
Abstract
Glucagon, a peptide hormone secreted from the α-cells of the pancreatic islets, is critical for blood glucose homeostasis. We reviewed the literature and employed a computational systems analysis of intracellular metabolic and electrical regulation of glucagon secretion to better understand these processes. The mathematical model of α-cell metabolic parameters is based on our previous model for pancreatic β-cells. We also formulated an ionic model for action potentials that incorporates Ca2+, K+, Na+ and Cl- currents. Metabolic and ionic models are coupled to the equations describing Ca2+ homeostasis and glucagon secretion that depends on activation of specific voltage-gated Ca2+ channels. Paracrine and endocrine regulations were analyzed with an emphasis on their effects on a hyperpolarization of membrane potential. This general model simulates and gives insight into the mechanisms of regulation of glucagon secretion under a wide range of experimental conditions. We also reviewed and analyzed dysfunctional mechanisms in α-cells to determine key pharmacological targets for modulating glucagon secretion in type 1 and 2 diabetes.
1. Introduction
Glucagon is the principal blood-glucose-increasing (counter-regulatory) hormone of mammals, with a variety of other effects as well. It is released from α-cells of the pancreatic islets, comprising only a few percent of the total cell mass of an islet. Electrophysiological studies on individual -cells have revealed that they are electrically excitable. Stimulation of glucagon secretion is secondary to increased action potential (AP) firing and also may be affected by paracrine signals (mediated by factors released from neighboring β- and δ-cells) including insulin, Zn2+, gamma amino butyric acid (GABA) and somatostatin, by endocrine signals such as glucagon-like peptide-1 (GLP-1) or leptin and by autonomic input (for a review, see refs.Citation1–Citation5). Glucagon plays a critical role in glucose homeostasis, diabetic ketoacidosis and its secretion is dysregulated in hypoglycemia associated with diabetes.Citation4,Citation6 Thus, it is important to elucidate the signals that trigger glucagon secretion and the transduction of these signals within the α-cell.
Pancreatic α- and β-cells show a completely different behavior depending on the serum glucose level. However, an inhibitory effect of glucose on the α-cell, like its stimulatory effect on the β-cell, requires metabolism of the sugar. Indeed, the α-cells are equipped with many of the proteins involved in the metabolic glucose sensing of the β-cell, including glucokinaseCitation7 and ATP-regulated K+-channels (KATP-channels).Citation2,Citation8 Glucose may also increase the intracellular concentration of ATP in α-cells.Citation9,Citation10
Electrical activity in α-cells is difficult to study within an intact islet and so this process is typically studied as isolated purified cells in vitro. Glucose concentrations modulate pancreatic α-cell electrical activity, Ca2+ signal and hormone secretion. At low glucose concentrations (3 mM or below) isolated α-cells can be electrically active and AP firing is required for glucagon secretion. Increased glucose concentration paradoxically suppresses electrical activity whereas activity in β-cells in increased.Citation1,Citation11,Citation12 This electrical activity usually involves a complex interaction between several different ion channels as in pancreatic β-cells. Pancreatic α-cells express voltage-gated Na+, K+ and Ca2+ channels that regulate AP firing at low glucose level.Citation1,Citation2,Citation8
Despite the recent increase in our knowledge of α-cell physiology and biochemistry, we still lack a coherent model of regulation in α-cells. Significant discrepancies exist among studies of glucagon secretion. It is at present unclear how increased glucose metabolism is coupled to the suppression of electrical activity in the α-cell, and the topic is subject to conflicting reports with the possibility of species differences (for review, see refs. Citation1 and Citation2). The present work attempts to bridge the gap between electrophysiological recordings, metabolic measurements, Ca2+ handling and glucagon secretion.
To achieve a better understanding of α-cell physiology and its role in diabetes we undertook an integrated consideration of data obtained on isolated α-cells, human and rodent islets and in clinical investigations. We used a computational systems biology approach with construction of a mathematical model of intracellular processes. We have previously focused on applying this approach to mechanisms of glucose sensing of insulin secretion, Ca2+ dynamics, electrophysiology events and exocytosis in pancreatic β-cells.Citation13-Citation18 Here we focus on pancreatic α-cells based on our computational models for β-cells. This allows also a comparison of the mechanisms of glucose sensing in α– and β-cells. Our aim was the investigation of the properties of rodent and human α–cells with a help of a novel computational model for metabolic processes, electrical activity, Ca2+ handling and glucagon secretion.
The mathematical model for α–cells was tested in various scenarios and the results of simulations were compared with the experimental data. The model gives a reasonable fit to important aspects of experimentally measured metabolic and AP properties of α–cells and provides a framework for analyzing the role of metabolic changes and individual ionic currents on glucagon secretion. In addition to a direct effect of glucose, the influence of several paracrine and endocrine mechanisms on AP firing and glucagon release were considered.
Defective regulation of glucagon secretion clearly contributes to type 1 and 2 diabetes in a variety of ways.Citation6,Citation19,Citation20 For this reason there is significant interest in understanding how α-cells regulate glucagon secretion in humans. We have attempted to analyze these processes and evaluate how particular impairments in the mechanisms of α-cell glucose sensing can lead to glucagon release changes in diabetes. We also discuss examples of potential therapeutic strategies.
2. Components of Glucagon Secretion Regulatory Mechanism and their Mathematical Description
Here we will briefly outline the different parts of the glucagon secretion regulatory mechanisms and describe the equations and parameters utilizing in the simulations of α-cell function. A schematic diagram of the biochemical steps, Ca2+ handling, channels and mechanisms of glucagon secretion regulation in α-cell is presented in . The data used to fit the computational model in our study were taken primarily from isolated rodent α-cells and islets. Since pancreatic α-cells remain incompletely understood, there remain gaps in any completely α-cell specific data set. For the purpose of making a convenient model we have chosen plausible suggestions based on mechanisms of regulation and parameters from β-cells or other cell types to close the gaps pending additional data.
Figure 1. Schematic diagram of the ionic current, metabolic processes, Ca2+ fluxes and glucagon secretion mechanisms in pancreatic α-cells. Transmembrane currents are: IKATP is the ATP-sensitive K+ channel current, IKDr is the rapid delayed-rectifier K+ current, IKA is the high voltage-activated A-type K+ current, IKGS is the K+ current through G protein K+-channel activated by somatostatin, ICaL and ICaNL and are high-voltage-activated L-type and non L-type Ca2+ currents, ICaT is the low-voltage-activated “T-type” Ca2+ current, IPCa is the plasma membrane Ca2+-pump current, INa is the voltage-gated Na+ current, INab is the Na+ background current, IClG is the GABA activated Cl- current. Glucose equilibrates across the plasma membrane and is phosphorylated by glucokinase to glucose 6-phosphate, which initiates glycolysis. Lactate dehydrogenase (LDH) converts a portion of pyruvate to lactate. Pyruvate produced by glycolysis enters the mitochondria and is metabolized in the tricarboxylic acid (TCA) cycle, which then yields reducing equivalents in the form of NADH and FADH2. kng is the coefficient describing a nonglycolitic source of NADH in mitochondria. The transfer of electrons from these reducing equivalents through the mitochondrial electron transport chain is coupled with the pumping of protons from the mitochondrial matrix to the intermembrane space. The resulting transmembrane electrochemical gradient drives the ATP synthesis at ATP-synthase. Part of the protons may leak back through uncoupling proteins (UCPs). The shuttle systems are required for the transfer of reducing equivalents from the cytoplasm to the mitochondrial matrix. ATP is transferred to the cytosol, raising the ATP/ADP ratio. This results in the closure of the ATP sensitive K+ channels, which in turn leads to depolarization of the cell membrane. In response, the voltage-sensitive Ca2+ channels open, promoting calcium entry and triggering exocytosis of glucagon granules. ATPc and ADPfree are the free cytosolic form of ATP and ADP, G3P is the glyceraldehydes 3-phosphate, PDH is the pyruvate dehydrogenase, ANT is the adenine nucleotide translocase, Ψm is the mitochondrial membrane potential. Solid lines indicate flux of substrates, and dashed lines indicate regulating effects, where (+) represents activation and (-) repression.
2.1. Intrinsic metabolic regulation
The cellular metabolic mechanisms leading to insulin secretion in pancreatic β-cells are fairly well understood (for reviews, see refs. Citation2 and Citation15). Similar machinery may operate in α-cells. Although both cell types possess different glucose transporters, it has been demonstrated that glucose transport is not a limiting factor in the metabolism of this sugar.Citation21,Citation22 Glucokinase was found in both cell typesCitation7 and may be also the rate-limiting step for glycolysis in α-cells.Citation23 α-cells are equipped with ATP-sensitive K+ channels of the same type as those constituting the resting conductance in β-cells (see below in Sec. 3.3.2). It seems that a metabolic mechanism of sensitivity to glucose is similar in α- and β-cells: mitochondrial metabolism generates ATP leading to an increase of the ATP/ADP concentration ratio, leading to closure of KATP channels. The resulting decrease in K+ efflux causes PM depolarization which changes the rate of exocytosis of glucagon granules.Citation2,Citation24
We have recently developed a mathematical model for regulated glucose sensing in pancreatic β-cellsCitation15,Citation25 and use it here to analyze α-cells. The cytoplasmic part of the model includes equations describing glucokinase, glycolysis, lactate dehydrogenase, NAD(P)H and ATP production and consumption (). The mitochondrial part includes production of NADH, which is regulated by pyruvate dehydrogenase. NADH serves as a source of electron transfer in the chain of oxidative reactions to establish a proton motive force, driving the F1F0 ATPase. Redox shuttles and mitochondrial Ca2+ handling were also modeled. We have performed a computational analysis of β-cell fuel metabolism to quantitatively assess how cytoplasmic ATP/ADP ratio can be controlled by mitochondrial and cytoplasmic processes.Citation15,Citation25
Key differences in the rates of energetic processes were found in α-cells vs. β-cells. Increased ATP/ADP ratios and NAD(P)H signals were found in rodent α-cells compared with β-cells at low glucose levels.Citation23,Citation26 In our previous β-cell model of glucose sensitivity no ATP production was generated without glucose consumption because only glucose served as a source of ATP production during glycolysis and NADH for oxidative phosphorylation. To describe a nonglycolitic source for ATP production in the absence of glucose or in low glucose we introduced a special term (other than pyruvate influx) in the equation for mitochondrial NADH ([NADH]m) dynamics in our model of glucose sensing. Now, the following equation describes the change in [NADH]m over time:
d [NADH]m /dt = (4.6 JPYR + JTNADH – 0.1 Jhres + kng)/ Vmmit – kNADHm [NADH]m (Eqn. 1)
where kng is the novel coefficient describing a nonglycolitic source of NADH in mitochondria, JPYR is the rate of pyruvate consumption in mitochondria, JTNADH is the flux through the NADH shuttles, Jhres is the rate of proton pumping through ETC, 4.6 and 0.1 are the stoichiometric coefficients, Vmmit is the relative mitochondrial matrix fraction of the cell volume, kNADHm is the rate constant of NADH consumption in mitochondria (see for detail Eqn. 14 in ref. Citation15).
Introducing Equation 1 instead of the corresponding equation for NADP dynamics (Eqn. 14 in the modelCitation15) we obtained a “complex” model for glucose sensing that can be used for α-cells and β-cells simultaneously even at low glucose levels (see below). This model is distinguished from the previous modelCitation15 only by kng coefficient in Equation 1 and at kng = 0 these models are identical.
2.2. Channels and plasma membrane potential regulation
Glucagon release, like insulin release, is influenced by physiological α-cell electrical activity that fundamentally resembles the excitation-secretion coupling seen in many secretory cell types (for reviews, see refs. Citation3 and Citation27). Like insulin-secreting β-cells, α-cells express different types of ion channels. There are at least four different types of K+-selective channels: the ATP-sensitive K+-channel (KATP-channel), the delayed rectifying K+-channel (KDr-channel), a transient K+-channel (A-type K+ channel) and a G protein-gated K+-channel activated by somatostatin (KGS-channel). Other channels include voltage-gated Na+-channels, at least four types of voltage-gated Ca2+-channel (VGCC) (T-, N-, P/Q- and L-type) and GABAA receptor chloride-channels (refs. Citation1,Citation2,Citation8,Citation28 see also below Sec. 3.3. for details). In the Supplemental Material we describe the properties (mostly similar to that described in other cells) of each type of ion channel and their mathematical descriptions.
A schematic diagram of the α-cell specific channels is presented (). The AP in a single α-cell can be described with the following current balance differential equation
− Cm (dVP /dt) = IKATP + IKDr + IKA + IKGS + ICaL + ICaNL + ICaT + IPCa + INa + INab + IClG (Eqn. 2)
where Vp is the plasma membrane potential, t is time, Cm is the whole-cell membrane capacitance, IKATP is the ATP-sensitive K+ channel current, IKDr is the rapid delayed-rectifier K+ current, IKA is the high voltage-activated A-type K+ current, IKGS is the K+ current through G protein-gated K+-channel activated by somatostatin, ICaL is the high-voltage-activated L-type Ca2+ current, ICaNL is the high-voltage-activated non-L-type Ca2+ currents, ICaT is the low-voltage-activated “T-type” Ca2+ current, IPCa is the plasma membrane Ca2+-pump current, INa is the voltage-gated Na+ current, INab is the Na+ background current, IClG is the GABA activated Cl- current.
2.3. Ca2+ handling
Ca2+ enters into cells through Ca2+ channels and is removed by Ca2+ pumps and exchangers. Ca2+ handling can also include ER and mitochondrial sequestration. Including only fluxes through Ca2+ channels and Ca2+ pumps on the PM, the equations for free cytoplasmic Ca2+ ([Ca2+]c) dynamics can be written:
d[Ca2+]c / dt = fi (−ICaL − ICaNL − ICaT − 2 ICap) /(2 F Vi) − ksg [Ca2+]c (Eqn. 3)
where fi is the fraction of free Ca2+ in cytoplasm, F is Faraday’s constant, Vi is the effective volumes of the cytosolic compartment, and ksg is a coefficient of the sequestration rate of Ca2+ at exocytosis
However, detailed data on Ca2+ dynamics during one spike in α-cells is lacking, so we used the coefficients we employed for mouse β–cells electrophysiological modelCitation14 ().
Table 1. Cell parameters
2.4. Role of Ca2+ channels in glucagon exocytosis
Capacitance measurements in α-cells have shown that the glucagon granule exocytosis is negligible at membrane potentials more negative than −20 mV but increases steeply at more depolarized voltages.Citation3,Citation8,Citation29,Citation30 This behavior may reflect a necessity for local calcium entry through high voltage-gated non-L-type Ca2+ channels in rodent and human α-cells because the process of release of primed granules in α-cells (as well as in β–cells) may need a local highly increased calcium in specific microdomains (for a review, see refs. Citation29 and Citation31). Microdomain Ca2+ may be proportional to the Ca2+ current through local Ca2+ channel,Citation32 and we model granule exocytosis to a first approximation as the dependence on Ca2+ influx through specific non-L-type Ca2+ channels.
Here we modeled the electrophysiological events inside one cell. However, in a single β-cell a few insulin granules are released per minuteCitation16,Citation33 and α-cells are likely to have similar characteristics.Citation5,Citation34 At present it is difficult to model α-cell exocytosis in single cells (see below, Sec. 4.5). Measured rates of glucagon secretion are also very variable and often represent only a comparison of relative rates. However, we found that an evaluation of the relative rates of glucagon secretion can be sufficient for our consideration.
For these reasons, we have adopted the convention of simulating one islet containing many α-cells with which to simulate a relative rate of glucagon secretion. We simulated a hypothetical perfusion experiment, where one islet is localized in a perfusion chamber. In this case the relative glucagon concentration in media surrounding this islet and relative glucagon output from the chamber can be written as
d[Glc]s / dt = fgs Na/Vpg− kpc [Glc]s (Eqn. 4)
where
fgs = kcg (-ICaNL/F) + krg (Eqn. 5)
Gs = kpg kpc [Glc]s (Eqn. 6)
[Glc]s is the relative glucagon concentration in medium, Na is the amount of α-cells in one islet, fgs is the rate of exocytosis in one cell, Vpg is the volume of the chamber, kcg is the stoichiometric coefficient, ICaNL/F is the Ca2+ influx through specific VGCCs (non-L-type Ca2+ channels in this work), krg is the rate coefficient for relative glucagon production in rest conditions (in hyperpolarized cells), Gs is the relative glucagon secretion rate in the perfusion experiment, kpc is the perfusion coefficient and kpg is the scaling coefficient.
To compare with the experimentally measured rates of glucagon secretion we also propose that α-cells in an islet account for approximately 20% of islet cells, i.e., about 200 α-cells in a mouse islet if an islet has about 1000 cellsCitation33 (coefficient Na in Eqn. 4). Other unknown coefficients in Equations 4–6 were evaluated according to the proposal that the mean relative glucagon concentration in the medium and the relative secretion rate were about 1 relative unit at low glucose in our hypothetical perfusion experiment ().
2.5. Computational model of regulation of glucagon secretion
In parallel with the metabolic model of glucose sensing, we have constructed a computational model of glucagon secretion in α-cells as the combination of the equations for the regulation of PM potential (Eqn. 2), Ca2+ handling (Eqn. 3), relative glucagon release (Eqns. 4–6) and the voltage-dependent gating variables for channels (see Supplemental Material). The influence of glucose was modeled as the changes in ATP/ADP ratio obtained from the model of glucose sensing that determines KATP channel opening.
However, precisely detailed mathematical models of electrophysiological, metabolic and exocytotic processes are impossible to construct at the present time (and in the immediate future) due to lack of both adequate experimental data and detailed models of separate processes. We also evaluated the model parameters from the literature, when possible, but they were also found by fitting a set of known experimental data as an important reality test (see Secs. 2.1–2.4 and Supp. Material).
and contain all the information necessary to carry out the simulations presented in this paper and are pointed out in the text as a simulation at basal level. In some cases, when the values of parameters differed from those at basal levels the values assigned are noted in the text or in the corresponding figure legends. To calculate the steady-state cellular parameters, the model was allowed to run up to steady values with no changes in coefficients. The units, except where indicated otherwise, are: time in milliseconds (ms), voltage in millivolts (mV), concentration in micromoles/liter (µM), current in femtoamperes (fA), fluxes in mmol s−1, conductance in picosiemens (pS), capacitance in femtofarads (fF). Numerical integration was performed using standard numerical methods.
Table 2. Membrane current parameters
These models are available for direct simulation on the website “Virtual Cell” (www.nrcam.uchc.edu) in “MathModel Database” on the “math workspace” in the library “Fridlyand” with the name: “Alpha and beta cells glucose sensing” and “Glucagon secretion” are represented. Visualization and graphical analysis were performed using “Excel.”
3. Review, Simulations and Analysis of Experimental Observations of α-cell Intracellular Regulatory Mechanisms
In this section we use computational simulations to critically review and analyze the experimental observations of intracellular regulatory mechanisms, where electrophysiological data were obtained for isolated α-cells. However, because our mathematical model of glucagon secretion has significant limitations (see Sec. 2.5) we will use it mainly to test and explain numerous seemingly mutually incompatible experimental observations and the development of hypotheses concerning the mechanisms of regulation of glucagon secretion, not for simulation of individual experiments.
Here, we define a “low glucose concentration” as approximately 1–2 mM (where glucagon secretion is maximal in isolated α-cells), “medium glucose concentration” is 3–5 mM (where glucagon secretion is minimal) and “high glucose concentration” is above 10 mM (where glucagon secretion begins to increase) (see below in Sec. 3.2).
3.1. Intrinsic metabolic regulation in α-cells
The ratio of lactate dehydrogenase to mitochondrial glycerol phosphate dehydrogenase activity is higher in α- compared with β-cells.Citation35,Citation36 (Low activity of lactate dehydrogenase is favoring the mitochondrial oxidation of glucose in β-cellsCitation15,Citation35). In contrast, pyruvate carboxylase, that is required for conversion of pyruvate to oxaloacetate for repleting the Krebs cycle with intermediates, has significantly higher expression in β-cells.Citation36 Interestingly in this regard, uncoupling protein 2 is expressed much more highly in α-cells compared with β-cells.Citation37
We examined how these differences can affect the metabolic responses of α-and β-cells to glucose, using the corrected model for glucose sensing (). To describe characteristic peculiarities of α-cells the activity of pyruvate dehydrogenase was decreased, and the activities of lactate dehydrogenase and uncoupling protein 2 were increased to compare with the β-cell model.Citation15 The rate of NADH production from nonglucose sources was assumed to be higher in α- compared with β-cells to take into account the increased ATP and NAD(P)H levels in α-cells at low glucose levels (see Sec. 2.1). We have found that in this case the rate of pyruvate consumption in mitochondria (JPYR) was significantly decreased in α-cells (). Mitochondrial membrane potential, ATP/ADP ratio and NADH in mitochondria were higher at low glucose and saturated sharply in α-cells unlike β-cells simulation ().
Figure 2. Effect of increasing glucose on α- and β-cells energetics. Extracellular glucose concentration was varied and the steady-state simulations of the model parameters are represented. Simulations were run with the basic set of parameters for β-cells (Tables 2 and 3 from ref. Citation15 and additionally with the coefficient describing the nonglycolitic source of NADH in mitochondria (kng, Eqn. 1). Corresponding parameters were changed for α-cells: kng was increased from 0.006 μmol ms−1 (for β-cells) to 0.05 μmol ms−1, VmLDH (maximal lactate dehydrogenase activity) was increased from 1.2 μmol ms−1 to 12 μmol ms−1, the maximal pyruvate dehydrogenase (PDH) activity (Vmpdh) was decreased from 0.3 μmol ms−1 to 0.08 μmol ms−1, the regulated leak coefficient (Plr) was increased from 0.0012 μmol ms−1 to 0.008 μmol ms−1, the glucokinase activity (Vglu) was taken as 0.01 μmol ms−1 for both α- and β-cells. (───) represents β- and (- - - -) α-cells. (A) (····) is the rate of the glucokinase reaction (close in α- and β-cells), JPYR is the rate of pyruvate consumption in mitochondria for β- and α-cells. (B) Ψm is the mitochondrial membrane potential. (C) [NADH]m is mitochondrial NADH. D. [ATP]c/[ADP]c is the ATP/ADP ratio in the cytoplasm.
![Figure 2. Effect of increasing glucose on α- and β-cells energetics. Extracellular glucose concentration was varied and the steady-state simulations of the model parameters are represented. Simulations were run with the basic set of parameters for β-cells (Tables 2 and 3 from ref. Citation15 and additionally with the coefficient describing the nonglycolitic source of NADH in mitochondria (kng, Eqn. 1). Corresponding parameters were changed for α-cells: kng was increased from 0.006 μmol ms−1 (for β-cells) to 0.05 μmol ms−1, VmLDH (maximal lactate dehydrogenase activity) was increased from 1.2 μmol ms−1 to 12 μmol ms−1, the maximal pyruvate dehydrogenase (PDH) activity (Vmpdh) was decreased from 0.3 μmol ms−1 to 0.08 μmol ms−1, the regulated leak coefficient (Plr) was increased from 0.0012 μmol ms−1 to 0.008 μmol ms−1, the glucokinase activity (Vglu) was taken as 0.01 μmol ms−1 for both α- and β-cells. (───) represents β- and (- - - -) α-cells. (A) (····) is the rate of the glucokinase reaction (close in α- and β-cells), JPYR is the rate of pyruvate consumption in mitochondria for β- and α-cells. (B) Ψm is the mitochondrial membrane potential. (C) [NADH]m is mitochondrial NADH. D. [ATP]c/[ADP]c is the ATP/ADP ratio in the cytoplasm.](/cms/asset/eb6a0185-8ef6-4032-85fc-5f6dcfea4236/kisl_a_10922193_f0002.gif)
The results of simulations are consistent with experimental data: glycolytic flux, measured as D-[53H]glucose utilization, was comparable in α- and β-cells. Rates of glucose utilization increased proportionally to the extracellular substrate concentration between 1 and 10 mM substrate and were virtually identical in non-β-cells (mainly α-cells) and β-cells.Citation22 The ATP/ADP ratio was significantly higher in α-cells compared with β-cells at low glucose levels.Citation26 For both α- and β-cells, increased glucose concentration leads to an elevation of the intracellular ATP/ADP-ratio.Citation1,Citation2,Citation38 However, experimental data on glucose metabolism agrees with the observation that ATP∕ADP changes in response to glucose are less variable in α-cells compared with β-cells.Citation9,Citation10,Citation26 The NAD(P)H signal was greater in mouse islet α-cells compared with β-cells at 1 mM glucose, however, the increase of this signal with glucose was less in α-cells.Citation23 Purified rat non-β cells (approximately 80% glucagon-producing α-cells) exhibit a much lower rate of glucose oxidation (only 30%) in response to glucose than in purified β-cells,Citation36 corresponding to the decreased rate of pyruvate consumption in mitochondria in our model (). Therefore, our systems analysis indicates that recorded differences in metabolic response between α-cells and β-cells can be explained in part by the variations in expression of the specific enzymes that are responsible for the glucose sensitivity mechanism. Based on this evaluation we can suggest that α-cells might have significantly suppressed mechanism of KATP channel inhibition with increased ATP/ADP ratio in comparison with β-cells.
3.2. Spontaneous AP firing in α-cells and metabolic mechanisms of regulation of glucagon secretion
Patch-clamp experiments on isolated mouse, rat, guinea pig and human α-cells have show n that, unlike β-cells, they produce spontaneous AP firing in the absence or low glucose levels (see refs. Citation1, Citation8, Citation39 and Citation40). We were able to simulate AP firing at low glucose levels (at the low [ATP]/[ADP] ratio) using the specified basal set of channels and coefficients (). Our model shows that the PM can be depolarized in α–cells up to a threshold for AP firing even with low glucose levels. Simulated AP spikes were in the range between -60 and 10 mV. Note, the model responds with depolarization and generation of AP firing in the same voltage range and frequency as was observed experimentally in mouse and human α-cells (see for example, refs. Citation8, Citation11 and Citation41).
Figure 3. Modeling of spontaneous spikes, glucagon secretion and the changes of intracellular Ca2+ concentration in response to a step increase in glucose concentration in α-cells. (A) PM voltage (VP) and corresponding detailed spike picture. (B) Relative glucagon secretion rate (Gs). (C) Cytoplasmic calcium ([Ca2+]c) transient. Glucose-induced KATP conductivity changes were simulated by a step increase of the [ATP]/[ADP] ratio at the arrow (1) from a low (ATP/ADP = 2, that corresponds to 1.3 mM glucose in ) to an intermediate glucose level (ATP/ADP = 6, that corresponds to 3.6 mM glucose in ) and at the arrow (2) from an intermediate to high glucose level (ATP/ADP = 19, that corresponds to 25 mM glucose in ). All other parameters were taken from the basic set of parameters ( and ). Changes in Vp were simulated using Eqn. 1, [Ca2+]c using Eqn. 3, and relative glucagon secretion using Eqn. 6.
![Figure 3. Modeling of spontaneous spikes, glucagon secretion and the changes of intracellular Ca2+ concentration in response to a step increase in glucose concentration in α-cells. (A) PM voltage (VP) and corresponding detailed spike picture. (B) Relative glucagon secretion rate (Gs). (C) Cytoplasmic calcium ([Ca2+]c) transient. Glucose-induced KATP conductivity changes were simulated by a step increase of the [ATP]/[ADP] ratio at the arrow (1) from a low (ATP/ADP = 2, that corresponds to 1.3 mM glucose in Fig. 2D) to an intermediate glucose level (ATP/ADP = 6, that corresponds to 3.6 mM glucose in Fig. 2D) and at the arrow (2) from an intermediate to high glucose level (ATP/ADP = 19, that corresponds to 25 mM glucose in Fig. 2D). All other parameters were taken from the basic set of parameters (Tables 1 and 2). Changes in Vp were simulated using Eqn. 1, [Ca2+]c using Eqn. 3, and relative glucagon secretion using Eqn. 6.](/cms/asset/ed907360-2166-485c-b009-223f814aabe3/kisl_a_10922193_f0003.gif)
Isolated mouse and rat pancreatic α-cells were electrically active in the absence of glucose but glucose inhibited electrical activity and suppressed glucagon secretion.Citation24,Citation40 We have also simulated the increase of [ATP]/[ADP] ratios following a stepwise increase of glucose from low to medium and high levels (arrows 1 and 2 in ). Increased [ATP]/[ADP] ratio resulted in the closure of KATP channels similarly as in the β-cell mathematical model (compare refs. Citation15 and Citation17) leading to PM depolarization. However, quite the opposite of the case in β-cells, PM depolarization can lead to suppression of existing AP firing in α-cells. This leads to reduced opening of the voltage-gated calcium channels, decreasing Ca2+ influx through specific VGCCs (non-L-type in mouse) and suppression of glucagon granule exocytosis with the increase of glucose from low to medium levels. The simulations correspond to the experimental observations discussed above.
We will denote this mechanism of glucose action as a metabolic mechanism of regulation of glucagon secretion. According to this mechanism decreased glucagon secretion with increased glucose occurs as a consequence of increased ATP/ADP and block of KATP channels leading to PM depolarization and suppression of AP firing that exists at low glucose level in α-cells.
In addition to the inhibitory effect of glucose seen at medium (3–5 mM) levels, glucose also exerts a stimulatory effect on glucagon release at higher concentration. For example, glucagon secretion at 20 mM was significantly higher than that observed at 3–5 mM glucose [but it was lower than at low glucose (1–2 mM)] in isolated mouse and human islets.Citation3,Citation8,Citation42,Citation43 Although the precise mechanism of this stimulatory effect is still obscure, our simulation shows that an additional suppression of KATP channels with increased glucose up to “high levels” results in PM depolarization and increased glucagon secretion. This may be a result of increasing voltage-dependent activation of non-L-type Ca2+ channels even without activation of AP firing ().
3.3. Channels as the modulators of PM potential, Ca2+ handling and glucagon secretion
We have evaluated here only the role of the most well-studied currents in α-cells. Taking into account this set of channels and currents we can reasonably describe the existing α-cell electrophysiological and Ca2+ handling properties together with glucagon secretion data.
3.3.1. Mechanism of AP spikes
A sequence of steps describing the mechanism of AP spikes in α-cells during stimulus-secretion coupling has been proposed.Citation2,Citation24,Citation44 According to this suggestion, KATP channels are responsible for changes in the membrane potential. At low glucose concentrations, the negative membrane potential in these cells is situated within a range that allows activation of T-type voltage-dependent Ca2+ channels. These channels drive the membrane potential to more positive values, allowing the activation of Na+ and VGCCs. Repolarization would take place following activation of A- and Kdr-type K+ channel. An increased extracellular glucose level would produce a rise in the ATP∕ADP ratio as a consequence of glucose intracellular metabolism, and block KATP channels. This inhibition would allow the PM to depolarize to potentials that change the entire panoply of currents sustaining the AP, consequently suppressing electrical activity.
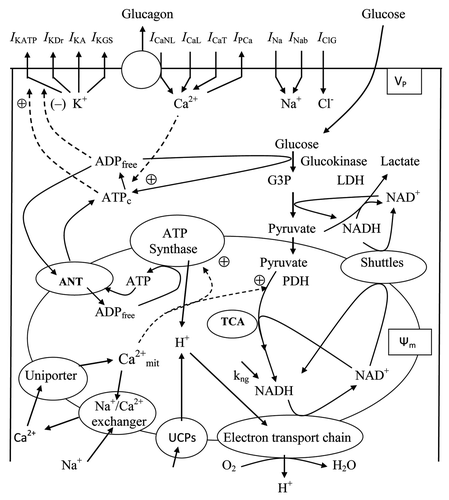
Our simulation confirms the possibility for this sequence of steps and allows a detailed analysis to be performed for the role of specific channels. The voltage-dependent currents during one cycle of spontaneous activity in the ionic model have been analyzed (). The upstroke phase is clearly initially generated by voltage-dependent Na+ channels (INa). When Na+ enters the cell, the membrane is further depolarized and high-voltage-activated Ca2+ channels open and activate transiently. The AP repolarization phase initially includes an activation of fast A-type K+ channels. After that the rapid delayed rectifier K+ channels also activate (IKDr current) leading to increased repolarization current. The repolarization phase also includes the effects of the time-dependent inactivation of Na+ and Ca2+ channels. This restores the resting potential and transiently hyperpolarises the α-cell (after-hyperpolarization). The after-hyperpolarization allows complete reactivation of the Na+-channels and Ca+-channels so that the next action potential is of full size.
Figure 4. Detailed simulated curves of currents during a single spike from . (A) Action potential (Vp); (B) Principal currents for action potential (INa, IKA and IKDr) are represented for one characteristic spike, where INa is main depolarizing and IKA and IKDr are main repolarizing factors. (C) Ca2+ currents:ICaL and ICaNL.
Voltage-dependent Na+ and K+ channels (the delayed-rectifier and A-type) together may define the spike-generation mechanism in α-cells. Other currents included in the model were relatively small during a spike generation and had an insignificant influence on AP shape. This simulated pattern corresponds to the experimental observation that the Na+-current amplitude in α-cells in freshly isolated mouse islets is 5–10-fold larger than combined Ca+ currents.Citation31 However, small currents can play an important role in total interspike current and influence on spike frequency and height (see below).
Glucagon secretion rate depends on activation of specific high voltage-gated calcium channels (non-L-type, see discussion above in Sec. 2.4). Simulated Ca2+ current through these channels during one spike is represented in . (However, the T-type Ca+ current was relative small and is not shown). A decrease in amplitude or complete inhibition of AP firing will therefore result in small openings of these specific VDCCs and this in turn means that fewer glucagon-containing granules undergo exocytosis.
3.3.2. KATP channels
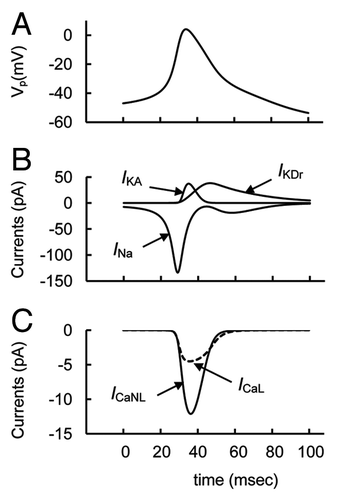
Glucose decreases KATP channel conductance through an increased ATP/ADP ratio that was simulated in our model (). However, AP firing in isolated human and rodent α–cells and glucagon release in intact rodent and human islets also can be suppressed both when KATP-channels are maximally activated by a high concentration of diazoxide (an activator of the KATP-channel) or when they are inhibited by a high concentration of tolbutamide (a blocker of the KATP-channel) at low glucose levels.Citation3,Citation8,Citation24,Citation27,Citation44,Citation45
We simulated these experimental conditions by changing the maximal conductivity of KATP-channels and have obtained the results similarly to the experiments described above. That is, AP firing and glucagon secretion was suppressed both under conditions of inhibited and activated KATP-channels (). Note, in our model based on the experimental data, KATP channels do not open fully at 0 mM glucose and close at high glucose because the ATP/ADP ratio has a sufficiently high level even at low glucose (as discussed above in Sec. 3.1) and the ATP/ADP (ratio) increase cannot fully block the KATP-channels (see Supp. Material, Eqns. A1–A3). For this reason, the inhibitory effect of diazoxide on glucagon secretion can be explained by an additional PM hyperpolarization (as a consequence of increased KATP-channel conductance) up to a level that is lower than the threshold for AP firing. Abolishing AP firing and the associated decreased glucagon secretion in a simulation of additional KATP-channel inhibition (that corresponds to the action of tolbutamide) can be explained similarly to the action of increased glucose as a consequence of PM depolarization (see above in Sec. 3.2).
Figure 5. Simulated parameters at activation and block of KATP channels. (A) PM voltage (VP) and (B) glucagon secretion rate transient in response to the changes in KATP channel conductance at low glucose level (ATP/ADP = 2). Initially the maximal conductance of KATP channel (gmKATP, Eqn. A1) was increased from basal level up to 60 nS to simulate KATP channels activation and PM repolarization. Then gmKATP was decreased to basal level (30 nS, ) at arrow 1 that leads to AP firing and an increase in relative glucagon secretion. For simulation of KATP channel block gmKATP was decreased from 30 nS to 10 nS at arrow 2 that leads to PM depolarization, AP firing suppression and decreased relative glucagon release. Then gmKATP was further decreased from 10 nS to 2 nS at arrow 3 leading to additional PM depolarization with an increased glucagon secretion as a consequence of supplementary activation of non-L type Ca2+ channels with increased PM potential. Other coefficients and initial conditions were as in .
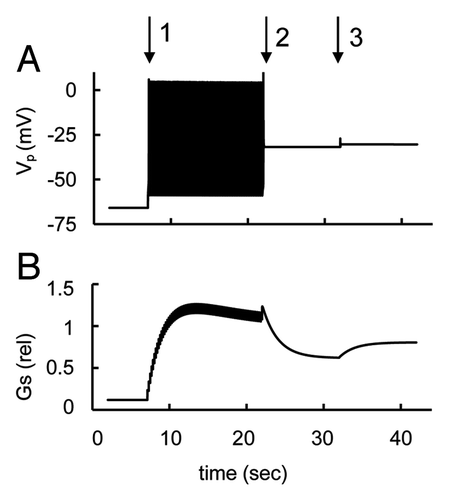
3.3.3. Delayed rectifying K+-channel
Inhibition of KDr-channels results in suppression of glucagon secretion at low, but not high, glucose levels in mouse and human isolated islets.Citation8,Citation46 We simulated these experiments by decreasing KDr channel conductance that corresponds to the effect of specific blockers of these channels (for example, stromatoxin) (). As suggestedCitation8,Citation46 a decrease in maximal KDr channels conductance suppressed AP firing and decreased glucagon secretion. This occurs because the voltage-gated KDr channels contribute significantly to the AP downstroke in α-cells (see above in Sec. 3.3.1) and after their inhibition there is no reactivation of the voltage-gated Na+ and Ca2+-channels. This paradoxically suppressed AP firing even though this leads to PM depolarization. At higher glucose levels AP firing is inhibited and block of KDr channel did not affect glucagon secretion (simulation not shown).
Figure 6. Simulation of block of rapid delayed rectified voltage-gated K+ (KDr) channels. (A) PM voltage (VP) and (B) glucagon secretion rate transient in response to a decrease of KDr channel conductance at low glucose level (ATP/ADP = 2). For simulation of block of KDr channel the maximal conductance (gmKr, Eqn. A4) was decreased from 18 nS (basal level) to 0 nS at arrow leading to PM depolarization, AP firing suppression and a decrease in relative glucagon secretion. Low glucose induced AP firing was simulated initially as in .
3.3.4. G protein-gated K+-channel and somatostatin action
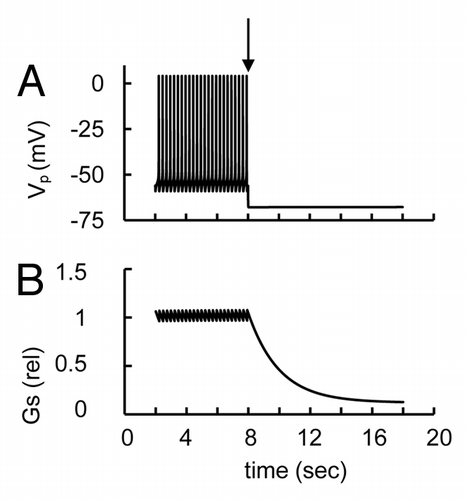
Somatostatin inhibits glucagon secretion in pancreatic α-cells.Citation39,Citation47-Citation49 Electrophysiological recordings showed that somatostatin activated a low-conductance G protein-coupled K+-channel in single rat and mouse α-cells, causing hyperpolarization of the PM and inhibition of electrical activity.Citation39,Citation47,Citation48 We have included this current in our model (IKGS, see and Supp. Material, Eqn. A13). This current acts on the PM potential similarly to the KATP channel current, because it can be described by similar equation (compare Eqns. A1 and A13 in Supp. Material), that is, its activation can lead to PM hyperpolarization. Similarly to activation of KATP channels this can result in inhibition of AP firing and a decrease in glucagon release. An example of a simulation of somatostatin action is shown in . This is consistent with the inhibitory effect of somatostatin on glucagon secretion.
Figure 7. Simulation of activation of G protein-gated K+-channel activated by somatostatin. (A) PM potential (VP) and (B) relative glucagon secretion (Gs). For simulation of G protein-gated K+-channel activation the maximal conductance (gmKGS, Eqn. A13) was increased from 0 nS (basal level) to 0.5 nS at arrow leading to PM repolarization, AP firing suppression and a decrease in glucagon secretion. Low glucose induced AP firing was simulated initially as in .
3.3.5. Voltage-gated Na+ channels
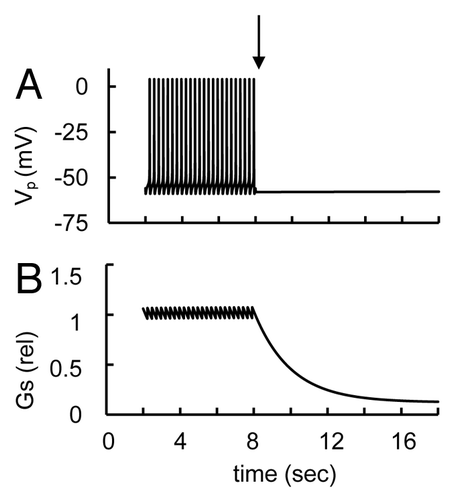
Tetrodotoxin, a specific blocker of certain voltage-gated Na+ channels, strongly inhibited glucagon secretion in mouse and human isolated islets at low but not high glucose levels.Citation8,Citation24,Citation30,Citation44 We utilized the mathematical model presented here to estimate the impact of Na+ channels on AP firing and glucagon secretion (). Blockade of Na+ channels induced partial PM repolarization. However, the main effect of Na+ channel block was a suppression AP firing and consequently glucagon secretion decreased. This occurs because the voltage-gated Na+-channels are critical for the development of the depolarizing phase of AP in α-cell (see above in Sec. 3.3.1). At high glucose levels AP firing was still inhibited and Na+ channel block did not change glucagon secretion significantly (not shown).
Figure 8. Simulated glucose induced spikes behavior and glucagon secretion at block of Na+ channel. (A) Low glucose induced AP firing (Vp) with the initial simulation as in . (B) relative glucagon secretion rate transients. For simulation Na+ channel block the maximal conductance gmNa (Eqn. A14) was decreased from 32 nS (basal level, ) to 0 nS at arrow that leads to AP firing suppression and a decrease in glucagon secretion.
3.3.6. Na+ background current
Closure of KATP-channels alone is not sufficient to cause membrane depolarization. Therefore, β-cells must be equipped with some sort of depolarizing inward current. However, the nature of the depolarizing current remains elusive.Citation50 For example this current in β-cells might result from forward operation of the Na-Ca exchanger, i.e., this can cause a Na+ inward current because one Ca2+ from the cytoplasm exchanges for three Na+ from the medium.Citation51 Another suggestion was activation of cationic (principally Na+) Trp-channels.Citation52,Citation53 In this article we used a model of some generalized inward Na+ current (without specifying exactly which one) to simulate this depolarizing current in α-cells (Eqn. 21 in Supp. Material). However, clearly additional experiments are required to define the properties of this current and its influence on α-cells in greater detail.
3.3.7. L-type Ca2+ channels
Glucagon secretion evoked by low glucose alone is “almost resistant” or only slightly affected by L-type Ca2+ channel blockers in isolated mouse islets.Citation30 Decreased L-type Ca2+ channel conductance also did not suppress AP firing and glucagon exocytosis in our model (not shown) because these channels have an insignificant influence on the AP upstroke (see above in Sec. 3.3.1).
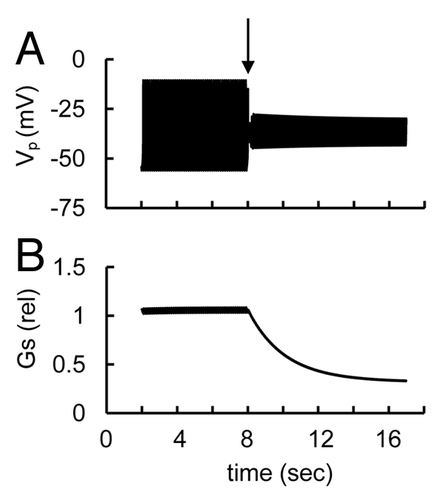
However, the L-type Ca2+ channel blocker isradipine suppressed the electrical activity induced by low glucose or tolbutamide in isolated α-cells and glucagon secretion in human islets.Citation8 This raises the question of whether the change in activity of L-type Ca2+ channels can lead to appropriate changes in AP firing. We found that suppression of AP firing with L-type Ca2+ channel block can be obtained by changing the basal conductivities of some channels in our model. For example, a decrease in conductivity of voltage-gated Na+ channels and increased conductivity of L-type Ca2+ channels in comparision with basal levels in can lead to an increased effect of L-type Ca2+ current on spike formation. In such conditions decreased conductivity of voltage-gated L-type Ca2+ current can suppress AP firing (). Based on this evaluation we can suggest that human α-cells might have increased L-type Ca2+ channel activity in comparison with mouse α-cells. Interestingly, small spikes were simulated in this case after block of L-type Ca2+ current (as well as in at block of KDr channels) that can also be found after suppression of AP firing in several experimental conditions (see for exampleCitation8,Citation54).
Figure 9. Simulated glucose induced spike pattern and glucagon secretion in response to the L-type Ca2+ channel blocker at increased L-type Ca2+ channel conductance (to compare with basal level) (illustration to Sec. 3.3.7). (A) PM voltage and (B) relative glucagon secretion rate transients. The maximal conductance (gmCaL, Eqn. A22) was decreased from 0.4 nS to 0.05 nS at arrow. gmNa (Eqn. A14) was decreased from 32 nS (basal level, ) to 22 nS, gmKATP (Eqn. A1) was decreased from basal level (30 nS, ) to 25 nS and PmCap (Eqn. A38) was increased from basal level (300 fA, ) to 500 fA to compensate for an influence of increased L-type Ca2+ channel current. These changes lead to a possibility that to AP firing may be blocked in response to the L-type Ca2+ channel blocker. Other coefficients and initial conditions were as in .
3.3.8. Non-L-type Ca2+ channels
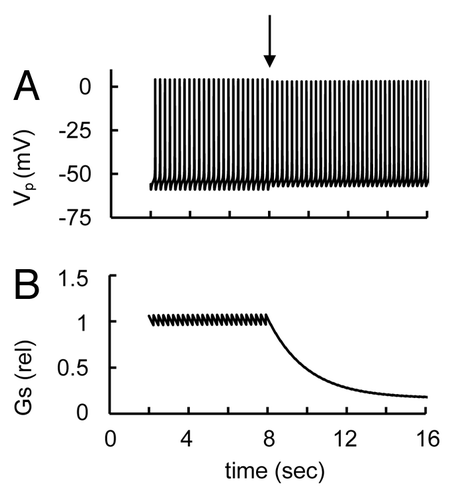
Non-L-type Ca2+ channels include N- and P/Q-type VGCCs. These channels have been linked to glucagon exocytosis and may be opened at PM potentials that are above that for opening L-type Ca2+ channels,Citation31 and we have modeled this high-voltage activation (Supp. Material, Eqn. A28 and ).
Figure 10. Simulation glucose-induced spikes activity in response to non-L-type Ca2+ channels blockers. (A) Action potential (Vp) and (B) relative glucagon secretion rate (Gs) transients. Block of non-L-type Ca2+ channels was simulated by a decrease of their maximal conductance at low glucose level (ATP/ADP = 2). At arrow the gmCaNL (Eqn. A28) was decreased from 0.3 nS (basal level, ) to 0.0015 nS. This leads to a decreased glucagon secretion at constant glucose-induced spike activity. Other coefficients were as in .
An N-type Ca2+ channel blocker (ω-conotoxin) inhibited glucagon release elicited by low glucose alone in mouse isletsCitation29,Citation39 and in single rat α-cells.Citation27 P/Q-type Ca2+ channels can serve as the main component of non-L-type Ca2+ channels in triggering exocytosis of glucagon granules in human α-cells.Citation8 This group found that a P/Q-type Ca2+ channel blocker (ω-agatoxin) did not inhibit spontaneous [Ca2+]c oscillations but suppressed exocytosis in isolated human α-cells and inhibited glucagon secretion in isolated human islets at low glucose levels.Citation8 Maintenance of spontaneous [Ca2+]c oscillations suggests that these channels do not have any important effect on AP firing (see below in Sec. 4.4). On the basis of these data we can suggest that block of P/Q-type Ca2+ channels in human α- cells leads to suppression of glucagon secretion but AP firing is maintained.
We simulated the action of an non-L-type Ca2+ channels blocker by reducing their maximal conductivity (). The results of simulation correspond to the suggestion that the non-L-type Ca2+ channel blocker can have only weak effects on electrical activity; however it can still significantly suppress glucagon secretion. This is because non-L-type Ca2+ channels play a small role in the AP upstroke but they are strongly responsible for glucagon release (see above in Secs. 2.4 and 3.3.1).
3.3.9. Cl- channels and γ-aminobutyric acid (GABA) action
GABA receptors have been identified in α-cells and glucagon secretion can be inhibited by GABA release from neighboring β-cells in rat.Citation3,Citation55,Citation56 The existence of Cl− currents activated by GABA in α-cells was described for cells isolated from guinea pig and rat islets, and application of GABA led to hyperpolarization and termination of AP firing.Citation28,Citation55 Interestingly, activation of GABAA receptors induces an inward Cl− current that inhibits neuronal firing in the CNS as consequence of PM hyperpolarization.Citation57 We have modeled the GABA-activated inward Cl− current ( and Supp. Material, Eqn. A39). Simulation of an activation of this current (as an increase in its maximal conductance) led to PM hyperpolarization that abolished AP firing and decreased glucagon secretion. For example, the simulated result (at gmCIG = 40 pS and above) was similar to (not shown). This mechanism is similar to a PM hyperpolarization and a glucagon secretion decrease by an activation of KATP channels (see for example left part of ).
3.4. Ca2+ dynamics and glucagon secretion
Increased glucagon secretion at low glucose levels is often associated with elevated [Ca2+]c in α-cells.Citation40,Citation58,Citation59 However, a glucose-induced drop in [Ca2+]c in α-cells likely contributes only partly to the strong glucose-induced inhibition of glucagon secretion in islets.Citation41 [Ca2+]c elevation was also found in α-cells of isolated mouse islets simultaneously with decreased glucagon secretion following exposure to increased glucose.Citation23
We have analyzed these contradictory experimental evidences using our model. Cytoplasmic Ca2+ concentration is a result of the interaction of the characteristic set of Ca2+ channels, Ca2+ pumps and electrical activity. Ca2+ enters cells through all types of open Ca2+ channels. On the other hand, glucagon release seems to be regulated mainly by specific high-voltage-activate Ca2+ channels (non-L-type, as discussed above in Secs. 2.4 and 3.3.8). This opens up the possibility for separate dynamics of [Ca2+]c and glucagon secretion, in contrast to β-cells (see ref. Citation31). Our simulations illustrate these cases. For example, [Ca2+]c and glucagon secretion have changed in one direction in the simulation of increased glucose using a chosen basal set of parameters (). However, the simulation in shows that PM depolarization following increased glucose can lead simultaneously to increased [Ca2+]c and decreased glucagon secretion when the conductivity for L-type channel was increased and the conductivity a non L-type Ca2+ channel was decreased (in comparison with the basal set in ).
Figure 11. Simulated simultaneous [Ca2+]c increase with decreased glucagon secretion due to changes in basal conductance of Ca2+ channels (illustration to Sec. 3.4). (A) Action potential (Vp), (B) relative glucagon secretion rate and (C) [Ca2+]c transients. Non-L-type Ca2+ channels conductance (gmCaNL, Eqn. A28) was decreased from basal level (0.3 nS, ) to 0.15 nS. L-type Ca2+ channels conductance (gmCaL, Eqn. A22) was increased from basal level (0.1 nS ) to 0.3 nS. Glucose-induced changes were simulated as in by a step increase of the [ATP]/[ADP] ratio from low (ATP/ADP = 2) to the intermediate glucose level (ATP/ADP = 10) at the arrow. All other parameters were taken from the basic set of parameters ( and ).
![Figure 11. Simulated simultaneous [Ca2+]c increase with decreased glucagon secretion due to changes in basal conductance of Ca2+ channels (illustration to Sec. 3.4). (A) Action potential (Vp), (B) relative glucagon secretion rate and (C) [Ca2+]c transients. Non-L-type Ca2+ channels conductance (gmCaNL, Eqn. A28) was decreased from basal level (0.3 nS, Table 2) to 0.15 nS. L-type Ca2+ channels conductance (gmCaL, Eqn. A22) was increased from basal level (0.1 nS Table 1) to 0.3 nS. Glucose-induced changes were simulated as in Figure 3 by a step increase of the [ATP]/[ADP] ratio from low (ATP/ADP = 2) to the intermediate glucose level (ATP/ADP = 10) at the arrow. All other parameters were taken from the basic set of parameters (Tables 1 and 2).](/cms/asset/8363a266-941f-4d46-a2c0-1a60523f4910/kisl_a_10922193_f0011.gif)
This result shows that when Ca2+ enters cells mainly through Ca+ channels which also promote glucagon release (non-L-type) the [Ca2+]c and glucagon secretion should change in the same direction with increased glucose (as in ), because the fraction of Ca2+ that penetrates into cell through L-type Ca+ channels during spikes is small. However, glucagon secretion can be decreased as a consequence of suppression of AP firing and closing non-L-type Ca+ channels, but [Ca2+]c can increase following an increase of Ca2+ influx through L-type Ca2+ channels following further PM depolarization, if the fractional Ca2+ entering via L-type Ca+ channels is increased. These examples show that Ca2+ dynamics may depend on the relationship between Ca+ channels that have different voltage-dependent characteristics.
However, [Ca2+]c and glucagon secretion should always decrease simultaneously with PM hyperpolarization that blocks all high voltage-gated Ca2+ channels (simulations not shown). This conclusion is consistent with the experimental data that opening of KATP channels with diazoxide that leads to PM repolarization lowered [Ca2+]c in isolated mouse α-cells.Citation24,Citation41
3.5. Experiments with activation of AP firing and glucagon secretion at high glucose levels
Isolated, purified α-cells are removed from any potential paracrine regulation from neighboring β- and δ-cells and only intrinsic glucose regulation can take place. Two different patterns were found in the investigations of isolated rodent α-cells. Isolated rat pancreatic α-cells were electrically active in the absence of glucose but glucose (20 mM) inhibited electrical activity.Citation40 Glucose reduced also AP firing leading to PM depolarization and an inhibition of glucagon secretion in isolated normal mouse α-cells.Citation24 Obviously, this pattern corresponds well to the metabolic mechanism of regulation of glucagon secretion (as discussed above in Sec. 3.2 and ).
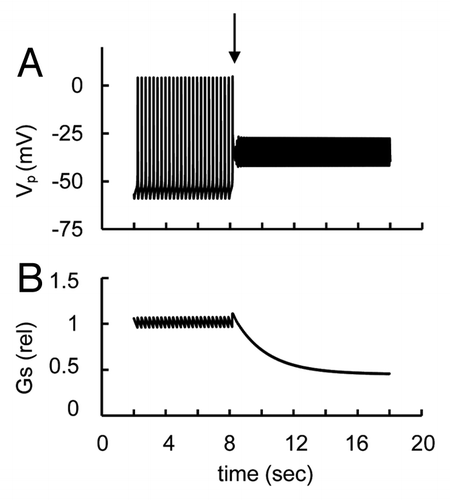
Other investigators found that glucose stimulated rather than inhibited glucagon release in isolated rat α-cells.Citation27,Citation54,Citation60,Citation61 Recent measurements of glucagon secretion from pure populations of flow-sorted mouse α-cells also have shown that contrary to its effect on intact islets, glucose stimulated glucagon secretion.Citation23 Indeed, the PM was repolarized and AP firing was suppressed at low glucose but AP firing appeared and glucagon secretion increased with increased glucose in isolated rat α-cells.Citation54 Depolarization from a hyperpolarized level and an appearance of AP firing were also observed in response to glucose in dispersed mouse α-cellsCitation11,Citation44
We were able to simulate this pattern using our model. For example, we found that AP firing is absent at low glucose levels but can be stimulated with increased glucose () if the maximal conductance of KATP channels was increased in comparison with the basal level (that we used for simulation in ), even though ATP/ADP ratio changes were the same as describing a simulation of α-cell glucose sensing in . This can be attributed to the additional hyperpolarization of PM with increased IKATP leading to suppression of AP firing and glucagon secretion at low glucose. [We have considered above the similar suppression of AP firing and glucagon release at the increase of maximal conductance of KATP channels at action of activators (left part of )]. It is noteworthy that any influence on ion currents leading to additional PM hyperpolarization at low glucose levels (other than voltage-gated currents) can lead to a similar effect. For example, the corresponding somatostatin activation of the G protein-gated K+ channel or GABA activation of Cl- current (that were suggested as zero for basal set of coefficients) can lead to the pattern in our model that is similar to (not shown).
Figure 12. Simulated glucose induced spike pattern and glucagon secretion at increased conductance of KATP channels (Illustration to Sec. 3.5). (A) Action potential (Vp) and (B) relative glucagon secretion (Gs) rate transients. Maximal KATP channels conductance (gmKATP in Eqn. A1) was increased from 30 nS (basal level) to 75 nS. Glucose-changes were simulated (as in ) by a step increase of the [ATP]/[ADP] ratio at the arrow from a low (ATP/ADP = 2) to an intermediate glucose level (ATP/ADP = 10, that corresponds to 4.7 mM glucose in ). All other parameters were taken from the basic set of parameters ( and ). AP firing was induced only at high glucose levels.
![Figure 12. Simulated glucose induced spike pattern and glucagon secretion at increased conductance of KATP channels (Illustration to Sec. 3.5). (A) Action potential (Vp) and (B) relative glucagon secretion (Gs) rate transients. Maximal KATP channels conductance (gmKATP in Eqn. A1) was increased from 30 nS (basal level) to 75 nS. Glucose-changes were simulated (as in Fig. 3) by a step increase of the [ATP]/[ADP] ratio at the arrow from a low (ATP/ADP = 2) to an intermediate glucose level (ATP/ADP = 10, that corresponds to 4.7 mM glucose in Fig. 2D). All other parameters were taken from the basic set of parameters (Tables 1 and 2). AP firing was induced only at high glucose levels.](/cms/asset/d1987a10-c12b-4e4f-88e7-bc55c4c6b78c/kisl_a_10922193_f0012.gif)
These data and our previous analysis show that increased glucagon secretion with increased glucose from a low to high level can take place if AP firing was suppressed at low glucose but can be induced by PM depolarization with increased glucose (for example as a consequence of closing KATP channels (see and Sec. 3.5). We suggest that the metabolic mechanism of regulation of glucagon secretion can act in both cases. This leads to PM depolarization with an increase of glucose leading to different patterns that can depend on initial PM repolarization with an associated increase in threshold potential in the second case that blocks AP firing at low glucose. Such differences in PM polarization in isolated α-cells are difficult to explain, but may be due in part to different methods of α-cells isolation leading to different conductivities of channels. However, these differences require further investigations.
4. Computational Systems Analysis of the Mechanisms of Glucagon Secretion in Pancreatic α-cells
We analyzed the mechanism of metabolic regulation of glucagon secretion in Sec. 3. In this section we will analyze other mechanisms for regulation of AP firing and glucagon secretion that were described in literature. Schematic diagrams for these discussed mechanisms are presented in .
Figure 13. Schematic diagram of metabolic and paracrine mechanisms at electrophisiologycal regulation of glucagon secretion. (A) Metabolic mechanism of regulation. This mechanism suggests that glucose metabolism leads to KATP channel inhibition at high glucose, which depolarizes the α-cell, inhibiting action potential firing and glucagon release (as in ). Glucose-changes were simulated (as in ) by a step increase of the [ATP]/[ADP] ratio at the arrow. Vp is PM potential, Gs is the relative rate of glucagon secretion. (B) Metabolic mechanism with additional PM hyperpolarization. Additional PM hyperpolarization (for example by an increase of KATP channels conductivity, see Sec. 3.5 and ) leads to suppression of AP firing and glucagon release at low glucose level. However, AP firing activates with increase of glucose as a consequence of KATP channel inhibition. Glucose-changes were simulated (as in ) by a step increase of the [ATP]/[ADP] ratio at the arrow. (C) Paracrine interaction in regulation of islet glucagon release. At low glucose level, where paracrine influence is insignificant, intrinsic α-cell mechanisms of channel interaction lead to AP firing and increased glucagon release. Increased glucose level causes insulin, zinc and GABA release from β-cells and somatostatin from δ-cells. This leads to membrane hyperpolarization, block of AP firing and inhibition of glucagon release (Sec. 4.2). Paracrine action leading to PM repolarisation was simulated as in at the arrow. (D) Glucagon secretion with suppressed both metabolic and paracrine regulatory mechanisms. Intrinsic α-cell mechanisms of channel interaction lead to AP firing and increased glucagon release independently on glucose concentration if both metabolic and paracrine regulatory mechanisms are suppressed or can compensate for each other. Glucose-change is represented by arrow.
![Figure 13. Schematic diagram of metabolic and paracrine mechanisms at electrophisiologycal regulation of glucagon secretion. (A) Metabolic mechanism of regulation. This mechanism suggests that glucose metabolism leads to KATP channel inhibition at high glucose, which depolarizes the α-cell, inhibiting action potential firing and glucagon release (as in Fig. 3). Glucose-changes were simulated (as in Fig. 3) by a step increase of the [ATP]/[ADP] ratio at the arrow. Vp is PM potential, Gs is the relative rate of glucagon secretion. (B) Metabolic mechanism with additional PM hyperpolarization. Additional PM hyperpolarization (for example by an increase of KATP channels conductivity, see Sec. 3.5 and Fig. 12) leads to suppression of AP firing and glucagon release at low glucose level. However, AP firing activates with increase of glucose as a consequence of KATP channel inhibition. Glucose-changes were simulated (as in Fig. 3) by a step increase of the [ATP]/[ADP] ratio at the arrow. (C) Paracrine interaction in regulation of islet glucagon release. At low glucose level, where paracrine influence is insignificant, intrinsic α-cell mechanisms of channel interaction lead to AP firing and increased glucagon release. Increased glucose level causes insulin, zinc and GABA release from β-cells and somatostatin from δ-cells. This leads to membrane hyperpolarization, block of AP firing and inhibition of glucagon release (Sec. 4.2). Paracrine action leading to PM repolarisation was simulated as in Fig. 7 at the arrow. (D) Glucagon secretion with suppressed both metabolic and paracrine regulatory mechanisms. Intrinsic α-cell mechanisms of channel interaction lead to AP firing and increased glucagon release independently on glucose concentration if both metabolic and paracrine regulatory mechanisms are suppressed or can compensate for each other. Glucose-change is represented by arrow.](/cms/asset/bcdd47bf-8af0-48d1-985e-434ced093411/kisl_a_10922193_f0013.gif)
4.1. Regulation of glucagon secretion through an effect on electrical activity
Numerous experimental studies and our simulations show that glucagon secretion can be regulated by electrical activity in α-cells if the opening of specific VGCCs is required for glucagon secretion (see above in Sec. 3). The ATP-sensitive K+ channel is considered to be the primary channel that can regulate PM potential. Experiments in which increasing concentrations of tolbutamide and diazoxide were used to titrate KATP-channels activity in α-cells in intact mouse, rat and human islets, indicate that an increase of glucagon secretion only occurs within a narrow window of KATP-channel activity where AP firing occurs.Citation3,Citation8,Citation30,Citation41 We were able to simulate these results by changing KATP-channel activity (). However, our simulation also shows that PM depolarization can increase glucagon release even without activation of AP firing simply by opening additional specific VGCCs (right part of ).
In general, these experiments and our simulations suggested that glucagon release may depend on membrane potential in a complex way that can lead to opposite results for the dependence on an initial PM potential. For example, PM depolarization can lead to AP firing and increased glucagon secretion if α-cells were hyperpolarized previously below threshold for AP firing and a depolarization activates this AP firing, or if there is an increased PM potential from medium to high level even though AP firing is absent (see ). On the other hand, a depolarization can block AP firing and decrease glucagon secretion if AP firing was pre-existent ( and ). There are many different mechanisms that can influence electrical activity and consequently glucagon secretion and we have attempted to analyze several of them.
4.2. Electrophysiological basis of paracrine regulation of glucagon secretion
Rodents form a characteristic α-cell mantle surrounding the core of β-cells, however, α- and β-cells show a more mixed distribution and the α-cells are frequently juxtaposed to the β-cells within human islets.Citation62,Citation63 Products released by β-cells can enter the immediate proximity of the α-cells. Thus, the α-cells can be exposed to high interstitial concentration of β- and δ-cells secretory products whenever they are secreted. This interplay between neighboring islet cells is a kind of “paracrine interaction” that may control glucagon release in α-cells. In this regard, in addition to a glucose, several paracrine inhibitors of glucagon secretion from non α-cells (i.e., insulin, zinc ions or GABA from β-cells or somatostatin from δ-cells) have been proposed (for a review see refs.Citation1,Citation3,Citation4,Citation64).
Modulation of ion channel activity would permit paracrine signals to have a rapid and precise effect on glucagon secretion.Citation65 We analyzed this proposal using our computational systems analysis approach. For example, our analysis showed that somatostatin and GABA can strongly inhibit glucagon secretion in pancreatic α-cells activating the corresponding channels that cause hyperpolarization of the PM, suppression of electrical activity and decreased glucagon secretion (see above in Secs. 3.3.4 and 3.3.9).
Insulin also has been proposed as an intraislet paracrine factor from neighboring β-cells that can suppress glucagon secretion in rodent and human islets.Citation1,Citation3,Citation4,Citation8,Citation64,Citation66-Citation68 Consistent with this role, the insulin receptor and the insulin–receptor downstream signaling molecules are highly expressed in pancreatic α-cells (see for Rev.Citation69). Recent investigations have provided some insight into the mechanism of insulin action. Insulin transiently inhibited electrical activity and glucagon secretion in isolated rat α-cells, most probably through a signaling pathway that triggered membrane hyperpolarization.Citation54 However, insulin may not modulate the specific channels as for GABA or somatostatin. Insulin can activate KATP-channels in mouse α-cells, an effect that may be mediated via the phosphatidylinositol 3-kinase signaling pathwayCitation70 leading to hyperpolarization. Interestingly, insulin can also act by modulating Akt, a critical downstream effector of the phosphatidylinositol 3-kinase, leading to a translocation of GABAA receptors to the cellular membrane to allow its ligand, GABA, also leading to membrane hyperpolarization.Citation71 We have analyzed the mechanisms of inhibition of glucagon secretion by PM hyperpolarisation by activation of KATP- and GABA-dependent Cl- channels (see above in left part of and Secs. 3.3.2 and 3.3.9).
Zinc is present in and co-crystallizes with insulin in β-cell granules and it is secreted from β-cells on exposure to high glucose, and thus could have an inhibitory effect on rat and mouse glucagon secretion (for a review, see refs. Citation54, Citation64 and Citation72). Zn2+ may reversibly activate KATP-channels concordantly reducing electrical activity and glucagon secretion in isolated rat α-cells.Citation54 Similarly, a decrease in Zn2+ may lead to closing KATP channels in isolated mouse islets.Citation72 The mechanism of inhibition of glucagon secretion as a consequence of an activation of KATP-channels and PM repolarization was analyzed above (see Sec. 3.3.2 and left part of ).
Based on the preceding analysis we can explain the manner in which paracrine mechanisms are able to inhibit glucagon secretion with increased glucose. Negative PM potential can exist in α-cells at low glucose level within a range that allows AP firing leading to glucagon release. Increased glucose leads to increased pancreatic insulin, GABA and zinc secretion from β-cells and somatostatin from δ-cells. These paracrine factors should lead to α-cell repolarization as a consequence of activation of corresponding channels. Decreased glucagon secretion occurs when α-cells are hyperpolarized lower than a threshold level for AP firing ().
4.3. Hormonal regulation
Several hormonal regulators were also investigated. Leptin receptor isoforms are expressed in mouse and human α-cells.Citation73 Application of leptin hyperpolarized the membrane potential in glucagon-secreting alphaTC1–9 cells and suppressed the electrical activity induced by low glucose. These effects were accompanied by decreased glucagon release from α TC1–9 cells and mouse islets.Citation73 According to these data and based on our analysis decreased glucagon secretion after the application of leptin can be explained by PM repolarization and suppress of AP firing as was suggested for paracrine mechanisms (see Sec. 4.2 and ). The action of leptin leading to PM hyperpolarization may be mediated by opening KATP channels in the mouse pancreatic β-cells.Citation74,Citation75 A similar process of KATP channels activation by leptin may also occur in the pancreatic α-cells (see alsoCitation73).
The incretin hormone GLP-1 is a powerful suppressor of glucagon secretion. The mechanism of suppression of glucagon secretion seems to include direct action of GLP-1 on α-cells and an indirect one through activation of β- and δ-cells.Citation39,Citation76,Citation77 Interestingly, GLP-1 can inhibit glucagon secretion without much of an effect on α-cell electrical activity in isolated mouse islets, and it was suggested that GLP-1 can selectively inhibit non-L-type Ca2+ channels in mouse which are responsible for glucagon release.Citation39 According to our simulation an inhibition of non-L-type Ca2+ channel can indeed lead to inhibition of glucagon secretion without suppression of AP firing (see above, and Sec. 3.3.8).
GLP-1 receptor mRNA was not detected in isolated rat α-cells nor did GLP-1 stimulate glucagon release in these cells.Citation54 It was proposed that the mechanism of action of GLP-1 might involve inhibition of the rat α-cells by somatostatin secreted from neighboring somatostatin-producing δ-cells that can be activated by GLP-1.Citation77 We considered this mechanism of somatostatin action above (see ). GLP-1 can also activate insulin secretion in β-cells,Citation78,Citation79 and insulin can inhibit glucagon secretion (see above in Sec. 4.2). However, the molecular details of GLP-1 action on α-cells remain elusive. For example, GLP-1 might also directly effects regulation of glucagon granule exocytosis (as it occurs in β-cells, see refs. Citation16 and Citation79). At present these mechanisms have not been investigated in α-cells and we were not able to analyze them using our computational approach.
4.4. [Ca2+]c handling in α-cells
A rise in the cytoplasmic Ca2+ is the important physiological trigger for fusion of the insulin-containing granules in β-cells (for a review see refs. Citation16 and Citation80). However, global [Ca2+]c may not directly determine glucagon release in α-cells even though a calcium influx through VGCCs located in the plasma membrane can play a decisive role in the fusion process for exocytosis of granules both in α- and β-cells (as discussed above in Secs. 2.4 and 3.4). We found that the difference in the voltage dependence of corresponding Ca2+ channels can explain contradictory data about cytoplasmic Ca2+ and glucagon secretion dynamics (see above in Sec. 3.4).
Isolated α-cells in intact mouse and human islets exposed to low glucose (or in the absence of glucose) exhibited spontaneous [Ca2+]c oscillations with period up to several minutes (for a review see refs.Citation8,Citation9,Citation23,Citation39,Citation41,Citation59). Spontaneous [Ca2+]c oscillations in isolated mouse α-cells result from synchronous spiking electrical activity.Citation41 The mechanisms that generate and terminate AP spikes are responsible for the periodical AP firing (bursting) in β-cells (see ref. Citation18). The periodical AP firing in β-cells provides an effective mechanism for rapid changes in extracellular Ca2+ influx through VDCCs that leads to cytosolic [Ca2+]c oscillations.Citation14,Citation81,Citation82 For these reasons, we also suggest that periodic AP firing (bursting) can also result in [Ca2+]c oscillations in α-cells. Then, an occurrence of [Ca2+]c oscillations reflects the existence of periodic AP firing. However, our analysis has shown that block of AP firing can occur both during PM repolarization and depolarization in α-cells (see discussion above in Sec. 3.3), and it may be difficult to recognize the direction of PM potential change (i.e., repolarization or depolarization) if only the block of [Ca2+]c oscillations is detected.
Subsequent generation and termination of AP firing may be determined by small cyclic changes in some voltage-gated and/or voltage independent gating variable currents in β-cells.Citation14,Citation18,Citation83 We have recently performed a detailed analysis of possible mechanisms of oscillatory processes in β-cells.Citation18 In light of this analysis, the mechanisms of [Ca2+]c oscillations and periodical hormonal secretion in the isolated α-cells compared with β-cells could be more closely examined in the future.
4.5. Regulation of exocytosis of glucagon–containing granules
Glucagon is secreted by exocytosis of large dense-core vesicles. Regulation of glucagon–containing granule secretion in α-cells is both complex and multifactorial and not limited to the effect of electrical activityCitation1,Citation5,Citation34,Citation84). Regulation of insulin–containing granule secretion in β-cells is also very complex, but the hierarchy and interplay between the different levels of control are understood well enough to form a broad outline (for a review see refs. Citation79 and Citation80). Mathematical modeling of the regulating pathway and its influence on insulin granule secretion in β-cells was discussed.Citation16,Citation85 Similarly, several proposals have been made for the influence of paracrine and endocrine factors on second messengers pathways (such as cAMP) and on glucagon–containing granule secretion,Citation1,Citation86,Citation87 and a kinetic model for glucagon secretion that describes Ca2+ triggered exocytosis in α-cells was recently developed.Citation59 However, in contrast to β-cells, our knowledge of the molecular regulation of exocytosis of glucagon-containing granules remains fragmentary. For this reason, we were not able to apply directly our computational approach to these mechanisms of regulation of glucagon release.
4.6. α-cells modeling approach
There are a large number of studies on computational modeling of β-cell function (for review see refs. Citation14,Citation15,Citation18 and Citation88). However, we have found only two computational theoretical models in the literature that consider intracellular regulatory electrophysiological mechanisms in α-cells. In one, a Hodgkin-Huxley-type ionic model for action potentials in α-cells was developed.Citation89 Simulation of AP firing was obtained in this model and it was shown that block of KATP channels can lead to a suppression of AP firing. However, no attempts were made for a simulation or analysis of metabolite changes, paracrine regulation, [Ca2+]c handling and glucagon secretion. Ion channels involved in the electrical activity in mouse α-cells were modeled using Monte Carlo algorithms to fit steady-state channel currents.Citation90 However, simulation of AP firing or glucagon secretion was not considered.
4.7. Mechanisms of regulation of glucagon secretion in non-human mammals
There is a significant difference in the mechanisms of metabolic and paracrine inhibition of glucagon secretion following increased glucose. According to our analysis both of these mechanisms of inhibition are based on a suppression of AP firing. However, PM depolarization should be initiated as an effect of the metabolic mechanism, but PM repolarization could play a decisive role for the paracrine and endocrine factors. This raises the question as to what mechanisms act in islets?
In this light, knowledge of both electrical activity and glucagon secretion is required to establish the mechanism for stimulus-secretion coupling in α-cell. However, electrical activity of α-cells was not measured in many studies performed on isolated islets so that analysis is restricted to only a few papers.
4.7.1. Isolated rodent islets
Glucose inhibited glucagon secretion in isolated mouse and rat islets.Citation3,Citation27,Citation39,Citation76 However, we were able to find only one article where AP firing was measured directly in α-cells inside rodent islets. PM depolarization and AP firing were found in α-cells in mouse and rat isolated islets after a change of glucose concentration from 11.1 to 2.8 mM and hyperpolarization and suppression of AP firing after return to 11.1 mm glucose.Citation12 Interestingly, other investigatorsCitation91 followed changes in membrane potential of individual cell types in whole mouse islets using a fluorescent membrane potential sensitive probe. They demonstrated that an increase in glucose concentration from 3 to 10 mM is paralleled by repolarization of α-cells and depolarization of β-cells. Our consideration suggests that the registration of PM repolarization with high glucose levels corresponds to the action of the paracrine mechanism of regulation (see and Sec. 4.2) (in compliance with suggestion in ref. Citation12).
Paracrine concept is difficult to reconcile for every condition, since several experiments have shown that the inhibition of glucagon secretion in isolated mouse islets already occurred at low glucose concentrations that have no effect on insulin or somatostatin secretion.Citation3 In this case the metabolic mechanism of regulation of glucagon secretion can explain these observations. However, AP firing was not measured directly in α-cells inside islets and this makes an explanation of these results problematic.
4.7.2. Diabetic animal models
Alloxan and streptozotozin treated rodent, dog and pig islets have been studied as islet injury or /β cell depletion models. When glucose was infused without insulin glucagon did not decrease but rose up to 50% in a diabetic dog. However, when large amounts of insulin was infused together with glucose, a prompt decline in glucagon (4-fold) was observed.Citation92 Postprandial insulin-driven suppression of glucagon secretion was also lost in the pigs treated with alloxan.Citation93 Similarly, glucagon secretion did not increase during hypoglycemia in streptozotocin treated rats, but it could be increased if the intrapancreatic insulin was switched off during hypoglycaemia.Citation94 A decisive role for endogenous insulin for the glucose-induced suppression of mouse glucagon secretion was found for streptozotozin treated mice.Citation68 Obviously, a metabolic mechanism could not explain the suppression of glucagon secretion in these cases but a paracrine mechanism may do so.
Importantly, our analysis of experimental observations obtained in vivo leads to the conclusion that paracrine mechanisms acting through PM repolarization may play a decisive role in suppression of glucagon secretion following increased glucose in non-human mammals.
Other proposals have also been made concerning mechanisms of regulation of glucagon release (such as direct effects on granule exocytosis, see above in Sec. 4.5) besides mechanisms acting through changes of PM potential. However, these proposals require further investigations and at present they are beyond the scope of our computational systems analysis.
4.8. Comparison of the mechanisms of regulation in α-and β-cells
It may seem puzzling that α-cells, having a similar metabolic pathways and ion channel set as the β-cells (at least in human, see refs. Citation8 and Citation95) can be regulated oppositely to β-cells. It was proposed that differences in ion channel expression contribute to the opposing regulation of insulin and glucagon secretion following changing levels of glucose.Citation2 Our computational analysis indicates how the variations in the conductivities of the channels can lead to a switch from characteristic α-cell AP firing at low glucose levels to AP firing only at increased glucose. We have shown in silico that an activation of the relatively strong hyperpolarizating currents in α-cells can perform such a shift (see above in Sec. 3.5). For example, the increase in KATP channel conductance is enough to transform AP firing from α- to β-cell type behavior (). Interestingly, a relatively higher density of KATP channels in β- cells than α-cells was reported.Citation11,Citation44,Citation96,Citation97 According to our analysis this can be sufficient to explain the differences in glucose dependent electrophysiological properties α- and β-cells. However, any influence on ion currents resulting in additional PM hyperpolarization at low glucose levels may lead to a similar effect (see Sec. 3.5.). For this reason, it is also possible that β-cells have decreased activity of Na+ background current or some other depolarizing factors in comparison with α-cells that also can lead to a corresponding shift in AP firing in region of increased glucose.
Another important difference is the possibility that over-all cytoplasmic calcium concentration may not play a decisive role in an activation of glucagon release in α-cells (as compared with microdomain sequestered calcium entry) whereas insulin secretion depends significantly on global cytoplasmic calcium (see discussion above in Sec. 3.4).
PM depolarization as well as a hyperpolarization can lead to block of AP firing and associated decrease of glucagon secretion in α-cells (see above in Secs. 3.3.2 and 4.1). This is a significant distinction from β-cells where only hyperpolarization leads to a decrease of insulin secretion in physiological conditions.
The molecular characterization of a metabolic glucose sensor is most worked out in pancreatic β-cells, with important roles for glucokinase and mitochondrial oxidative fluxes in the regulation of ATP-sensitive K+ channels. We suggest that the metabolic mechanism of PM depolarization by KATP channel inhibition with increased ATP/ADP ratio is significantly suppressed in α-cells (see above in Sec. 3.1). Specific components of this metabolic mechanism which are well expressed in β-cells (such as a low activity of lactate dehydrogenase and uncoupling protein 2 and increased activity of pyruvate dehydrogenase) are compromised in α-cells (see above in Secs. 2.1 and 3.1). In this case the metabolic mechanism can serve to maintain a sufficient ATP/ADP ratio at low glucose levels that leads to regulation of the PM potential within narrow limits where AP firing can occur. However, the increase in ATP/ADP ratio following a further increase in glucose is supressed in α-cells (see discussion above in Sec. 3.1). An important component of metabolic glucose sensing such as KATP channels may play an important role in regulation of α-cell function through a change in PM potential. For example, a repolarization of PM through a paracrine mechanism action may occur through opening of KATP channels (see Sec. 4.2).
We suggest that such α-cell behavior is not surprising, if an aim of this system is a suppression of glucagon secretion following increased glucose. Indeed PM depolarization is the mechanism leading to insulin secretion in β–cells. For this reason we suggest that PM repolarization should be responsible for decreased glucagon secretion that according to our analysis may be a consequence of paracrine mechanisms. Block of a metabolic mechanism can open the way for this kind of mechanism for PM repolarization (see above in Sec. 4.2).
We hypothesize that the suppression of a glucose sensitive mechanism in the α-cell can also lead to different fates of α- and β-cells during the developing of type 2 diabetes mellitus. The existence of specific mechanisms for β-cell glucose sensitivity itself, and particularly the increase of mitochondrial membrane potential with increased glucose, may underlie the increased sensitivity of these cells to injury (see for details, ref. Citation25). According to this hypothesis the absence of a significant increase of mitochondrial potential with glucose, as simulated in our model (), may prevent α-cells from metabolic injury, keeping them intact when β-cells can be damaged.
5. Human α-cell Function and Diabetes
The growing intense focus on the role of the α-cell in diabetes (for a review see refs. Citation6, Citation19 and Citation20) has increased the importance of understanding how electrical activity induced by metabolism of sugar and other agonists is established and how it is altered in various pathological states.
5.1. Human isolated α-cells and islets
Human islets contain a substantially higher proportion of α-cells than rodent islets, normal α-cell mass in the whole human pancreas amounting to ~40% of β-cell mass.Citation98 Comprehensive electrophysiological characterization of the ion channels was made recently for isolated human α-cells.Citation8 The same set of channels exist in mouse and human α-cells (see refs. Citation8 and Citation31).
Regenerative electrical activity was observed in the majority of isolated human α-cells analyzed at low glucose levels. Tolbutamide (100–400 μmol/l) depolarized the cells, and was associated with a reduction of AP peak voltage.Citation8 These results can be explained by some role of KATP channels as a factor that regulates PM potential similarly to isolated mouse α-cells (see Sec. 3.3.2). However, electrical activity of α-cells inside human islets was not measured.
Intracellular Ca2+ oscillations in α-cells inside isolated human islets were registered at low glucose levels and these oscillations were suppressed with increased glucose.Citation99 This pattern can correspond to existence of AP firing at low glucose and its suppression with increased glucose level (see discussion above in Sec. 4.3).
Increased glucose concentration inhibited significantly glucagon secretion at 6 mM in comparison with 1 mM, and insulin inhibited glucagon secretion at low glucose level (1mM) in isolated human islets.Citation8 Insulin secretion increased also sharply in isolated human islets (other than in isolated mouse islets at increased glucose level from 1 to 5 mM.Citation3,Citation8 Inhibition of GABAA receptor increased glucagon secretion significantly at high glucose levels, however this inhibition did not effect insulin secretion in isolated human islets.Citation56 These data can be interpreted as evidence of suppression of glucagon secretion by insulin and GABA at elevated glucose. β-cell-mediated paracrine signaling may predominate over intrinsic signals in the regulation of glucagon secretion in human and there are numerous indirect data and speculations that intrinsic insulin can be major paracrine factor for α-cells in human islets.Citation4,Citation6,Citation64
Distinct paracrine factors may play a different role in rodent and human islets. For example, it was found that Zn2+ stimulated rather than inhibited glucagon secretion in isolated human islets.Citation8 Zn2+ did not play a significant role in the regulation of glucagon release in type 1 diabetic humans where only a few β-cells exist.Citation100
Based on these limited date and our previous consideration we can suggest that α-cells in normal human islets (as well as in non-human mammals) have AP firing at low glucose levels with an associated increase in glucagon secretion. AP firing and glucagon secretion are suppressed by paracrine factors (possible by insulin) with increased glucose and a metabolic regulatory mechanism may not play a significant role. However, further studies are necessary to investigate this important question.
5.2. Abnormal α-cells function and diabetes
Normally, the postprandial increase in plasma glucose leads to increased insulin secretion and the moderate suppression of glucagon release. On the other hand, the physiological defense against falling plasma glucose includes decreased pancreatic insulin, Zn2+ and GABA secretion from the β-cell and somatostatin from δ-cells, as well as an increased glucagon secretion that prevent a sharp decrease of glucose concentration in blood.Citation6,Citation20
These defense mechanisms are compromised in type 1 and type 2 diabetes where β-cell function is significantly inhibited. Glucose sensing in glucagon release becomes defective. Diabetes is associated with aberrant secretion of glucagon at high plasma glucose levels (normally inhibited at such levels).Citation6,Citation19,Citation101,Citation102 In patients with type 2 diabetes it has been shown that the increased glucagon concentration postprandially contributes to hyperglycemia because hyperglucagonemia results in increased hepatic glucose production. This adds an endogenous source of glucose to the exogenous glucose from the meal.Citation103,Citation104 However, mean fasting plasma glucagon concentration may not differ significantly between nondiabetic and type 2 (often and type 1) diabetic subjects.Citation6,Citation102,Citation105
Although the precise mechanisms of relative hyperglucagonemia with increased plasma glucose level in the diabetic state is still obscure, this behavior can be caused by loss of the decrement in intraislet paracrinic factors from β-cells (insulin, GABA and/or zinc) that normally results in suppressing of glucagon secretion at high glucose level (as discussed above in Sec. 4.7.2).
AP firing is required to maintain a significant rate of glucagon secretion (see discussion above in Secs. 3.2 and 5.1). Maintenance of AP firing and the associated high rate of glucagon release at elevated glucose level can readily be obtained in our model by blocking paracrine mechanisms of glucose sensing (Fig. Thirteen D). It is also possible that the metabolic mechanism leading to PM depolarization under conditions of low paracrine activity, that usually leads to hyperpolarization, can compensate for each other at high glucose levels to continue AP firing.
Interestingly, elevated plasma glucagon levels are associated in type 1 diabetes with severe diabetic ketoacidosis.Citation102,Citation106 While mechanisms underlying hyperglucagonemia in these cases are still obscure this may be due to the combination of low insulin concentrations and β-cell contents in the islet. For example, insulin can regulate gene expression in α-cells through several transcription factors (see ref. Citation69).
In islets isolated from donors with type-2 diabetes insulin release was increased (however significantly less than for nondiabetic donors) and glucagon release was decreased (however this decrease was less than for nondiabetic donors) at transitions from low (1 mM) to high (16.7 mM) glucose level.Citation56 These data correspond to an expected decrease in paracrine inhibition glucagon release with decreased activity of β-cells.
However, glucagon release from islets isolated from donors with type-2 diabetes was reduced at low glucose and that glucose significantly stimulated rather than inhibited secretion in these islets in work.Citation3 Based on our foregoing analysis we can explain this observation. For example, at suppressed paracrine mechanisms this behavior resembles the second case for isolated rodent α-cells (see and Sec. 3.5) and can be explained in the same manner, that is the initial PM potential could be lower than the threshold for AP firing in these islets and AP firing is then activated when glucose is increased leading to PM depolarization. However, further studies, including electrophysiological recordings with secretion measurements, are necessary to determine the mechanisms of regulation of glucagon secretion in diabetic human islets.
5.3. Potential therapeutic approaches
New therapeutic approaches targeting glucagon secretion may prove useful in the treatment of diabetes.Citation19,Citation20 According to our analysis the paracrine mechanism acting through PM repolarization may play a decisive role in a suppression of glucagon secretion following increased glucose in human as well as in non-human mammals. For this reason we can suggest that agents inducing PM hyperpolarization with increased glucose may be useful as a therapeutic approach. For example, somatostatin infusion inducing PM repolarization (see above in Sec. 4.2) was used to suppress hyperglucagonemia in type 1 diabetes. However, several significant side effects of somatostatin and its long acting analogs preclude its chronic use (see refs. Citation20 and Citation107).
Leptin can also hyperpolarize the PM in α-cells (see above in Sec. 4.3). For this reason, the inhibitory effect of leptin on the α-cells may be a plausible therapeutic strategy for suppression of glucagon secretion in Type 1 diabetes.Citation20,Citation108 However, leptin could also suppress insulin secretionCitation74 so this strategy may have complex effects on both glucagon and insulin secretion if β-cells maintain activity (see also refs. Citation20,Citation73).
Amylin (IAPP, or islet-associated polypeptide) is a β-cell hormone that is co-secreted with insulin. Clinical studies, in which a commercially available amylin analog (pramlintide) was used, have shown that amylin can decrease circulating glucagon levels.Citation20,Citation109 The mechanism of amylin analog action, as well as other paracrine factors, may lead to PM hyperpolarization in α-cells. However, further studies, including electrophysiological investigations, are necessary.
Interestingly, KATP channels openers (e.g., diazoxide) can hyperpolarize the α-cell PM, prevent AP firing and blocking glucagon secretion (as discussed above in Secs. 3.3.2 and 4.1). However, KATP channel activation will also suppress insulin secretion. Application of a KATP channel opener before or with meals may decrease both glucagon secretion and hepatic glucose production in Type 1 and advanced type 2 diabetes, where insulin secretion is insignificant. We suggest that KATP channels openers can also be potential therapeutic agents in treatment of diabetes. However, this has not been directly addressed in clinical studies.
Finding specific agents that lead to PM hyperpolarization only in α-cells rather than β-cells may be helpful for treatment of type 2 diabetes where intact β-cells remain active. This can include specific activators of K+ and Cl- outward currents or blockers of inward Na+ current in α-cells. Paracrine factors may also lead to α-cell repolarization as a consequence of activation of hyperpolarizing ion channels (see discussion above in Sec. 4.2) and an activation of specific receptors can increase the associated currents even though the amount of paracrine factors from β-cells may be greatly reduced in diabetes.
PM depolarization in α-cells can suppress AP firing and glucagon secretion (see above in Sec. 3.2). Sulphonylurea drugs (like tolbutamide and glibenclamide) block KATP-channels leading to depolarization. However, sulphonylurea therapy has in fact been reported to stimulate glucagon secretion in type-1 diabetesCitation105,Citation110,Citation111 and in contrast to inhibit glucagon secretion in both healthyCitation66,Citation105 and type-2 diabetic subjects.Citation112 Our model suggests that the effect of sulphonylureas depends on initial PM potential conditions (see Sec. 4.1) and, in principle, can even increase glucagon release if AP firing is initially suppressed and PM depolarization leads to initiation of APs as shown in . Increased PM depolarization from a medium level (in medium glucose) even at suppressed AP firing can also increase glucagon secretion (see discussion above in Sec. 3.2 and right part of ). This may explain the contradictory data on the effect of sulphonylureas on glucagon secretion.
The incretin hormone GLP-1 is a powerful suppressor of glucagon secretion (see above in Sec. 4.3). GLP-1 based therapies target both insulin and glucagon secretion, and inhibition of glucagon accounts for a large part of the glucose-lowering effect in type 2 diabetic patients.Citation19,Citation77 There is reason to believe that indirect reciprocal β-cell-mediated paracrine signaling (partially the GLP-1 mediated increase in insulin secretion) may predominate in the suppression of glucagon secretion in type 2 diabetic patients.Citation105 Interestingly, the use of a GLP-1 analog, liraglutide, may help in controlling hyperglycemia with a concomitant reduction in insulin dose in type 1 patientsCitation113 indicating a potential action of GLP-1 analogs directly on α-cells. However, the mechanisms behind the possible direct inhibitory effect of GLP-1 on glucagon granule exocytosis have not been elucidated and at present additional studies are needed to apply the computational approach to this effect.
Conclusion
Here we have reviewed intrinsic glucose-sensing, metabolic, paracrine and endocrine mechanisms that control glucagon secretion in α-cells and proposed a computational model for these processes. This model was developed to gain further insights into coupling of metabolic processes, PM potential, [Ca2+]c handling and glucagon release in α-cells, that remains incompletely understood. We have shown seemingly contradictory experimental data in this field may be synthesized into a coherent mathematical model. In this model the metabolic features and major ionic currents can be fitted to recent experimental data and expression experiments. The key feature of the model is that it can be used to explore mechanisms underlying AP spikes that regulate glucagon secretion. The model reproduces a number of experimental observations ranging from metabolic changes and voltage-clamp current traces to Ca2+ and glucagon secretion dynamics. This model can be used as the groundwork for in silico examination of the regulation of glucagon secretion through a change in the metabolic processes, channels function and [Ca2+]c dynamics. Understanding the cellular and molecular regulation of glucagon release can lead to refinement of current therapies and to the discovery of new targets that improve treatment of both major types of diabetes and also give additional insight in several types of monogenic diabetes. However, further studies supported by improved mathematical models are necessary to understand the stimulus-secretion coupling in the α-cell.
| Abbreviations | ||
| AP | = | action potential |
| [Ca2+]c | = | free cytoplasmic Ca2+ |
| GABA | = | gamma amino butyric acid |
| GLP-1 | = | glucagon-like peptide-1 |
| KATP-channel | = | ATP-sensitive K+-channel |
| KDr | = | rapid delayed rectified voltage-gated K+ channels |
| KGS | = | G protein-gated K+-channel activated by somatostatin |
| Kv | = | voltage-gated K+ channels |
| [NADH]m | = | NADH concentration in mitochondria |
| PM | = | plasma membrane |
| Vp | = | plasma membrane potential |
| VGCC | = | voltage-gated Ca2+ channel |
Additional material
Download Zip (247.8 KB)Acknowledgments
Funding for this study was provided in part by the National Institute of Diabetes and Digestive and Kidney Diseases Grants DRTC P60DK020595, DK-48494, DK-063493, a Research Grant from the Keck foundation and the Blum-Kovler Foundation and the Kovler Diabetes Center.
Supplemental Materials
For the appendix and references Citation114–Citation125, please see the supplemental materials, which may be found here: www.landesbioscience.com/journals/islets/article/22193
References
- Gromada J, Franklin I, Wollheim CB. Alpha-cells of the endocrine pancreas: 35 years of research but the enigma remains. Endocr Rev 2007; 28:84 - 116; http://dx.doi.org/10.1210/er.2006-0007; PMID: 17261637
- Rorsman P, Salehi SA, Abdulkader F, Braun M, MacDonald PEK. K(ATP)-channels and glucose-regulated glucagon secretion. Trends Endocrinol Metab 2008; 19:277 - 84; http://dx.doi.org/10.1016/j.tem.2008.07.003; PMID: 18771934
- Walker JN, Ramracheya R, Zhang Q, Johnson PR, Braun M, Rorsman P. Regulation of glucagon secretion by glucose: paracrine, intrinsic or both?. Diabetes Obes Metab 2011; 13:Suppl 1 95 - 105; http://dx.doi.org/10.1111/j.1463-1326.2011.01450.x; PMID: 21824262
- Unger RH, Orci L. Paracrinology of islets and the paracrinopathy of diabetes. Proc Natl Acad Sci U S A 2010; 107:16009 - 12; http://dx.doi.org/10.1073/pnas.1006639107; PMID: 20798346
- Quesada I, Tudurí E, Ripoll C, Nadal A. Physiology of the pancreatic alpha-cell and glucagon secretion: role in glucose homeostasis and diabetes. J Endocrinol 2008; 199:5 - 19; http://dx.doi.org/10.1677/JOE-08-0290; PMID: 18669612
- Cryer PE. Minireview: Glucagon in the pathogenesis of hypoglycemia and hyperglycemia in diabetes. Endocrinology 2012; 153:1039 - 48; http://dx.doi.org/10.1210/en.2011-1499; PMID: 22166985
- Heimberg H, De Vos A, Moens K, Quartier E, Bouwens L, Pipeleers D, et al. The glucose sensor protein glucokinase is expressed in glucagon-producing alpha-cells. Proc Natl Acad Sci U S A 1996; 93:7036 - 41; http://dx.doi.org/10.1073/pnas.93.14.7036; PMID: 8692940
- Ramracheya R, Ward C, Shigeto M, Walker JN, Amisten S, Zhang Q, et al. Membrane potential-dependent inactivation of voltage-gated ion channels in alpha-cells inhibits glucagon secretion from human islets. Diabetes 2010; 59:2198 - 208; http://dx.doi.org/10.2337/db09-1505; PMID: 20547976
- Ravier MA, Rutter GA. Glucose or insulin, but not zinc ions, inhibit glucagon secretion from mouse pancreatic alpha-cells. Diabetes 2005; 54:1789 - 97; http://dx.doi.org/10.2337/diabetes.54.6.1789; PMID: 15919801
- Ishihara H, Maechler P, Gjinovci A, Herrera PL, Wollheim CB. Islet beta-cell secretion determines glucagon release from neighbouring alpha-cells. Nat Cell Biol 2003; 5:330 - 5; http://dx.doi.org/10.1038/ncb951; PMID: 12640462
- Barg S, Galvanovskis J, Göpel SO, Rorsman P, Eliasson L. Tight coupling between electrical activity and exocytosis in mouse glucagon-secreting alpha-cells. Diabetes 2000; 49:1500 - 10; http://dx.doi.org/10.2337/diabetes.49.9.1500; PMID: 10969834
- Manning Fox JE, Gyulkhandanyan AV, Satin LS, Wheeler MB. Oscillatory membrane potential response to glucose in islet beta-cells: a comparison of islet-cell electrical activity in mouse and rat. Endocrinology 2006; 147:4655 - 63; http://dx.doi.org/10.1210/en.2006-0424; PMID: 16857746
- Fridlyand LE, Harbeck MC, Roe MW, Philipson LH. Regulation of cAMP dynamics by Ca2+ and G protein-coupled receptors in the pancreatic beta-cell: a computational approach. Am J Physiol Cell Physiol 2007; 293:C1924 - 33; http://dx.doi.org/10.1152/ajpcell.00555.2006; PMID: 17928534
- Fridlyand LE, Jacobson DA, Kuznetsov A, Philipson LH. A model of action potentials and fast Ca2+ dynamics in pancreatic beta-cells. Biophys J 2009; 96:3126 - 39; http://dx.doi.org/10.1016/j.bpj.2009.01.029; PMID: 19383458
- Fridlyand LE, Philipson LH. Glucose sensing in the pancreatic beta cell: a computational systems analysis. Theor Biol Med Model 2010; 7:15; http://dx.doi.org/10.1186/1742-4682-7-15; PMID: 20497556
- Fridlyand LE, Philipson LH. Coupling of metabolic, second messenger pathways and insulin granule dynamics in pancreatic beta-cells: a computational analysis. Prog Biophys Mol Biol 2011; 107:293 - 303; http://dx.doi.org/10.1016/j.pbiomolbio.2011.09.001; PMID: 21920379
- Fridlyand LE, Tamarina N, Philipson LH. Modeling of Ca2+ flux in pancreatic beta-cells: role of the plasma membrane and intracellular stores. Am J Physiol Endocrinol Metab 2003; 285:E138 - 54; PMID: 12644446
- Fridlyand LE, Tamarina N, Philipson LH. Bursting and calcium oscillations in pancreatic beta-cells: specific pacemakers for specific mechanisms. Am J Physiol Endocrinol Metab 2010; 299:E517 - 32; http://dx.doi.org/10.1152/ajpendo.00177.2010; PMID: 20628025
- Dunning BE, Gerich JE. The role of alpha-cell dysregulation in fasting and postprandial hyperglycemia in type 2 diabetes and therapeutic implications. Endocr Rev 2007; 28:253 - 83; http://dx.doi.org/10.1210/er.2006-0026; PMID: 17409288
- Unger RH, Cherrington AD. Glucagonocentric restructuring of diabetes: a pathophysiologic and therapeutic makeover. J Clin Invest 2012; 122:4 - 12; http://dx.doi.org/10.1172/JCI60016; PMID: 22214853
- Gorus FK, Malaisse WJ, Pipeleers DG. Differences in glucose handling by pancreatic A- and B-cells. J Biol Chem 1984; 259:1196 - 200; PMID: 6141162
- Heimberg H, De Vos A, Pipeleers D, Thorens B, Schuit F. Differences in glucose transporter gene expression between rat pancreatic alpha- and beta-cells are correlated to differences in glucose transport but not in glucose utilization. J Biol Chem 1995; 270:8971 - 5; http://dx.doi.org/10.1074/jbc.270.15.8971; PMID: 7721807
- Le Marchand SJ, Piston DW. Glucose suppression of glucagon secretion: metabolic and calcium responses from alpha-cells in intact mouse pancreatic islets. J Biol Chem 2010; 285:14389 - 98; http://dx.doi.org/10.1074/jbc.M109.069195; PMID: 20231269
- Gromada J, Ma X, Høy M, Bokvist K, Salehi A, Berggren PO, et al. ATP-sensitive K+ channel-dependent regulation of glucagon release and electrical activity by glucose in wild-type and SUR1-/- mouse alpha-cells. Diabetes 2004; 53:Suppl 3 S181 - 9; http://dx.doi.org/10.2337/diabetes.53.suppl_3.S181; PMID: 15561909
- Fridlyand LE, Phillipson LH. Mechanisms of glucose sensing in the pancreatic β-cell: A computational systems-based analysis. Islets 2011; 3:224 - 30; http://dx.doi.org/10.4161/isl.3.5.16409; PMID: 21814042
- Detimary P, Dejonghe S, Ling Z, Pipeleers D, Schuit F, Henquin JC. The changes in adenine nucleotides measured in glucose-stimulated rodent islets occur in beta cells but not in alpha cells and are also observed in human islets. J Biol Chem 1998; 273:33905 - 8; http://dx.doi.org/10.1074/jbc.273.51.33905; PMID: 9852040
- Olsen HL, Theander S, Bokvist K, Buschard K, Wollheim CB, Gromada J. Glucose stimulates glucagon release in single rat alpha-cells by mechanisms that mirror the stimulus-secretion coupling in beta-cells. Endocrinology 2005; 146:4861 - 70; http://dx.doi.org/10.1210/en.2005-0800; PMID: 16081632
- Rorsman P, Berggren PO, Bokvist K, Ericson H, Möhler H, Ostenson CG, et al. Glucose-inhibition of glucagon secretion involves activation of GABAA-receptor chloride channels. Nature 1989; 341:233 - 6; http://dx.doi.org/10.1038/341233a0; PMID: 2550826
- Göpel S, Zhang Q, Eliasson L, Ma XS, Galvanovskis J, Kanno T, et al. Capacitance measurements of exocytosis in mouse pancreatic alpha-, beta- and delta-cells within intact islets of Langerhans. J Physiol 2004; 556:711 - 26; http://dx.doi.org/10.1113/jphysiol.2003.059675; PMID: 14966302
- MacDonald PE, De Marinis YZ, Ramracheya R, Salehi A, Ma X, Johnson PR, et al. A K ATP channel-dependent pathway within alpha cells regulates glucagon release from both rodent and human islets of Langerhans. PLoS Biol 2007; 5:e143; http://dx.doi.org/10.1371/journal.pbio.0050143; PMID: 17503968
- Rorsman P, Braun M, Zhang Q. Regulation of calcium in pancreatic α- and β-cells in health and disease. Cell Calcium 2012; 51:300 - 8; http://dx.doi.org/10.1016/j.ceca.2011.11.006; PMID: 22177710
- Sherman A, Keizer J, Rinzel J. Domain model for Ca2(+)-inactivation of Ca2+ channels at low channel density. Biophys J 1990; 58:985 - 95; http://dx.doi.org/10.1016/S0006-3495(90)82443-7; PMID: 2174274
- Rorsman P, Renström E. Insulin granule dynamics in pancreatic beta cells. Diabetologia 2003; 46:1029 - 45; http://dx.doi.org/10.1007/s00125-003-1153-1; PMID: 12879249
- Andersson SA, Pedersen MG, Vikman J, Eliasson L. Glucose-dependent docking and SNARE protein-mediated exocytosis in mouse pancreatic alpha-cell. Pflugers Arch 2011; 462:443 - 54; http://dx.doi.org/10.1007/s00424-011-0979-5; PMID: 21643653
- Sekine N, Cirulli V, Regazzi R, Brown LJ, Gine E, Tamarit-Rodriguez J, et al. Low lactate dehydrogenase and high mitochondrial glycerol phosphate dehydrogenase in pancreatic beta-cells. Potential role in nutrient sensing. J Biol Chem 1994; 269:4895 - 902; PMID: 8106462
- Schuit F, De Vos A, Farfari S, Moens K, Pipeleers D, Brun T, et al. Metabolic fate of glucose in purified islet cells. Glucose-regulated anaplerosis in beta cells. J Biol Chem 1997; 272:18572 - 9; http://dx.doi.org/10.1074/jbc.272.30.18572; PMID: 9228023
- Diao J, Allister EM, Koshkin V, Lee SC, Bhattacharjee A, Tang C, et al. UCP2 is highly expressed in pancreatic alpha-cells and influences secretion and survival. Proc Natl Acad Sci U S A 2008; 105:12057 - 62; http://dx.doi.org/10.1073/pnas.0710434105; PMID: 18701716
- Ostenson CG, Agren A, Brolin SE, Petersson B. Adenine nucleotide concentrations in A2-cell rich and normal pancreatic islets of the guinea pig. Diabete Metab 1980; 6:5 - 11; PMID: 6989662
- De Marinis YZ, Salehi A, Ward CE, Zhang Q, Abdulkader F, Bengtsson M, et al. GLP-1 inhibits and adrenaline stimulates glucagon release by differential modulation of N- and L-type Ca2+ channel-dependent exocytosis. Cell Metab 2010; 11:543 - 53; http://dx.doi.org/10.1016/j.cmet.2010.04.007; PMID: 20519125
- Gromada J, Bokvist K, Ding WG, Barg S, Buschard K, Renström E, et al. Adrenaline stimulates glucagon secretion in pancreatic A-cells by increasing the Ca2+ current and the number of granules close to the L-type Ca2+ channels. J Gen Physiol 1997; 110:217 - 28; http://dx.doi.org/10.1085/jgp.110.3.217; PMID: 9276750
- Quoix N, Cheng-Xue R, Mattart L, Zeinoun Z, Guiot Y, Beauvois MC, et al. Glucose and pharmacological modulators of ATP-sensitive K+ channels control [Ca2+]c by different mechanisms in isolated mouse alpha-cells. Diabetes 2009; 58:412 - 21; http://dx.doi.org/10.2337/db07-1298; PMID: 19008345
- Braun M, Ramracheya R, Bengtsson M, Clark A, Walker JN, Johnson PR, et al. Gamma-aminobutyric acid (GABA) is an autocrine excitatory transmitter in human pancreatic beta-cells. Diabetes 2010; 59:1694 - 701; http://dx.doi.org/10.2337/db09-0797; PMID: 20413510
- Salehi A, Vieira E, Gylfe E. Paradoxical stimulation of glucagon secretion by high glucose concentrations. Diabetes 2006; 55:2318 - 23; http://dx.doi.org/10.2337/db06-0080; PMID: 16873696
- Göpel SO, Kanno T, Barg S, Weng XG, Gromada J, Rorsman P. Regulation of glucagon release in mouse -cells by KATP channels and inactivation of TTX-sensitive Na+ channels. J Physiol 2000; 528:509 - 20; http://dx.doi.org/10.1111/j.1469-7793.2000.00509.x; PMID: 11060128
- Hirose H, Maruyama H, Kido K, Ito K, Koyama K, Tashiro Y, et al. Alpha- and beta-cell function in obese Zucker (fa/fa) rats: a study with the isolated perfused pancreas. Clin Sci (Lond) 1994; 86:311 - 6; PMID: 8156742
- Spigelman AF, Dai X, MacDonald PE. Voltage-dependent K(+) channels are positive regulators of alpha cell action potential generation and glucagon secretion in mice and humans. Diabetologia 2010; 53:1917 - 26; http://dx.doi.org/10.1007/s00125-010-1759-z; PMID: 20446079
- Gromada J, Høy M, Olsen HL, Gotfredsen CF, Buschard K, Rorsman P, et al. Gi2 proteins couple somatostatin receptors to low-conductance K+ channels in rat pancreatic alpha-cells. Pflugers Arch 2001; 442:19 - 26; http://dx.doi.org/10.1007/s004240000474; PMID: 11374064
- Yoshimoto Y, Fukuyama Y, Horio Y, Inanobe A, Gotoh M, Kurachi Y. Somatostatin induces hyperpolarization in pancreatic islet alpha cells by activating a G protein-gated K+ channel. FEBS Lett 1999; 444:265 - 9; http://dx.doi.org/10.1016/S0014-5793(99)00076-9; PMID: 10050772
- Hauge-Evans AC, King AJ, Carmignac D, Richardson CC, Robinson IC, Low MJ, et al. Somatostatin secreted by islet delta-cells fulfills multiple roles as a paracrine regulator of islet function. Diabetes 2009; 58:403 - 11; http://dx.doi.org/10.2337/db08-0792; PMID: 18984743
- Rorsman P, Eliasson L, Kanno T, Zhang Q, Gopel S. Electrophysiology of pancreatic β-cells in intact mouse islets of Langerhans. Prog Biophys Mol Biol 2011; 107:224 - 35; http://dx.doi.org/10.1016/j.pbiomolbio.2011.06.009; PMID: 21762719
- Gall D, Gromada J, Susa I, Rorsman P, Herchuelz A, Bokvist K. Significance of Na/Ca exchange for Ca2+ buffering and electrical activity in mouse pancreatic beta-cells. Biophys J 1999; 76:2018 - 28; http://dx.doi.org/10.1016/S0006-3495(99)77359-5; PMID: 10096898
- Colsoul B, Schraenen A, Lemaire K, Quintens R, Van Lommel L, Segal A, et al. Loss of high-frequency glucose-induced Ca2+ oscillations in pancreatic islets correlates with impaired glucose tolerance in Trpm5-/- mice. Proc Natl Acad Sci U S A 2010; 107:5208 - 13; http://dx.doi.org/10.1073/pnas.0913107107; PMID: 20194741
- Jacobson DA, Philipson LH. TRP channels of the pancreatic beta cell. Handb Exp Pharmacol 2007:409-24.
- Franklin I, Gromada J, Gjinovci A, Theander S, Wollheim CB. Beta-cell secretory products activate alpha-cell ATP-dependent potassium channels to inhibit glucagon release. Diabetes 2005; 54:1808 - 15; http://dx.doi.org/10.2337/diabetes.54.6.1808; PMID: 15919803
- Wendt A, Birnir B, Buschard K, Gromada J, Salehi A, Sewing S, et al. Glucose inhibition of glucagon secretion from rat alpha-cells is mediated by GABA released from neighboring beta-cells. Diabetes 2004; 53:1038 - 45; http://dx.doi.org/10.2337/diabetes.53.4.1038; PMID: 15047619
- Taneera J, Jin Z, Jin Y, Muhammed SJ, Zhang E, Lang S, et al. γ-Aminobutyric acid (GABA) signalling in human pancreatic islets is altered in type 2 diabetes. Diabetologia 2012; 55:1985 - 94; http://dx.doi.org/10.1007/s00125-012-2548-7; PMID: 22538358
- Stein V, Nicoll RA. GABA generates excitement. Neuron 2003; 37:375 - 8; http://dx.doi.org/10.1016/S0896-6273(03)00056-4; PMID: 12575946
- Johansson H, Gylfe E, Hellman B. The actions of arginine and glucose on glucagon secretion are mediated by opposite effects on cytoplasmic Ca2+. Biochem Biophys Res Commun 1987; 147:309 - 14; http://dx.doi.org/10.1016/S0006-291X(87)80122-5; PMID: 3307774
- González-Vélez V, Dupont G, Gil A, González A, Quesada I. Model for glucagon secretion by pancreatic α-cells. PLoS One 2012; 7:e32282; http://dx.doi.org/10.1371/journal.pone.0032282; PMID: 22412861
- Pipeleers DG, Schuit FC. in't Veld PA, Maes E, Hooghe-Peters EL, Van de Winkel M, et al. Interplay of nutrients and hormones in the regulation of insulin release. Endocrinology 1985; 117:824-33.
- Bokvist K, Olsen HL, Høy M, Gotfredsen CF, Holmes WF, Buschard K, et al. Characterisation of sulphonylurea and ATP-regulated K+ channels in rat pancreatic A-cells. Pflugers Arch 1999; 438:428 - 36; http://dx.doi.org/10.1007/s004240051058; PMID: 10519134
- Bosco D, Armanet M, Morel P, Niclauss N, Sgroi A, Muller YD, et al. Unique arrangement of alpha- and beta-cells in human islets of Langerhans. Diabetes 2010; 59:1202 - 10; http://dx.doi.org/10.2337/db09-1177; PMID: 20185817
- Cabrera O, Berman DM, Kenyon NS, Ricordi C, Berggren PO, Caicedo A. The unique cytoarchitecture of human pancreatic islets has implications for islet cell function. Proc Natl Acad Sci U S A 2006; 103:2334 - 9; http://dx.doi.org/10.1073/pnas.0510790103; PMID: 16461897
- Robertson RP, Zhou H, Slucca M. A role for zinc in pancreatic islet β-cell cross-talk with the α-cell during hypoglycaemia. Diabetes Obes Metab 2011; 13:Suppl 1 106 - 11; http://dx.doi.org/10.1111/j.1463-1326.2011.01448.x; PMID: 21824263
- Ashcroft F, Rorsman P. Type 2 diabetes mellitus: not quite exciting enough?. Hum Mol Genet 2004; 13:Spec No 1 R21 - 31; http://dx.doi.org/10.1093/hmg/ddh066; PMID: 14734629
- Banarer S, McGregor VP, Cryer PE. Intraislet hyperinsulinemia prevents the glucagon response to hypoglycemia despite an intact autonomic response. Diabetes 2002; 51:958 - 65; http://dx.doi.org/10.2337/diabetes.51.4.958; PMID: 11916913
- Greenbaum CJ, Havel PJ, Taborsky GJ Jr., Klaff LJ. Intra-islet insulin permits glucose to directly suppress pancreatic A cell function. J Clin Invest 1991; 88:767 - 73; http://dx.doi.org/10.1172/JCI115375; PMID: 1679440
- Meier JJ, Ueberberg S, Korbas S, Schneider S. Diminished glucagon suppression after β-cell reduction is due to impaired α-cell function rather than an expansion of α-cell mass. Am J Physiol Endocrinol Metab 2011; 300:E717 - 23; http://dx.doi.org/10.1152/ajpendo.00315.2010; PMID: 21285404
- Bansal P, Wang Q. Insulin as a physiological modulator of glucagon secretion. Am J Physiol Endocrinol Metab 2008; 295:E751 - 61; http://dx.doi.org/10.1152/ajpendo.90295.2008; PMID: 18647881
- Leung YM, Ahmed I, Sheu L, Gao X, Hara M, Tsushima RG, et al. Insulin regulates islet alpha-cell function by reducing KATP channel sensitivity to adenosine 5′-triphosphate inhibition. Endocrinology 2006; 147:2155 - 62; http://dx.doi.org/10.1210/en.2005-1249; PMID: 16455778
- Xu E, Kumar M, Zhang Y, Ju W, Obata T, Zhang N, et al. Intra-islet insulin suppresses glucagon release via GABA-GABAA receptor system. Cell Metab 2006; 3:47 - 58; http://dx.doi.org/10.1016/j.cmet.2005.11.015; PMID: 16399504
- Slucca M, Harmon JS, Oseid EA, Bryan J, Robertson RP. ATP-sensitive K+ channel mediates the zinc switch-off signal for glucagon response during glucose deprivation. Diabetes 2010; 59:128 - 34; http://dx.doi.org/10.2337/db09-1098; PMID: 19808893
- Tudurí E, Marroquí L, Soriano S, Ropero AB, Batista TM, Piquer S, et al. Inhibitory effects of leptin on pancreatic alpha-cell function. Diabetes 2009; 58:1616 - 24; http://dx.doi.org/10.2337/db08-1787; PMID: 19401420
- Kieffer TJ, Heller RS, Leech CA, Holz GG, Habener JF. Leptin suppression of insulin secretion by the activation of ATP-sensitive K+ channels in pancreatic beta-cells. Diabetes 1997; 46:1087 - 93; http://dx.doi.org/10.2337/diabetes.46.6.1087; PMID: 9166685
- Ning K, Miller LC, Laidlaw HA, Watterson KR, Gallagher J, Sutherland C, et al. Leptin-dependent phosphorylation of PTEN mediates actin restructuring and activation of ATP-sensitive K+ channels. J Biol Chem 2009; 284:9331 - 40; http://dx.doi.org/10.1074/jbc.M806774200; PMID: 19208634
- de Heer J, Rasmussen C, Coy DH, Holst JJ. Glucagon-like peptide-1, but not glucose-dependent insulinotropic peptide, inhibits glucagon secretion via somatostatin (receptor subtype 2) in the perfused rat pancreas. Diabetologia 2008; 51:2263 - 70; http://dx.doi.org/10.1007/s00125-008-1149-y; PMID: 18795252
- Holst JJ, Christensen M, Lund A, de Heer J, Svendsen B, Kielgast U, et al. Regulation of glucagon secretion by incretins. Diabetes Obes Metab 2011; 13:Suppl 1 89 - 94; http://dx.doi.org/10.1111/j.1463-1326.2011.01452.x; PMID: 21824261
- Leech CA, Dzhura I, Chepurny OG, Kang G, Schwede F, Genieser HG, et al. Molecular physiology of glucagon-like peptide-1 insulin secretagogue action in pancreatic β cells. Prog Biophys Mol Biol 2011; 107:236 - 47; http://dx.doi.org/10.1016/j.pbiomolbio.2011.07.005; PMID: 21782840
- Seino S, Takahashi H, Fujimoto W, Shibasaki T. Roles of cAMP signalling in insulin granule exocytosis. Diabetes Obes Metab 2009; 11:Suppl 4 180 - 8; http://dx.doi.org/10.1111/j.1463-1326.2009.01108.x; PMID: 19817800
- Eliasson L, Abdulkader F, Braun M, Galvanovskis J, Hoppa MB, Rorsman P. Novel aspects of the molecular mechanisms controlling insulin secretion. J Physiol 2008; 586:3313 - 24; http://dx.doi.org/10.1113/jphysiol.2008.155317; PMID: 18511483
- Beauvois MC, Merezak C, Jonas JC, Ravier MA, Henquin JC, Gilon P. Glucose-induced mixed [Ca2+]c oscillations in mouse beta-cells are controlled by the membrane potential and the SERCA3 Ca2+-ATPase of the endoplasmic reticulum. Am J Physiol Cell Physiol 2006; 290:C1503 - 11; http://dx.doi.org/10.1152/ajpcell.00400.2005; PMID: 16381799
- Martín F, Soria B. Glucose-induced [Ca2+]i oscillations in single human pancreatic islets. Cell Calcium 1996; 20:409 - 14; http://dx.doi.org/10.1016/S0143-4160(96)90003-2; PMID: 8955555
- Göpel SO, Kanno T, Barg S, Eliasson L, Galvanovskis J, Renström E, et al. Activation of Ca(2+)-dependent K(+) channels contributes to rhythmic firing of action potentials in mouse pancreatic beta cells. J Gen Physiol 1999; 114:759 - 70; http://dx.doi.org/10.1085/jgp.114.6.759; PMID: 10578013
- Gustavsson N, Wei SH, Hoang DN, Lao Y, Zhang Q, Radda GK, et al. Synaptotagmin-7 is a principal Ca2+ sensor for Ca2+ -induced glucagon exocytosis in pancreas. J Physiol 2009; 587:1169 - 78; http://dx.doi.org/10.1113/jphysiol.2008.168005; PMID: 19171650
- Pedersen MG, Cortese G, Eliasson L. Mathematical modeling and statistical analysis of calcium-regulated insulin granule exocytosis in β-cells from mice and humans. Prog Biophys Mol Biol 2011; 107:257 - 64; http://dx.doi.org/10.1016/j.pbiomolbio.2011.07.012; PMID: 21839108
- De Marinis YZ, Zhang E, Amisten S, Taneera J, Renström E, Rorsman P, et al. Enhancement of glucagon secretion in mouse and human pancreatic alpha cells by protein kinase C (PKC) involves intracellular trafficking of PKCalpha and PKCdelta. Diabetologia 2010; 53:717 - 29; http://dx.doi.org/10.1007/s00125-009-1635-x; PMID: 20020096
- Tian G, Sandler S, Gylfe E, Tengholm A. Glucose- and hormone-induced cAMP oscillations in α- and β-cells within intact pancreatic islets. Diabetes 2011; 60:1535 - 43; http://dx.doi.org/10.2337/db10-1087; PMID: 21444924
- Bertram R, Sherman A, Satin LS. Electrical bursting, calcium oscillations, and synchronization of pancreatic islets. Adv Exp Med Biol 2010; 654:261 - 79; http://dx.doi.org/10.1007/978-90-481-3271-3_12; PMID: 20217502
- Diderichsen PM, Göpel SO. Modelling the electrical activity of pancreatic alpha-cells based on experimental data from intact mouse islets. J Biol Phys 2006; 32:209 - 29; http://dx.doi.org/10.1007/s10867-006-9013-0; PMID: 19669464
- Gonzalez-Velez V, Gil A, Quesada I. Minimal state models for ionic channels involved in glucagon secretion. Math Biosci Eng 2010; 7:793 - 807; http://dx.doi.org/10.3934/mbe.2010.7.793; PMID: 21077708
- Hjortoe GM, Hagel GM, Terry BR, Thastrup O, Arkhammar PO. Functional identification and monitoring of individual alpha and beta cells in cultured mouse islets of Langerhans. Acta Diabetol 2004; 41:185 - 93; http://dx.doi.org/10.1007/s00592-004-0164-9; PMID: 15660202
- Braaten JT, Faloona GR, Unger RH. The effect of insulin on the alpha-cell response to hyperglycemia in long-standing alloxan diabetes. J Clin Invest 1974; 53:1017 - 21; http://dx.doi.org/10.1172/JCI107638; PMID: 4592597
- Meier JJ, Kjems LL, Veldhuis JD, Lefèbvre P, Butler PC. Postprandial suppression of glucagon secretion depends on intact pulsatile insulin secretion: further evidence for the intraislet insulin hypothesis. Diabetes 2006; 55:1051 - 6; http://dx.doi.org/10.2337/diabetes.55.04.06.db05-1449; PMID: 16567528
- Zhou H, Tran PO, Yang S, Zhang T, LeRoy E, Oseid E, et al. Regulation of alpha-cell function by the beta-cell during hypoglycemia in Wistar rats: the “switch-off” hypothesis. Diabetes 2004; 53:1482 - 7; http://dx.doi.org/10.2337/diabetes.53.6.1482; PMID: 15161752
- Braun M, Ramracheya R, Bengtsson M, Zhang Q, Karanauskaite J, Partridge C, et al. Voltage-gated ion channels in human pancreatic beta-cells: electrophysiological characterization and role in insulin secretion. Diabetes 2008; 57:1618 - 28; http://dx.doi.org/10.2337/db07-0991; PMID: 18390794
- Huang YC, Rupnik M, Gaisano HY. Unperturbed islet α-cell function examined in mouse pancreas tissue slices. J Physiol 2011; 589:395 - 408; http://dx.doi.org/10.1113/jphysiol.2010.200345; PMID: 21078586
- Quesada I, Nadal A, Soria B. Different effects of tolbutamide and diazoxide in alpha, beta-, and delta-cells within intact islets of Langerhans. Diabetes 1999; 48:2390 - 7; http://dx.doi.org/10.2337/diabetes.48.12.2390; PMID: 10580428
- Henquin JC, Rahier J. Pancreatic alpha cell mass in European subjects with type 2 diabetes. Diabetologia 2011; 54:1720 - 5; http://dx.doi.org/10.1007/s00125-011-2118-4; PMID: 21465328
- Quesada I, Todorova MG, Alonso-Magdalena P, Beltrá M, Carneiro EM, Martin F, et al. Glucose induces opposite intracellular Ca2+ concentration oscillatory patterns in identified alpha- and beta-cells within intact human islets of Langerhans. Diabetes 2006; 55:2463 - 9; http://dx.doi.org/10.2337/db06-0272; PMID: 16936194
- Cooperberg BA, Cryer PE. Insulin reciprocally regulates glucagon secretion in humans. Diabetes 2010; 59:2936 - 40; http://dx.doi.org/10.2337/db10-0728; PMID: 20811038
- Shah P, Basu A, Basu R, Rizza R. Impact of lack of suppression of glucagon on glucose tolerance in humans. Am J Physiol 1999; 277:E283 - 90; PMID: 10444424
- Unger RH, Aguilar-Parada E, Müller WA, Eisentraut AM. Studies of pancreatic alpha cell function in normal and diabetic subjects. J Clin Invest 1970; 49:837 - 48; http://dx.doi.org/10.1172/JCI106297; PMID: 4986215
- Baron AD, Schaeffer L, Shragg P, Kolterman OG. Role of hyperglucagonemia in maintenance of increased rates of hepatic glucose output in type II diabetics. Diabetes 1987; 36:274 - 83; http://dx.doi.org/10.2337/diabetes.36.3.274; PMID: 2879757
- Cervera A, Wajcberg E, Sriwijitkamol A, Fernandez M, Zuo P, Triplitt C, et al. Mechanism of action of exenatide to reduce postprandial hyperglycemia in type 2 diabetes. Am J Physiol Endocrinol Metab 2008; 294:E846 - 52; http://dx.doi.org/10.1152/ajpendo.00030.2008; PMID: 18334612
- Cooperberg BA, Cryer PE. Beta-cell-mediated signaling predominates over direct alpha-cell signaling in the regulation of glucagon secretion in humans. Diabetes Care 2009; 32:2275 - 80; http://dx.doi.org/10.2337/dc09-0798; PMID: 19729529
- Müller WA, Faloona GR, Unger RH. Hyperglucagonemia in diabetic ketoacidosis. Its prevalence and significance. Am J Med 1973; 54:52 - 7; PMID: 4629972
- Raskin P, Unger RH. Hyperglucagonemia and its suppression. Importance in the metabolic control of diabetes. N Engl J Med 1978; 299:433 - 6; http://dx.doi.org/10.1056/NEJM197808312990901; PMID: 683275
- Yu X, Park BH, Wang MY, Wang ZV, Unger RH. Making insulin-deficient type 1 diabetic rodents thrive without insulin. Proc Natl Acad Sci U S A 2008; 105:14070 - 5; http://dx.doi.org/10.1073/pnas.0806993105; PMID: 18779578
- Nyholm B, Orskov L, Hove KY, Gravholt CH, Møller N, Alberti KG, et al. The amylin analog pramlintide improves glycemic control and reduces postprandial glucagon concentrations in patients with type 1 diabetes mellitus. Metabolism 1999; 48:935 - 41; http://dx.doi.org/10.1016/S0026-0495(99)90232-9; PMID: 10421239
- Bohannon NV, Lorenzi M, Grodsky GM, Karam JH. Stimulatory effects of tolbutamide infusion on plasma glucagon in insulin-dependent diabetic subjects. J Clin Endocrinol Metab 1982; 54:459 - 62; http://dx.doi.org/10.1210/jcem-54-2-459; PMID: 7054228
- Østergård T, Degn KB, Gall MA, Carr RD, Veldhuis JD, Thomsen MK, et al. The insulin secretagogues glibenclamide and repaglinide do not influence growth hormone secretion in humans but stimulate glucagon secretion during profound insulin deficiency. J Clin Endocrinol Metab 2004; 89:297 - 302; http://dx.doi.org/10.1210/jc.2003-031011; PMID: 14715864
- Landstedt-Hallin L, Adamson U, Lins PE. Oral glibenclamide suppresses glucagon secretion during insulin-induced hypoglycemia in patients with type 2 diabetes. J Clin Endocrinol Metab 1999; 84:3140 - 5; http://dx.doi.org/10.1210/jc.84.9.3140; PMID: 10487677
- Varanasi A, Bellini N, Rawal D, Vora M, Makdissi A, Dhindsa S, et al. Liraglutide as additional treatment for type 1 diabetes. Eur J Endocrinol 2011; 165:77 - 84; http://dx.doi.org/10.1530/EJE-11-0330; PMID: 21646283
- Rajan AS, Aguilar-Bryan L, Nelson DA, Nichols CG, Wechsler SW, Lechago J, et al. Sulfonylurea receptors and ATP-sensitive K+ channels in clonal pancreatic alpha cells. Evidence for two high affinity sulfonylurea receptors. J Biol Chem 1993; 268:15221 - 8; PMID: 8325894
- Jacobson DA, Philipson LH. Ion Channels and Insulin secretion. In: Seino S, Bell G, eds. Pancreatic Beta Cell in Health and Desease. Japan: Springer, 2008:91- 110.
- Yang SN, Berggren PO. Beta-cell CaV channel regulation in physiology and pathophysiology. Am J Physiol Endocrinol Metab 2005; 288:E16 - 28; http://dx.doi.org/10.1152/ajpendo.00042.2004; PMID: 15585596
- Drews G, Krippeit-Drews P, Düfer M. Electrophysiology of islet cells. Adv Exp Med Biol 2010; 654:115 - 63; http://dx.doi.org/10.1007/978-90-481-3271-3_7; PMID: 20217497
- Philipson LH. Beta-cell ion channels: keys to endodermal excitability. Horm Metab Res 1999; 31:455 - 61; http://dx.doi.org/10.1055/s-2007-978774; PMID: 10494870
- Leung YM, Ahmed I, Sheu L, Tsushima RG, Diamant NE, Hara M, et al. Electrophysiological characterization of pancreatic islet cells in the mouse insulin promoter-green fluorescent protein mouse. Endocrinology 2005; 146:4766 - 75; http://dx.doi.org/10.1210/en.2005-0803; PMID: 16109783
- Vignali S, Leiss V, Karl R, Hofmann F, Welling A. Characterization of voltage-dependent sodium and calcium channels in mouse pancreatic A- and B-cells. J Physiol 2006; 572:691 - 706; PMID: 16513675
- Beeler GW, Reuter H. Reconstruction of the action potential of ventricular myocardial fibres. J Physiol 1977; 268:177 - 210; PMID: 874889
- Göpel S, Kanno T, Barg S, Galvanovskis J, Rorsman P. Voltage-gated and resting membrane currents recorded from B-cells in intact mouse pancreatic islets. J Physiol 1999; 521:717 - 28; http://dx.doi.org/10.1111/j.1469-7793.1999.00717.x; PMID: 10601501
- Rorsman P. Two types of Ca2+ currents with different sensitivities to organic Ca2+ channel antagonists in guinea pig pancreatic alpha 2 cells. J Gen Physiol 1988; 91:243 - 54; http://dx.doi.org/10.1085/jgp.91.2.243; PMID: 2453604
- Chabwine JN, Talavera K, Verbert L, Eggermont J, Vanderwinden JM, De Smedt H, et al. Differential contribution of the Na(+)-K(+)-2Cl(-) cotransporter NKCC1 to chloride handling in rat embryonic dorsal root ganglion neurons and motor neurons. FASEB J 2009; 23:1168 - 76; http://dx.doi.org/10.1096/fj.08-116012; PMID: 19103648
- Hille B. Ion channels of exitable membranes. Sunderland: Sinauer Associates, Inc, 2001.

