Abstract
DNA methylation is a key epigenetic mechanism involved in the developmental regulation of gene expression. Alterations in DNA methylation are established contributors to inter-individual phenotypic variation and have been associated with disease susceptibility. The degree to which changes in loci-specific DNA methylation are under the influence of heritable and environmental factors is largely unknown. In this study, we quantitatively measured DNA methylation across the promoter regions of the dopamine receptor 4 gene (DRD4), the serotonin transporter gene (SLC6A4/SERT) and the X-linked monoamine oxidase A gene (MAOA) using DNA sampled at both ages 5 and 10 years in 46 MZ twin-pairs and 45 DZ twin-pairs (total n=182). Our data suggest that DNA methylation differences are apparent already in early childhood, even between genetically identical individuals, and that individual differences in methylation are not stable over time. Our longitudinal-developmental study suggests that environmental influences are important factors accounting for interindividual DNA methylation differences, and that these influences differ across the genome. The observation of dynamic changes in DNA methylation over time highlights the importance of longitudinal research designs for epigenetic research.
Introduction
Epigenetics refers to the reversible regulation of various genomic functions mediated principally through changes in DNA methylation and chromatin structure.Citation1 Epigenetic processes are essential for normal cellular development and tissue differentiation, and allow the long-term regulation of gene function through non-mutagenic mechanisms.Citation2 Unlike the DNA sequence which is stable and strongly conserved, epigenetic processes are developmentally dynamic, and are known to be influenced by numerous factors including the environment,Citation3 DNA sequence variationCitation4,Citation5 and stochastic events in the cell.Citation6
DNA methylation is the best understood epigenetic modification modulating transcriptional plasticity. The addition of a methyl group at CpG dinucleotides, over-represented in CpG-islands in the promoter regulatory regions of many genes, displaces the binding of transcription factors and attracts methylbinding proteins that instigate chromatin compaction and gene silencing.Citation1 In addition to regulating gene expression, DNA methylation plays a critical role in mediating X-chromosome inactivation in females,Citation7 the maintenance of genomic imprintingCitation8 and protecting the genome from pathogenic retroviral elements.Citation9 Because epigenetic processes are integral for cellular development and function, aberrant DNA methylation signatures are hypothesized to be involved in diverse human pathologiesCitation10 including cancer,Citation11 imprinting disordersCitation12 and a range of complex neuropsychiatric phenotypes such as psychosis,Citation13 depression,Citation14 drug addiction,Citation15 ADHDCitation16 and autism.Citation17
The factors influencing dynamic epigenetic processes are complex and not yet fully understood. Two questions have yet to be fully answered. First, to what extent is DNA methylation influenced by heritable versus environmental factors? Second, what is the stability of DNA methylation over time? A better understanding of the factors influencing variation in DNA methylation and their role in mediating DNA methylation dynamics is important. This will not only elucidate the role of these factors in producing DNA methylation differences that may underlie complex multifactorial disorders, but will also inform future epigenetic studies that undertake a cross-sectional approach.
The most powerful method to estimate the contribution of heritable and environmental factors to variation in a quantitative trait such as DNA methylation is to compare the degree of within-pair twin concordance between monozygotic (MZ) and dizygotic (DZ) twin pairs.Citation18 To date, only a few studies have used this approach to estimate the heritability of DNA methylation. One study reported significantly higher genome-wide epigenetic differences in buccal cells obtained from DZ co-twins compared to MZ co-twins, suggesting that epigenetic signatures may be heritable.Citation19 Another study detected high levels of heritability for a small proportion (23%) of CpG sites analysed in a genome-wide study,Citation20 although the majority of CpG sites showed little evidence of heritability. A number of other studies have uncovered DNA methylation differences between inbred animalsCitation21,Citation22 and between MZ twins,Citation19,Citation20,Citation23–Citation25 suggesting that environmental factors are likely to influence DNA methylation.
The most powerful method to estimate the extent of DNA methylation change over time is to carry out an intra-individual longitudinal study tracking developmental changes in the same individuals. Despite evidence that dynamic changes in DNA methylation occur in conjunction with normal developmental processes and aging, or in response to environmental stimuli,Citation26 little empirical work has assessed intra-individual changes over time across specific regions of the genome. Two studies are of note. The first compared the epigenetic profiles of young and old MZ twins, finding that epigenetic discordance increased with age,Citation23 although this study was cross-sectional and did not assess developmental change in the same individuals. A second study assessed developmental changes in DNA methylation over time using singleton and family-based cohorts.Citation27 Interestingly, this study found evidence for familial clustering of DNA methylation change over time, suggesting that the maintenance of DNA methylation may be under some genetic control, at least at a global level.
Although findings from these recent studies have shed light on the complex nature of DNA methylation, to our knowledge no study has assessed the degree to which changes in loci-specific DNA methylation over time are under the influence of heritable, environmental or stochastic factors. Here we report findings from a longitudinal twin study involving DNA sampled at two time points from both MZ and DZ twin-pairs. We address three questions: (1) Are individual differences in DNA methylation stable over time? (2) Is DNA methylation in childhood familial and if so, is it heritable? (3) Is change in DNA methylation between ages 5 and 10 years heritable or environmentally regulated?
To date, most research on longitudinal changes in DNA methylation has focused on adults, with suggestion that the epigenome becomes more variable with age, a phenomenon described as ‘epigenetic drift’.Citation23 Previous work from our group highlighted significant DNA methylation differences within MZ-twin pairs detectable even in very young children.Citation24 In this study we therefore focus on epigenetic changes occurring during the first decade of life. The epigenome is particularly labile during early development, especially in utero and through childhood when the complex patterns of DNA methylation and histone modifications required for normal tissue differentiation and development are being established.Citation3 As epigenetic patterns are inherited mitotically in somatic cells, they provide a possible mechanism by which the effects of external environmental factors at specific stages in development can be propagated through development, producing long-term phenotypic changes.
In this study we assess the extent to which heritable and environmental factors contribute to individual differences and developmental changes in DNA methylation at specific regions of the genome during childhood. We focus on DNA methylation across the promoter regions of three key genes studied widely in the field of biological psychiatry: the dopamine receptor 4 gene (DRD4), the serotonin transporter gene (SLC6A4, also referred to as SERT) and the monoamine oxidase A gene (MAOA). Genetic variation in all three genes has been implicated in the etiology of psychiatric disorders including attention deficit hyperactivity disorder (DRD4), depression (SERT) and antisocial behavior (MAOA).Citation28–Citation30 However, these genetic associations are inconsistent and are sometimes only present in the context of environmental influences, suggesting that risk may be mediated by gene-environment interactions.Citation31–Citation33 Since epigenetic factors are known to operate at the interface between ‘nature’ and ‘nurture’, we selected these genes as the initial focus for our longitudinal twin analyses of DNA methylation.
Results
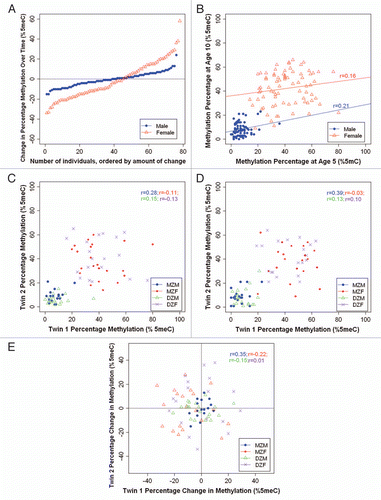
The average DNA methylation levels across DRD4, SERT and MAOA amplicons are described in , with DNA methylation at individual CpG units shown in Supplementary Figure 2. As expected, there were no significant group differences in average DNA methylation level in the genes investigated (DRD4, SERT and MAOA) between MZ and DZ twins, between the two time points (ages 5 and 10 years), or between sexes (except for MAOA). The significant sex differences in MAOA DNA methylation at ages 5 (F(1,124) = 344.81, p < 0.001 for all twins; F(1,125) = 151.15, p < 0.001 for MZ twins; and F(1,124) = 195.42, p < 0.001 for DZ twins) and 10 years (F(1,123) = 392.96, p < 0.001 for all twins; F(1,225) = 104.07, p < 0.001 for MZ twins; and F(1,225) = 94.51, p < 0.001 for DZ twins) are expected and can be explained by the allele-specific silencing of one X chromosome by X-inactivation in females.Citation34 Next, we review the stability and heritability of DNA methylation for each gene, in turn.
DRD4.
Stability. We observed a wide range of changes in average DRD4 DNA methylation between ages 5–10 years (). While DNA methylation across the DRD4 amplicon increased in some children (negative change value), it decreased in other children (positive change value) over time. The methylation change ranged from −19% to +21%; 43% of children (n = 68) showed a change of at least 5% and 15% of children (n = 24) showed a change of at least 10% in DRD4 DNA methylation between age 5–10 years. Interestingly, despite these mean level changes, we observed significant inter-individual stability in DRD4 DNA methylation from age 5 to age 10 years (r = 0.43; p < 0.001) ().
Heritability. and D show the average DRD4 DNA methylation percentage of each twin versus his/her co-twin at ages 5 and 10 years, respectively. We observed strong twin correlations for DRD4 DNA methylation. At age 5, the within-pair MZ correlation was 0.61 (p < 0.001) and the within-pair DZ correlation was 0.43 (p < 0.001). The MZ and DZ twin correlations did not differ significantly from each other (Z = 1.10; p = 0.27). At age 10, the within-pair MZ correlation was 0.67 (p < 0.001) and the within-pair DZ correlation was 0.64 (p < 0.001), and did not differ significantly from each other (Z = 0.22; p = 0.82). shows the intraindividual change in DRD4 methylation from age 5–10 years for each twin versus his/her co-twin. We observed strong twin correlations for change in DRD4 DNA methylation over time. Interestingly, the within-pair correlation in the change in DRD4 DNA methylation was similar among MZ and DZ twins (r = 0.52, p < 0.001 and r = 0.45, p = 0.003, respectively). These results suggest that the familial correlations in DRD4 methylation change are attributable to environmental factors that are shared by children growing up in the same family, and are not necessarily heritable.
SERT.
Stability. We observed a wide range of changes in average SERT DNA methylation between ages 5–10 years (). The DNA methylation change ranged from −41% to +23%, with 32% of children (n = 50) showing a change of at least 5% and 14% of children (n = 22) showing a change of at least 10% in SERT DNA methylation. Moreover, we observed no significant inter-individual stability in SERT DNA methylation over time (r = 0.046; p = 0.539) ().
Heritability. and D show the average SERT DNA methylation percentage of each twin versus his/her co-twin at ages 5 and 10 years, respectively. Twin correlations for SERT DNA methylation were small to medium in magnitude. At age 5, the within-pair MZ correlation was 0.21 (p = 0.165) and the within-pair DZ correlation was 0.34 (p = 0.023). The MZ and DZ twin correlations did not differ significantly from each other (Z = −0.64; p = 0.52). At age 10, the within-pair MZ correlation was 0.35 (p = 0.02) and the within-pair DZ correlation was 0.24 (p = 0.139), and did not differ significantly from each other (Z = 0.54; p = 0.59). These relatively low correlations suggest that variation in SERT DNA methylation is mostly attributable to unique environmental factors experienced by each child in the family, not to shared environmental events or to heritable factors. shows the intraindividual change in SERT DNA methylation from age 5–10 years for each twin versus his/her co-twin. We observed low and similar within-pair correlations in SERT DNA methylation change among MZ and DZ twins (r = 0.22, p = 0.167 and r = 0.15, p = 0.385, respectively). This suggests that variation in SERT DNA methylation change amongst children is attributable to unique environmental factors experienced by each child in the family and is not heritable.
MAOA.
As expected, because most genes on one X-chromosome are epigenetically silenced by X-chromosome inactivation,Citation34 we observed significant sex differences in DNA methylation across the MAOA amplicon, with males showing very low levels of average methylation and little inter-individual variability (7.6% ± 5.5% at age 5; 8.3% ± 6.3% at age 10) compared to females who had much higher and more variable levels of MAOA DNA methylation (40.3% ± 14.7% at age 5; 41.6% ± 13.9% at age 10). As such, we carried out all analyses separately for females and males. Because we stratified the sample by sex, our sample size is small and does not permit meaningful statistical comparisons between MZ and DZ twins. However, the pattern of results merits note.
Stability. We observed a wide range of changes in average MAOA DNA methylation between ages 5–10 years (). For male children, between ages 5–10 years, MAOA DNA methylation change ranged from −15% to +24%; 51% of children (n = 39) showed a change of at least 5 and 18% of children (n = 14) showed a change of at least 10%. For female children, between ages 5–10 years, MAOA DNA methylation change ranged from −34% to +58%, with 74% of children (n = 58) showing a change of at least 10 and 32% of children (n = 25) showing a change of at least 20%. In addition, we observed low interindividual stability in MAOA DNA methylation over time, for both males (r = 0.21; p = 0.162) and females (r = 0.16; p = 0.139) (), suggesting that children do not maintain their rank order of MAOA DNA methylation over time.
Heritability. and D show the average MAOA DNA methylation percentage of each twin versus his/her co-twin at ages 5 and 10 years, respectively. For males, we observed weak-to-modest twin correlations for MAOA DNA methylation. At age 5, the within-pair MZ correlation was 0.28 (p = 0.254) and the within-pair DZ correlation was 0.15 (p = 0.507). At age 10, the within-pair MZ correlation was 0.39 (p = 0.088) and the within-pair DZ correlation was 0.13 (p = 0.558). It is of interest that the MZ correlations are about two times the magnitude of the DZ correlations, suggestive of a heritable contribution to individual differences in MAOA DNA methylation among male children. shows the intraindividual change in MAOA DNA methylation from age 5–10 years for each twin versus his co-twin. The within-pair MZ correlation was 0.35 (p = 0.164) and the within-pair DZ correlation was −0.15 (p = 0.525). These correlations suggest that while MZ twins are changing in the same direction as their co-twins over time, DZ twins are actually changing in distinct directions over time. For females, we observed very low twin correlations for MAOA DNA methylation at ages 5 and 10 years (rs = −0.11 and −0.03 for MZ twins, and rs = −0.13 and 0.1 for DZ twins, respectively), as well as for intraindividual change from age 5–10 years (r = −0.22 for MZ twins and r = 0.01 for DZ twins), suggesting very little heritability and no common environmental influences on MAOA DNA methylation.
Discussion
DNA methylation is an important epigenetic mechanism operating at the interface between the genome and the environment to regulate phenotypic plasticity. In this study, we assessed the contribution of heritable and environmental factors to variation in DNA methylation across two ages (5 and 10 years) during childhood development. Quantitative DNA methylation profiling across the promoter/regulatory regions of three neuropsychiatric candidate genes (DRD4, SERT and MAOA) was performed in DNA samples from MZ and DZ twins obtained at two time points. In all three of the analysed regions, we observed high levels of MZ twin discordance in DNA methylation and dynamic changes in individual DNA methylation between ages 5 and 10 years that could largely be attributed to environmental influences.
For DRD4, we observed significant familiality for average DNA methylation with little evidence of heritability when comparing MZ- and DZ-twin concordance rates. These data suggest that variation in DRD4 DNA methylation is mostly attributable to environmental factors that are shared among children growing up in the same family. For SERT, we observed low levels of familiality for average DNA methylation and little evidence of heritability. This suggests that variation in SERT DNA methylation is primarily attributable to unique environmental factors experienced by each child in the family, and not to shared events or heritable factors. Our finding is of interest since the expression of SERT is known to be responsive to unique external psychosocial factorsCitation35 and polymorphisms in the gene have been shown to moderate susceptibility for depression in the context of severe childhood stress.Citation32 It has been speculated that epigenetic processes may mediate such gene-environment interactions.Citation36 An alternative explanation is that random stochastic epigenetic changes, unique to each child, influence DNA methylation in the SERT promoter. DNA methylation is known to be influenced by stochastic events; experiments tracking the inheritance of epigenetic marks through generations of genetically-identical cells in tissue-culture, for example, have indicated that there is considerable infidelity in the maintenance of DNA methylation patterns in mammalian cells, and that de novo methylation events are fairly common during mitosis.Citation6,Citation37
For MAOA we observed a more complex pattern of results, with noticeable sex differences in twin concordance at both ages and for change over time. Like most genes on the X-chromosome, MAOA demonstrates epigenetically mediated allele-specific expression in females resulting from X-chromosome inactivation.Citation38 X-chromosome inactivation silences genes on one X-chromosome to ensure dosage compensation with males via a process involving hypermethylation of CpG islands. X-inactivation in any given cell is typically random, and is maintained once established so that the inactivated allele is transcriptionally silenced for the lifetime of that cell.Citation39 Interestingly, several reports highlight considerable variability in the degree of inactivation of several loci on the X-chromosome in females, including MAOA.Citation38,Citation40,Citation41 Our data showing high between-individual variation in female MAOA DNA methylation concur with these observations and indicate variably incomplete inactivation at this locus. Another contributor to interindividual variation in allelic expression across the X-chromosome in females is skewed X-inactivation, where either the maternally- or paternally-inherited X-chromosome is preferentially silenced.Citation42 In the normal population of females without a family history of X-linked disorders, 5–20% of women have constitutional skewing of X-inactivation.Citation43 Mounting evidence indicates that the degree and direction of X-chromosome skewing is highly variable between individuals, with changes occurring over timeCitation44 and considerable variation reported between female MZ twins.Citation45
It is thus not surprising that, given the stochastic nature of monoallelic expression on the X-chromosome and evidence that X-inactivation can be randomly skewed, we see considerable variation regardless of zygosity within female twin-pairs. It is interesting that in males, but not females, MAOA DNA methylation appears to be moderately influenced by heritable factors; by definition males inherit their X-chromosome maternally with MZ twin-pairs always sharing the same X-chromosome. This is not the case for DZ male twin-pairs, who could variably inherit either of their mothers' X-chromosomes. Epigenetic marks can appear heritable either if they are directly influenced by the DNA sequence, a phenomenon supported by recent reports of widespread allele-specific DNA methylation across the genome,Citation4,Citation5 or more controversially, if epigenetic information can be transmitted across generations through meiosis. The latter is supported by a limited but growing body of evidence.Citation22,Citation46–Citation48 It is thus plausible that DNA sequence variation or inherited epigenetic marks on the X-chromosome influence DNA methylation levels at this locus in males.
Our findings have a number of implications for understanding the factors influencing epigenetic variation. First, the findings document that there are DNA methylation differences between genetically identical individuals (i.e., MZ twins). This concurs with findings from previous twin studies of DNA methylationCitation19,Citation20,Citation23–Citation25 and highlights the potential role of variable DNA methylation in explaining non-complete phenotypic concordance between genetically identical individuals. Second, the findings suggest that environmental influences (both shared and non-shared) are important factors accounting for interindividual DNA methylation differences. This supports the notion that DNA methylation may act as a biological index of environmental influence as suggested by both animalCitation3,Citation49 and human studies.Citation26 Third, the findings suggest that DNA methylation differences that are predominantly attributable to environmental factors are apparent already in early childhood. This is an important observation—although previous animal studies have shown that environmental factors experienced early in life can lead to long-lasting phenotypic change underlined by alteration in DNA methylation,Citation50 our study is the first to show that variation in DNA methylation in specific genomic regions during childhood is attributable to non-heritable factors. Third, our findings suggest that there is high variability in DNA methylation profiles across different regions and that change in DNA methylation over time can be influenced by a range of shared and non-shared environmental factors, depending on genomic location. This has implications for epigenetic studies that undertake a genome-wide approach as it may be difficult to draw conclusions on a global scale. Finally, the findings suggest that cross-sectional studies of DNA methylation may be problematic. Our observation of changes in individual DNA methylation between ages 5 and 10 years adds to the limited body of evidence supporting the notion that epigenetic changes are developmentally dynamic. A previous study used DNA samples from the same individuals to assess changes in DNA methylation over time,Citation27 although it did not use a genetically-informative twin design. Our findings highlight the importance of standardising sample collection and the care required in interpreting epigenetic findings in cross-sectional designs.
Our study has several strengths. First, to our knowledge, the study is unique in combining the classical twin study with a longitudinal design in order to test hypotheses about heritable and environmental contributions to DNA methylation. Second, by undertaking a loci-specific approach, we were able to perform an extensive investigation of DNA methylation across specific genomic regions. We utilised a highly sensitive and reliable method to detect levels of DNA methylation across the promoter/regulatory regions of three genes previously implicated in neuropsychiatric disorders (DRD4, SERT and MAOA); the Sequenom EpiTYPER system is a highly accurate method for quantifying small differences in DNA methylation,Citation51 and each of the assays utilised in this study are highly reproducible across duplicate samples.Citation52 Third, our sample size of 46 MZ twin-pairs and 45 DZ twin-pairs, assessed at two time-points, is larger than other twin studies of DNA methylation to dateCitation19,Citation20 and represents the most thorough twin study of epigenetic variation across specific regions of the genome.
Our study also has limitations. First, our focus on three specific genomic regions means we are unable to extrapolate our findings to other regions of the genome. Our data highlight a high level of heterogeneity across different genomic regions, but future studies will need to employ genome-wide profiling methods to establish the extent of this heterogeneity. Whilst genome-wide profiling of DNA methylation would be an extremely valuable approach, current approaches to perform such analyses are either economically restrictive for the number of samples included in our study or preclude the detailed investigation of numerous CpG sites across any given gene promoter (e.g., the Illumina Infinium 27K methylation assay only assays 1 or 2 CpG sites per gene). Using a highly quantitative loci-specific approach, we were able to perform an extensive investigation of DNA methylation across specific genomic regions, giving a level of detail not achievable using current genome-wide assays. Second, we studied genomic DNA from buccal cells and it is not known whether the findings will generalize to genomic DNA from other tissue sources, such as blood or brain, given the tissue-specific nature of the epigenome.Citation53–Citation55 Third, it is possible that the DNA samples taken at different ages were derived from different cellular populations, resulting in epigenetic differences across samples. Fourth, the present study was designed to test hypotheses about heritable, environmental and developmental influences on DNA methylation during the course of childhood development. Our design does not permit us to link epigenetic variation to specific environmental experiences. Finally, although this study is the largest twin study of DNA methylation to date, our sample size is relatively small for a twin study and this has precluded the use of quantitative twin model fitting.
This is the first study to examine longitudinal changes in DNA methylation in both MZ- and DZ-twins across specific genomic regions during the course of childhood development. High levels of MZ twin differences in DNA methylation and changes in DNA methylation over time were observed at the promoter/regulatory regions of the DRD4, SERT and MAOA genes. Most of these dynamic changes in DNA methylation were attributable to environmental influences and were not heritable. These observations highlight the complex nature of epigenetic variation across the genome during the first decade of life, but also emphasize the utility of DNA methylation as a biomarker of environmental influences. Research investigating dynamic changes in the epigenome is in its infancy, but is one of the fastest growing fields in biological and medical research and supported by rapid technological and methodological developments. Understanding the contribution of heritable and environmental factors to epigenetic processes may facilitate the development of better molecular tools that improve the diagnosis, prognosis and treatment of common complex diseases.
Materials and Methods
Subjects.
Participants were children enrolled in the Environmental Risk (E-Risk) Longitudinal Twin Study, which has been described in detail elsewhere.Citation56 In brief, E-Risk investigates how genetic and environmental factors shape children's development. The E-Risk sampling frame comprised two consecutive birth cohorts (1994 and 1995) in the Twins' Early Development Study, a birth register of twins born in England and Wales.Citation57 The study follows an epidemiological sample of families with young twins who were interviewed in the home when the twins were aged 5, 7, 10 and 12 years. We collected DNA from the children when they were 5 years old and again when they were 10 years old. For the present DNA methylation analysis, we randomly selected 46 Caucasian MZ twin-pairs (23 male pairs, 23 female pairs) and 45 Caucasian DZ twin pairs (23 male pairs, 22 female pairs), totaling 182 children. All DNA was extracted from buccal cells using an established method that yields high molecular weight genomic DNA.Citation58 All DNA samples were tested for degradation and purity using spectrophotometry and gel electrophoresis; any degraded or impure samples were excluded from analysis.
Ethics statement.
Ethical approval was granted by the Joint South London and Maudsley and the Institute of Psychiatry Research Ethics Committee.
DNA methylation analysis.
We performed DNA methylation analysis on three candidate genes: DRD4, SERT and MAOA. Assays were designed using the online Sequenom EpiDesigner software (see Web Resources). The oligo sequences and the location of the amplicons across which DNA methylation was assessed in this study are given in Supplementary Table 1 and Supplementary Figure 1.
Genomic DNA (375 ng) was treated with sodium bisulfite using the EZ-96 DNA Methylation Kit (Zymo Research, CA, USA) following the manufacturers' standard protocol. Bisulfite-PCR amplification was conducted using Hot Star Taq DNA polymerase (Qiagen, UK) and cycling conditions of 45 cycles with an annealing temperature of 56°C for all amplicons. Subsequent to bisulfite-PCR amplification, DNA methylation analysis was conducted using the Sequenom EpiTYPER system (Sequenom Inc., CA, USA) as described previously.Citation51 The Sequenom EpiTYPER system is a highly reliable and quantitative technology for determining the density of methylated cytosines across specific genomic loci.Citation51 It utilises base-specific cleavage followed by MALDI-TOF mass spectrometry in which the size ratio of the cleaved products provides quantitative methylation estimates for CpG sites within a target region. We observed a strong correlation (≥0.95) between experimental duplicates for the three assays utilized in this study, suggesting that our data are highly reliable.Citation52 Positive controls, including both artificially methylated and artificially unmethylated samples were included in all experimental procedures to ensure unambiguous PCR amplification of bisulfite-treated samples. Finally, to avoid any potential observation bias, all samples were randomized in the experiment and processed blind to sample identification.
Statistical analysis.
The level of DNA methylation of DRD4, SERT and MAOA was examined using the Sequenom EpiTYPER system that provides a quantitative methylation score (the higher the methylation score, the more methylated is the DNA template). DNA methylation scores are expressed as percentage methylation (% 5 meC). Data generated from the EpiTYPER software were treated with stringent quality control analysis where CpG units with low calling rates and individuals with a high number of missing CpG units were removed. Each amplicon was designed to span numerous CpG sites (18 CpG units (30 CpG sites) for DRD4, 15 CpG units (27 CpG sites) for SERT and 6 CpG units (7 CpG sites) for MAOA) across the 5′ promoter region of each gene (see Suppl. Fig. 1 and Suppl. Table 1 for location of amplicons). Outside of specific transcription-factor binding sites, the proportion of methylated cytosines across a region, rather than at any specific position, is thought to control the transcriptional potential of the gene by attracting methyl-binding proteins and altering chromatin conformation.Citation37 Because we had no a priori reason to focus our analyses on specific CpGs, we calculated the average DNA methylation level for each genomic region by taking the mean of the multiple CpG sites in the amplicon. To ensure no bias in twin analyses or longitudinal analyses, we only included CpG sites that were reliably quantified in both twins within a pair and in both twins across time. The total number of twin-pairs included in the analyses for each gene can be seen in Supplementary Table 1.
Results specific to each candidate gene are presented in three parts. First, to assess stability of individual differences, we calculated correlations between children's average DNA methylation at age 5 years and their average DNA methylation at age 10 years. Second, to assess heritable and environmental influences on variation in DNA methylation, we calculated correlations between children's DNA methylation, separately within MZ twin pairs and within DZ twin pairs at ages 5 and 10 years, respectively. Third, to assess heritable and environmental influences on change in DNA methylation across childhood, we first calculated intraindividual change scores for each child (i.e., age 5–age 10) and then calculated correlations between change in children's DNA methylation within MZ twin pairs and DZ twin pairs.
Figures and Tables
Figure 1 Average DNA methylation level in DRD4, SERT and MAOA amplicons at ages 5 and 10 years. * Significant differences (p < 0.001) in average MAOA DNA methylation level between male and female children. MZ, monozygotic; DZ, dizygotic; MZM, monozygotic male; MZF, monozygotic female; DZM, dizygotic male; DZF, dizygotic female.
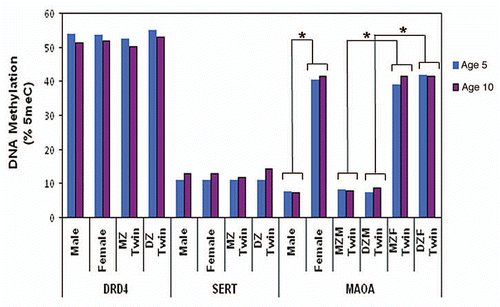
Figure 2 Longitudinal analysis of DRD4 DNA methylation in MZ and DZ twins. (A) Individual changes in average DRD4 DNA methylation between ages 5 and 10 years. (B) Inter-individual stability correlations for DRD4 DNA methylation, between ages 5 and 10 years. (C) MZ and DZ twin correlations for average DRD4 DNA methylation at age 5. (D) MZ and DZ twin correlations for average DRD4 DNA methylation at age 10. (E) MZ and DZ twin correlations for intraindividual change in DRD4 DNA methylation from age 5 to age 10 years.
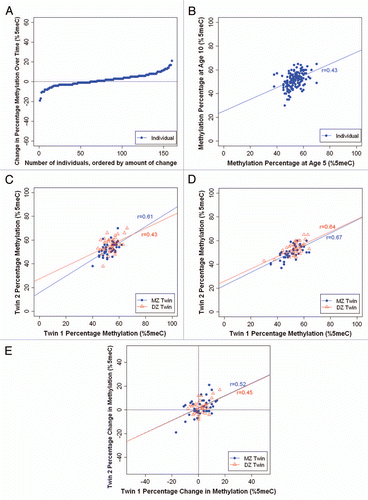
Figure 3 Longitudinal analysis of SERT DNA methylation in MZ and DZ twins. (A) Individual changes in average SERT DNA methylation between ages 5 and 10 years. (B) Inter-individual stability correlations for SERT DNA methylation, between ages 5 and 10 years. (C) MZ and DZ twin correlations for average SERT DNA methylation at age 5. (D) MZ and DZ twin correlations for average SERT DNA methylation at age 10. (E) MZ and DZ twin correlations for intraindividual change in SERT DNA methylation from age 5 to age 10 years.
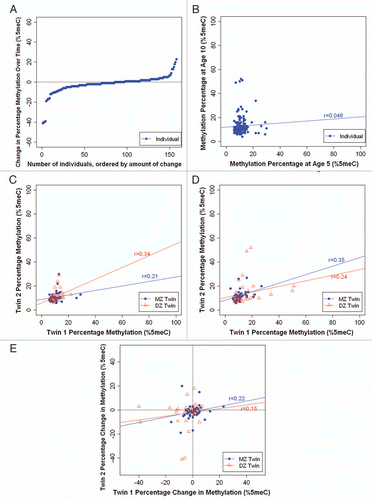
Figure 4 Longitudinal analysis of MAOA DNA methylation in MZ and DZ twins, stratified by sex. (A) Individual changes in average MAOA DNA methylation between ages 5 and 10 years. (B) Inter-individual stability correlations for MAOA DNA methylation, between ages 5 and 10 years. (C) MZ and DZ twin correlations for average MAOA DNA methylation at age 5. (D) MZ and DZ twin correlations for average MAOA DNA methylation at age 10. (E) MZ and DZ twin correlations for intraindividual change in MAOA DNA methylation from age 5 to age 10 years. MZM, monozygotic male; MZF, monozygotic female; DZM, dizygotic male; DZF, dizygotic female.
Additional material
Download Zip (45.2 KB)Acknowledgements
This research received support from UK Medical Research Council grants G9806489, G0100527 and G0601483, US-NIH grants MH077874 and HD061298, and the London University Central Research Fund. Avshalom Caspi is a Royal Society-Wolfson Merit Award holder. Chloe Wong is a PhD student funded by the UK Medical Research Council. The authors declare no conflicts of interest.
References
- Jaenisch R, Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet 2003; 33:245 - 254
- Henikoff S, Matzke MA. Exploring and explaining epigenetic effects. Trends Genet 1997; 13:293 - 295
- Dolinoy DC, Weidman JR, Jirtle RL. Epigenetic gene regulation: linking early developmental environment to adult disease. Reprod Toxicol 2007; 23:297 - 307
- Kerkel K, Spadola A, Yuan E, Kosek J, Jiang L, Hod E, et al. Genomic surveys by methylation-sensitive SNP analysis identify sequence-dependent allele-specific DNA methylation. Nat Genet 2008; 40:904 - 908
- Schalkwyk LC, Meaburn EL, Smith R, Dempster EL, Jeffries AR, Davies MN, et al. Allelic Skewing of DNA Methylation Is Widespread across the Genome. Am J Hum Genet 2010; 86:196 - 212
- Ushijima T, Watanabe N, Okochi E, Kaneda A, Sugimura T, Miyamoto K. Fidelity of the methylation pattern and its variation in the genome. Genome Res 2003; 13:868 - 874
- Chow JC, Yen Z, Ziesche SM, Brown CJ. Silencing of the mammalian X chromosome. Annu Rev Genomics Hum Genet 2005; 6:69 - 92
- Delaval K, Feil R. Epigenetic regulation of mammalian genomic imprinting. Curr Opin Genet Dev 2004; 14:188 - 195
- Jahner D, Stuhlmann H, Stewart CL, Harbers K, Lohler J, Simon I, et al. De novo methylation and expression of retroviral genomes during mouse embryogenesis. Nature 1982; 298:623 - 628
- Hatchwell E, Greally JM. The potential role of epigenomic dysregulation in complex human disease. Trends Genet 2007; 23:588 - 595
- Jones PA, Baylin SB. The epigenomics of cancer. Cell 2007; 128:683 - 692
- Feinberg AP. Phenotypic plasticity and the epigenetics of human disease. Nature 2007; 447:433 - 440
- Mill J, Tang T, Kaminsky Z, Khare T, Yazdanpanah S, Bouchard L, et al. Epigenomic profiling reveals DNAmethylation changes associated with major psychosis. Am J Hum Genet 2008; 82:696 - 711
- Mill J, Petronis A. Molecular studies of major depressive disorder: the epigenetic perspective. Mol Psychiatry 2007; 12:799 - 814
- Renthal W, Nestler EJ. Epigenetic mechanisms in drug addiction. Trends Mol Med 2008; 14:341 - 350
- Mill J, Petronis A. Pre- and peri-natal environmental risks for attention-deficit hyperactivity disorder (ADHD): the potential role of epigenetic processes in mediating susceptibility. J Child Psychol Psychiatry 2008; 49:1020 - 1030
- Schanen NC. Epigenetics of autism spectrum disorders. Hum Mol Genet 2006; 15:138 - 150
- Plomin R, DeFries JC, McClearn GE, McGuffin P. Behavioral Genetics 2008; New York Worth Publishers
- Kaminsky ZA, Tang T, Wang SC, Ptak C, Oh GH, Wong AH, et al. DNA methylation profiles in monozygotic and dizygotic twins. Nat Genet 2009; 41:240 - 245
- Boks MP, Derks EM, Weisenberger DJ, Strengman E, Janson E, Sommer IE, et al. The relationship of DNA methylation with age, gender and genotype in twins and healthy controls. PLoS One 2009; 4:6767
- Duhl DM, Vrieling H, Miller KA, Wolff GL, Barsh GS. Neomorphic agouti mutations in obese yellow mice. Nat Genet 1994; 8:59 - 65
- Morgan HD, Sutherland HG, Martin DI, Whitelaw E. Epigenetic inheritance at the agouti locus in the mouse. Nat Genet 1999; 23:314 - 318
- Fraga MF, Ballestar E, Paz MF, Ropero S, Setien F, Ballestar ML, et al. Epigenetic differences arise during the lifetime of monozygotic twins. Proc Natl Acad Sci USA 2005; 102:10604 - 10609
- Mill J, Dempster E, Caspi A, Williams B, Moffitt T, Craig I. Evidence for monozygotic twin (MZ) discordance in methylation level at two CpG sites in the promoter region of the catechol-O-methyltransferase (COMT) gene. Am J Med Genet B Neuropsychiatr Genet 2006; 141:421 - 425
- Petronis A, Gottesman II, Kan P, Kennedy JL, Basile VS, Paterson AD, et al. Monozygotic twins exhibit numerous epigenetic differences: clues to twin discordance?. Schizophr Bull 2003; 29:169 - 178
- Heijmans BT, Tobi EW, Stein AD, Putter H, Blauw GJ, Susser ES, et al. Persistent epigenetic differences associated with prenatal exposure to famine in humans. Proc Natl Acad Sci USA 2008; 105:17046 - 17049
- Bjornsson HT, Sigurdsson MI, Fallin MD, Irizarry RA, Aspelund T, Cui H, et al. Intra-individual change over time in DNA methylation with familial clustering. Jama 2008; 299:2877 - 2883
- Craig IW. The importance of stress and genetic variation in human aggression. Bioessays 2007; 29:227 - 236
- Li D, Sham PC, Owen MJ, He L. Meta-analysis shows significant association between dopamine system genes and attention deficit hyperactivity disorder (ADHD). Hum Mol Genet 2006; 15:2276 - 2284
- Lotrich FE, Pollock BG. Meta-analysis of serotonin transporter polymorphisms and affective disorders. Psychiatr Genet 2004; 14:121 - 129
- Thapar A, Langley K, Asherson P, Gill M. Gene-environment interplay in attention-deficit hyperactivity disorder and the importance of a developmental perspective. Br J Psychiatry 2007; 190:1 - 3
- Caspi A, Sugden K, Moffitt TE, Taylor A, Craig IW, Harrington H, et al. Influence of life stress on depression: moderation by a polymorphism in the 5-HTT gene. Science 2003; 301:386 - 389
- Caspi A, McClay J, Moffitt TE, Mill J, Martin J, Craig IW, et al. Role of genotype in the cycle of violence in maltreated children. Science 2002; 297:851 - 854
- Plath K, Mlynarczyk-Evans S, Nusinow DA, Panning B. Xist RNA and the mechanism of X chromosome inactivation. Annu Rev Genet 2002; 36:233 - 278
- Caspi A, Hariri AR, Holmes A, Uher R, Moffitt TE. Genetic sensitivity to the environment: The case of the serotonin transporter gene and its implications for studying complex diseases and traits. Am J Psychiatry 2010; 167:509 - 527
- Docherty S, Mill J. Epigenetic mechanisms as mediators of environmental risks for psychiatric disorders. Psychiatry 2008; 7:500 - 506
- Riggs AD, Xiong Z, Wang L, LeBon JM. Methylation dynamics, epigenetic fidelity and X chromosome structure. Novartis Found Symp 1998; 214:214 - 225
- Hendriks RW, Chen ZY, Hinds H, Schuurman RK, Craig IW. An X chromosome inactivation assay based on differential methylation of a CpG island coupled to a VNTR polymorphism at the 5′ end of the monoamine oxidase A gene. Hum Mol Genet 1992; 1:187 - 194
- Avner P, Heard E. X-chromosome inactivation: counting, choice and initiation. Nat Rev Genet 2001; 2:59 - 67
- Carrel L, Willard HF. X-inactivation profile reveals extensive variability in X-linked gene expression in females. Nature 2005; 434:400 - 404
- Pinsonneault JK, Papp AC, Sadee W. Allelic mRNA expression of X-linked monoamine oxidase a (MAOA) in human brain: dissection of epigenetic and genetic factors. Hum Mol Genet 2006; 15:2636 - 2649
- Craig IW, Harper E, Loat CS. The genetic basis for sex differences in human behaviour: role of the sex chromosomes. Ann Hum Genet 2004; 68:269 - 284
- Belmont JW. Genetic control of X inactivation and processes leading to X-inactivation skewing. Am J Hum Genet 1996; 58:1101 - 1108
- Hatakeyama C, Anderson CL, Beever CL, Penaherrera MS, Brown CJ, Robinson WP. The dynamics of X-inactivation skewing as women age. Clin Genet 2004; 66:327 - 332
- Rosa A, Picchioni MM, Kalidindi S, Loat CS, Knight J, Toulopoulou T, et al. Differential methylation of the X-chromosome is a possible source of discordance for bipolar disorder female monozygotic twins. Am J Med Genet B Neuropsychiatr Genet 2008; 147:459 - 462
- Anway MD, Skinner MK. Epigenetic transgenerational actions of endocrine disruptors. Endocrinology 2006; 147:43 - 49
- Pembrey ME, Bygren LO, Kaati G, Edvinsson S, Northstone K, Sjostrom M, et al. Sex-specific, maleline transgenerational responses in humans. Eur J Hum Genet 2006; 14:159 - 166
- Rakyan VK, Chong S, Champ ME, Cuthbert PC, Morgan HD, Luu KV, et al. Transgenerational inheritance of epigenetic states at the murine Axin(Fu) allele occurs after maternal and paternal transmission. Proc Natl Acad Sci USA 2003; 100:2538 - 2543
- Weaver IC, Cervoni N, Champagne FA, D'Alessio AC, Sharma S, Seckl JR, et al. Epigenetic programming by maternal behavior. Nat Neurosci 2004; 7:847 - 854
- Cooney CA, Dave AA, Wolff GL. Maternal methyl supplements in mice affect epigenetic variation and DNA methylation of offspring. J Nutr 2002; 132:2393 - 2400
- Coolen MW, Statham AL, Gardiner-Garden M, Clark SJ. Genomic profiling of CpG methylation and allelic specificity using quantitative high-throughput mass spectrometry: critical evaluation and improvements. Nucleic Acids Res 2007; 35:119
- Docherty SJ, Davis OS, Haworth CM, Plomin R, Mill J. Bisulfite-based epityping on pooled genomic DNA provides an accurate estimate of average group DNA methylation. Epigenetics Chromatin 2009; 2:3
- Illingworth R, Kerr A, Desousa D, Jorgensen H, Ellis P, Stalker J, et al. A novel CpG island set identifies tissuespecific methylation at developmental gene loci. PLoS Biol 2008; 6:22
- Rakyan VK, Down TA, Thorne NP, Flicek P, Kulesha E, Graf S, et al. An integrated resource for genomewide identification and analysis of human tissue-specific differentially methylated regions (tDMRs). Genome Res 2008; 18:1518 - 1529
- Kitamura E, Igarashi J, Morohashi A, Hida N, Oinuma T, Nemoto N, et al. Analysis of tissue-specific differentially methylated regions (TDMs) in humans. Genomics 2007; 89:326 - 337
- Moffitt TE. Teen-aged mothers in contemporary Britain. J Child Psychol Psychiatry 2002; 43:727 - 742
- Trouton A, Spinath FM, Plomin R. Twins early development study (TEDS): a multivariate, longitudinal genetic investigation of language, cognition and behavior problems in childhood. Twin Res 2002; 5:444 - 448
- Freeman B, Smith N, Curtis C, Huckett L, Mill J, Craig IW. DNA from buccal swabs recruited by mail: evaluation of storage effects on long-term stability and suitability for multiplex polymerase chain reaction genotyping. Behav Genet 2003; 33:67 - 72