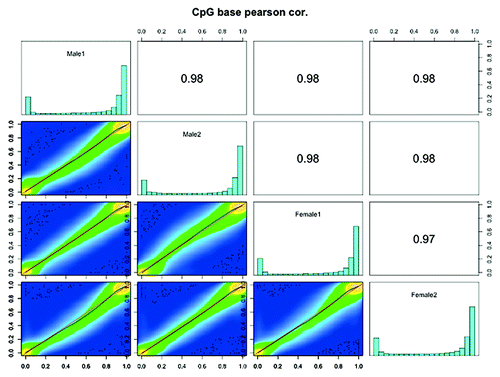
One reason for performing RRBS experiments with four pools of DNA (each containing six individuals) was to minimize the effects of biological variability. and Figure S5-S8 show that the overall global methylation profile is similar between all the pooled samples. We constructed scatter plots of DNA methylation of CpG10 between four zebrafish RRBS methylomes (). We observed very high positive correlation between Male1 vs. Male2 (Pearson’s correlation coefficient = 0.98), Female1 vs. Female2 (Pearson’s correlation coefficient = 0.97), demonstrating minimal variation between the pooled samples. We also observed high positive correlation between male and female methylomes () suggesting that globally, sex specific differences in methylation are low in adult zebrafish. However, there might be several CpG site-specific methylation differences between the male and female brain samples. Detailed analysis of sex-specific differential methylation is beyond the scope of this study and will be the subject of future research. Taken together, our results illustrate that use of RRBS in zebrafish can produce reproducible reference methylomes.
Figure 5. Scatter plot and correlation of CpG methylation between zebrafish RRBS methylomes. Scatter plots of percentage methylation values for each pair in four zebrafish libraries (Male1, Male2, Female1 and Female2). Numbers on the upper right corner denote pair-wise Pearson’s correlation scores. The histograms on the diagonal are methylation distribution of CpG sites for each sample.
Relationship of CpG10 with CpG features
Since we found a high level of methylation of CpG10 in the zebrafish brain, we sought to determine whether the mapped CpG10 correspond to CpG islands. The list of CpG islands from the SeqMonk feature table was used for this analysis. Regions 2 kb either side of CpG islands were defined as CpG island shores, and regions a further 2 kb from the CpG shores were defined as CpG island shelves. This analysis revealed CpG10 are highly enriched in CpG islands shores (46.8% of the total CpG10), while only 9.25% of the total CpG10 are in the core CpG islands (). Furthermore, 42.7% of CpG10 are distant from any CpG features, i.e., more than 4 kb distant from either side of a core CpG islands. The CpG10 were exported to the UCSC genome browser as a custom track and visualized in comparison with CpG island track of the browser for Zv9 assembly. The whole genome chromosome-wide view enabled genome-wide visualization of CpG10 and showed several regions where core CpG islands are absent but CpG10 were densely mapped.
Table 6. Distribution of CpG10 in CpG feature context*
In contrast, a recent RRBS study on humans (MspI digested and size selection of 40–220 bp fragments) showed 47.5% and 19.5% of the investigated CpG dinucleotides were in core CpG island and CpG island shores respectively.Citation43 Our data indicate that the mapped CpG dinucleotides are more prevalent in CpG island shores of the zebrafish RR genome and less frequent in core CpG islands compared with the equivalent analysis in human.
Distribution of CpG10 in relation with genes
We investigated the distribution of the CpG10 relative to the location of genes in the zebrafish genome (gene location information was taken from the SeqMonk feature table information). Interestingly, we found that 45% of CpG10 mapped to gene bodies () and a much smaller percentage (7%) mapped to gene promoters (promoters were defined as regions up to –5 kb from the transcription start site (TSS) of the gene). These results contrasted with human RRBS data where 32% of the investigated CpG sites were in promoters.Citation43 Fifty one percent of the CpG sites were distant, i.e., further than 5 kb upstream of the gene. describes the detailed distribution of CpG10 in relation to TSS and gene bodies. Analysis of gene body-associated CpG10 revealed that 59% reside within introns and 41% within exons ().
Figure 6. Distribution of CpG10 in genome and in gene body. A. Shows the overall distribution of CpG10 of Male1 sample in the genome. B. Shows the distribution of gene body associated CpG10.
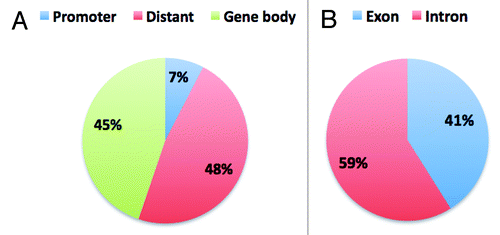
Table 7. Distribution of CpG10 in relation to genes*
Discussion
Zebrafish are an ideal animal model for studying developmental biology since their embryos are transparent, easily accessed from the 1-cell stage, and available in large numbers. Therefore, it is of interest to determine how global methylation patterns in zebrafish compare with those in humans. For this comparison, methylation analysis must be applied in the same detail to zebrafish as it is in humans. RRBS is a tractable method for generating base-pair resolution methylation profile to generate data that could inform biological processes in development and disease.
Here, we have evaluated RRBS in zebrafish and, to our knowledge, we have provided the first single-base resolution DNA methylation map for brain tissue in this organism. The zebrafish genome is almost half the size of the human genome, and considering the relative size of RR genome of both organisms (31 Mb vs. 74 Mb), the number of CpG sites covered in the zebrafish RR genome (40–220 bp) was proportional (1.43 million CpG sites compared with almost 4 million in humans). The overall frequency of CpG sites in the zebrafish genome is much higher than that in human and mice (1.77, 1.04 and 1.15 CpGs per 100 bp respectively). However, the fold enrichment of CpG sites obtained for the zebrafish RR genome was lower than human and mice (2.4-fold compared with 5.7-fold in humans and 5-fold in mice). Although the zebrafish genome has a higher density of CpG sites, CCGG (MspI site) motifs are not as prevalent as in humans. Therefore, in future studies it may be desirable to explore the use of other restriction enzymes that might cut more frequently at the CpG motif. Recently, a double digest of MspI with another methylation-insensitive enzyme was shown to improve the CpG coverage in RRBS experiments in human and mice.Citation43 A similar approach could be applied to the zebrafish genome for enhanced CpG coverage. For human and mice, as MspI already provides good coverage of core CpG islands, a non CG containing enzyme (ApeKI, recognition site G^CWGC) was used in combination with MspI to improve coverage in CpG poor regions. However, to apply the double restriction digest method for zebrafish, consideration could be given to the use of another CG motif containing enzyme (for example TaqαI, recognition site T^CGA or BssSI, recognition site C^ACGAG) with MspI as it is likely to improve the coverage within the core CpG islands (we already showed that MspI provides higher coverage for CpG island shore and distant CpGs compared with the core CpG island).
The zebrafish genome is substantially polymorphic in nature and the quality of the reference genome is not optimal (Sanger Institute release notes for Zv9). In addition, the recently published zebrafish genome indicates that zebrafish repetitive DNA constitutes 52.2% of the genome, the highest repeat content so far recorded for a vertebrate. Increased levels of repetitive DNA almost certainly contribute to the observation of increased global methylation in zebrafish relative to human.Citation44,Citation45 Further, repetitive sequences and the incomplete genome (the zebrafish genome sequence is 83% complete) provide an explanation for increased multiple mapping in zebrafish compared with human (almost 5-fold higher than in humans). We observed that increasing the sequenced read length to 100 bp improved the unique mapping efficiency to > 50% in zebrafish (unpublished data).
The true reflection of CpG distribution in the RR genome should be obtained after alignment and filtering by coverage and not on the initial output from the sequencing, since for subsequent analysis and interpretation only high coverage CpGs will be included. We obtained 0.3 to 0.42 million CpG sites with high coverage in our libraries (CpG10). Considering the higher CpG density of the zebrafish genome, these numbers are low compared with the human RRBS data. Nevertheless, RRBS allowed us to generate a genome-wide nucleotide resolution methylation profile of 0.39 million high coverage CpG sites (on average) in the zebrafish samples. Increased read length or deeper sequencing would significantly increase the number of high coverage CpG sites.
Investigation of CpG10 distribution lead to the interesting observation of higher enrichment of CpG island shores in zebrafish compared with core CpG islands, unlike similar human RRBS libraries. There are multiple methods for defining CpG islands and, as noted by Saxanov et al., definitions of CpG islands are based on “ad hoc thresholds”Citation46. For humans, Takai and Jones’s method of predicting CpG island, which is widely used, states the minimum length of CpG island should be 500 bp,Citation47 whereas for example Gardiner and Frommer defined a minimum 200-bp stretch of CpG rich DNA.Citation48 Within the SeqMonk feature table (based on Ensembl annotation), the length of the shortest CpG island in zebrafish is 399 bp. Therefore, the definition of CpG islands affects the distribution of mapped CpGs within different CpG features, i.e., if CpG islands are defined as short regions, then more sequenced CpGs will fall outside the defined CpG islands. As the definition of CpG islands is arbitrary, we propose that inclusion of different CpG features (including shore and shelf) provides a more comprehensive approach for methylation profiling in enrichment-based methylation analysis (e.g., RRBS).
For decades, the methylation status of CpG islands received much attention from investigators. However, recent studies showed CpG island shores as crucial elements where DNA methylation status was highly variable between diseased and matched normal tissues. Furthermore, differential methylation of CpG island shores is often tissue specific, and the methylation status of shores correlates strongly with gene expression.Citation49,Citation50 In this respect, our results on zebrafish RRBS are promising, as the CpG10 were enriched in CpG island shores which might facilitate further investigation of the functional role of DNA methylation status outside CpG islands, including the role of methylation in gene regulatory regions and long-range chromosome interactions.
Tissue-specific differences in methylation are frequently associated with specific sites in the genome, whereas global methylation profiles between tissues appear not to vary (e.g., in mammalian genomes most CpG islands largely remain unmethylated regardless of tissue type). The comparative analysis of methylation distribution in brain and liver samples demonstrated that the differences in global methylation patterns we observed between zebrafish and other mammalian RRBS were not due to the choice of tissue. Therefore, high levels of methylation present in the zebrafish brain likely reflect global methylation differences between teleosts and mammals.
Recent studies detailing WGBS analysis of methylation in oocyte, sperm, mid-blastula embryo and muscle in zebrafish have provided detailed information about global methylation of the zebrafish genome.Citation19,Citation20 It is notable that the RRBS genomes generated here broadly reflect the global methylation profiles observed using the WGBS method, in that both indicate a much higher percentage methylation in zebrafish than in mammalian genomes. The similarity between RRBS and WGBS generated data indicates that the RRBS method provides a reliable snapshot of methylation in the zebrafish genome. The RRBS method can therefore be used as an economical substitute for WGBS where indicated.
Another feature of interest is the high proportion of mapped CpGs in the gene bodies compared with the promoter regions. The functional role of gene body methylation remains unclear. Recent studies showed sharp transitions in methylation status at exon-intron boundaries.Citation51 It has been suggested that site-specific occupancy of CTCF pauses RNA polymerase II activity, and that the interaction of CTCF and RNA polymerase II plays role in alternate splicing of genes.Citation52 It has also been suggested that specific histone modifications mark gene bodies of different classes of genes based on CpG density and methylation status.Citation53 Furthermore, it is possible that DNA methylation levels are generally higher in gene bodies than in non-genic regionsCitation54,Citation55 and that gene-body methylation facilitates transcriptional elongation by blocking non-specific or intragenic transcription.Citation56 However, a recent large-scale meta-analysis called in question the notion that gene body methylation is associated with transcriptional activity, and instead proposed that gene body methylation is determined by accessibility to DNA methyltransferases.Citation57 The high CpG coverage in gene bodies provided by zebrafish RRBS will allow future exploration of the function and consequences of gene body methylation.
It is likely that, similar to other species, variations in zebrafish methylation regulate tissue-specific gene expression. Modifications of chromatin proteins (such as histones) at enhancers were shown to be associated with tissue-specific cell differentiation in zebrafish.Citation58 However, little is known about the role of methylation at such enhancers. Further, DNA methylation can determine binding of chromatin architecture proteins such as CTCF, to give cell type-specific chromatin configurations that may instruct gene expression.Citation59 Zebrafish represent a powerful model for determining the in vivo function of non-coding DNA elements.
This study is limited by its use of a heterogeneous tissue type (brain) and by the poorer quality of the zebrafish genome compared with the human genome. Further, a known limitation of bisulfite sequencing based methods is that the protocol is unable to distinguish difference between 5-methylcytosine and 5-hydroxymethylcytosine. Recent studies indicated presence of 5-hydroxymethylcytosine in human and mouse brainCitation60; however, determining the status of potential demethylating modifications such as 5-hydroxymethylcytosine in zebrafish is warranted. Our analysis demonstrates differences in the global methylation signatures and distributions of CpG sites between zebrafish and other mammalian genomes. The base-pair resolution reference methylome of zebrafish provides a resource for future studies to document the functional role of DNA methylation in this organism.
Materials and Methods
Ethics statement
Animal handling and manipulations were conducted in accordance with the guidelines of the University of Otago Animal Ethics Committee under protocol 48–11.
Sample preparation
Adult zebrafish AB strains used for this study were maintained at the Otago Zebrafish Facility, Department of Pathology, University of Otago. Brains were dissected from 12 males and 12 females, and then each brain was halved through the sagittal plane. Six halved male brains were combined into a pooled sample referred to as Male1. Similarly, Male2, Female1 and Female2 were comprised of a pool of six halved brain. Livers (n = 10) were harvested by dissection from wild type male and female fish, and pooled. Genomic DNA was extracted from each sample using the PureLink Genomic DNA Mini Kit (Invitrogen) according to the manufacturer’s instructions.
RRBS library preparation and sequencing
Bisulfite-converted genomic DNA libraries were prepared according to the previously described methods.Citation37,Citation61 Briefly, genomic DNA was digested overnight with MspI (New England Biolabs), followed by end-repair and addition of 3′ A overhangs. Methylated adaptors (Illumina) with a 3′ T overhang were ligated to the A tailed DNA fragments. For reduced representation, 40 to 220 bp (pre-adaptor-ligation size) fragments were excised from 3% Nusieve agarose gels (Lonza) and bisulfite-converted with EZ DNA methylation Gold kit (Zymo Research). Bisulfite converted libraries were amplified by PCR and sequenced on an Illumina HiSeq2000 sequencer with a single-ended, 49 bp run (Beijing Genomics Institute). FASTQ sequence files were obtained containing sequenced reads for each sample (Fig. S1). For zebrafish liver, 9 million single-ended, 100 bp reads were sequenced (New Zealand Genomics Limited).
Sequence quality check and alignment
For sequenced reads obtained for each individual sample, quality checks of the reads, processing and alignments were performed according to our previously published pipeline.Citation37 The quality of the sequenced reads in all zebrafish libraries was high with a median Phred score of > 30 till the end of last sequencing cycle (see representative FastQC quality plot in Figure S2; FastQC software package is distributed from Babraham Institute). As a result, trimming of the 3′ end of the reads was not necessary for these libraries. However, for the zebrafish liver RRBS library, we hard-trimmed the sequenced reads from 100 bp to 65 bp as the Phred score values dropped significantly after 65 sequencing cycle. Adaptor sequences were removed from the reads using our in-house cleanadaptors program.Citation37 For the 49 bp sequenced reads, traces of adaptor sequences in the reads were minimal as confirmed by both cleanadaptors and FastQC (from Illumina sequenced reads FastQC searches for known Illumina adaptor sequences). The sequenced reads were aligned against the zebrafish reference genome Zv9 using the bisulfite alignment program Bismark v0.6.4.Citation62 The alignments were performed on a Mac Pro with 64 bit duo quad core Intel Xeon processors and with 22 Gb RAM running MacOS 10.6.
RRBS data analysis
From the zebrafish whole genome assembly (Zv9), an in silico reduced representation (RR) genome based on MspI cleavage sites (C^CGG) and fragment sizes of 40–220 bp was generated by the mkrrgenome program.Citation37 Custom written UNIX and awk (an interpreted programming language) scripts and commands were used to describe the distribution of fragments in the genome. Methylation analysis was performed using the R package of methylKit.Citation63 Briefly, after alignment by Bismark, the SAM files containing uniquely aligned reads were numerically sorted and then processed in R studio (version 0.97.312) using the methylKit package. CpG sites covered by at least 10 sequenced reads (termed as CpG10) were retained to generate the reference methylome. Each sequenced and filtered CpG site was assigned a percentage methylation score. Coverage and correlation plots were generated by methylKit using sorted SAM file for the samples. Human and mouse brain whole genome bisulfite sequencing data for control samples were downloaded from MethylomeDBCitation64 and processed with UNIX and awk scripts (see Table S1).
To investigate CpG10 positions, in relation to the gene and CpG features, the SeqMonk feature table information for Zv9 was used. SeqMonk (freely distributed from Babraham Institute) provide .DAT files containing information on CpG islands and genes in zebrafish. These files were parsed by a purpose-written program (identgeneloc), which then identified proximal genes and CpG islands for the CpG10 sites. The resulting information was further processed with awk scripts to generate the distribution of CpG10 positions.
Additional material
Download Zip (341.8 KB)Acknowledgments
We gratefully acknowledge the contribution of Dr. Gwenn Le Mée. of the Department of Pathology, University of Otago in Zebrafish liver DNA sample preparation and for helpful discussions. We thank Dr Euan Rodger for assistance and discussions during the preparation of the manuscript. This work is supported by University of Otago research grant and Gravida: National Centre for Growth and Development research grant. AC is supported by a scholarship from Gravida. SN is supported by the Marsden Fund and the Rutherford Discovery Fellowship.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Materials
Supplemental materials may be found here: http://www/landesbioscience.com/journals/epigenetics/article/25797
References
- Elango N, Yi SV. DNA methylation and structural and functional bimodality of vertebrate promoters. Mol Biol Evol 2008; 25:1602 - 8; http://dx.doi.org/10.1093/molbev/msn110; PMID: 18469331
- Bock C, Beerman I, Lien WH, Smith ZD, Gu H, Boyle P, et al. DNA methylation dynamics during in vivo differentiation of blood and skin stem cells. Mol Cell 2012; 47:633 - 47; http://dx.doi.org/10.1016/j.molcel.2012.06.019; PMID: 22841485
- Carrel L, Willard HF. X-inactivation profile reveals extensive variability in X-linked gene expression in females. Nature 2005; 434:400 - 4; http://dx.doi.org/10.1038/nature03479; PMID: 15772666
- Rollins RA, Haghighi F, Edwards JR, Das R, Zhang MQ, Ju J, et al. Large-scale structure of genomic methylation patterns. Genome Res 2006; 16:157 - 63; http://dx.doi.org/10.1101/gr.4362006; PMID: 16365381
- Suzuki MM, Bird A. DNA methylation landscapes: provocative insights from epigenomics. Nat Rev Genet 2008; 9:465 - 76; http://dx.doi.org/10.1038/nrg2341; PMID: 18463664
- Igarashi J, Muroi S, Kawashima H, Wang X, Shinojima Y, Kitamura E, et al. Quantitative analysis of human tissue-specific differences in methylation. Biochem Biophys Res Commun 2008; 376:658 - 64; http://dx.doi.org/10.1016/j.bbrc.2008.09.044; PMID: 18805397
- Chatterjee A, Morison IM. Monozygotic twins: genes are not the destiny?. Bioinformation 2011; 7:369 - 70; http://dx.doi.org/10.6026/97320630007369; PMID: 22355239
- Baylin S, Bestor TH. Altered methylation patterns in cancer cell genomes: cause or consequence?. Cancer Cell 2002; 1:299 - 305; http://dx.doi.org/10.1016/S1535-6108(02)00061-2; PMID: 12086841
- Mirbahai L, Williams TD, Zhan H, Gong Z, Chipman JK. Comprehensive profiling of zebrafish hepatic proximal promoter CpG island methylation and its modification during chemical carcinogenesis. BMC Genomics 2011; 12:3; http://dx.doi.org/10.1186/1471-2164-12-3; PMID: 21205313
- Bailey GS, Williams DE, Hendricks JD. Fish models for environmental carcinogenesis: the rainbow trout. Environ Health Perspect 1996; 104:Suppl 1 5 - 21; PMID: 8722107
- Wardle FC, Odom DT, Bell GW, Yuan B, Danford TW, Wiellette EL, et al. Zebrafish promoter microarrays identify actively transcribed embryonic genes. Genome Biol 2006; 7:R71; http://dx.doi.org/10.1186/gb-2006-7-8-r71; PMID: 16889661
- Berghmans S, Jette C, Langenau D, Hsu K, Stewart R, Look T, et al. Making waves in cancer research: new models in the zebrafish. Biotechniques 2005; 39:227 - 37; http://dx.doi.org/10.2144/05392RV02; PMID: 16116796
- Zon LI. Zebrafish: a new model for human disease. Genome Res 1999; 9:99 - 100; PMID: 10022974
- Lam SH, Gong Z. Modeling liver cancer using zebrafish: a comparative oncogenomics approach. Cell Cycle 2006; 5:573 - 7; http://dx.doi.org/10.4161/cc.5.6.2550; PMID: 16582610
- Lam SH, Wu YL, Vega VB, Miller LD, Spitsbergen J, Tong Y, et al. Conservation of gene expression signatures between zebrafish and human liver tumors and tumor progression. Nat Biotechnol 2006; 24:73 - 5; http://dx.doi.org/10.1038/nbt1169; PMID: 16327811
- Lieschke GJ, Currie PD. Animal models of human disease: zebrafish swim into view. Nat Rev Genet 2007; 8:353 - 67; http://dx.doi.org/10.1038/nrg2091; PMID: 17440532
- Wu SF, Zhang H, Hammoud SS, Potok M, Nix DA, Jones DA, et al. DNA methylation profiling in zebrafish. Methods Cell Biol 2011; 104:327 - 39; http://dx.doi.org/10.1016/B978-0-12-374814-0.00018-5; PMID: 21924171
- Goll MG, Halpern ME. DNA methylation in zebrafish. Prog Mol Biol Transl Sci 2011; 101:193 - 218; http://dx.doi.org/10.1016/B978-0-12-387685-0.00005-6; PMID: 21507352
- Jiang L, Zhang J, Wang JJ, Wang L, Zhang L, Li G, et al. Sperm, but not oocyte, DNA methylome is inherited by zebrafish early embryos. Cell 2013; 153:773 - 84; http://dx.doi.org/10.1016/j.cell.2013.04.041; PMID: 23663777
- Potok ME, Nix DA, Parnell TJ, Cairns BR. Reprogramming the maternal zebrafish genome after fertilization to match the paternal methylation pattern. Cell 2013; 153:759 - 72; http://dx.doi.org/10.1016/j.cell.2013.04.030; PMID: 23663776
- Mhanni AA, McGowan RA. Global changes in genomic methylation levels during early development of the zebrafish embryo. Dev Genes Evol 2004; 214:412 - 7; http://dx.doi.org/10.1007/s00427-004-0418-0; PMID: 15309635
- Macleod D, Clark VH, Bird A. Absence of genome-wide changes in DNA methylation during development of the zebrafish. Nat Genet 1999; 23:139 - 40; http://dx.doi.org/10.1038/13767; PMID: 10508504
- Feng S, Cokus SJ, Zhang X, Chen PY, Bostick M, Goll MG, et al. Conservation and divergence of methylation patterning in plants and animals. Proc Natl Acad Sci U S A 2010; 107:8689 - 94; http://dx.doi.org/10.1073/pnas.1002720107; PMID: 20395551
- Andersen IS, Reiner AH, Aanes H, Aleström P, Collas P. Developmental features of DNA methylation during activation of the embryonic zebrafish genome. Genome Biol 2012; 13:R65; http://dx.doi.org/10.1186/gb-2012-13-7-r65; PMID: 22830626
- Laird PW. Principles and challenges of genomewide DNA methylation analysis. Nat Rev Genet 2010; 11:191 - 203; http://dx.doi.org/10.1038/nrg2732; PMID: 20125086
- Meissner A, Mikkelsen TS, Gu H, Wernig M, Hanna J, Sivachenko A, et al. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature 2008; 454:766 - 70; PMID: 18600261
- Gertz J, Varley KE, Reddy TE, Bowling KM, Pauli F, Parker SL, et al. Analysis of DNA methylation in a three-generation family reveals widespread genetic influence on epigenetic regulation. PLoS Genet 2011; 7:e1002228; http://dx.doi.org/10.1371/journal.pgen.1002228; PMID: 21852959
- Bock C, Kiskinis E, Verstappen G, Gu H, Boulting G, Smith ZD, et al. Reference Maps of human ES and iPS cell variation enable high-throughput characterization of pluripotent cell lines. Cell 2011; 144:439 - 52; http://dx.doi.org/10.1016/j.cell.2010.12.032; PMID: 21295703
- Steine EJ, Ehrich M, Bell GW, Raj A, Reddy S, van Oudenaarden A, et al. Genes methylated by DNA methyltransferase 3b are similar in mouse intestine and human colon cancer. J Clin Invest 2011; 121:1748 - 52; http://dx.doi.org/10.1172/JCI43169; PMID: 21490393
- Iwamoto K, Bundo M, Ueda J, Oldham MC, Ukai W, Hashimoto E, et al. Neurons show distinctive DNA methylation profile and higher interindividual variations compared with non-neurons. Genome Res 2011; 21:688 - 96; http://dx.doi.org/10.1101/gr.112755.110; PMID: 21467265
- Lubin FD, Roth TL, Sweatt JD. Epigenetic regulation of BDNF gene transcription in the consolidation of fear memory. J Neurosci 2008; 28:10576 - 86; http://dx.doi.org/10.1523/JNEUROSCI.1786-08.2008; PMID: 18923034
- Ma DK, Marchetto MC, Guo JU, Ming GL, Gage FH, Song H. Epigenetic choreographers of neurogenesis in the adult mammalian brain. Nat Neurosci 2010; 13:1338 - 44; http://dx.doi.org/10.1038/nn.2672; PMID: 20975758
- Migliore L, Coppedè F. Genetics, environmental factors and the emerging role of epigenetics in neurodegenerative diseases. Mutat Res 2009; 667:82 - 97; http://dx.doi.org/10.1016/j.mrfmmm.2008.10.011; PMID: 19026668
- Samaco RC, Neul JL. Complexities of Rett syndrome and MeCP2. J Neurosci 2011; 31:7951 - 9; http://dx.doi.org/10.1523/JNEUROSCI.0169-11.2011; PMID: 21632916
- Mill J, Tang T, Kaminsky Z, Khare T, Yazdanpanah S, Bouchard L, et al. Epigenomic profiling reveals DNA-methylation changes associated with major psychosis. Am J Hum Genet 2008; 82:696 - 711; http://dx.doi.org/10.1016/j.ajhg.2008.01.008; PMID: 18319075
- Guo Y, Monahan K, Wu H, Gertz J, Varley KE, Li W, et al. CTCF/cohesin-mediated DNA looping is required for protocadherin α promoter choice. Proc Natl Acad Sci U S A 2012; 109:21081 - 6; http://dx.doi.org/10.1073/pnas.1219280110; PMID: 23204437
- Chatterjee A, Stockwell PA, Rodger EJ, Morison IM. Comparison of alignment software for genome-wide bisulphite sequence data. Nucleic Acids Res 2012; 40:e79; http://dx.doi.org/10.1093/nar/gks150; PMID: 22344695
- Hartung T, Zhang L, Kanwar R, Khrebtukova I, Reinhardt M, Wang C, et al. Diametrically opposite methylome-transcriptome relationships in high- and low-CpG promoter genes in postmitotic neural rat tissue. Epigenetics 2012; 7:421 - 8; http://dx.doi.org/10.4161/epi.19565; PMID: 22415013
- Shimoda N, Yamakoshi K, Miyake A, Takeda H. Identification of a gene required for de novo DNA methylation of the zebrafish no tail gene. Dev Dyn 2005; 233:1509 - 16; http://dx.doi.org/10.1002/dvdy.20455; PMID: 15937923
- Goll MG, Anderson R, Stainier DY, Spradling AC, Halpern ME. Transcriptional silencing and reactivation in transgenic zebrafish. Genetics 2009; 182:747 - 55; http://dx.doi.org/10.1534/genetics.109.102079; PMID: 19433629
- Rai K, Huggins IJ, James SR, Karpf AR, Jones DA, Cairns BR. DNA demethylation in zebrafish involves the coupling of a deaminase, a glycosylase, and gadd45. Cell 2008; 135:1201 - 12; http://dx.doi.org/10.1016/j.cell.2008.11.042; PMID: 19109892
- Xie W, Barr CL, Kim A, Yue F, Lee AY, Eubanks J, et al. Base-resolution analyses of sequence and parent-of-origin dependent DNA methylation in the mouse genome. Cell 2012; 148:816 - 31; http://dx.doi.org/10.1016/j.cell.2011.12.035; PMID: 22341451
- Wang J, Xia Y, Li L, Gong D, Yao Y, Luo H, et al. Double restriction-enzyme digestion improves the coverage and accuracy of genome-wide CpG methylation profiling by reduced representation bisulfite sequencing. BMC Genomics 2013; 14:11; http://dx.doi.org/10.1186/1471-2164-14-11; PMID: 23324053
- Kettleborough RN, Busch-Nentwich EM, Harvey SA, Dooley CM, de Bruijn E, van Eeden F, et al. A systematic genome-wide analysis of zebrafish protein-coding gene function. Nature 2013; 496:494 - 7; http://dx.doi.org/10.1038/nature11992; PMID: 23594742
- Howe K, Clark MD, Torroja CF, Torrance J, Berthelot C, Muffato M, et al. The zebrafish reference genome sequence and its relationship to the human genome. Nature 2013; 496:498 - 503; http://dx.doi.org/10.1038/nature12111; PMID: 23594743
- Saxonov S, Berg P, Brutlag DL. A genome-wide analysis of CpG dinucleotides in the human genome distinguishes two distinct classes of promoters. Proc Natl Acad Sci U S A 2006; 103:1412 - 7; http://dx.doi.org/10.1073/pnas.0510310103; PMID: 16432200
- Takai D, Jones PA. Comprehensive analysis of CpG islands in human chromosomes 21 and 22. Proc Natl Acad Sci U S A 2002; 99:3740 - 5; http://dx.doi.org/10.1073/pnas.052410099; PMID: 11891299
- Gardiner-Garden M, Frommer M. CpG islands in vertebrate genomes. J Mol Biol 1987; 196:261 - 82; http://dx.doi.org/10.1016/0022-2836(87)90689-9; PMID: 3656447
- Irizarry RA, Ladd-Acosta C, Wen B, Wu Z, Montano C, Onyango P, et al. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat Genet 2009; 41:178 - 86; http://dx.doi.org/10.1038/ng.298; PMID: 19151715
- Doi A, Park IH, Wen B, Murakami P, Aryee MJ, Irizarry R, et al. Differential methylation of tissue- and cancer-specific CpG island shores distinguishes human induced pluripotent stem cells, embryonic stem cells and fibroblasts. Nat Genet 2009; 41:1350 - 3; http://dx.doi.org/10.1038/ng.471; PMID: 19881528
- Laurent L, Wong E, Li G, Huynh T, Tsirigos A, Ong CT, et al. Dynamic changes in the human methylome during differentiation. Genome Res 2010; 20:320 - 31; http://dx.doi.org/10.1101/gr.101907.109; PMID: 20133333
- Shukla S, Kavak E, Gregory M, Imashimizu M, Shutinoski B, Kashlev M, et al. CTCF-promoted RNA polymerase II pausing links DNA methylation to splicing. Nature 2011; 479:74 - 9; http://dx.doi.org/10.1038/nature10442; PMID: 21964334
- Hahn MA, Wu X, Li AX, Hahn T, Pfeifer GP. Relationship between gene body DNA methylation and intragenic H3K9me3 and H3K36me3 chromatin marks. PLoS One 2011; 6:e18844; http://dx.doi.org/10.1371/journal.pone.0018844; PMID: 21526191
- Aran D, Toperoff G, Rosenberg M, Hellman A. Replication timing-related and gene body-specific methylation of active human genes. Hum Mol Genet 2011; 20:670 - 80; http://dx.doi.org/10.1093/hmg/ddq513; PMID: 21112978
- Lister R, Pelizzola M, Dowen RH, Hawkins RD, Hon G, Tonti-Filippini J, et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 2009; 462:315 - 22; http://dx.doi.org/10.1038/nature08514; PMID: 19829295
- Bird A, Tate P, Nan X, Campoy J, Meehan R, Cross S, et al. Studies of DNA methylation in animals. J Cell Sci Suppl 1995; 19:37 - 9; http://dx.doi.org/10.1242/jcs.1995.Supplement_19.5; PMID: 8655645
- Jjingo D, Conley AB, Yi SV, Lunyak VV, Jordan IK. On the presence and role of human gene-body DNA methylation. Oncotarget 2012; 3:462 - 74; PMID: 22577155
- Bogdanovic O, Fernandez-Miñán A, Tena JJ, de la Calle-Mustienes E, Hidalgo C, van Kruysbergen I, et al. Dynamics of enhancer chromatin signatures mark the transition from pluripotency to cell specification during embryogenesis. Genome Res 2012; 22:2043 - 53; http://dx.doi.org/10.1101/gr.134833.111; PMID: 22593555
- Marsman J, Horsfield JA. Long distance relationships: enhancer-promoter communication and dynamic gene transcription. Biochim Biophys Acta 2012; 1819:1217 - 27; http://dx.doi.org/10.1016/j.bbagrm.2012.10.008; PMID: 23124110
- Kriukienė E, Liutkevičiūtė Z, Klimašauskas S. 5-Hydroxymethylcytosine–the elusive epigenetic mark in mammalian DNA. Chem Soc Rev 2012; 41:6916 - 30; http://dx.doi.org/10.1039/c2cs35104h; PMID: 22842880
- Chatterjee A, Rodger EJ, Stockwell PA, Weeks RJ, Morison IM. Technical considerations for reduced representation bisulfite sequencing with multiplexed libraries. J Biomed Biotechnol 2012; 2012:741542; http://dx.doi.org/10.1155/2012/741542; PMID: 23193365
- Krueger F, Andrews SR. Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 2011; 27:1571 - 2; http://dx.doi.org/10.1093/bioinformatics/btr167; PMID: 21493656
- Akalin A, Kormaksson M, Li S, Garrett-Bakelman FE, Figueroa ME, Melnick A, et al. methylKit: a comprehensive R package for the analysis of genome-wide DNA methylation profiles. Genome Biol 2012; 13:R87; http://dx.doi.org/10.1186/gb-2012-13-10-r87; PMID: 23034086
- Xin Y, Chanrion B, O’Donnell AH, Milekic M, Costa R, Ge Y, et al. MethylomeDB: a database of DNA methylation profiles of the brain. Nucleic Acids Res 2012; 40:Database issue D1245 - 9; http://dx.doi.org/10.1093/nar/gkr1193; PMID: 22140101
- Smith ZD, Gu H, Bock C, Gnirke A, Meissner A. High-throughput bisulfite sequencing in mammalian genomes. Methods 2009; 48:226 - 32; http://dx.doi.org/10.1016/j.ymeth.2009.05.003; PMID: 19442738