Abstract
Helicobacter pylori is an adapted gastric pathogen that colonizes the human stomach, causing severe gastritis and gastric cancer. A hallmark of infection is the ability of this organism to evade detection by the human immune system. H. pylori has evolved a number of features to achieve this, many of which involve glyco-conjugates including the lipopolysaccharide, peptidoglycan layer, glycoproteins, and glucosylated cholesterol. These major bacterial components possess unique features from those of other gram-negative organisms, including differences in structure, assembly, and modification. These defining characteristics of H. pylori glycobiology help the pathogen establish a long-lived infection by providing camouflage, modulating the host immune response, and promoting virulence mechanisms. In this way, glyco-conjugates are essential for H. pylori pathogenicity and survival, allowing it to carve out a niche in the formidable environment of the human stomach.
Introduction
Helicobacter pylori is a gram-negative Epsilonproteobacterium that chronically colonizes half the world’s population.Citation1 So far, an environmental reservoir for H. pylori has not been identified and the bacterium is thought to be transmitted directly from person-to-person, often early in life. As an adapted gastric pathogen, H. pylori can cause life-long infection, which may manifest itself in a range of outcomes from remaining asymptomatic, to causing mild gastritis, to severe disease including peptic ulcers and gastric cancer.Citation2 In a feat for the bacterial kingdom, this pathogen is adapted specifically to survive and flourish in the unforgiving caustic environment of the human stomach. H. pylori must strike a balance between causing disease and staying below the radar of the host immune system. To do this, it has evolved a refined set of virulence factors, many of which allow persistence by evasion of host immune detection and modulation of immune response.
H. pylori is unique from its gram-negative relatives not only in its evolved environmental niche, but in the assembly and biochemical structures of several major macromolecules. In this review, we address the biochemical and pathogenic properties of these bacterial components, specifically surface-associated glyco-conjugates. Lipopolysaccharide, peptidoglycan, glycoproteins, and glucosylated cholesterol represent important glyco-conjugates with structural properties designed to promote H. pylori colonization, evasion of immune defenses, and establishment of long-term infection. Development of an effective vaccine to protect against infection has eluded the scientific community and remains the holy grail for the field. Outer membrane and surface-exposed bacterial components are of critical importance in vaccine research as they are accessible targets for protective antibodies and are often immunogenic. Accordingly, many of the glyco-conjugates discussed in this review possess immune modulating and pathogenic properties, and thus are relevant in the quest for a vaccine. Therefore, understanding the complexities of their assembly and structure may push the field toward novel therapeutics that can prevent the numerous sequelae and life-long burden associated with this gastric pathogen.
Lipopolysaccharide
Synthesis and transport
Lipopolysaccharide is a primary component of the bacterial outer membrane. It consists of three parts: the lipid A domain, a set of core sugars, and a long chain of oligosaccharide repeats known as the O-antigen.Citation3 LPS contributes to bacterial virulence in a number of ways. For example, the lipid A domain is a potent endotoxin, which can generate a powerful inflammatory response via interaction with the innate immune Toll-like receptor 4/myeloid differentiation factor 2 (TLR4/MD2) complex. The core sugars help to maintain bacterial outer membrane stability, a characteristic required for virulence traits including detergent resistance and expression of flagellar structures.Citation3 Extending into the environment, the O-antigen chain shields the bacterium from host complement deposition and may contain sugar subunits that mimic human sugars, as discussed in detail later in this review.Citation4,Citation5
In H. pylori, the enzymes responsible for LPS biosynthesis are scattered throughout the genome instead of being organized in a single cluster as in other bacteria. This may reflect the fact that H. pylori synthesizes its O-antigen by single addition of monosaccharides and does not have a group of genes devoted to building a repeating oligosaccharide unit.Citation6 Lipid A/core and O-antigen are assembled separately at the cytoplasmic side of the inner membrane and independently translocated across the membrane to the periplasmic leaflet.Citation7 As shown in , O-chain synthesis begins with the activity of WecA, an enzyme that transfers an initiating N-acetylglucosamine (GlcNAc)-1-phosphate residue from an activated nucleotide donor to the lipid carrier undecaprenyl phosphate (UndP), generating UndPP-GlcNAc.Citation8 In most H. pylori strains, the O-antigen is extended by sequential addition of galactose and GlcNAc residues by respective glycosyltransferases. This activity generates a sugar backbone to which fucose residues are appended, creating Lewis antigens. The completed O-antigen chain is translocated across the inner membrane by Wzk, a protein that is not homologous to any known O-antigen translocases.Citation8 Unexpectedly, Wzk is a homolog of the flippase PglK in Campylobacter jejuni, which is involved in translocation of UndPP-linked heptasaccharide during protein N-glycosylation, a process that has not been observed in H. pylori.Citation8,Citation9 Wzk is necessary and sufficient for translocation of UndPP-linked glycans in H. pylori and displays relaxed substrate specificity, able to transport saccharide chains of various lengths and compositions.Citation8 It is unclear whether the H. pylori genome once contained a gene cluster producing proteins required for N-glycosylation and retained only wzk, or whether other Epsilonproteobacteria, e.g., C. jejuni, adapted the gene to produce N-glycoproteins. Regardless, Wzk in H. pylori provides a novel link between LPS biosynthesis and protein glycosylation systems. Once across the inner membrane, the glycan chain is separated from its lipid carrier and joined to the lipid A/core complex by O-antigen ligase, WaaL.Citation3 Subsequently, UndPP is mono-dephosphorylated by an undecaprenyl pyrophosphate phosphatase and recycled back to the cytoplasmic face of the inner membrane to repeat the cycle.Citation10
Figure 1. LPS transport from cytoplasm to periplasm in H. pylori. O-antigen assembly begins with the activity of WecA, which transfers an initiating GlcNAc-1-phosphate residue from an activated UDP donor to the lipid carrier undecaprenyl phosphate (UndP) at the cytosplasmic face of the inner membrane. The O-chain is extended by the alternating activities of galactosyltransferases and GlcNAc-transferases (purple), which receive their respective sugar substrates from a UDP-sugar donor. This generates a GlcNAc (green circles) and galactose (blue circles) sugar backbone to which fucosyltransferases A, B, and C transfer fucose residues (orange triangles) from an activated GDP-fucose donor. The Fut protein ruler mechanism is illustrated by length of protein corresponding to length of O-chain sugar that is fucosylated.Citation38 The completed O-chain containing Lewis antigens (patterned fucose residues linked to GlcNAc and galactose sugars) is translocated to the periplasmic face of the inner membrane by Wzk. Finally, it is removed from the UndPP carrier and transferred onto the lipid A/core by WaaL, generating a mature LPS species. UndPP is recycled to the cytoplasmic leaflet, releasing one phosphate group to reform the UndP acceptor.
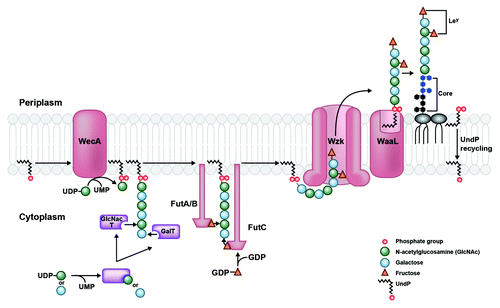
Mature LPS must then be transported across the periplasm and shuttled across the outer membrane for final surface presentation; these steps are performed by the lipopolysaccharide transport (Lpt) machinery.Citation11 H. pylori possesses homologs to several Lpt proteins characterized in E. coli; however, it also seems to lack obvious homologs to several critical components of the system.Citation12 For example, H. pylori shows strong homology to only one of three inner membrane Lpt proteins (LptB; not LptF or LptG), the complex of which is thought to fuel LPS movement. Further, H. pylori is missing obvious homologs to three key Lpt proteins that bridge LPS transport as it traverses the periplasmic space. One of these proteins is the periplasmic chaperone LptA, which only exists at low similarity to its E. coli equivalent. The expected loci for the two remaining periplasmic Lpt components (LptC and LptE) encode proteins sharing minor domain characteristics with their E. coli counterparts.Citation13 Interestingly, the proteins encoded in these H. pylori loci are found across Epsilonproteobacteria, suggesting that this class of bacteria may contain unique conserved functional equivalents to E. coli LptC and LptE proteins.Citation13 Finally, H. pylori does contain a strong homolog of LptD, which delivers LPS to the outer leaflet of the outer membrane, completing the transport process. All in all, H. pylori seems to possess most of the machinery that E. coli uses to transport LPS, though genetic loci, sequence, and structure of several proteins are not perfectly conserved.
Lipid A biosynthesis
The lipid A domain is the bioactive center of lipopolysaccharide due to its interaction with the TLR4/MD2 complex. When stimulated by lipid A, this conserved pattern recognition receptor initiates a potent signaling cascade resulting in production of pro-inflammatory cytokines and a potent immune response.Citation3 Alterations in lipid A structure including acyl chain number and length, phosphate groups, and presence of polar functional groups can all impact the strength of the TLR4 response.Citation14 For example, H. pylori lipid A shows low immunostimulatory activity relative to other gram-negative bacteria, up to 1000-times lower than that of E. coli.Citation15-Citation17 This immune-dampening effect is attributed to the highly modified structure of H. pylori lipid A, which is structurally unique from that of E. coli.
Lipid A is synthesized via the Raetz pathway, a nine-step enzymatic process conserved in most gram-negative bacteria.Citation3,Citation18 The result of the Raetz pathway in E. coli is a β(1,6)-linked disaccharide of glucosamine that is bis-phosphorylated and hexa-acylated. This structure is attached to 3-deoxy-D-manno-octulosonic acid (Kdo) sugars that connect it to the core and O-antigen.Citation3 H. pylori produces a similar structure through the Raetz pathway (), which is then heavily modified during transport to the outer bacterial membrane (). H. pylori possesses only eight homologs to the nine Raetz pathway enzymes, with the exception in the last step of the pathway.Citation19 In E. coli, the last two acylation events are catalyzed by LpxL and LpxM ( and ), which each add an acyl chain to the existent 2′ and 3′ hydroxyacyl chains, respectively. H. pylori only contains a homolog of LpxL encoded by hp0280 (strain 26695), while an obvious homolog of LpxM was not found despite demonstration of enzymatic activity corresponding to an identical function.Citation20 Detailed bioinformatic analysis led to identification of the LpxM functional equivalent in H. pylori, encoded by hp0270, and homologs in closely related species including Campylobacter jejuni (cjj81176_0482) and Wolinella succinogenes (ws1775). Hp0270 and homologs restore a hexa-acylated phenotype in a penta-acylated E. coli LpxM mutant. Further, assays using radiolabeled lipid A substrate and purified membranes expressing each protein demonstrate their enzymatic activities in vitro (Rubin and Trent, unpublished data).
Figure 2. The Raetz pathway of lipid A biosynthesis in H. pylori. H. pylori synthesizes a bis-phosphorylated hexa-acylated lipid A via the conserved nine-step Raetz pathway using eight homologs of E. coli enzymes, and one enzyme (Hp0270) that is genetically distinct from its E. coli counterpart. The final product of this pathway, shown at the bottom left, is heavily modified during transport to the outer membrane.
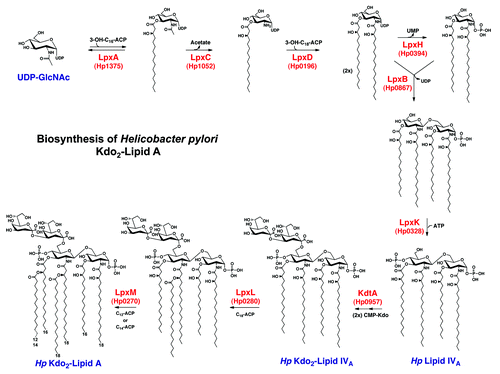
Figure 3. Modification of H. pylori lipid A generates its unique structure. Kdo2-lipid A is flipped to the periplasmic leaflet of the inner membrane by the conserved ABC-transporter MsbA. The 1-phosphate group is removed by LpxE (Hp0021), and a phosphorylethanolamine (pEtN) group is added in its place by EptA (Hp0022). The latter activity generates diacylglycerol (DAG), as phosphatidylethanolamine (PE) is likely the donor molecule for the pEtN group. Next, a two-protein Kdo-hydrolase complex, Kdo H1/H2 (encoded by hp0579 and hp0580), removes the terminal Kdo group. The last modification to occur before transport across the periplasm is the removal of the 4′-phosphate group by LpxF (Hp1580). Finally, the outer-membrane β-barrel LpxR (Hp0694) catalyzes removal of the 3′-linked acyl chains, generating the final lipid A structure in the outer membrane of H. pylori (inset). The core oligosaccharide and O-antigen chain with fucose additions are only shown in the inset for simplicity.
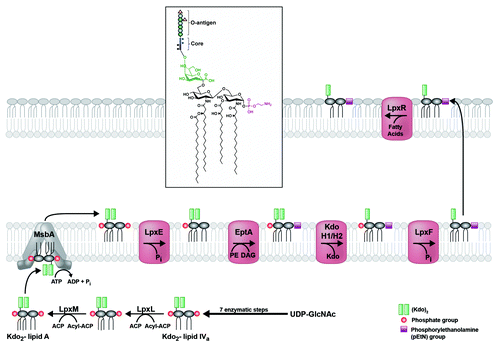
The hexa-acylated lipid A, conjugated to core sugars, is flipped to the periplasmic face of the inner membrane by MsbA, an essential ABC transporter that is well-conserved in H. pylori with its E. coli homolog (). Genetic interruption of hp0270 is lethal, suggesting that in H. pylori, MsbA requires hexa-acylated lipid A as substrate (Rubin and Trent, unpublished data). After flipping across the inner membrane, the lipid A domain of LPS is modified by several enzymes before transport across the periplasm (). First, the 1-phosphate group is removed by LpxE (Hp0021) and a phosphorylethanolamine (pEtN) is added in its place by EptA (Hp0022).Citation21 These successive modifications reduce the net negative charge of the LPS, thereby promoting resistance to cationic antimicrobial peptides (CAMPs) like polymyxin B.Citation19 Next, the terminal Kdo sugar is removed by the two-protein Kdo-hydrolase complex, Kdo H1/H2 (Hp0579 and Hp0580).Citation22 Kdo hydrolysis is required for expression of Lewis antigens (addressed in next section) and fully extended O-antigen. It also promotes CAMP resistance indirectly, in that Kdo hydrolysis must occur before the next modification: removal of the 4′-phosphate group by LpxF (Hp1580), which also decreases the net negative charge of LPS.Citation23 Removal of both 1- and 4′-phosphate groups is critical for many aspects of H. pylori pathology including CAMP resistance, limitation of TLR4 mediated immune recognition, and colonization of the mammalian gastric mucosa.Citation23 Before traversing the periplasm, the lipid A molecule is joined with the core sugars and O-antigen, forming a mature LPS molecule (). Once the LPS is transported to the outer leaflet of the outer membrane, the lipid A moiety undergoes a final modification: removal of the 3′-linked acyl chains by LpxR (Hp0694), producing the final tetra-acylated lipid A structure characteristic of the H. pylori outer membrane (, inset).Citation20 The LpxR-catalyzed modification is thought to contribute to evasion of the innate immune system as acyl chain number is a key factor in determining lipid A toxicity in other bacteria.Citation24-Citation26
Overall, the highly-modified end product of lipid A biosynthesis in H. pylori is unique among gram negative bacteria, far removed from its hexa-acylated E. coli precursor in structure and immunogenicity. It is the result of a stringently adapted system, finely tuned to allow establishment of a low immunogenic profile for H. pylori in the host.
Unlike H. pylori, close relative C. jejuni did not evolve an extensive lipid A modification network and therefore produces a structure closer to that of E. coli. C. jejuni does, however, encode a homolog of H. pylori EptA, which transfers a pEtN group to the lipid A backbone. While still possessing pEtN transferase activity for the lipid A domain, this protein (Cj0256, or EptC) shows divergent substrate specificity when compared with its H. pylori homolog. Cullen and TrentCitation27 elucidated a novel link between LPS and flagellar assembly pathways by demonstrating that Cj0256 transfers pEtN to both the lipid A backbone and flagellar rod protein FlgG; without pEtN modification of FlgG, production of flagella is blocked. Adding to the list of EptC targets, Scott et al.Citation28 showed that this promiscuous enzyme also targets the terminal sugar of N-linked heptasaccharides on various periplasmic proteins. Most recently, Cullen et al.Citation29 added a fourth target to the list: the first heptose of the lipooligosaccharide inner core. In general, these pEtN decorations seem to be required for effective mammalian and avian host colonization, and appear to influence TLR4/MD2 signaling in the case of the inner core modification.Citation29 The multi-functional nature of C. jejuni EptC may be the result of genomic refinement or adaptation to the unique environmental demands that C. jejuni encounters. Conversely, H. pylori EptA may also have additional targets that have yet to be discovered; research in this field is ongoing.
Lewis antigen expression in the O-antigen of LPS
LPS of H. pylori is commonly characterized by the integration of Lewis (Le) and related blood group antigens into the O-chain sugar backbone. Le antigens are expressed on various cell surfaces of the human host. Specifically in the stomach, these cells include: mucous-producing foveolar epithelial cells lining the stomach walls, and multiple cells of the gastric glands found in the pits and depressions of the mucosal layer.Citation5 Structurally, Le antigens closely resemble the ABO blood groups present on the surface of circulating human red blood cells, constituted from a sugar backbone of galactose (Gal) and GlcNAc with peripheral fucose additions.
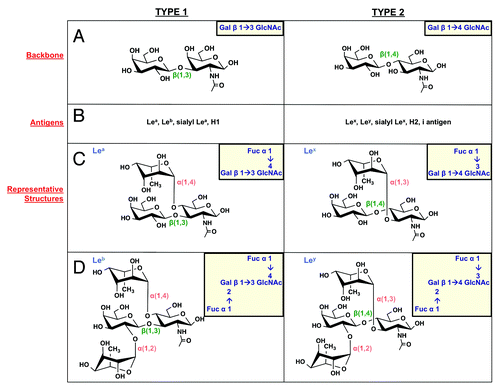
There are two types of precursor backbone chains that underlie the full antigen structures. Type 1 chains give rise to Lea and Leb, while type 2 chains build Lex and Ley antigens. The backbone chains differ only in configuration of the glycosidic linkage between Gal and GlcNAc, which form a β(1,3) linkage in type 1 chains and a β(1,4) linkage in type 2 chains. These frameworks can then be decorated with lateral fucose residues to generate the complete Le antigen structures (). For example, transfer of fucose to carbon 4 of the GlcNAc sugar in the type 1 backbone generates Lea activity, while the additional presence of a fucose residue linked to carbon 2 of galactose is a Leb structure. A similar process underlies construction of type 2 Le antigens, except that fucose residues are appended to the β(1,4) Gal-GlcNAc framework and the Lex precursor structure involves addition of fucose to carbon 3 of the GlcNAc sugar, instead of carbon 4.
Figure 4. Lewis antigen structures. H. pylori can produce type 1 (left column) and type 2 (right column) Lewis antigens. (A) Type 1 Le antigens are based on a β(1,3)-linked galactose-GlcNAc sugar backbone, while type 2 antigens show a β(1,4) linkage. These backbones give rise to the Le antigens listed in panel (B). (C) Lea and Lex are built by α(1,4) or α(1,3) addition of a fucose residue to the GlcNAc sugar of the type 1 and type 2 backbone, respectively. (D) Leb and Ley are built by α(1,2) addition of a fucose residue to Lea and Lex structures, respectively.
In H. pylori, Lex and Ley antigens are prevalent relative to other Le structures, found in 80–90% of strains across the world; however, a and b structures have been detected at low frequencies in both Asian and European isolates.Citation30,Citation31 Some strains seem not to express Le antigens at all, instead possessing O-antigen composed of rare configurations of mannose and rhamnose sugars.Citation32 The type 1 and 2 backbones compose the O-antigen chain itself, with the initiating GlcNAc residue eventually ligated directly onto a heptose residue of the core region. As noted, polymerization of the O-antigen chain takes place in the cytoplasm by the sequential activities of glycosyl- and fucosyltransferases (detailed below). The completed chain is flipped to the periplasm by Wzk and ligated to the lipid A/core structure by WaaL ().Citation8
The genome of H. pylori encodes all of the machinery necessary for construction and integration of Le antigens to the bacterial LPS. Both type 1 and 2 backbones are synthesized by the alternating activities of galactosyl (GalT)- and glucosaminyltransferases (GlcNAcT) (). First, a galactose sugar is added to the initiating GlcNAc residue by one of two distinct GalT proteins, which form either a β(1,3) (type 1) or a β(1,4) (type 2) linkage ().Citation33,Citation34 Next, the β(1,3)-GlcNAcT adds a second GlcNAc residue to the growing chain.Citation35 From there, alternating monomeric sugar additions by the GalTs and GlcNAcT build the full O-antigen backbone.Citation6,Citation36,Citation37
Once the backbone chain is in place, then peripheral fucose residues are added by fucosyltransferases, FutA, FutB, and FutC with various specificities to generate the Le antigen structures just described. Fucose is synthesized via a two-step mechanism and attached to an activated GDP nucleotide, generating a donor for each fucosyltransfer reaction. Depending on the strain, FutA and FutB may be mono- or bifunctional fucosyltransferases, producing α(1, 4) and/or α(1, 3)-linkages between fucose and GlcNAc, thereby generating Lea (type 1) and/or Lex (type 2), respectively (). FutA and FutB are characterized by C-terminal heptad-repeat regions of variable length that allow homo- and heterodimerization. Further, Nilsson et al.Citation38 presented a molecular ruler mechanism by which heptad-repeat number determines the length of FutA and FutB proteins, and therefore length of O-chain to be fucosylated (). In explanation, they show that one heptad-repeat corresponds to one Gal-GlcNAc backbone unit in the glycan chain, such that the location of the active site and recipient sugar residue are finely controlled.Citation38 A second fucose residue is added to Lea or Lex by FutC, which produces Leb (type 1) or Ley (type 2) by way of an α(1,2)-linkage between fucose and galactose (). A typical O-antigen chain in H. pylori contains multiple internal Lex residues and a terminal Lex or Ley. Decoration of the O-antigen chain with fucose residues is an ordered process; for example, the α(1,3)-fucosyltransferase first attaches fucose to internal GlcNAc residues to form Lex antigens, then the α(1,2)-enzyme fucosylates the terminal galactose residue, generating a Ley antigen.Citation5
Lewis antigen expression in the H. pylori O-chain is mosaic, as a single O-chain may contain various Le identities, including mixtures of type 1 and type 2 structures.Citation5 In addition, the fucosyltransferase and β(1,3) GalT genes are subject to phase variation, or random switching on and off of expression during the lifetime of a given bacterial cell.Citation34,Citation39 This leads to an expansive diversity of possible Le antigen patterns across a population of H. pylori cells during the course of infection. Phase variation in the α(1,3)-fucosyltransferase genes (futA, futB) and in the β(1,3) GalT gene is controlled by the presence of 5′ poly-A/C, and poly-C only nucleotide repeat tracts, respectively. These repeat stretches can cause slipped strand mispairing during DNA synthesis, resulting in alternate and/or aberrant reading frames that may change or inactivate the gene.Citation40 The α(1,2)-enzyme (futC) contains a similar poly-C tract as well as several other features that lead to highly variable expression. For example, the gene sequence contains imperfect TAA-, GAA-, and AAA- repeats that can likewise lead to slipped strand mispairing. An internal Shine-Dalgarno sequence and putative stem-loop structure also serve to shift the reading frame in and out of proper coding orientation.Citation41
Lewis antigens in pathogenesis
As an adapted gastric pathogen, H. pylori employs a number of strategies to promote survival and avoid detection in the host. Le antigen expression can be counted under that umbrella, as these host-like patterned sugars have been shown to allow immune evasion, mediate adherence and colonization, and modulate the immune response against H. pylori.
To subvert host defenses, the bacterium camouflages itself in sugars that closely resemble host cell surface structures. This process is known as molecular mimicry and can promote pathogenesis in two ways. First, it offers a path of immune escape since the host is unlikely to react to self-epitopes. However, this depends on the Lewis antigen type expressed by the host. For example, a Lex-positive H. pylori strain that infects a Lex-positive host could theoretically escape immune attack, while this would not necessarily be the case for a Ley-positive strain in the same host. Conversely, the host may raise antibodies to Lewis antigens on the bacterial surface and in so doing generate cross-reacting antibodies that cause damage to self-tissue, giving H. pylori an opportunity to invade.Citation42 In support of the former mechanism, Pohl et al.Citation34 demonstrated that H. pylori can change its Le pattern to adapt to the Le antigen expression profile of the host. For example, H. pylori expressing Lex was used to infect wild-type and Leb-transgenic mice. After eight months, bacteria isolated from several of the Leb-transgenic but not wild-type mice had switched on Leb expression and, in these mice, colonization levels were higher than in transgenics that did not show Leb expression. This suggests that H. pylori that expressed Leb was better able to survive in its self-tolerant Leb host. Sequence analysis showed that phase variation and an inherited increase in number of poly-C tract repeats in the type 1 β(1,3) GalT was responsible for switching on Leb expression in certain H. pylori strains.Citation34 Interestingly, the β(1,3) GalT gene can recombine with its upstream neighbor to create a chimeric product required for synthesis of all Lewis antigens. This suggests a novel finding that cross-talk may exist between type 1 and type 2 antigen synthesis pathways in the form of a recombined type 1 enzyme.Citation43
Further, recombination events within and between the two α(1,3/4) fucosyltransferase genes (futA, futB) are thought to contribute to O-antigen evolution within the host, producing a wide array of diverse structures even from a single-cell colony inoculum. The host environment is thought to exert selective pressure on LPS phenotypes, forcing H. pylori communities to evolve variety in their Le antigen and O-chain structures that may promote survival in the current host and provide an advantage upon transmission to a new host.Citation44
As surface-exposed entities, Le antigens can also contribute to pathogenesis by mediating adherence to and colonization of the host gastric epithelium. In fact, strains that produce asymptomatic infections often do not express Lewis antigens, suggesting that presence of these structures contributes to establishment of disease.Citation32,Citation45 Further, interruption of Lex expression reduces colonization and limits adherence of H. pylori to the gastric mucosa by preventing interaction of Lex with host receptor galectin-3.Citation46,Citation47 However, Le antigens are also found in asymptomatic infections, and can even dampen the host immune response to H. pylori. Bergman et al.Citation48 demonstrated that Le-positive H. pylori strains were able to interact with the C-type lectin DC-SIGN on the surface of gastric dendritic cells. This interaction blocks maturation of T-helper 1 cells and leads to general immune suppression and reduced production of pro-inflammatory cytokines. Contrarily, Le-negative strains do not show specific binding to dendritic cells through DC-SIGN, promoting maturation of T-helper 1 cells and thereby inducing a stronger immune response.Citation48 The marked diversity of Lewis antigen expression makes it difficult to define their role in infection and disease progression. However, evidence abounds to suggest that the unique ability of H. pylori to produce Le antigens affords the bacterium a number of advantages inside the host.
On the whole, LPS is a multi-faceted molecule that plays a central role in establishment and maintenance of H. pylori infection. It is different from that of other gram-negative bacteria due to the presence of Le antigens in the O-chain and a distinctly non-immunostimulatory lipid A domain. These characteristics define H. pylori as a host-adapted pathogen capable of long-term infection.
Peptidoglycan
Cell shape
Another glycan that contributes to H. pylori pathogenesis is the thin, mesh-like peptidoglycan (PG) sacculus, found in the periplasmic space between inner and outer membranes.Citation49 Compared with the extensive, surface-exposed PG lattice of gram-positive organisms, the gram-negative PG serves less of a protective function and instead contributes more to cell shape and integrity.Citation50 The typical PG monomer is an amino sugar backbone of β(1,4)-linked GlcNAc and N-acetylmuramic acid (MurNAc) connected by the 3-carboxyl group of each MurNAc to a short peptide chain (). This peptide chain varies in composition, length, and degree of cross-linking from species to species. Glycan chains are cross-linked by transpeptidases that form bonds between certain amino acids within the peptide chains of individual PG monomers. Cross-linking between glycan chains causes the PG to adopt its characteristic lattice-like structure.
Figure 5.H. pylori peptidoglycan structure and modification. (A) The H. pylori PG monomer consists of an extended backbone chain of β(1,4)-linked GlcNAc-MurNAc sugars, linked to a short peptide chain by the 3-carboxyl group of the MurNAc residue. (B and C)H. pylori encodes a number of PG-modifying enzymes. Enzymes, genes, and activities are listed in table B, and the impact on the PG layer for each enzyme is illustrated in C.
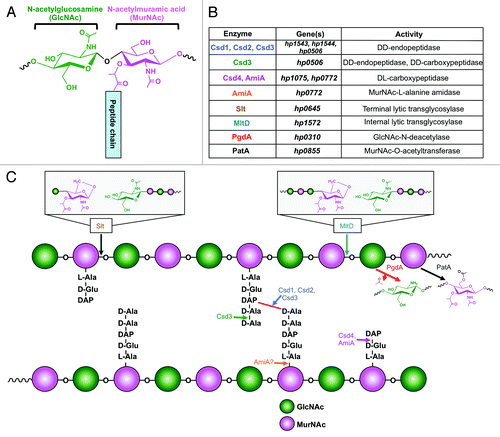
H. pylori PG is structurally unique from other bacteria with regard to extent of cross-linking and lengths of glycan and peptide chains. For example, it shows a remarkably low level of cross-linking (17%) compared with E. coli (25–30%) and other gram-negative bacteria.Citation51 This is evidenced by the predominance of monomer and dimer glycan chains and absence of higher order polymers.Citation52 It also possesses a high proportion of non-reducing (1,6)-anhydro MurNAc residues, which terminate extension of the glycan chain (, insets). As a result, H. pylori PG contains very short glycan chains of 9 to 11 disaccharides on average compared with 21 in E. coli.Citation53 Further, bacteria may exhibit muropeptide chains of assorted lengths from di- to pentapeptides depending on expression of various carboxy- and endopeptidases that break terminal and internal peptide bonds, respectively (). H. pylori is similar to many other gram-negative organisms in terms of the composition of its PG muropeptides; it initially produces mostly pentapeptides consisting of l-alanine, d-glutamic acid, L-di-aminopimelic acid (DAP), d-alanine, and a final d-alanine. Cross-links most often occur between DAP and alanine residues ().Citation52
The PG sacculus is instrumental in determining cell shape.Citation52 As a helical organism, H. pylori falls into a group of bacteria with a complex cellular morphology that is finely controlled by multiple proteins. The PG meshwork must be cut to make room for new fragments during cell elongation, division, or establishment of cell shape. A number of periplasmic hydrolases act to trim and refine the macromolecular structure. For example, endopeptidases cleave cross-links between PG fragments; carboxypeptidases trim the final amino acid in a peptide chain; amidases remove peptides from glycan chains; and lytic transglycosylases terminate the glycan chain by cleaving between MurNAc and GlcNAc residues and converting MurNAc into the (1,6)-anhydro non-reducing form. Endo- and carboxypeptidases come in several flavors; the former are able to cut between DD- and DL-amino acids, and the latter cut between DD-, LD-, and DL-amino acids. shows the effects of several H. pylori hydrolases on the PG structure.
The helical shape of H. pylori is theorized to promote a type of fast “cork-screw” motility in the viscous gastric mucus layer where the pathogen resides in its human host. Recent research by Sycuro et al.Citation54 has made a novel connection between PG cross-linking and modification with generation of the characteristic helical shape of H. pylori. Specifically, four proteins encoded by genes cell shape determinant 1 (csd1), csd2, csd3, and csd4 have been found individually necessary for the curvature and twist associated with the helical pitch of single H. pylori cells. Single mutants of each gene show various curved as opposed to helical morphologies and a dramatic increase in tetra-pentapeptide cross-linked dimers. As Csd1–3 contain endopeptidase domains, this evidence suggests that the proteins function in cleaving cross-links between muropeptides of associated glycan monomers ().Citation54 Csd3 also seems to possess carboxypeptidase activity, cleaving pentapeptides to generate tetrapeptides.Citation55 Carboxypeptidase Csd4 trims tripeptides into dipeptides, which can also sever connections between linked PG fragments without directly cleaving the peptide cross-bridge (). Single csd4 mutants show a straight rod morphology, suggesting that even seemingly minor trimming of the PG layer can lead to dramatic effects in overall cell morphology.Citation56 The authors suggest that these changes in PG structure contribute to generation of helical shape in the following ways: localized removal of cross-links between glycan chains is thought to relax connectivity of the rigid glycan framework at critical points and possibly along multiple axes, creating an offset curvature and twist that generates the helix. Trimming by Csd3 and Csd4 may contribute by limiting cross-linking between shortened muropeptides, or potentially signaling the bacteria to insert new PG fragments as dipeptide indicates aging PG.Citation54,Citation56 Importantly, all Csd mutants show defects in colonization of a mouse gastric infection model, but only Csd4 mutants display defects in motility. This phenotype is likely due to the complete lack of curvature in this particular mutant as opposed to the curved Csd1–3 mutants. These results suggest that helical shape is essential for effective colonization but this effect is not imparted by particularly effective motility.Citation54 Therefore, curvature seems more essential to effective motility than helical shape. Csd3 has been independently identified by Bonis et al.Citation55 to possess endo- and carboxypeptidase activity, leading to its annotation as HdpA (Helicobacter DD-peptidase A). However, this group found that HdpA controls additional aspects of cell morphology beyond modifying PG, including cell diameter, length, and formation of a single pole. For example, a portion of the cell population lacking this gene adopts a stocky branched phenotype with multiple poles of irregular length containing functional flagella. Observation of this additional interesting phenotype may be explained by the significant morphological diversity of H. pylori and by the use of different experimental strains. Ultimately, HdpA mutants were also defective in colonization, potentially owing to poor motility or adhesion to host cells.Citation55
The Csd proteins are conserved across Epsilonproteobacteria and in helical Gammaproteobacteria. For example, Campylobacter jejuni expresses Pgp1, a homolog of Csd4 in H. pylori.Citation57 Like csd4, deletion of pgp1 results in a straight, rod-shaped cell with defects in motility and colonization of the gastric epithelium. Enzymatically, Pgp1 possesses activity identical to that of Csd4, trimming PG tripeptides to dipeptides ().Citation57 Clearly, remodeling of the PG layer on both large and small scales majorly impacts cell shape and allows the helical bacteria to adopt their characteristic shape.
Morphology transition and remodeling
Interestingly, the PG peptide chain changes in composition depending on growth phase; during early log phase, a pentapeptide chain is prevalent, but when cells transition into stationary phase, changes occur both in peptide composition and overall PG structure. These changes are correlated with a morphology transition unique to H. pylori that occurs with the switch from log to stationary phase: the helical-to-coccoid transition.Citation52
One such change in PG structure is an increase in the amount of dipeptide structures within the meshwork.Citation52 During the wild-type H. pylori morphology transition, the PG structure accumulates high levels of dipeptides from the activity of PG hydrolases that trim the third amino acid from tripeptide motifs.Citation52 Chaput et al.Citation58 identified AmiA as one such PG hydrolase, and the first genetic factor required for the helix-to-coccus transition in H. pylori. The authors suggest that H. pylori AmiA possesses carboxypeptidase activity, cleaving the bond between the second and third amino acids in a tripeptide PG chain (). This is in contrast to its E. coli homolog, a MurNAc-l-alanine amidase that cleaves the bond between N-acetylmuramic acid and the first amino acid of the peptide moiety, l-alanine.Citation59 Loss of AmiA in H. pylori leads to impaired accumulation of the dipeptide motif and stable levels of tripeptide. It is possible that AmiA also has amidase activity, due to C-terminal MurNAc-l-alanine amidase homology; however this activity has not yet been demonstrated. Importantly, the PG tripeptide motif is a known agonist of human Nod1, which leads to NF- κB activation and immune response when stimulated. It follows that the decrease in PG tripeptide resulting from AmiA activity allows coccoid-transitioned H. pylori to avoid IL-8 production and NF-κB activation.Citation58 These results indicate that the coccoid transition in H. pylori may be another mechanism of immune escape rather than unregulated deterioration of the cell.
H. pylori also encodes two lytic transglycosylases, Slt and MltD, enzymes that cleave between MurNAc and GlcNAc residues of the glycan backbone and generate (1,6)-anhydro MurNAc.Citation60 Loss of Slt and MltD leads to excessively long glycan chains distinct from the typically short chains (9–11 units) of wild-type H. pylori. The effects of single mutants are additive, suggesting that the two enzymes have non-overlapping functions. This may be due to differences in substrate preference; Slt seems to cleave tripeptides, which are found predominantly at the non-reducing ends of glycan chains and MltD cleaves within a glycan chain (). The extended-chain phenotype can be attributed to decreased levels of (1,6)-anhydro MurNAc, which is normally found at high levels in H. pylori and terminates further glycan chain elongation. The tripeptide motif released by Slt activity is a typical PG turnover product, while MltD activity instead introduces gaps into the PG macromolecular structure, making space for additions to and remodeling of the layer.
In E. coli, PG turnover products are transported back into the cytoplasm for salvaging and reuse.Citation59 Interestingly, H. pylori does not seem to recycle its PG; it lacks an inner membrane transporter required for uptake of PG turnover products and cytoplasmic amidases and ligases that break down and rebuild these products into newly functional PG precursors.Citation61 As a result, H. pylori contains a high concentration of PG degradation products in its periplasm. A pivotal study from Viala et al.Citation62 showed that Slt is necessary for induction of IL-8 secretion and, further, is induced in vivo in human gastric biopsies. It seems that the PG degradation products generated by Slt, specifically PG-tripeptide (), are recognized by the Nod1 receptor on the host cell surface. Upon recognition of bacterial PG, Nod1 initiates an intracellular signaling cascade that leads to production of pro-inflammatory cytokines and mounting of an immune response. Interestingly, delivery of PG was shown to be dependent on the presence of a type 4 secretion system (T4SS) encoded by the Cag pathogenicity island, suggesting that H. pylori PG may pass through the needle-like apparatus to gain access to the host cell.Citation62 Therefore, these processes occur only in particularly virulent strains of H. pylori containing the Cag island. H. pylori relies on an arsenal of hydrolases both to establish its helical shape, to allow it to change, and in certain cases, to indirectly promote pathogenesis.
PG modifications in pathogenesis
Beyond trimming sugars and peptides on a macro-scale, H. pylori also modifies its PG layer in a number of ways that help protect it from environmental insult. For example, PG deacetylation seems to be important in modulating the host immune response to allow persistence of infection.Citation63-Citation65 PgdA, a GlcNAc-N-deacetylase, was identified in H. pylori to promote resistance to lysozyme, a host innate immune molecule that cleaves between GlcNAc and MurNAc residues of the PG glycan chain (). Interestingly, pgdA expression is induced under conditions of oxidative stress, including in vitro during direct contact with reactive oxygen species-producing macrophages.Citation64 Overall, expression of PgdA and resultant PG deacetylation seems to mitigate the immune response, limiting elicitation of IL-8, IL-10, and tumor necrosis factor-α production from host cells. Thus, oxidative stress serves as a signal to induce PgdA expression, allowing H. pylori to colonize and survive in the gastric mucosa more effectively.Citation64 Another enzyme, PatA, is responsible for O-acetylation of MurNAc in H. pylori PG (), which confers protection against lysozyme that is additive with that of PgdA. Many examples exist for bacteria that use either N-deacetylation or O-acetylation to modify their PG and thereby resist lysozyme. However, H. pylori is unique in that some strains demonstrate both modifications, which contribute independently to lysozyme resistance and persistence in the host.Citation64-Citation66
Protein Glycosylation
Protein glycosylation systems are abundant in eukaryotic organisms, but are also being discovered in increasing numbers of bacterial species. Helicobacter and Campylobacter spp are prime examples of bacteria that contain the machinery necessary for glycosylating proteins, which ultimately contributes to pathogenicity in various ways. In H. pylori, O-linked glycosylation is used to decorate serine and threonine residues of flagellin proteins with nine-carbon sugars; these modifications are required for flagellar filament assembly. C. jejuni possesses a similar O-linked system, but is also capable of attaching sugars to the amide nitrogen of asparagine, generating N-linked glycans. This activity in C. jejuni is encoded by the 12-gene pgl locus, which contains all the machinery necessary for building and transporting upwards of 70 N-linked glycoproteins.
The major targets of O-linked glycosylation in H. pylori include the two flagellar filament proteins, FlaA and FlaB (). Major flagellin FlaA is glycosylated on 7 serine or threonine residues, while minor FlaB subunits show 10 modified residues. In C. jejuni, only FlaA is glycosylated, with 16–19 serine and threonine residues showing modification.Citation67 These sugar additions are concentrated in regions of the protein that are thought to be surface-exposed once integrated into the greater filament. A precise consensus sequence for incorporation of these glycosylations has not yet been identified, and instead may be controlled by nearby hydrophobic residues.Citation68
Figure 6. O-linked glycosylation of flagellar proteins in H. pylori. (A) Pseudaminic acid (PseAc) is synthesized in a five-step enzymatic process from a UDP-GlcNAc precursor. The structure of PseAc is shown (inset). It is then linked to a CMP nucleotide donor by PseF, and transferred by PseE from this donor to a serine or threonine residue on a FlaA or FlaB protein before the protein folds. (B) Glycosylated FlaA and FlaB proteins fold in the cytoplasm and display surface-exposed PseAc residues. Modified FlaA and FlaB are then translocated through the flagellar machinery to compose the flagellar filament. PseAc additions are apparent on both proteins within the filament.
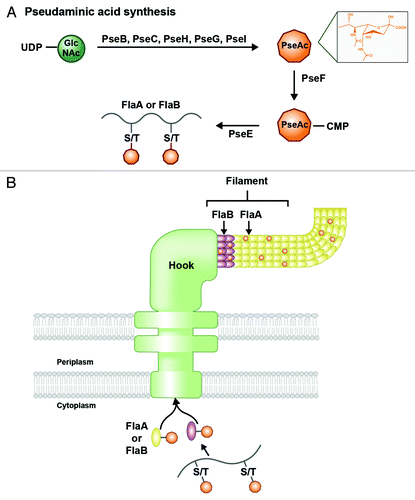
The nine-carbon sugar known to decorate the flagellar filaments of H. pylori and C. jejuni is pseudaminic acid (PseAc).Citation68 C. jejuni also produces modified versions of PseAc with chemical substitutions or additions, while H. pylori was thought to display only the unmodified form of the sugar until recently (see Höpf et al. explanation below). The PseAc biosynthesis pathway has been characterized predominantly in C. jejuni; the sugar is synthesized in the cytoplasm and then transferred from an activated nucleotide (CMP-PseAc) to the hydroxyl group of a serine or threonine residue on the flagellin protein, all by the sequential activities of seven enzymes encoded by the pse genes ().Citation67-Citation69 H. pylori uses homologs of the pse genes to generate PseAc. The glycosylated flagellin subunits are then exported from the cytoplasm by the flagellar secretory machinery for integration into the emerging filament (). In the absence of glycosylation, an external flagellar filament does not form, rendering the bacterium non-motile.Citation67,Citation68,Citation70 This suggests that O-linked glycosylation is either required to stabilize individual flagellin proteins or may facilitate filament polymerization.Citation68
Beyond the canonical pse genes, Asakura et al.Citation71 characterized a protein involved in O-linked glycosylation that negatively affects H. pylori pathogenicity. When hp0518 is inactivated, mutants show a hyper-pathogenic phenotype with faster motility, more efficient colonization of host cells, and increased NF-kB activation. Hp0518 was demonstrated to be a deglycosylase, removing O-linked PseAc residues from FlaA subunits in the cytoplasm before export through the flagellar machinery. The mutant strain possesses excessively glycosylated flagella, which are linked with its rapid motility. Further, hp0518 mutants show increased phosphorylation of CagA toxin, which is encoded on the Cag pathogenicity island and injected into host cells by the T4SS.Citation71 In wild-type bacteria, Hp0518 may provide a balance for establishing optimal levels of glycosylated filament subunits. It is also possible that this deglycosylase can be inactivated or downregulated at certain stages of growth, allowing the bacterium to switch to a more invasive and pathogenic phenotype, resulting in more severe disease. However, the physiological relevance of Hp0518 remains to be determined in the context of infection.
Although C. jejuni generates a sizable population of glycoproteins, it was historically thought that H. pylori only glycosylates flagellin proteins. However, recent research suggests that protein glycosylation in H. pylori extends beyond the flagellins, and further that a pathway independent of PseAc synthesis exists, suggesting greater heterogeneity in identity of sugars conjugated to proteins in H. pylori. Höpf et al.Citation72 found novel glycoproteins in H. pylori by extracting a total protein fraction and staining with glyco-specific dyes that react with oxidized carbohydrate groups. The group identifies 9 candidate glycoproteins that are either cytoplasmic or inner membrane-associated, including two subunits of the F0F1 ATPase, as well as catalase. Further, they demonstrate that H. pylori may decorate its proteins with sugars besides unmodified PseAc, including Kdo groups, which suggests a connection between LPS assembly and glycosylation machineries. They identify PseAc with an acetamidino substitution, legionaminic acid, and bacillosamine, the latter two of which are involved in C. jejuni protein glycosylation but have never before been seen in H. pylori glycoproteins.Citation72
In opposition to its well-developed O-linked glycosylation system, H. pylori does not seem to generate N-linked glycoproteins. In the pgl locus, close relative C. jejuni encodes all the machinery necessary to generate a defined heptasaccharide chain for protein N-conjugation. PglB, a periplasmic oligosaccharyltransferase, determines the fate of the heptasaccharide by either releasing it or transferring it to an asparagine residue on an acceptor protein. Interestingly, release of the heptasaccharide into the periplasm seems to be osmoregulated, declining with increasing environmental solute concentrations. This is thought to be a mechanism of coping with salt stress and an evolutionary measure to ensure maintenance of N-linked protein glycosylation.Citation73 Of the proteins encoded by the pgl locus, H. pylori only seems to possess a homolog to PglK, which it encodes in the wzk gene. In C. jejuni, PglK flips the lipid-linked heptasaccharide donor to the periplasmic face of the inner membrane; from there, the heptasaccharide can be N-conjugated to a protein in the periplasm.Citation74 However, instead of transporting a glycan chain to be used in N-glycosylation, H. pylori Wzk is involved in LPS transport, moving the lipid-linked O-antigen glycan chain of H. pylori across the inner bacterial membrane ().Citation8
Glycoengineering
Glycoproteins can be exploited for a number of biotechnological applications, from treatment of autoimmune disease to vaccine development. The field of glycoengineering has grown significantly in the last decade, as researchers have discovered how to harness various bacterial glycosylation enzymes for development of designer sugars and glycoproteins. Recently, focus has switched from intricate chemical synthesis to utilization of living bacteria to mass-produce customized glycoproteins.
For example, Lewis antigens are an attractive candidate for treating autoimmune disorders due to their immunosuppressant effect upon interaction with DC-SIGN.Citation48 Hug et al.Citation75 described an all-enzymatic system that generates a tetrasaccharide chain composed of galactose and GlcNAc, N-links it to an acceptor protein in vivo, and then decorates the conjugated glycan with a terminal Lex fucose residue in vitro. To achieve this, E. coli is functionally reconstituted with enzymes from Hemophilus influenza, C. jejuni, and H. pylori, resulting in a cell that produces a glycosylated protein none of the four contributing bacterial species can create on their own. The enzymes work together to build the backbone tetrasaccharide, with E. coli WecA () transferring the initiating GlcNAc residue and the glycosyltransferase from Hemophilus influenza finishing out the three remaining sugar residues. The chain is then transferred by the C. jejuni oligosaccharyltransferase, PglB to the acceptor protein AcrA, a C. jejuni protein that carries two N-glycosylation acceptor sites. The final step involves addition of a fucose residue by the H. pylori α(1, 3) fucosyltransferase, which occurs in vitro due to the inability of E. coli to produce the GDP-fucose donor required for the reaction. The authors suggest that the system provides a foundation for synthesis of an expansive variety of glyco-conjugates besides Lex, as the tetrasaccharide may be modified to produce many different human antigens. With Le antigens as a starting point, the multi-faceted and diverse glycosylation machineries of different bacteria may be used together to produce a wide array of glyco-conjugates with extensive potential therapeutic applications.
Altman et al.Citation76 took a different route, proposing a glycoengineering vaccine development strategy that involves various forms of truncated LPS conjugated to tetanus toxoid (TT) or bovine serum albumin (BSA). Briefly, these researchers use a β(1,4)-GalT H. pylori mutant that synthesizes an extended α(1,6) core glycan in lieu of an O-antigen chain. From this mutant, they generate both delipidated and deacylated LPS and link each to TT and/or BSA through a Kdo sugar and chemical spacer. They find the delipidated conjugates to be immunogenic in rabbits and mice, eliciting strong IgG responses. Excitingly, the rabbit post-immune sera prove to be broadly cross-reactive with whole-cell H. pylori and bactericidal against α(1,6)-glycan-expressing strains. The bottom line of this study is the demonstration of partial protection against H. pylori challenge by one of the engineered conjugates, suggesting that delipidated LPS from this mutant strain and others should be pursued as a highly promising vaccine development approach.
Helicobacter pylori and Cholesterol
H. pylori has the unique ability to incorporate the steroid hormone cholesterol into its membrane as a surprisingly large constituent, up to 25% of the lipid membrane content.Citation77,Citation78 However, H. pylori lacks the genes required for de novo sterol synthesis, and therefore must obtain cholesterol from its environment.Citation40,Citation77,Citation79 Indeed, H. pylori extracts membrane-bound cholesterol from host cells, which it then glucosylates and incorporates into the membrane. Cholesterol affords the bacterium a number of advantages during the infection process; this steroid hormone has been shown to promote colonization, innate immune evasion, and antibiotic resistance.Citation80,Citation81 It may also reinforce the bacterial membrane, promoting resistance to toxic host compounds.Citation82,Citation83
Wunder et al.Citation80 delivered a seminal set of data that helped define the role of membrane-incorporated cholesterol in promoting pathogenesis. They find that H. pylori can sense and move toward cholesterol in its environment at levels as low as 250 μM, extract it from the cholesterol-rich lipid rafts of epithelial cells, glucosylate it by the action of a cholesterol-α-glucosyltransferase, Hp0421, and integrate it into the membrane.Citation80 In the absence of Hp0421, H. pylori is more susceptible to phagocytosis and therefore triggers a strong T cell-mediated immune response.Citation84,Citation85 Conversely, wild-type bacteria are phagocytosed to a limited extent and generate a weak immune response. Further, high concentrations of cholesterol encourage phagocytosis, suggesting that cholesterol is steadily converted to cholesteryl-α-glucoside and this is required to evade phagocytosis.Citation80 Mice infected with the hp0421 mutant strain were able to rapidly clear the infection, suggesting that α-glucosylation of cholesterol is required for survival in vivo via evasion of phagocytosis and T-cell activation.
H. pylori has been shown to associate with cholesterol-rich host cell membranes, specifically lipid rafts.Citation80 Further, infection with wild-type H. pylori is known to cause clustering of lipid rafts at sites of host-pathogen contact, thereby concentrating local cholesterol levels.Citation86 Some of this cholesterol is extracted from the host cell membrane and converted into cholesteryl-α-glucosides. However, the pathogen seems to maintain a balance with the host membrane, as host cell cholesterol-rich domains are also required for proper assembly and function of the bacterial T4SS. Host membrane cholesterol most likely contributes by providing a sturdy rigid membrane for docking of the needle-like apparatus, which can then inject toxin CagA into the cytoplasm of host cells. In support of this, Lai et al.Citation86 showed that cholesterol depletion in host cells prevents translocation and phosphorylation of the CagA toxin, as well as its cellular functions including induction of the motile “hummingbird” phenotype and IL-8 secretion. Interestingly, the same phenotypes are seen with the hp0421 mutant, suggesting that α-glucosylation of cholesterol is also required for T4SS functionality and lipid raft clustering.Citation85 As part of this process, the host receptor for the bacterial T4SS has been identified as α5β1 integrin, which binds protein CagL at the periphery of the apparatus.Citation87 The integrin is raft-associated and therefore also recruited to sites of H. pylori-host cell contact, most likely by association with cholesterol. This important host integrin additionally contributes to recognition of bacterial peptidoglycan by cooperation with the Nod1 receptor; this makes sense, as peptidoglycan is thought to be delivered to the host cell via the T4SS.Citation62,Citation88 These lines of evidence suggest that adherence of H. pylori to the host cell leads to significant cholesterol-dependent membrane reorganization, promoting proper function of the T4SS and its downstream effectors. Overall, cholesterol is an important host-derived component utilized by H. pylori to reinforce its membrane, evade phagocytosis, and ensure assembly and function of vital virulence factors.
Conclusions
H. pylori has evolved into a professional gastric pathogen, able to survive and thrive in environmental conditions that kill most other bacteria. Clearly, glyco-conjugates are essential in this multi-layered pathogenic strategy that involves evading the host immune response so that it can remain hidden in the gastric epithelium. Several questions remain at the forefront of the field; for example, the full H. pylori LPS transport pathway remains to be elucidated, as well as determination of how H. pylori senses the Lewis antigen profile of the host. In the same vein, it is still unclear how or whether H. pylori remodels its LPS in the host as it does in vitro. Additionally, the repertoire of O-glycoproteins continues to expand, requiring innovative techniques for detailed characterization. Mechanisms for sensing cholesterol remain undefined, as well as how cholesteryl α-glucosides prevent phagocytosis. Finally, with the advent of glycoengineering, it may be possible in the near future to design saccharides and/or glycoproteins that elicit immune responses appropriate for vaccine development. Continuing to study the myriad ways in which H. pylori uses glyco-conjugates to stay one step ahead of the host is the best approach to close these gaps in the field and lay the groundwork for development of novel therapeutics.
| Abbreviations: | ||
| GlcNAc | = | N-acetylglucosamine |
| UndP/PP | = | undecaprenyl phosphate/pyrophosphate |
| Kdo | = | 3-deoxy-D-manno-octulosonic acid |
| pEtN | = | phophorylethanolamine |
| Le | = | Lewis |
| PG | = | peptidoglycan |
| MurNAc | = | N-acetylmuramic acid |
| T4SS | = | type 4 secretion system |
Acknowledgments
This work was supported by Grant AI064184 from the National Institutes of Health (NIH) to MST.
Submitted
06/10/2013
Revised
07/05/2013
Accepted
07/11/2013
Disclosure of Potential Conflicts of Interest
No potential conflict of interest was disclosed.
Related Research Data
References
- Blaser MJ. Helicobacter pylori and gastric diseases. BMJ 1998; 316:1507 - 10; http://dx.doi.org/10.1136/bmj.316.7143.1507; PMID: 9582144
- Brown LM. Helicobacter pylori: epidemiology and routes of transmission. Epidemiol Rev 2000; 22:283 - 97; http://dx.doi.org/10.1093/oxfordjournals.epirev.a018040; PMID: 11218379
- Raetz CRH, Whitfield C. Lipopolysaccharide endotoxins. Annu Rev Biochem 2002; 71:635 - 700; http://dx.doi.org/10.1146/annurev.biochem.71.110601.135414; PMID: 12045108
- Lerouge I, Vanderleyden J. O-antigen structural variation: mechanisms and possible roles in animal/plant-microbe interactions. FEMS Microbiol Rev 2002; 26:17 - 47; http://dx.doi.org/10.1111/j.1574-6976.2002.tb00597.x; PMID: 12007641
- Moran AP. Relevance of fucosylation and Lewis antigen expression in the bacterial gastroduodenal pathogen Helicobacter pylori.. Carbohydr Res 2008; 343:1952 - 65; http://dx.doi.org/10.1016/j.carres.2007.12.012; PMID: 18279843
- Berg DE, Hoffman PS, Appelmelk BJ, Kusters JG. The Helicobacter pylori genome sequence: genetic factors for long life in the gastric mucosa. Trends Microbiol 1997; 5:468 - 74; http://dx.doi.org/10.1016/S0966-842X(97)01164-5; PMID: 9447657
- Hug I, Feldman MF. Analogies and homologies in lipopolysaccharide and glycoprotein biosynthesis in bacteria. [cited 2013 May 7 ] Glycobiology 2011; 21:138 - 51; http://dx.doi.org/10.1093/glycob/cwq148; PMID: 20871101
- Hug I, Couturier MR, Rooker MM, Taylor DE, Stein M, Feldman MF. Helicobacter pylori lipopolysaccharide is synthesized via a novel pathway with an evolutionary connection to protein N-glycosylation. PLoS Pathog 2010; 6:e1000819; http://dx.doi.org/10.1371/journal.ppat.1000819; PMID: 20333251
- Alaimo C, Catrein I, Morf L, Marolda CL, Callewaert N, Valvano MA, et al. Two distinct but interchangeable mechanisms for flipping of lipid-linked oligosaccharides. EMBO J 2006; 25:967 - 76; http://dx.doi.org/10.1038/sj.emboj.7601024; PMID: 16498400
- El Ghachi M, Derbise A, Bouhss A, Mengin-Lecreulx D. Identification of multiple genes encoding membrane proteins with undecaprenyl pyrophosphate phosphatase (UppP) activity in Escherichia coli.. J Biol Chem 2005; 280:18689 - 95; http://dx.doi.org/10.1074/jbc.M412277200; PMID: 15778224
- Sperandeo P, Dehò G, Polissi A. The lipopolysaccharide transport system of Gram-negative bacteria. Biochim Biophys Acta 2009; 1791:594 - 602; http://dx.doi.org/10.1016/j.bbalip.2009.01.011; PMID: 19416651
- Liechti G, Goldberg JB. Outer membrane biogenesis in Escherichia coli, Neisseria meningitidis, and Helicobacter pylori: paradigm deviations in H. pylori.. Front Cell Infect Microbiol 2012; 2:29; http://dx.doi.org/10.3389/fcimb.2012.00029; PMID: 22919621
- Bos MP, Tommassen J. The LptD chaperone LptE is not directly involved in lipopolysaccharide transport in Neisseria meningitidis.. J Biol Chem 2011; 286:28688 - 96; http://dx.doi.org/10.1074/jbc.M111.239673; PMID: 21705335
- Raetz CRH, Reynolds CM, Trent MS, Bishop RE. Lipid A modification systems in gram-negative bacteria. Annu Rev Biochem 2007; 76:295 - 329; http://dx.doi.org/10.1146/annurev.biochem.76.010307.145803; PMID: 17362200
- Muotiala A, Helander IM, Pyhälä L, Kosunen TU, Moran AP. Low biological activity of Helicobacter pylori lipopolysaccharide. Infect Immun 1992; 60:1714 - 6; PMID: 1548097
- Ogawa T, Suda Y, Kashihara W, Hayashi T, Shimoyama T, Kusumoto S, et al. Immunobiological activities of chemically defined lipid A from Helicobacter pylori LPS in comparison with Porphyromonas gingivalis lipid A and Escherichia coli-type synthetic lipid A (compound 506). Vaccine 1997; 15:1598 - 605; http://dx.doi.org/10.1016/S0264-410X(97)00102-3; PMID: 9364689
- Ogawa T, Asai Y, Sakai Y, Oikawa M, Fukase K, Suda Y, et al. Endotoxic and immunobiological activities of a chemically synthesized lipid A of Helicobacter pylori strain 206-1. FEMS Immunol Med Microbiol 2003; 36:1 - 7; http://dx.doi.org/10.1016/S0928-8244(03)00093-2; PMID: 12727359
- Raetz CR. Biochemistry of endotoxins. Annu Rev Biochem 1990; 59:129 - 70; http://dx.doi.org/10.1146/annurev.bi.59.070190.001021; PMID: 1695830
- Tran AX, Whittimore JD, Wyrick PB, McGrath SC, Cotter RJ, Trent MS. The lipid A 1-phosphatase of Helicobacter pylori is required for resistance to the antimicrobial peptide polymyxin. J Bacteriol 2006; 188:4531 - 41; http://dx.doi.org/10.1128/JB.00146-06; PMID: 16740959
- Stead CM, Beasley A, Cotter RJ, Trent MS. Deciphering the unusual acylation pattern of Helicobacter pylori lipid A. J Bacteriol 2008; 190:7012 - 21; http://dx.doi.org/10.1128/JB.00667-08; PMID: 18757539
- Tran AX, Karbarz MJ, Wang X, Raetz CRH, McGrath SC, Cotter RJ, et al. Periplasmic cleavage and modification of the 1-phosphate group of Helicobacter pylori lipid A. J Biol Chem 2004; 279:55780 - 91; http://dx.doi.org/10.1074/jbc.M406480200; PMID: 15489235
- Stead CM, Zhao J, Raetz CRH, Trent MS. Removal of the outer Kdo from Helicobacter pylori lipopolysaccharide and its impact on the bacterial surface. Mol Microbiol 2010; 78:837 - 52; http://dx.doi.org/10.1111/j.1365-2958.2010.07304.x; PMID: 20659292
- Cullen TW, Giles DK, Wolf LN, Ecobichon C, Boneca IG, Trent MS. Helicobacter pylori versus the host: remodeling of the bacterial outer membrane is required for survival in the gastric mucosa. PLoS Pathog 2011; 7:e1002454; http://dx.doi.org/10.1371/journal.ppat.1002454; PMID: 22216004
- Kawasaki K, Ernst RK, Miller SI. 3-O-deacylation of lipid A by PagL, a PhoP/PhoQ-regulated deacylase of Salmonella typhimurium, modulates signaling through Toll-like receptor 4. J Biol Chem 2004; 279:20044 - 8; http://dx.doi.org/10.1074/jbc.M401275200; PMID: 15014080
- Coats SR, Pham T-TT, Bainbridge BW, Reife RA, Darveau RP. MD-2 mediates the ability of tetra-acylated and penta-acylated lipopolysaccharides to antagonize Escherichia coli lipopolysaccharide at the TLR4 signaling complex. J Immunol 2005; 175:4490 - 8; PMID: 16177092
- Teghanemt A, Zhang D, Levis EN, Weiss JP, Gioannini TL. Molecular basis of reduced potency of underacylated endotoxins. J Immunol 2005; 175:4669 - 76; PMID: 16177114
- Cullen TW, Trent MS. A link between the assembly of flagella and lipooligosaccharide of the Gram-negative bacterium Campylobacter jejuni.. Proc Natl Acad Sci U S A 2010; 107:5160 - 5; http://dx.doi.org/10.1073/pnas.0913451107; PMID: 20194750
- Scott NE, Nothaft H, Edwards AVG, Labbate M, Djordjevic SP, Larsen MR, et al. Modification of the Campylobacter jejuni N-linked glycan by EptC protein-mediated addition of phosphoethanolamine. J Biol Chem 2012; 287:29384 - 96; http://dx.doi.org/10.1074/jbc.M112.380212; PMID: 22761430
- Cullen TW, O’Brien JP, Hendrixson DR, Giles DK, Hobb RI, Thompson SA, et al. EptC of Campylobacter jejuni mediates phenotypes involved in host interactions and virulence. Infect Immun 2013; 81:430 - 40; http://dx.doi.org/10.1128/IAI.01046-12; PMID: 23184526
- Simoons-Smit IM, Appelmelk BJ, Verboom T, Negrini R, Penner JL, Aspinall GO, et al. Typing of Helicobacter pylori with monoclonal antibodies against Lewis antigens in lipopolysaccharide. J Clin Microbiol 1996; 34:2196 - 200; PMID: 8862584
- Heneghan MA, McCarthy CF, Moran AP. Relationship of blood group determinants on Helicobacter pylori lipopolysaccharide with host lewis phenotype and inflammatory response. Infect Immun 2000; 68:937 - 41; http://dx.doi.org/10.1128/IAI.68.2.937-941.2000; PMID: 10639467
- Kocharova NA, Knirel YA, Widmalm G, Jansson PE, Moran AP. Structure of an atypical O-antigen polysaccharide of Helicobacter pylori containing a novel monosaccharide 3-C-methyl-D-mannose. Biochemistry 2000; 39:4755 - 60; http://dx.doi.org/10.1021/bi992635k; PMID: 10769132
- Logan SM, Conlan JW, Monteiro MA, Wakarchuk WW, Altman E. Functional genomics of Helicobacter pylori: identification of a beta-1,4 galactosyltransferase and generation of mutants with altered lipopolysaccharide. Mol Microbiol 2000; 35:1156 - 67; http://dx.doi.org/10.1046/j.1365-2958.2000.01784.x; PMID: 10712696
- Pohl MA, Romero-Gallo J, Guruge JL, Tse DB, Gordon JI, Blaser MJ. Host-dependent Lewis (Le) antigen expression in Helicobacter pylori cells recovered from Leb-transgenic mice. J Exp Med 2009; 206:3061 - 72; http://dx.doi.org/10.1084/jem.20090683; PMID: 20008521
- Logan SM, Altman E, Mykytczuk O, Brisson J-R, Chandan V, Schur MJ, et al. Novel biosynthetic functions of lipopolysaccharide rfaJ homologs from Helicobacter pylori.. Glycobiology 2005; 15:721 - 33; http://dx.doi.org/10.1093/glycob/cwi057; PMID: 15814825
- Aspinall GO, Monteiro MA, Moran AP, Pang H, Penner JL, Shaver RT. Lipopolysaccharides from Helicobacter pylori.. Prog Clin Biol Res 1995; 392:93 - 101; PMID: 8524968
- Aspinall GO, Monteiro MA, Pang H, Walsh EJ, Moran AP. Lipopolysaccharide of the Helicobacter pylori type strain NCTC 11637 (ATCC 43504): structure of the O antigen chain and core oligosaccharide regions. Biochemistry 1996; 35:2489 - 97; http://dx.doi.org/10.1021/bi951852s; PMID: 8652593
- Nilsson C, Skoglund A, Moran AP, Annuk H, Engstrand L, Normark S. An enzymatic ruler modulates Lewis antigen glycosylation of Helicobacter pylori LPS during persistent infection. Proc Natl Acad Sci U S A 2006; 103:2863 - 8; http://dx.doi.org/10.1073/pnas.0511119103; PMID: 16477004
- Appelmelk BJ, Monteiro MA, Martin SL, Moran AP, Vandenbroucke-Grauls CM. Why Helicobacter pylori has Lewis antigens. Trends Microbiol 2000; 8:565 - 70; http://dx.doi.org/10.1016/S0966-842X(00)01875-8; PMID: 11115753
- Alm RA, Ling LS, Moir DT, King BL, Brown ED, Doig PC, et al. Genomic-sequence comparison of two unrelated isolates of the human gastric pathogen Helicobacter pylori.. Nature 1999; 397:176 - 80; http://dx.doi.org/10.1038/16495; PMID: 9923682
- Wang G, Ge Z, Rasko DA, Taylor DE. Lewis antigens in Helicobacter pylori: biosynthesis and phase variation. Mol Microbiol 2000; 36:1187 - 96; http://dx.doi.org/10.1046/j.1365-2958.2000.01934.x; PMID: 10931272
- Appelmelk BJ, Negrini R, Moran AP, Kuipers EJ. Molecular mimicry between Helicobacter pylori and the host. Trends Microbiol 1997; 5:70 - 3; http://dx.doi.org/10.1016/S0966-842X(96)10084-6; PMID: 9108933
- Pohl MA, Kienesberger S, Blaser MJ. Novel functions for glycosyltransferases Jhp0562 and GalT in Lewis antigen synthesis and variation in Helicobacter pylori.. Infect Immun 2012; 80:1593 - 605; http://dx.doi.org/10.1128/IAI.00032-12; PMID: 22290141
- Nilsson C, Skoglund A, Moran AP, Annuk H, Engstrand L, Normark S. Lipopolysaccharide diversity evolving in Helicobacter pylori communities through genetic modifications in fucosyltransferases. PLoS One 2008; 3:e3811; http://dx.doi.org/10.1371/journal.pone.0003811; PMID: 19043574
- Monteiro MA, St Michael F, Rasko DA, Taylor DE, Conlan JW, Chan KH, et al. Helicobacter pylori from asymptomatic hosts expressing heptoglycan but lacking Lewis O-chains: Lewis blood-group O-chains may play a role in Helicobacter pylori induced pathology. Biochem Cell Biol 2001; 79:449 - 59; PMID: 11527214
- Edwards NJ, Monteiro MA, Faller G, Walsh EJ, Moran AP, Roberts IS, et al. Lewis X structures in the O antigen side-chain promote adhesion of Helicobacter pylori to the gastric epithelium. Mol Microbiol 2000; 35:1530 - 9; http://dx.doi.org/10.1046/j.1365-2958.2000.01823.x; PMID: 10760152
- Fowler M, Thomas RJ, Atherton J, Roberts IS, High NJ. Galectin-3 binds to Helicobacter pylori O-antigen: it is upregulated and rapidly secreted by gastric epithelial cells in response to H. pylori adhesion. Cell Microbiol 2006; 8:44 - 54; http://dx.doi.org/10.1111/j.1462-5822.2005.00599.x; PMID: 16367865
- Bergman MP, Engering A, Smits HH, van Vliet SJ, van Bodegraven AA, Wirth H-P, et al. Helicobacter pylori modulates the T helper cell 1/T helper cell 2 balance through phase-variable interaction between lipopolysaccharide and DC-SIGN. J Exp Med 2004; 200:979 - 90; http://dx.doi.org/10.1084/jem.20041061; PMID: 15492123
- Ghuysen J-M, Hakenbeck R. Bacterial Cell Wall. Elsevier; 1994.
- Höltje JV. Growth of the stress-bearing and shape-maintaining murein sacculus of Escherichia coli.. Microbiol Mol Biol Rev 1998; 62:181 - 203; PMID: 9529891
- Takebe I. Extent of cross linkage in the murein sacculus of Escherichia coli B cell wall. Biochim Biophys Acta 1965; 101:124 - 6; http://dx.doi.org/10.1016/0926-6534(65)90038-2; PMID: 14329278
- Costa K, Bacher G, Allmaier G, Dominguez-Bello MG, Engstrand L, Falk P, et al. The morphological transition of Helicobacter pylori cells from spiral to coccoid is preceded by a substantial modification of the cell wall. J Bacteriol 1999; 181:3710 - 5; PMID: 10368145
- Harz H, Burgdorf K, Höltje JV. Isolation and separation of the glycan strands from murein of Escherichia coli by reversed-phase high-performance liquid chromatography. Anal Biochem 1990; 190:120 - 8; http://dx.doi.org/10.1016/0003-2697(90)90144-X; PMID: 2285138
- Sycuro LK, Pincus Z, Gutierrez KD, Biboy J, Stern CA, Vollmer W, et al. Peptidoglycan crosslinking relaxation promotes Helicobacter pylori’s helical shape and stomach colonization. Cell 2010; 141:822 - 33; http://dx.doi.org/10.1016/j.cell.2010.03.046; PMID: 20510929
- Bonis M, Ecobichon C, Guadagnini S, Prévost M-C, Boneca IGA. A M23B family metallopeptidase of Helicobacter pylori required for cell shape, pole formation and virulence. Mol Microbiol 2010; 78:809 - 19; http://dx.doi.org/10.1111/j.1365-2958.2010.07383.x; PMID: 20815828
- Sycuro LK, Wyckoff TJ, Biboy J, Born P, Pincus Z, Vollmer W, et al. Multiple peptidoglycan modification networks modulate Helicobacter pylori’s cell shape, motility, and colonization potential. PLoS Pathog 2012; 8:e1002603; http://dx.doi.org/10.1371/journal.ppat.1002603; PMID: 22457625
- Frirdich E, Biboy J, Adams C, Lee J, Ellermeier J, Gielda LD, et al. Peptidoglycan-modifying enzyme Pgp1 is required for helical cell shape and pathogenicity traits in Campylobacter jejuni.. PLoS Pathog 2012; 8:e1002602; http://dx.doi.org/10.1371/journal.ppat.1002602; PMID: 22457624
- Chaput C, Ecobichon C, Cayet N, Girardin SE, Werts C, Guadagnini S, et al. Role of AmiA in the morphological transition of Helicobacter pylori and in immune escape. PLoS Pathog 2006; 2:e97; http://dx.doi.org/10.1371/journal.ppat.0020097; PMID: 17002496
- van Heijenoort J. Peptidoglycan hydrolases of Escherichia coli.. [cited 2013 May 22 ] Microbiol Mol Biol Rev 2011; 75:636 - 63; http://dx.doi.org/10.1128/MMBR.00022-11; PMID: 22126997
- Scheurwater E, Reid CW, Clarke AJ. Lytic transglycosylases: bacterial space-making autolysins. Int J Biochem Cell Biol 2008; 40:586 - 91; http://dx.doi.org/10.1016/j.biocel.2007.03.018; PMID: 17468031
- Helicobacter pylori: physiology and genetics. Washington, DC: ASM Press; 2001.
- Viala J, Chaput C, Boneca IG, Cardona A, Girardin SE, Moran AP, et al. Nod1 responds to peptidoglycan delivered by the Helicobacter pylori cag pathogenicity island. Nat Immunol 2004; 5:1166 - 74; http://dx.doi.org/10.1038/ni1131; PMID: 15489856
- Wang G, Olczak A, Forsberg LS, Maier RJ. Oxidative stress-induced peptidoglycan deacetylase in Helicobacter pylori.. J Biol Chem 2009; 284:6790 - 800; http://dx.doi.org/10.1074/jbc.M808071200; PMID: 19147492
- Wang G, Maier SE, Lo LF, Maier G, Dosi S, Maier RJ. Peptidoglycan deacetylation in Helicobacter pylori contributes to bacterial survival by mitigating host immune responses. Infect Immun 2010; 78:4660 - 6; http://dx.doi.org/10.1128/IAI.00307-10; PMID: 20805339
- Wang G, Lo LF, Forsberg LS, Maier RJ. Helicobacter pylori peptidoglycan modifications confer lysozyme resistance and contribute to survival in the host. MBio 2012; 3:e00409 - 12; http://dx.doi.org/10.1128/mBio.00409-12; PMID: 23221800
- Vollmer W. Structural variation in the glycan strands of bacterial peptidoglycan. FEMS Microbiol Rev 2008; 32:287 - 306; http://dx.doi.org/10.1111/j.1574-6976.2007.00088.x; PMID: 18070068
- Schirm M, Soo EC, Aubry AJ, Austin J, Thibault P, Logan SM. Structural, genetic and functional characterization of the flagellin glycosylation process in Helicobacter pylori.. Mol Microbiol 2003; 48:1579 - 92; http://dx.doi.org/10.1046/j.1365-2958.2003.03527.x; PMID: 12791140
- Thibault P, Logan SM, Kelly JF, Brisson JR, Ewing CP, Trust TJ, et al. Identification of the carbohydrate moieties and glycosylation motifs in Campylobacter jejuni flagellin. J Biol Chem 2001; 276:34862 - 70; http://dx.doi.org/10.1074/jbc.M104529200; PMID: 11461915
- McNally DJ, Hui JPM, Aubry AJ, Mui KKK, Guerry P, Brisson J-R, et al. Functional characterization of the flagellar glycosylation locus in Campylobacter jejuni 81-176 using a focused metabolomics approach. J Biol Chem 2006; 281:18489 - 98; http://dx.doi.org/10.1074/jbc.M603777200; PMID: 16684771
- Gilbreath JJ, Cody WL, Merrell DS, Hendrixson DR. Change is good: variations in common biological mechanisms in the epsilonproteobacterial genera Campylobacter and Helicobacter. Microbiol Mol Biol Rev 2011; 75:84 - 132; http://dx.doi.org/10.1128/MMBR.00035-10; PMID: 21372321
- Asakura H, Churin Y, Bauer B, Boettcher JP, Bartfeld S, Hashii N, et al. Helicobacter pylori HP0518 affects flagellin glycosylation to alter bacterial motility. Mol Microbiol 2010; 78:1130 - 44; http://dx.doi.org/10.1111/j.1365-2958.2010.07393.x; PMID: 21091500
- Höpf PS, Ford RS, Zebian N, Merkx-Jacques A, Vijayakumar S, Ratnayake D, et al. Protein glycosylation in Helicobacter pylori: beyond the flagellins?. PLoS One 2011; 6:e25722; http://dx.doi.org/10.1371/journal.pone.0025722; PMID: 21984942
- Nothaft H, Liu X, McNally DJ, Li J, Szymanski CM. Study of free oligosaccharides derived from the bacterial N-glycosylation pathway. [cited 2013 Jun 5 ] Proc Natl Acad Sci U S A 2009; 106:15019 - 24; http://dx.doi.org/10.1073/pnas.0903078106; PMID: 19706478
- Szymanski CM, Yao R, Ewing CP, Trust TJ, Guerry P. Evidence for a system of general protein glycosylation in Campylobacter jejuni.. Mol Microbiol 1999; 32:1022 - 30; http://dx.doi.org/10.1046/j.1365-2958.1999.01415.x; PMID: 10361304
- Hug I, Zheng B, Reiz B, Whittal RM, Fentabil MA, Klassen JS, et al. Exploiting bacterial glycosylation machineries for the synthesis of a Lewis antigen-containing glycoprotein. J Biol Chem 2011; 286:37887 - 94; http://dx.doi.org/10.1074/jbc.M111.287755; PMID: 21878645
- Altman E, Chandan V, Harrison BA, Veloso-Pita R, Li J, KuoLee R, et al, Regional Helicobacter pylori Study Group. Design and immunological properties of Helicobacter pylori glycoconjugates based on a truncated lipopolysaccharide lacking Lewis antigen and comprising an α-1,6-glucan chain. Vaccine 2012; 30:7332 - 41; http://dx.doi.org/10.1016/j.vaccine.2012.04.035; PMID: 22534169
- Ansorg R, Müller KD, von Recklinghausen G, Nalik HP. Cholesterol binding of Helicobacter pylori.. Zentralbl Bakteriol 1992; 276:323 - 9; http://dx.doi.org/10.1016/S0934-8840(11)80538-4; PMID: 1576402
- Haque M, Hirai Y, Yokota K, Oguma K. Lipid profiles of Helicobacter pylori and Helicobacter mustelae grown in serum-supplemented and serum-free media. Acta Med Okayama 1995; 49:205 - 11; PMID: 7502681
- Testerman TL, McGee DJ, Mobley HL. Helicobacter pylori growth and urease detection in the chemically defined medium Ham’s F-12 nutrient mixture. J Clin Microbiol 2001; 39:3842 - 50; http://dx.doi.org/10.1128/JCM.39.11.3842-3850.2001; PMID: 11682496
- Wunder C, Churin Y, Winau F, Warnecke D, Vieth M, Lindner B, et al. Cholesterol glucosylation promotes immune evasion by Helicobacter pylori.. Nat Med 2006; 12:1030 - 8; http://dx.doi.org/10.1038/nm1480; PMID: 16951684
- McGee DJ, George AE, Trainor EA, Horton KE, Hildebrandt E, Testerman TL. Cholesterol enhances Helicobacter pylori resistance to antibiotics and LL-37. Antimicrob Agents Chemother 2011; 55:2897 - 904; http://dx.doi.org/10.1128/AAC.00016-11; PMID: 21464244
- Shimomura H, Hosoda K, Hayashi S, Yokota K, Oguma K, Hirai Y. Steroids mediate resistance to the bactericidal effect of phosphatidylcholines against Helicobacter pylori.. FEMS Microbiol Lett 2009; 301:84 - 94; http://dx.doi.org/10.1111/j.1574-6968.2009.01807.x; PMID: 19843309
- Shimomura H, Hosoda K, McGee DJ, Hayashi S, Yokota K, Hirai Y. Detoxification of 7-dehydrocholesterol fatal to Helicobacter pylori is a novel role of cholesterol glucosylation. J Bacteriol 2013; 195:359 - 67; http://dx.doi.org/10.1128/JB.01495-12; PMID: 23144252
- Beigier-Bompadre M, Moos V, Belogolova E, Allers K, Schneider T, Churin Y, et al. Modulation of the CD4+ T-cell response by Helicobacter pylori depends on known virulence factors and bacterial cholesterol and cholesterol α-glucoside content. J Infect Dis 2011; 204:1339 - 48; http://dx.doi.org/10.1093/infdis/jir547; PMID: 21921201
- Wang H-J, Cheng W-C, Cheng H-H, Lai C-H, Wang W-C. Helicobacter pylori cholesteryl glucosides interfere with host membrane phase and affect type IV secretion system function during infection in AGS cells. Mol Microbiol 2012; 83:67 - 84; http://dx.doi.org/10.1111/j.1365-2958.2011.07910.x; PMID: 22053852
- Lai C-H, Chang Y-C, Du S-Y, Wang H-J, Kuo C-H, Fang S-H, et al. Cholesterol depletion reduces Helicobacter pylori CagA translocation and CagA-induced responses in AGS cells. Infect Immun 2008; 76:3293 - 303; http://dx.doi.org/10.1128/IAI.00365-08; PMID: 18443091
- Kwok T, Zabler D, Urman S, Rohde M, Hartig R, Wessler S, et al. Helicobacter exploits integrin for type IV secretion and kinase activation. Nature 2007; 449:862 - 6; http://dx.doi.org/10.1038/nature06187; PMID: 17943123
- Hutton ML, Kaparakis-Liaskos M, Turner L, Cardona A, Kwok T, Ferrero RL. Helicobacter pylori exploits cholesterol-rich microdomains for induction of NF-kappaB-dependent responses and peptidoglycan delivery in epithelial cells. Infect Immun 2010; 78:4523 - 31; http://dx.doi.org/10.1128/IAI.00439-10; PMID: 20713621