Abstract
The methylated DNA immunoprecipitation method (MeDIP) is a genome-wide, high-resolution approach that detects DNA methylation with oligonucleotide tiling arrays or high throughput sequencing platforms. A simplified high-throughput MeDIP assay will enable translational research studies in clinics and populations, which will greatly enhance our understanding of the human methylome. We compared three commercial kits, MagMeDIP Kit TM (Diagenode), Methylated-DNA IP Kit (Zymo Research) and Methylamp™ Methylated DNA Capture Kit (Epigentek), in order to identify which one has better reliability and sensitivity for genomic DNA enrichment. Each kit was used to enrich two samples, one from fresh tissue and one from a cell line, with two different DNA amounts. The enrichment efficiency of each kit was evaluated by agarose gel band intensity after Nco I digestion and by reaction yield of methylated DNA. A successful enrichment is expected to have a 1:4 to 10:1 conversion ratio and a yield of 80% or higher. We also evaluated the hybridization efficiency to genome-wide methylation arrays in a separate cohort of tissue samples. We observed that the MagMeDIP kit had the highest yield for the two DNA amounts and for both the tissue and cell line samples, as well as for the positive control. In addition, the DNA was successfully enriched from a 1:4 to 10:1 ratio. Therefore, the MagMeDIP kit is a useful research tool that will enable clinical and public health genome-wide DNA methylation studies.
Introduction
A variety of platforms have emerged for genome-wide and whole-genome DNA methylation profiling.Citation1-Citation3 The large number of platforms available for methylation analysis can be divided into three large groups, according to how they modify the DNA before interrogating it with microarrays or next generation sequencing platforms: i) bisulfite converted DNA (bisulfite pyrosequencingCitation4 and BeadChip analysis);Citation5 ii) affinity assays that precipitate methylated DNA (MeDIPCitation6 and MBD)Citation7 and; iii) restriction enzyme methods that recognize methylated and unmethylated sequences (CHARM,Citation8 LUMA,Citation9 HELPCitation10). After the initial DNA enrichment or chemical modification step, several technologies are available for genome-wide analyses: i) array hybridization systems, such as the Illumina Infinium and GoldenGate systems, or oligonucleotide tiling arrays, such as the Nimblegen and Agilent CpG Islands plus Promoters arrays; ii) pyrosequencers and; iii) next generation sequencing platforms such as Illumina/Solexa, ABI/SOLiD, Roche454 and Helicos/Single molecule sequencing.Citation11 In contrast to tumor samples, cell lines can be evaluated systematically for methylation and expression by using demethylating agents plus HDAC inhibitors for the pharmacologic unmasking of genes silenced by methylation, followed by RNA microarray expression analysisCitation12,Citation13
The methylated DNA immunoprecipitation method (MeDIP) is a genome-wide, high-resolution approach to detect DNA methylation.Citation14 The method utilizes anti-methyl-cytosine antibody to immunoprecipitate DNA that contains highly methylated CpG sites. Briefly, genomic DNA is sheared by sonication to produce random fragments. The generation of small fragments is the key to guaranteeing efficient immunoprecipitation and the level of resolution that is necessary for further characterization. Sonicated DNA is subsequently immunocaptured with a monoclonal antibody specific for 5‑mC, purified, labeled and hybridized to oligonucleotide tiling arrays or used for high-throughput sequencing.Citation15 MeDIP is a fast and simple approach to determine DNA methylation on a genome-wide scale and compare DNA methylation patterns between two samples with diversely different DNA methylation status.Citation16
This powerful molecular biology assay has mostly being used in basic science research labs that have access to clinical samples with either microarrays or deep sequencing technologies.Citation17,Citation18 A simplified high-throughput MeDIP assay will enable translational research studies in clinics and populations, which will greatly enhance our understanding of the human methylome.
The aim of this study was to compare three commercial MeDIP protocols that can be used for high-throughput DNA enrichment: MagMeDIP KITTM (Diagenode), Methylated-DNA IP KIT (Zymo Research), and Methylamp™ Methylated DNA capture kit (Epigentek). Commercial MeDIP kits can increase the speed of translation of complex genome-wide assays from the bench to the clinic, and to population based studies.
Materials and Methods
Patients
Patients for this study were consented at hospitals in Spain, Chile and USA. Normal bronchial wash was obtained from patients consented in Boston University Medical School. Normal and cervical tumor tissue was collected from patients who visited the outpatient clinics of Doctor Hernán Henríquez Aravena (HHHA) tertiary care regional hospital in Temuco, Chile. OSCC tumor and normal oral cavity tissue samples were collected at Hospital Gregorio Marañón in Madrid, Spain. All participants signed a consent form that clearly explained the risks and benefits of the study. The study was approved by the Ethics Committee of each participating hospital, as well as by the Johns Hopkins Institutional Review Board.
DNA extraction: DNA was extracted from frozen tissue samples and H1299 cell lines that were digested with 1% SDS and 50 μg/mL proteinase K (Boehringer Mannheim) at 48°C overnight, followed by phenol/chloroform extraction and ethanol precipitation of DNA as previously described.Citation19
DNA sonication: Samples with two different amounts of DNA, 0.5 μg and 1 μg, were used as input DNA. The DNA was sheared using a water bath sonicator (Bioruptor UCD-200, Diagenode) at “LOW” power setting (alternating 5 min sonication and 2 min in ice) for a total sonication time of 15 min. Sonicated DNA was then analyzed on a 1.5% agarose gel to ensure that sonicated fragments had an optimal size of 200–1000 bp.
Methylated DNA Immunoprecipitation (MeDIP): We performed MeDIP on the same samples using three different commercial kits. For each sample we had two different starting DNA amounts: 0.5 μg and 1 μg. Every sample was processed in triplicate.
MagMeDIP KIT (Diagenode)
IP incubation mix (45 μL Water, 24 μL MagBuffer A, 6 μl MagBuffer B, 1.5 μl methylated DNA positive control, 1.5 μl unmethylated DNA negative control) was prepared in triplicate per sample. Sonicated DNA was added to two tubes: input DNA (10% of sonicated DNA) and Immunoprecipitated (IP) sample DNA. IP incubation mix was added to both tubes, heated at 95°C for three minutes and briefly centrifuged at 4°C. DNA was then stored at 4°C.
Bead wash buffer was prepared by diluting 1:5 the MagBuffer A (100 μl/IP). Then we resuspended the provided Magbeads and transferred the amount of Magbeads needed into a new tube (11 μl/sample). The tubes were placed on a magnet or centrifuge to eliminate the supernatant. Magbeads were washed twice with ice-cold Bead wash buffer, resuspended in Bead wash buffer and kept on ice. In a new tube, the provided antibody was used to prepare the Diluted Antibody mix 4°C for 4 h.
The MagWash buffers (1 and 2) and Magnetic Rack were placed on ice. The tubes were placed into the ice-cold Magnetic Rack and the buffer was discarded. The DNA Samples were washed three times with 100 μl ice-cold MagWash Buffer‑1 and ice-cold MagWash Buffer‑2. After the last wash, the last traces of Wash buffer were discarded. The bead pellets were kept on ice to proceed to the next step.
Input samples were centrifuged briefly and from this point on treated in parallel with IP samples. Complete DNA isolation buffer (DIB) was prepared adding 1 μL of Proteinase K per 100 μL of DIB. The tubes were removed from the Magnetic Rack complete DIB was added to each sample and incubated at 55°C for 15 min and then at 100°C for 15 min. The samples were then centrifuged at 14,000 rpm for 5 min at 4°C. The supernatants were transferred into new labeled tubes and stored at -20°C.
MethylAmp Methylated DNA Capture Kit
The first step of the IP procedure was to add 100 μl of antibody buffer to each well and then add the following antibodies: 1 μl normal mouse IgG to the negative control well and 0.5–1 μl anti-5-methylcytosine to every IP well. The IP procedure was done in triplicate for each sample. The strip wells were covered with parafilm M and incubated at room temperature for 60 min. Previously sonicated DNA was incubated at 95°C for 2 min with antibody buffer and immediately put it on ice. Five μl of the sonicated DNA solution was placed in a 0.5 mL vial as “input” DNA and put it on ice. The incubated antibody solution was removed and the strip wells were washed with wash buffer and with antibody buffer. 100 μl of the sonicated DNA solution was added to each well and covered with parafilm M to incubate at room temperature for 90–120 min on an orbital shaker (50–100 rpm). The supernatant was removed, the wells were washed 3 times and 60 μl of DNA release buffer containing 1 μl of proteinase K was added to each sample (including “input” vials). The wells were covered with strip caps and incubated at 65°C in a water bath for 60 min. After incubation 180 μl of 100% ethanol was added and the solution was transferred to a spin column, containing 100 μl of binding buffer, and centrifuged at 12,000 rpm for 20 sec. The column was washed with 90% ethanol twice. Subsequently, 20 μl of elution buffer was added and the column was centrifuged at 12,000 rpm for 20 sec. Input and IP DNA were then stored at –20°C.
Methylated-DNA IP KIT (Zymo Research)
DNA wash buffer was prepared by adding 24 mL of 100% ethanol to 6 mL DNA Wash Buffer concentrate. DNA wash buffer was added to protein A-sepharose slurry beads. The following reagents were then added to a Zymo-Spin IVM (ZS-IVM) column for each sample. Each sample was processed in triplicate: 250 μl MIP Buffer, 50 μl of Protein A-Sepharose Slurry and 4 μl Mouse Anti-5-Methylcytosine. The ZS-IVM column was capped tightly and the tube was inverted 2–4 times to mix the sample. The sample was then placed on a rocker at room temperature for 30 min. Input DNA samples were diluted and denatured in the DNA Denaturing Buffer to a final volume of 50 μl. The input DNA was denatured at 98°C for 5 min and immediately transferred to ice. The denatured DNA was added to the antibody/protein A mixture after Step 2 above was complete. The antibody/protein A/DNA mixture was incubated at 37°C for 0.5–2 h on a rotator or rocker. After breaking off the bottom tip of the ZS-IVM sample column, the column was placed into a collection tube and centrifuged at 800 x g (~3,000 rpm) for one minute. The flow through was discarded. The wash was repeated 3 times, discarding the flow through and saving the pellet in the column after each wash. The ZS-IVM column was transferred into a new collection tube. 400 μl of IP DNA Binding Buffer were added to the ZS-IVM column to completely re-suspend the pellet. After centrifuging at 10,000 x g (~13,000 rpm) for 30 sec the flow through was transferred into a Zymo-Spin™ IC (ZS‑IC) column inside a new collection tube and centrifuged at 10,000 x g for 30 sec. The flow through was discarded. 200 μl of DNA Wash Buffer were added to the ZS-IC column, the sample was centrifuged at 10,000 x g for 1 min, and the flow through was discarded. This wash was done twice. The column was placed into a new 1.5 mL microcentrifuge tube and 10 μl DNA Elution Buffer were placed directly to the column matrix, before centrifuging briefly at 10,000 x g to elute the DNA, which was subsequently stored at -20°C.
Comparison of enrichment efficiency
Enrichment efficiency was defined by agarose gel band intensity after Nco I digestion and by reaction yield. Control DNA was added to every sample at the beginning of each experiment for easy monitoring of the methylated DNA IP process. The Control DNA contains both in vitro methylated DNA and non-methylated DNA at a ratio of 1:4, respectively. Methylated DNA IP enrichment efficiency can be determined following PCR with Control Primers I (5′-GGTTAATGAATCGGCCAACGCGCG-3′) and II (5′-GAGGGAGCTTCCAGGGGGAAA-3′) and then Nco I digestion of the PCR products to differentiate methylated from non-methylated DNA template. A successful enrichment should invert the ratio from 1:4 to 10:1 or higher. The Control primers will generate a 350 bp PCR amplicon, which once digested with Nco I, will produce two 175 bp fragments for methylated pUC19 min and an intact 350 bp fragment for non-methylated pUC19. Reaction yield was determined by comparing the amount of DNA used at the beginning and at the end of each reaction.
Hybridization efficiency to promoter methylation microarrays
To compare the hybridization efficiency to promoter methylation microarrays of the kit that had the highest yield, MagMeDIP, we hybridized 14 samples (7 normal and 7 tumors) to genome-wide methylation arrays and measured the total number of significant methylation signals. Briefly, after methylated DNA enrichment with MagMeDIP and genome-wide amplification (WGA2, Sigma-Aldrich) the immunoprecipitated DNA fraction was labeled with Cy5 fluorophere and the input genomic DNA was labeled with Cy3 fluorophere. Labeled DNA from the enriched and the input pools were combined and hybridized to the 385K Human CpG Island-Plus-Promoter Array (Roche-NimbleGen), which represents 28K UCSC-annotated CpG islands and promoter regions for 17K RefSeq genes from the HG18 build. Methylation peak scores were obtained for each probe using the ACME algorithm.Citation20 Nexus software (Biodiscovery) was used to summarize the results across 14 samples enriched with MagMeDIP. For comparison purposes we used Nexus software to summarize the results of 14 samples from normal bronchial tissues that were enriched with MethylAmp and hybridized to the same Roche/Nimblegen 385K Human CpG Island-Plus-Promoter Array for a prior experiment. The number of chromosomal regions and genes methylated across the genome were the parameters used to evaluate the hybridization efficiency to 385K Nimblegen methylation arrays.
Results
The optimal sonication time was defined as the time needed to obtain DNA fragments between 300 and 1500 bp. The optimal sonication time for frozen fresh tissue samples and H1299 cell lines was 10 and 15 min, respectively (). The Methylated-DNA IP had the most intense electrophoresis bands when followed by the MagMeDIP kit and the MethylAmp kit ().
Figure 1. (A) Sonication times and fragments lengths in base pairs (bp) for cell line DNA at three different time periods: a:5 min; b:10 min; c:15 min; (B) Sonication times and fragments lengths in base pairs (bp) for fresh tissue DNA at three different time periods: a:5min; b:10min.
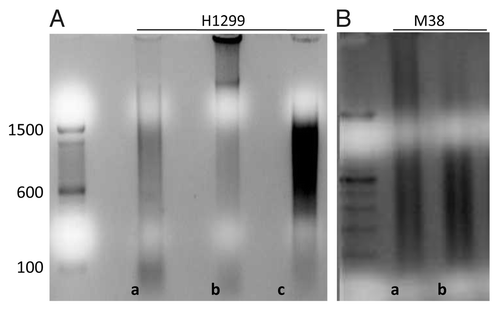
Figure 2. Agarose gel intensity after Nco I digestion for the two different aliquots of the same samples processed by three different MeDIP kits: Methylamp™ Methylated DNA Capture KIT (Epigentek), Methylated-DNA IP KIT (Zymo Research) and MagMeDIP KIT TM (Diagenode). T: Fresh Tissue DNA. C: Cell line DNA. MC: Methylated DNA Control. C+: Methylated/Non-methylated Control DNA
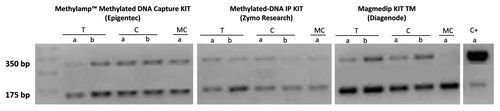
The three kits also had different reaction yields. () The MagMeDIP Kit had higher yields, followed by the MethylAmp and the Methylated-DNA IP kit. The 500 ng aliquot of fresh tissue DNA gave a yield of 90.1% with the MagMeDIP, and the 1 μg aliquot gave a 98.8% yield. The 1 μg of DNA extracted from cell lines gave a yield of 81.78% and the 0.5 μg aliquot a yield of 75.84%. The positive control had a yield of 90.3%. ()
Table 1. Efficiency and consistency of the experiments performed in triplicate using two different amounts of initial DNA: 0.5ug and 1ug. DNA was isolated from fresh tissue (M38) cell lines (H1299) and fully methylated controls (C+) for the: MagMeDIP kit (Diagenode); Methylated-DNA IP Kit (Zymo Research); and MethylAmp™ Kit (Epigentek)
Figure 3. Reaction yield for different amounts of initial DNA, 0.5 μg and 1 μg of both, fresh tissue (T) and cell lines (C), for the: (A) MagMeDIP kit (Diagenode); (B) Methylated-DNA IP Kit (Zymo Research); and (C) Methylamp™ Kit (Epigentek).
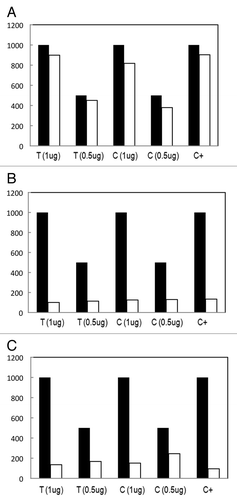
The Methylamp Kit gave a higher yield with the 500 ng aliquot (33.6%) when compared with the 1 μg aliquot (13.7%) for the oral cancer tissue. The yield given by the 500 ng aliquot of cell line DNA was higher (48.9%) than the yield given by 1 μg of cell line DNA (15.1%). The positive control gave a yield of 9.5%. ()
The Methylated-DNA IP Kit gave a yield of 22.7% for the 500 ng tissue sample and 10.1% for the 1 μg aliquot. The cell line DNA of the 500 ng sample had a 25.9% yield and the 1 μg aliquot gave a 21.5% yield. The positive control yield was 13.4%. ()
The hybridization efficiency to 385K Nimblegen methylation arrays was better for DNA enriched with the MagMeDIP kit when compared with the DNA enriched with the MethylAmp kit. The number of genes methylated and the methylation frequency of the genes, how many samples across the genome were methylated, were both higher for the samples enriched with the MagMeDIP kit, when compared with the MethylAmp kit (See ).
Table 2. Hybridization efficiency of MeDIP kits to methylation arrays
The DNA enriched with MagMeDIP kit () not only hybridized to a larger number of regions when compared with the MethylAmp MeDIP kit (), but also had higher hybridization peaks. Table S1 (MagMeDIP) and Table S2 (MethylAmp MeDIP) summarize the genomic coordinates of methylated events by showing the chromosomal region, the region length, the cytoband location, the number of methylated genes and the frequency of methylated events for these genes across arrays.
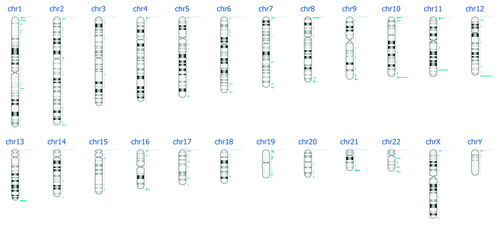
Figure 4. DNA methylation events per chromosome after DNA enrichment with MagMeDIP kit, hybridization to Nimblegen 385K Human Promoter plus CpG Islands arrays and bioinformatics analysis.
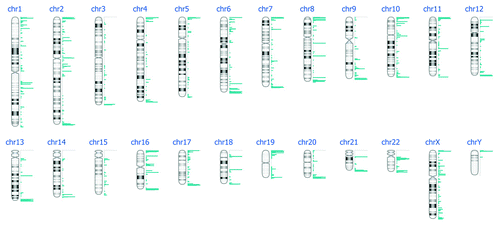
Figure 5. DNA methylation events per chromosome after DNA enrichment with MethylAmp MeDIP kit, hybridization to Nimblegen 385K Human Promoter plus CpG Islands arrays and bioinformatics analysis.
Discussion
The MagMeDIP kit had the best enrichment efficiency of all three kits tested. It had the highest yield for the 500 ng and 1 μg tissue sample, cell line samples, and for the positive control, as can be observed in . In addition, the band produced after the Nco I digestion showed that the DNA was successfully enriched from a 1:4 to 10:1 ratio. Finally, the hybridization efficiency to methylation arrays was better for the MagMeDIP kit. The number of chromosomal regions and genes methylated across the genome were higher for the samples enriched with MagMeDIP, which is what we expected after testing the enrichment efficiency of each kit.
MeDIP-chip and MeDIP-seq (MeDIP prior to deep sequencing) have become important tools in genome-wide epigenomics discovery research. Several research groups have used this DNA enrichment tool for their basic science and discovery research projects.Citation21 Given the widespread acceptance and availability of immunoassays in clinical medicine today, the use of antibodies to detect methylated DNA in the clinic may be readily accepted by clinicians. The MagMeDIP kit is a useful tool that can enable clinical and public health translational genome-wide DNA methylation studies, as we move forward to validate the markers identified in discovery efforts.
There are other methylated DNA enrichment kits available today that do not use antibodies against 5‑mC to capture methylated DNA. Alternatively, double stranded methylated DNA capture can be achieved with methyl-CpG binding domain (MBD) protein capture, which facilitates ligation of double-stranded adaptors for next-generation sequencing. Both methylated sequences enrichment methods have been found to be 99% concordant (their difference did not exceed a given threshold), when assessing methylation levels at CpGs and non-CpGs cytosines with deep sequencing platforms.Citation17 Interestingly, the enrichment efficiency of 5‑mC antibodies and MBD proteins differs when using methylation microarrays platforms. MeDIP enriches for methylated regions with a low CpG density, while MBD favors regions of higher CpG density and identifies the greatest proportion of CpG islands represented in oligonucleotide methylation microarrays.Citation14 The commercial kits available that use MBD proteins to enrich for methylated DNA are listed in Table S3.
Differential shearing methods can increase the enrichment efficiency of all the kits we tested; however, the recommended sheared size to hybridize DNA to Nimblegen tiling arrays is 200–1000 bp. When we tried to use DNA with smaller sheared sizes (200–600 bp), the labeling reaction had to be performed with shorter primers, which in turn limits the binding efficiency of the labeled DNA to the tiling arrays. Since the goal of our MeDIP experiment is to hybridize the DNA to oligonucleotide tiling arrays with long probes (50–73 mers), we have used a shearing size of 200–1000 bp, as recommended by the array manufacturer.
This study has some limitations. We tested the three kits only in a limited number of lung cancer cell lines and oral cancer tissue samples. Hypermethylation in cancer is tissue specific, a fact that may account for some of the observed differences. Furthermore, we hybridized to genome-wide array methylation arrays samples from different tissue types. The samples that were enriched with MagMeDIP prior to hybridization to methylation arrays were cervical tissue samples (7 normal and 7 cervical tumor tissue samples), while the samples that were enriched with MethylAmp MeDIP prior to hybridization were normal bronchial wash samples (14).
It is widely known that CpG island hypermethylation in the promoter region increases in tumor tissue and could thus account for the observed differences between these two experiments. But the number and magnitude of methylation events in normal tissue, regardless of tissue type, is not expected to vary to a great extent. However, when we compared only hybridization efficiency of the normal bronchial and cervical tissue samples, we observed a similar difference in methylation magnitude and frequency between the normal cervical and bronchial samples as those seen in and (data not shown). This led us to conclude that the observed difference in the number and magnitude of methylated events between the two methylation array experiments shown is more than what you would normally expect to see across different tissue samples and disease types, as is due to the efficiency of DNA enrichment prior to hybridization to the genome-wide methylation arrays.
Additional material
Download Zip (150.6 KB)Disclosure of Potential Conflicts of Interest
D. Sidransky owns Oncomethylome Sciences, SA stock, which is subject to certain restrictions under University policy. D. Sidransky is a paid consultant to Oncomethylome Sciences, SA, and is a paid member of the company's Scientific Advisory Board.
Financial support
This research was supported in part by the following grant awards: National Cancer Institute (NCI) 1K01CA164092–01; NCI Early Detection Research Network grant U01 CA84986; an NCI Supplement to Promote Diversity Award to U01 CA84986; a National Institute of Dental and Craniofacial Research (NIDCR) and NIH Specialized Program of Research Excellence grant (SPORE) P50DE019032; and NIDCR grant RC2 DE20957. The funding agencies had no role in the design of the study, data collection or analysis, the interpretation of the results, the preparation of the manuscript, or the decision to submit the manuscript for publication.
E. Soudry is recipient of a fellowship grant from the American Physicians Fellowship for Medicine in Israel.
Related Research Data
References
- Down TA, Rakyan VK, Turner DJ, Flicek P, Li H, Kulesha E, et al. A Bayesian deconvolution strategy for immunoprecipitation-based DNA methylome analysis. Nat Biotechnol 2008; 26:779 - 85; http://dx.doi.org/10.1038/nbt1414; PMID: 18612301
- Vucic EA, Wilson IM, Campbell JM, Lam WL. Methylation analysis by DNA immunoprecipitation (MeDIP). Methods Mol Biol 2009; 556:141 - 53; http://dx.doi.org/10.1007/978-1-60327-192-9_10; PMID: 19488876
- Bock C, Tomazou EM, Brinkman AB, Muller F, Simmer F, Gu H, et al. Quantitative comparison of genome-wide DNA methylation mapping technologies. Nat Biotechnol 2010; 28:1106 - 14; http://dx.doi.org/10.1038/nbt.1681; PMID: 20852634
- Reed K, Poulin ML, Yan L, Parissenti AM. Comparison of bisulfite sequencing PCR with pyrosequencing for measuring differences in DNA methylation. Anal Biochem 2010; 397:96 - 106; http://dx.doi.org/10.1016/j.ab.2009.10.021; PMID: 19835834
- Shen R, Fan JB, Campbell D, Chang W, Chen J, Doucet D, et al. High-throughput SNP genotyping on universal bead arrays. Mutat Res 2005; 573:70 - 82; http://dx.doi.org/10.1016/j.mrfmmm.2004.07.022; PMID: 15829238
- Mohn F, Weber M, Schubeler D, Roloff TC. Methylated DNA immunoprecipitation (MeDIP). Methods Mol Biol 2009; 507:55 - 64; http://dx.doi.org/10.1007/978-1-59745-522-0_5; PMID: 18987806
- Serre D, Lee BH, Ting AH. MBD-isolated Genome Sequencing provides a high-throughput and comprehensive survey of DNA methylation in the human genome. Nucleic Acids Res 2010; 38:391 - 9; http://dx.doi.org/10.1093/nar/gkp992; PMID: 19906696
- Irizarry RA, Ladd-Acosta C, Carvalho B, Wu H, Brandenburg SA, Jeddeloh JA, et al. Comprehensive high-throughput arrays for relative methylation (CHARM). Genome Res 2008; 18:780 - 90; http://dx.doi.org/10.1101/gr.7301508; PMID: 18316654
- Karimi M, Johansson S, Stach D, Corcoran M, Grander D, Schalling M, et al. LUMA (LUminometric Methylation Assay)–a high throughput method to the analysis of genomic DNA methylation. Exp Cell Res 2006; 312:1989 - 95; http://dx.doi.org/10.1016/j.yexcr.2006.03.006; PMID: 16624287
- Oda M, Greally JM. The HELP assay. Methods Mol Biol 2009; 507:77 - 87; http://dx.doi.org/10.1007/978-1-59745-522-0_7; PMID: 18987808
- Ammerpohl O, Martín-Subero JI, Richter J, Vater I, Siebert R. Hunting for the 5th base: Techniques for analyzing DNA methylation. Biochim Biophys Acta 2009; 1790:847-62.
- Schuebel KE, Chen W, Cope L, Glockner SC, Suzuki H, Yi JM, et al. Comparing the DNA hypermethylome with gene mutations in human colorectal cancer. PLoS Genet 2007; 3:1709 - 23; http://dx.doi.org/10.1371/journal.pgen.0030157; PMID: 17892325
- Yamashita K, Upadhyay S, Osada M, Hoque MO, Xiao Y, Mori M, et al. Pharmacologic unmasking of epigenetically silenced tumor suppressor genes in esophageal squamous cell carcinoma. Cancer Cell 2002; 2:485 - 95; http://dx.doi.org/10.1016/S1535-6108(02)00215-5; PMID: 12498717
- Nair SS, Coolen MW, Stirzaker C, Song JZ, Statham AL, Strbenac D, et al. Comparison of methyl-DNA immunoprecipitation (MeDIP) and methyl-CpG binding domain (MBD) protein capture for genome-wide DNA methylation analysis reveal CpG sequence coverage bias. Epigenetics 2011; 6:34 - 44; http://dx.doi.org/10.4161/epi.6.1.13313; PMID: 20818161
- Jacinto FV, Ballestar E, Esteller M. Methyl-DNA immunoprecipitation (MeDIP): hunting down the DNA methylome. Biotechniques 2008; 44:35, 7, 9 passim.
- Weng YI, Huang TH, Yan PS. Methylated DNA immunoprecipitation and microarray-based analysis: detection of DNA methylation in breast cancer cell lines. Methods Mol Biol 2009; 590:165 - 76; http://dx.doi.org/10.1007/978-1-60327-378-7_10; PMID: 19763503
- Harris RA, Wang T, Coarfa C, Nagarajan RP, Hong C, Downey SL, et al. Comparison of sequencing-based methods to profile DNA methylation and identification of monoallelic epigenetic modifications. Nat Biotechnol 2010; 28:1097 - 105; http://dx.doi.org/10.1038/nbt.1682; PMID: 20852635
- Robinson MD, Stirzaker C, Statham AL, Coolen MW, Song JZ, Nair SS, et al. Evaluation of affinity-based genome-wide DNA methylation data: effects of CpG density, amplification bias, and copy number variation. Genome Res 2010; 20:1719 - 29; http://dx.doi.org/10.1101/gr.110601.110; PMID: 21045081
- Hoque MO, Lee CC, Cairns P, Schoenberg M, Sidransky D. Genome-wide genetic characterization of bladder cancer: a comparison of high-density single-nucleotide polymorphism arrays and PCR-based microsatellite analysis. Cancer Res 2003; 63:2216 - 22; PMID: 12727842
- Scacheri PC, Crawford GE, Davis S. Statistics for ChIP-chip and DNase hypersensitivity experiments on NimbleGen arrays. Methods Enzymol 2006; 411:270 - 82; http://dx.doi.org/10.1016/S0076-6879(06)11014-9; PMID: 16939795
- Myers RM, Stamatoyannopoulos J, Snyder M, Dunham I, Hardison RC, Bernstein BE, et al. A user's guide to the encyclopedia of DNA elements (ENCODE). PLoS Biol 2011; 9:e1001046; http://dx.doi.org/10.1371/journal.pbio.1001046; PMID: 21526222