
Abstract
Pro-inflammatory cytokines are important mediators of β-cell demise in type 1 diabetes, and similar mechanisms are increasingly implicated in type 2 diabetes, where a state of chronic inflammation may persist. It is likely that the actions of anti-inflammatory cytokines are also altered in diabetes. Cytokines are released from immune cells, which may be recruited to the islets in diabetes, but they can also be produced by islet endocrine cells in response to environmental factors, including enteroviral infection. Since enteroviral infection of islet cells may influence the development of diabetes in humans, we examined the actions of two cytokines, IL-13 and IL-6, whose expression are reported to be altered in β-cells during enteroviral infection. Human and rodent islet cells were shown to express receptors for both IL-13 and IL-6, and treatment with either cytokine resulted in the rapid phosphorylation of STAT3 and STAT6. However, while β-cells were protected against a range of cytotoxic insults during exposure to IL-13, treatment with IL-6 enhanced cytotoxicity and western blotting revealed that IL-13 induced one specific isoform of phospho-STAT6 preferentially. Upon incubation with both cytokines together, the isoform of STAT6 that was upregulated by IL-13 alone was again induced, and the effects of IL-6 on β-cell viability were attenuated. Overall, the results suggest that induction of specific isoforms of STAT family transcription factors may underlie the cytoprotective actions of IL-13, and they imply that selective targeting of specific STAT-mediated signaling components could provide a means to ameliorate the loss of β-cell viability in diabetes.
Introduction
Increasing evidence implies that alterations in the balance between anti- and pro-inflammatory cytokines may play a role in the development of both type 1 and type 2 diabetes in humans. In type 1 diabetes, pro-inflammatory cytokines are secreted from CD4+ and CD8+ lymphocytes and from macrophages during the process of insulitisCitation1,Citation2 whereas the secretion of anti-inflammatory cytokines (e.g., IL-4, IL-10 and IL-13) is reduced.Citation3,Citation4 As such, the potentially beneficial effects of these molecules on β-cell viability is minimised.Citation5-Citation7 Type 2 diabetes is associated with a chronic low-grade islet inflammation, and although the numbers of infiltrating immune cells are reduced by comparison with type 1 diabetes, the islet cells may still be exposed to elevated levels of inflammatory mediators such as IL-6, TNFα and IL-1β.Citation8,Citation9 Thus, in both diseases, islet cells encounter a complex inflammatory milieu, the exact composition of which will be critical in determining the fate of the β-cells. Additionally, it is known that islet endocrine cells are themselves capable of producing certain cytokines (both pro- and anti-inflammatory molecules),Citation10,Citation11 and it seems probable that agents or conditions that alter the profile of islet cell cytokine production may influence their viability. In particular, it has been shown that this profile is changed during persistent enteroviral infection of islet cells,Citation12 and this is of significance because histological evidence implies that human islet cells can sustain a persistent enteroviral infection in patients with diabetes.Citation13,Citation14 On this basis, it seems possible that the presence of enterovirus could influence the sensitivity of the β-cells to cytotoxic stimuli.
Cytokines exert their actions via specific cell surface receptors, which when activated, promote signaling cascades that culminate in altered gene expression via transcription factors such as NFκB or the signal transducer and activator of transcription (STAT) family. The role of NFκB in cytokine mediated β-cell toxicity is increasingly well established,Citation2 but the influence of STAT family members on β-cell viability has received less attention. Currently, seven members of this family are recognized, and many cytokines are capable of inducing the activation of more than one isoform.Citation15,Citation16 STAT1 and STAT2 have been considered as key targets for activation by pro-inflammatory cytokines whereas STAT6 is more frequently stimulated by anti-inflammatory cytokines, including IL-4 and IL-13.Citation15,Citation17,Citation18 STAT3 phosphorylation is also sometimes associated with anti-inflammatory activity, but STAT3 is activated by a range of stimuli, including IL-6,Citation19 leptinCitation20 and prolactin,Citation21 not all of which are associated with anti-inflammatory responses. Therefore, a first aim in the current study was to evaluate the impact of IL-6 and IL-13 on STAT signaling in the pancreatic β-cell.
IL-13 is known to mediate its effects by interaction with its cognate cell surface receptor [comprising the IL-4 receptor α (IL-4Rα) chain and the IL-13 receptor α 1 (IL-13Rα1) chain (reviewed in refs. Citation22 and Citation23)] whereas IL-6 binds to a different receptor comprising a unique protein binding subunit (IL-6R), which recruits two glycoprotein 130 (GP130) monomers to mediate signal transduction (reviewed in ref. Citation24). Upon interaction of these cytokines with their relevant receptors, Janus kinases (Jak) become associated with the cytoplasmic tail of each complex and are phosphorylated at defined sites. Activation of the Jak kinases then induces the phosphorylation of key tyrosine residues on the cytoplasmic tail of either IL-4Rα or GP130, leading to recruitment and phosphorylation of STAT proteins, which then dissociate from the receptor complex, homodimerize and translocate to the nucleus to regulate the expression of target genes.Citation25 Furthermore, other signaling pathways can be induced by IL-6 and IL-13, including the activation of PI-3 kinase and phosphorylation of Akt.Citation26,Citation27 Therefore, a further aim was to establish the impact of IL-13 and IL-6 on β-cell viability and to assess which signal transduction cascades are important for mediating these effects. A rodent β-cell line (INS-1E) was employed as a model system for the study, with key data further validated using a newly established human insulin-secreting cell line and/or human islets.
Results
Expression of IL-13 and IL-6 receptor components
Expression of components of the IL-13 receptor was examined by RT-PCR in RNA extracted from INS-1E cells or isolated human islets. An appropriately sized amplicon was generated for both receptor subunits from each source (Fig. S1) and their identities confirmed by direct sequencing.
Expression of IL-6 receptor components was also observed in INS-1E cells (Fig. S2A and B) or human islets (Fig. S2C). It is known that both membrane bound and soluble forms of IL-6R (sIL-6R) exist; the latter being generated by alternative splicing of the transcript to yield a molecule which lacks the C-terminal transmembrane domain encoded within exon 10.Citation28 Therefore, primers were designed to amplify a region around exon 10 and RT-PCR analysis revealed that human islets express both forms of the IL-6 receptor ().
Figure 1. Human islets express both membrane-bound and soluble isoforms of the IL-6R. (A) cDNA was generated from human islet RNA and amplified by RT-PCR using primers spanning exon 10 of the IL-6R [membrane-bound form (IL-6R): 265 bp; soluble form (sIL-6R): 171 bp]. Bands were separated by electrophoresis and visualized under long-wave UV illumination. (B) The identity of amplicons was confirmed by the direct sequencing of each excised band. The sequence of each amplicon is presented, with exon 10 emboldened.
![Figure 1. Human islets express both membrane-bound and soluble isoforms of the IL-6R. (A) cDNA was generated from human islet RNA and amplified by RT-PCR using primers spanning exon 10 of the IL-6R [membrane-bound form (IL-6R): 265 bp; soluble form (sIL-6R): 171 bp]. Bands were separated by electrophoresis and visualized under long-wave UV illumination. (B) The identity of amplicons was confirmed by the direct sequencing of each excised band. The sequence of each amplicon is presented, with exon 10 emboldened.](/cms/asset/2b2ab393-80f8-465c-af09-e5ad66f9f24f/kisl_a_10924249_f0001.gif)
IL-13 is cytoprotective in INS-1E cells
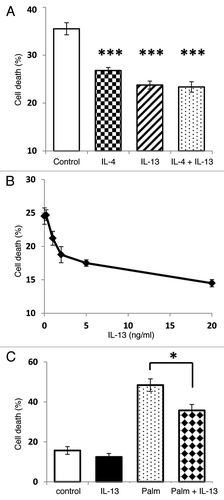
IL-13 is cytoprotective to islet cells treated with pro-inflammatory cytokines,Citation7 but there are few data indicating whether IL-13 also influences cell viability in response to other cytotoxic insults. Therefore, INS-1E cells were cultured for 96 h under serum-deprived conditions as a means to reduce viability. Incubation of serum-deprived cells with IL-13 (or a related cytokine, IL-4) significantly improved their viability compared with that of untreated controls (), but co-incubation with both IL-13 and IL-4 did not enhance cell viability further (). The effects of IL-13 (0–20 ng/ml) were dose-dependent, with an EC50 of ~2 ng/ml (), and it also significantly improved the viability of cells treated with the saturated fatty acid, palmitate (250 μM) ().
Figure 2. IL-13 treatment protects INS-1E cells against a range of cytotoxic stimuli. Cell death was induced either by incubation of INS-1E cells under serum-free conditions for 96 h (A and B) or with 250 μM palmitate for 48 h (C). (A) Serum deprived cells were either untreated (control) or incubated with IL-13 (20 ng/ml), IL-4 (2 ng/ml) or both cytokines together. (B) Serum deprived cells were treated with increasing concentrations of IL-13 (0–20 ng/ml). (C) For fatty acid experiments, cells were treated with IL-13 (20 ng/ml) prior to incubation with palmitate. In all cases, the viability of cells was measured by flow cytometry using PI staining. (A and C) Data represent mean values ± SEM (n = 3), ***p < 0.001, **p < 0.01 as indicated. (B) Data are presented as mean values ± SEM from a representative experiment (n = 6) which was repeated with similar results.
Activation of the PI-3K/Akt pathway is unlikely to be responsible for the cytoprotective effect of IL-13 in INS-1E cells
Upon the binding of IL-13 to its receptor a number of Jak kinases have been reported to be phosphorylated,Citation29 and among these, Jak2 is frequently a major substrate. Therefore, in the current study Jak2 phosphorylation was monitored. Western blotting analysis revealed a basal level of Jak2 phosphorylation in untreated INS-1E cells, and as expected, this was increased significantly during exposure to IL-13 ().
Figure 3. The PI-3K/Akt signal transduction pathway is not involved in IL-13 mediated cytoprotection. INS-1E cells were either untreated or stimulated with IL-13 (20 ng/ml). Following treatment, total protein was extracted, and the protein concentration equalised to 50 μg. The levels of pJak2 and β-actin were determined by Western blotting. (A) Blots are representative of three separate experiments. (B) The intensity of bands was measured by densitometry, with pJak2 expressed relative to β-actin. Data are presented as the mean values from three separate experiments, **p < 0.01 relative to control. (C) INS-1E cells were incubated under serum starved conditions for 96 h alone (control) or in the presence of IL-13 (20 ng/ml), wortmannin (500 nM) or IL-13 + wortmannin. After treatment, the viability of cells was measured by flow cytometry following PI staining. Data represent mean values ± SEM (n = 3), ***p < 0.001 relative to wortmannin alone.
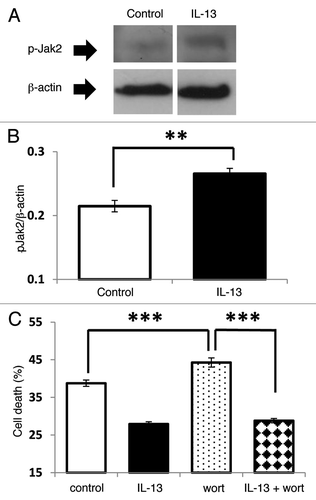
Subsequent to Jak phosphorylation, IL-13 can induce the phosphorylation of Akt, and in some cell types the PI-3K/Akt pathway may contribute to cytoprotection.Citation26 To establish whether this pathway plays a role in the cytoprotective response induced by IL-13 in INS-1E cells, the PI-3K inhibitor wortmannin was employed. As expected, IL-13 improved the viability of serum deprived cells, but treatment with wortmannin did not influence this cytoprotective response ().
The protective effects of IL-13 involve Jak/STAT signaling in INS-1E cells
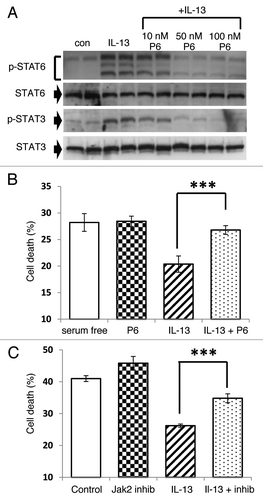
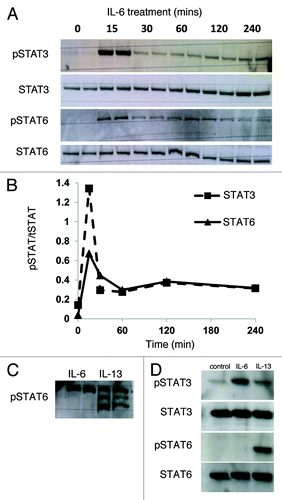
In some cell types it has been reported that both STAT6 and STAT3 can become phosphorylated in response to IL-13,Citation15 and we examined whether this happens in INS-1E () cells and human islets (). Following treatment with IL-13, Western blotting with selective antisera for pSTAT6 detected an intensely stained band migrating at a molecular weight ~105 kDa, corresponding with the expected size of STAT6. Surprisingly, however, the antibody also labeled two additional bands running at apparently higher molecular weights in INS-1E cells under these conditions (). A similar pattern was also detected in response to IL-4 or after treatment of either a different rodent β-cell line (BRIN-BD11) or the recently developed human insulin secreting cell line, 1.1B4 with IL-13 (not shown). Importantly, pre-incubation of the pSTAT6 antiserum with a specific blocking peptide corresponding to the epitope employed for antibody generation, abolished the appearance of all three bands, thereby confirming the specificity to the antibody (not shown).
Figure 4. STAT6 and STAT3 are phosphorylated in response to IL-13 in both INS-1E cells and human islets. (A and C) INS-1E cells were treated with IL-13 (20 ng/ml) over a time-course of 240 min. (B and D) Human islets were also exposed to IL-13 (20 ng/ml) for 30 min. After treatment, total protein was extracted, and the protein concentration equalized to either (A) 50 μg, (B) 10 μg, (C) 100 μg or (D) 30 μg per well. The expression levels of (A and B) pSTAT6 and total STAT6 or (C and D) pSTAT3 and total STAT3 were determined by western blot. (A) Lower dotted arrow indicates 105 kDa, middle is approximately 150 kDa and upper is around 200 kDa. (A–D) Data are representative of at least three separate experiments. (E and F) A series of blots comparing untreated (control) INS-1E cells with those exposed to IL-13 (20 ng/ml) for 30 min were analyzed by densitometry and pSTAT expressed relative to total STAT [(E) STAT6, n = 7; (F) STAT3, n = 6]. (G) Representative blots were also analyzed by densitometry and pSTAT expressed relative to total STAT over time. **p < 0.01, *p < 0.05 as indicated.
![Figure 4. STAT6 and STAT3 are phosphorylated in response to IL-13 in both INS-1E cells and human islets. (A and C) INS-1E cells were treated with IL-13 (20 ng/ml) over a time-course of 240 min. (B and D) Human islets were also exposed to IL-13 (20 ng/ml) for 30 min. After treatment, total protein was extracted, and the protein concentration equalized to either (A) 50 μg, (B) 10 μg, (C) 100 μg or (D) 30 μg per well. The expression levels of (A and B) pSTAT6 and total STAT6 or (C and D) pSTAT3 and total STAT3 were determined by western blot. (A) Lower dotted arrow indicates 105 kDa, middle is approximately 150 kDa and upper is around 200 kDa. (A–D) Data are representative of at least three separate experiments. (E and F) A series of blots comparing untreated (control) INS-1E cells with those exposed to IL-13 (20 ng/ml) for 30 min were analyzed by densitometry and pSTAT expressed relative to total STAT [(E) STAT6, n = 7; (F) STAT3, n = 6]. (G) Representative blots were also analyzed by densitometry and pSTAT expressed relative to total STAT over time. **p < 0.01, *p < 0.05 as indicated.](/cms/asset/108fe2e0-6eb5-4772-8e45-0834eb63f117/kisl_a_10924249_f0004.gif)
In addition to enhanced phosphorylation of STAT6, an increase in the phosphorylation of STAT3 was also detected upon IL-13 stimulation of INS-1E cells () or human islets (), although in these cases only a single phosphorylated protein was detected. More detailed examination of the time-course of STAT6 and STAT3 phosphorylation revealed a temporally similar pattern (). Phosphorylation of each protein occurred within 30 min of stimulation before then declining over the remainder of the time-course. Additional experiments revealed, however, that INS-1E cells transfected with a STAT3-specific reporter construct exhibited only a modest increase in luciferase activity when treated with IL-13 (2.7 ± 1.4 fold change from control) despite the clear increase in STAT3 phosphorylation seen under these conditions. To confirm that the phosphorylation of STAT proteins occurred via Jak activation, P6, a high affinity competitive global inhibitor of Jak kinasesCitation30 was used. Cells were preincubated with 10–100 nM P6 for 2 h prior to the addition of IL-13 for a further 30 min, and this effectively antagonised IL-13 induced phosphorylation of STAT6 and STAT3 (). The same inhibitor was then employed to determine whether the Jak/STAT pathway could be implicated in the cytoprotection mediated by IL-13. The viability of serum starved INS-1E cells treated with P6 (100 nM) alone was unchanged from that of untreated cells (). However, when cells were exposed to IL-13 in the presence of P6, the inhibitor abrogated the cytoprotective response (). To further support these data, a selective inhibitor of Jak2 activation (hexabromocyclohexane) was also employed. This agent exerted little direct effect on the viability of serum starved INS-1E cells, although it significantly antagonized the improvement in cell viability seen upon IL-13 treatment ().
Figure 5. IL-13 mediates cytoprotection via a Jak/STAT dependent pathway. (A) INS-1E cells were pre-incubated with increasing concentrations of P6 (0–100 nM) prior to a 30 min exposure to IL-13 (20 ng/ml). Protein was extracted, and the concentration of each sample equalised (25 μg) before expression of pSTAT6, STAT6, pSTAT3 and STAT3 were determined by Western blotting. Data are representative of two separate experiments. (B and C) Cells were grown under serum deprived conditions for 96 h and left untreated (serum free) or exposed to either (B) 100 nM P6, 20 ng/ml IL-13 or IL-13 + P6 or (C) 1 μM Jak2 inhibitor, 20 ng/ml IL-13 or IL-13 + Jak2 inhibitor. The viability of cells was then monitored by flow cytometry after PI staining. Data represent mean values ± SEM (n = 3), ***p < 0.001 relative to IL-13 alone.
IL-6 enhances STAT phosphorylation but reduces INS-1E cell viability
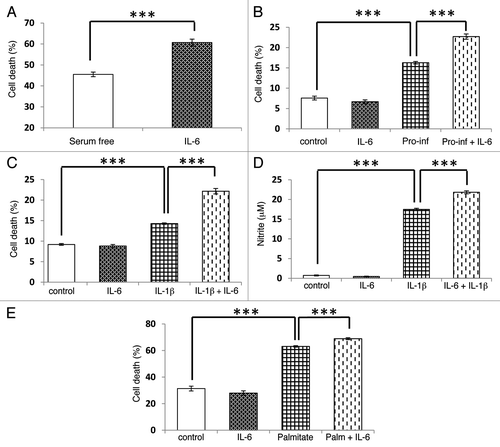
For comparative purposes, the influence of IL-6 on cell viability was then investigated by incubation of INS-1E cells under serum deprived conditions for 96 h. By contrast with the response seen during treatment with IL-13, exposure to IL-6 significantly worsened the viability of the cells under these conditions (). However, IL-6 did not directly influence the viability of INS-1E cells cultured in serum-replete medium (). Treatment of INS-1E cells for 48 h with a pro-inflammatory cytokine mix (containing TNFα, IFNγ and IL-1β) significantly reduced the viability of β-cells, and exposure to IL-6 again potentiated the loss of viability seen under these conditions (). Additionally, the cytotoxic effect of a single cytokine, IL-1β, was also enhanced by IL-6 treatment (). Since IL-1β may exert its effects via the induction of iNOS and subsequent production of nitric oxide (NO), we examined whether IL-6 influenced NO synthesis. IL-6 alone did not alter nitrite levels (an index of NO production) above the control value, whereas IL-1β dramatically increased nitrite production from INS-1E cells (). However, when IL-6 and IL-1β were present in combination, nitrite levels were potentiated above that seen with IL-1β alone (). Similarly, in 1.1B4 cells IL-6 potentiated IL-β induced cytotoxicity (Fig. S3B) and nitrite production (Fig. S3C). Finally, IL-6 also significantly potentiated the cytotoxic effects of palmitate treatment in INS-1E cells ().
Figure 6. IL-6 potentiates the cytotoxic effects of pro-inflammatory cytokines, serum deprivation and a saturated fatty acid. The viability of INS-1E cells or the formation of nitrite were induced by (A) 96 h serum deprivation; (B) incubation with a pro-inflammatory cytokine mix (pro-inf: 20 ng/ml of IL-1β, TNFα and IFNγ) for 48 h; (C and D) treatment with IL-1β (20 ng/ml) for 48 h; or (E) exposure to palmitate (250 μM) for 48 h. In all cases, cells were also incubated in the absence or presence of IL-6 (20 ng/ml) as shown. Cell viability was determined by flow cytometry after PI staining while nitrite was measured by the Griess assay. Data represent mean values ± SEM (n = 3–4), ***p < 0.001 as indicated.
IL-6 has been reported to induce STAT3 phosphorylation in human and mouse islets,Citation19 although the role of STAT6 in IL-6-induced responses has not been investigated. Therefore we assessed the profile of STAT activation following treatment of INS-1E with IL-6. As anticipated, STAT3 was rapidly phosphorylated in response to the cytokine (). This response peaked within 15 min, and levels of the phosphorylated protein remained elevated over the 4-h time-course (). IL-6 treatment also dramatically enhanced STAT3-responsive luciferase activity following transfection of INS-1E cells with a reporter construct (IL-6: 59.9 ± 24.6 fold increase from control, p < 0.05; n = 4). Unlike the situation seen with IL-13, however, probing of the protein extract with a pSTAT6 antiserum revealed a single immunopositive band upon IL-6 treatment (). The time-course followed a similar pattern to that seen with pSTAT3. Incubation of cells with both IL-6 and IL-13 together, followed by western blotting of the protein extracts with the pSTAT6 antiserum, resulted in an identical pattern of bands compared with that seen with IL-13 alone. Similar data were generated with 1.1B4 cells (Fig. S3A). Intriguingly, the pSTAT6 band induced by IL-6 migrated in parallel with the uppermost band regulated by IL-13 (). Treatment of human islets with IL-6 revealed a different pattern of STAT phosphorylation such that a rapid increase in STAT3 phosphorylation was seen but STAT6 phosphorylation was unchanged from control ().
Figure 7. STAT3 and STAT6 become phosphorylated in response to IL-6 treatment. (A and B) INS-1E cells were treated with IL-6 (20 ng/ml) over a 4-h time-course or alternatively (C) with IL-6 for 15 min or IL-13 (20 ng/ml) for 30 min. (D) Human islets were treated with IL-6 or IL-13 (20 ng/ml) for 30 min. After treatment total protein was extracted, and the protein concentration equalized to either (A) 50 μg, (C) 40 μg or (D) 15 μg per well. Levels of pSTAT3, STAT3, pSTAT6 and STAT6 were determined by western blotting. (A, C and D) Data are representative of at least two separate experiments. (B) The intensity of immunoreactive bands was determined by densitometry and the generation of pSTAT expressed relative to total STAT for each isoform.
The potentiating effects of IL-6 on viability and nitrite synthesis are reduced by incubation of cells with IL-13
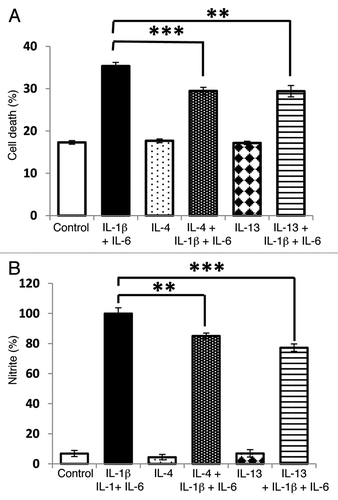
Finally, we explored whether IL-13 (or IL-4) can improve the viability of INS-1E cells under conditions under when they were also exposed to the usually cytotoxic combination of IL-1β and IL-6. As expected, IL-1β and IL-6 caused a net loss of cell viability but pre-incubation of the cells with IL-13 (or IL-4) for 48 h attenuated this response (). Culture with either of these anti-inflammatory cytokines also reduced the nitrite concentration achieved in response to the combination of IL-1β and IL-6 ().
Figure 8. Potentiation of cytokine induced cytotoxicity by IL-6 is sensitive to IL-13. INS-1E cells were pre-incubated with IL-13 (20 ng/ml) or IL-4 (20 ng/ml) for 48 h before exposure to IL-1β (20 ng/ml) and IL-6 (20 ng/ml) for a further 48 h. Following treatment, cell viability was measured by flow cytometry (A), and nitrite synthesis was assessed by the Griess assay (B). Data represent mean values ± SEM (n = 3), **p < 0.01, ***p < 0.001 as indicated.
The phosphorylated forms of STAT6 that were generated within the cells under these conditions were also investigated. As described previously, incubation of cells with IL-13 resulted in the induction of three bands that were labeled by the pSTAT6 antisera, whereas treatment with IL-6 yielded only a single band. When both cytokines were added together (either simultaneously or following a 48-h pre-incubation with IL-13) the pattern of bands detected was identical to that seen during IL-13 treatment alone (not shown).
Discussion
Pro-inflammatory cytokines are well established as important mediators of β-cell loss during the pathogenesis of type 1 diabetes. However, such molecules may also be important in the progression of type 2 diabetes as this condition is increasingly recognized as being characterized by a chronic, low grade inflammation within pancreatic islets.Citation31 On this basis, it seems likely that the progression of both forms of diabetes is influenced by pro-inflammatory mediators operating at the islet level, and by extension, this implies that they might be similarly regulated by the presence (or absence) of anti-inflammatory cytokines within the islet milieu. In support of this, an anti-inflammatory cytokine mix was shown to significantly improve the viability of human islets exposed to a cocktail of pro-inflammatory cytokines in culture.Citation5 Furthermore, in the NOD mouse, treatment with anti-inflammatory cytokines reduced and delayed the onset of spontaneous diabetes,Citation32-Citation34 while gene transfer of IL-4 into the β-cells of these animals both prevented the onset of diabetes and reversed disease progression.Citation35,Citation36 Some cytokines (such as IL-6) have been reported to exert either pro- or anti-inflammatory functions depending on the precise conditions,Citation37 suggesting that there may be a plasticity of response, which determines the final outcome at the cellular level.
Against this background, we have investigated the signaling pathways utilized in β-cells by IL-13 and IL-6, and we show that cognate receptors for each are present in rodent and human β-cells and that activation of these can promote Jak/STAT signaling. We also report that human islets may express two distinct forms of the IL-6 receptor, representing the membrane-bound form and a smaller isoform which lacks the transmembrane domain and as a consequence, may be released from the cell.Citation28,Citation38 The latter is not thought to act as a decoy receptor (which is frequently the case for other soluble receptors), but rather, at least in other contexts, it promotes IL-6 trans-signaling in neighboring cells that express GP130.Citation38 Therefore, within the islet, release of IL-6R from β-cells might lead to the induction of IL-6 signaling in infiltrating immune cells (as seen in the airways of patients during allergic asthmaCitation39) or alternatively, to enhanced IL-6 signaling in other islet endocrine cell subtypes.
We also show that binding of IL-6 and IL-13 to their respective receptors leads to opposite effects on β-cell viability. In particular, IL-13 protected β-cells against a range of cytotoxic stimuli, whereas IL-6 accentuated the deleterious effects of these agents. The principal difference appears to correlate with an altered pattern of phosphorylation of STAT6 in response to IL-13 and IL-6 suggesting that the status of this transcription factor may be a critical determinant of β-cell viability.
Previously, we have reported that IL-4 protects β-cells from the cytotoxic effects of pro-inflammatory cytokines,Citation6 and in the current study, it has been revealed that IL-13, which often shares a common down-stream signaling pathway with IL-4, similarly improves the viability of INS-1E cells cultured in the presence of pro-inflammatory cytokines. However, we also show that IL-13 provides cytoprotection against the loss of viability associated with serum withdrawal (i.e., upon the removal of pro-survival factors) and that caused by incubation of cells with the saturated fatty acid palmitate (a condition resembling lipotoxicity). Therefore, rather than simply antagonizing the detrimental effects of pro-inflammatory cytokines, IL-13 appears to induce a more general “cytoprotective phenotype” in the β-cell. As such, these considerations imply that IL-13 activates a critical down-stream signaling event that sits at, or beyond, the point of convergence of various pro-apoptotic pathways operating within the β-cell.
Mechanistically, the protective effects of IL-13 are expected to be mediated via its cognate cell surface receptor and, in support of this, the expression of both subunits of this receptor (IL-13Rα1 and IL-4Rα) were detected at the mRNA level in INS-1E cells and in human islets. Experiments using the PI-3K inhibitor wortmannin failed to influence the cytoprotective response, suggesting that cell viability is not regulated principally via changes in Akt phosphorylation in INS-1E cells during exposure to IL-13. This result differs from conclusions arising during experiments investigating the cytoprotective actions of IL-4 in β-cells,Citation17 implying that subtle differences may exist in the signal transduction pathways utilized by IL-4 and IL-13 despite the apparent commonalities they share. In our studies, no additivity in response was seen when both cytokines were provided to the cells together, implying that each can elicit a maximal cytoprotective response independently of the other.
Among the potential down-stream targets for the IL-13 and IL-4 receptors are members of the STAT family of transcription factors, with increases in STAT6 phosphorylation being frequently reported during exposure of cells to either IL-13 or IL-4.Citation17,Citation25,Citation40 However, additional STAT isoforms are also phosphorylated in response to these cytokines,Citation15,Citation37 suggesting a complexity of distal signaling events.
In the present work, increased phosphorylation of both STAT6 and STAT3 was observed during exposure of rodent β-cells and human islets to IL-13. Therefore, it seems possible that either (or both) of these transcription factors might contribute to IL-13 induced cytoprotection. However, additional studies with IL-6 imply that altered phosphorylation of STAT3 is unlikely to mediate cytoprotection in β-cells since application of this cytokine caused a temporally similar increase in STAT3 phosphorylation to that observed with IL-13, but unlike the situation with IL-13, this was associated with enhanced cytotoxicity. Furthermore, the activity of a STAT3 reporter construct was only marginally elevated by IL-13, whereas IL-6 dramatically increased reporter activity. Taken together, these data imply that changes in STAT3 phosphorylation are not directly and necessarily associated with alterations in β-cell viability during exposure to cytokines. By contrast, different conclusions emerge from consideration of the phosphorylation of STAT6 in response to IL-6 and IL-13. In the case of this transcription factor, it was again evident that each cytokine could promote phosphorylation, but a markedly different pattern of bands was seen. Cells exposed to IL-13 displayed a triple banding pattern when immunoblots were probed with anti-phospho-STAT6 whereas only a single isoform was induced by IL-6. This implies that the critical cytoprotective actions of IL-13 may result from generation of one of the two differentially phosphorylated isoforms of STAT6. Quantitatively, the lowest molecular weight (i.e., fastest migrating) isoform of STAT6 appears to be more abundant in the cells since this was detected most readily using an antibody that recognizes total and phosphorylated STAT6. By contrast, the higher molecular weight (more slowly migrating) band was detected only weakly by this antibody. Moreover, it was the faster migrating of the two isoforms whose phosphorylation was enhanced selectively in response to IL-13, suggesting that this may be responsible for mediating cytoprotection.
Although the precise identity of the three phospho-STAT6 bands has not been defined, it is noteworthy that they were also detected in rat BRIN-BD11 cells and in the newly described human insulin secreting cell line, 1.1B4, suggesting that they are not restricted to rodent β-cells. However, in human islets only two bands were readily detected by anti-STAT6 antiserum, one of which (the more rapidly migrating form) was preferentially upregulated in response to IL-13. The antibody used in our study detects preferentially the phosphorylation of tyrosine 641 in STAT6, which is essential for activation of the transcription factor. Therefore, all of the STAT6 isoforms detected here are likely to contain this residue, but it seems probable that some must also be subject to additional post-translational modification to mediate their altered migration characteristics on SDS gels. While various STAT6 isoforms have been identified in other studies, these are usually smaller than the full length protein (~105 kDa)Citation40 and are unlikely to be represented within the more slowly migrating phosphorylated proteins detected here. However, a number of additional post-translational modifications of STAT6 have been described, including altered patterns of serine phosphorylation, methylation, acetylation and O-linked N-acetylglucosaminylation.Citation40 We hypothesize, therefore, that the upper bands seen in cells exposed to IL-6 and IL-13 are likely to represent tyrosine phosphorylated isoforms displaying such modifications. More importantly for the present work, we suggest that the isoform which is likely to mediate cytoprotection, is the most rapidly migrating isoform of pSTAT6 since this was induced selectively by IL-13 in cell lines and human islets.
We also report that exposure of β-cells to IL-6 resulted in potentiation of nitrite production (an index of NO generation) in response to pro-inflammatory cytokines, whereas IL-13 inhibited this effect. Similar observations have been made after IL-6 treatment,Citation41 and elevations in NO synthesis caused by IL-6 have been attributed previously to STAT3 activation in INS-1 cells.Citation42 Furthermore, IL-13 has also been shown to inhibit NO production in colonic mucosaCitation43 and macrophages.Citation44 Although NO has been implicated in the cytotoxicity induced by pro-inflammatory cytokines, it is unlikely to be of relevance to the loss of viability induced by palmitate or serum withdrawal. Therefore, we conclude that changes in NO generation are unlikely to underlie the cytoprotection afforded by IL-13.
The observations reported here assume particular significance when considered in the context of immunohistochemical evidence that human islet cells can sustain an enteroviral infection in patients with diabetesCitation13,Citation14 and the finding that enteroviral infection leads to upregulation of islet IL-6 generation and a reduction in IL-13.Citation12 On this basis, it seems probable that in either type 1 or type 2 diabetes, there may be an altered inflammatory milieu within the islets generated both by influent immune cells and by changing patterns of cytokine secretion by the endocrine cells themselves, which favors β-cell loss. Since we now identify possible down-stream signaling pathways that selectively mediate the maintenance of cell viability, targeting of these mechanisms may offer a means to slow the rate of β-cell loss, especially under conditions where enteroviral infections contribute to this process.
Materials and Methods
Cell culture
The rat insulinoma cell line, INS-1E,Citation45 was cultured in RPMI-1640 medium containing 11 mM glucose, 10% FCS, 2 mM L-glutamine, 100 μg/ml streptomycin, 100 U/ml penicillin and 50 μM β-mercaptoethanol. The human insulin secreting cell line, 1.1B4,Citation46 was cultured in similar medium but without mercaptoethanol. Cells were cultured in 5% CO2 at 37°C and 100% humidity and were sub-cultured upon reaching 70–80% confluence.
Isolated human islets were obtained with ethical approval via the Oxford Centre for Islet Transplantation and the Worcestershire Clinical Research Unit. Upon arrival, islet preparations were visually assessed for purity using dithizone stainingCitation47 and then divided into non-adherent culture dishes where they were maintained overnight before treatment.
Cytokines
IL-6 and IL-13 were purchased from R and D Systems (506-RL-010 and 1945-RL-025 respectively), while all other cytokines were obtained from Sigma-Aldrich. Stock solutions were prepared in PBS supplemented with 0.1% BSA.
Fatty acid treatments
A stock solution of palmitate was prepared in 50% ethanol and solubilized by heating for 10 min at 70°C. Palmitate was bound to 10% fatty-acid free BSA (MP Biomedicals, 100152) by incubation at 37°C for 1 h, and the complex was added to the cells to yield a 1% solution of BSA. Control cells received a final concentration of 1% BSA and 0.5% ethanol with no added fatty acid.
RT-PCR
Total RNA was extracted from INS-1E cells or human islets with Trizol reagent. cDNA was generated by reverse transcription and amplified by PCR in single tube reactions using custom designed primers for rat and human IL-13Rα1, IL-4Rα, IL-6R or GP130 (Table S1). Typically, the PCR consisted of 35 cycles at 95°C for 30 sec, then 30 sec at an appropriate annealing temperature (Table S1) and finally at 72°C for 30 sec. PCR products were separated on 1% agarose gels by electrophoresis and viewed under long-wave UV illumination. Bands were excised and purified, and their identities were confirmed by direct sequencing (Eurofins MWG Operon).
Cell viability
INS-1E cells were seeded into 6-well plates at a density of 105 cells/well. In all experiments, cells were incubated with appropriate reagents for up to 96 h. Reagents were replaced after 48 h to ensure their efficacy did not diminish during the incubation period. To measure viability, cells were harvested and resuspended in propidium iodide (10 μg/ml) before analysis by flow cytometry.Citation48 All viability experiments were performed at least three separate times (indicated by n = 3), with between 3 and 6 repeats for each condition within any given experiment.
Western blotting
INS-1E cells were grown to confluence in 25 cm2 growth area flasks before cytokine treatment for appropriate time periods. Whole cell proteins were extracted after lysis.Citation49 The protein concentration of samples was equalized prior to the sample being denatured and then loaded onto pre-cast TRIS-HCl buffered 12.5% poly-acrylamide gels. Proteins were separated by electrophoresis and then electrotransferred onto a polyvinylidene difluoride membrane. Membranes were blocked in a Tris-buffered saline solution containing 0.05% Tween and 5% dried milk powder and then probed with primary antibody diluted in blocking solution at 4°C overnight, with the exception of the β-actin antibody, which was incubated at room temperature (20°C) for 2 h. The primary antibodies were raised against β-actin (1:10000, Sigma, A5441), p-JAK2 (1:500, Cell Signaling, 3776S), STAT3 (1:1000, Cell Signaling, 9132S), p-STAT3 (1:500, Cell Signaling, 9131S), STAT6 (1:200, Santa Cruz, sc-981) and p-STAT6 (1:200, Santa Cruz, sc-1162R). After washing, membranes were probed with appropriate alkaline phosphatase (AP)-conjugated secondary antibodies for 1h (1:25000, Sigma, A3687). Bands were detected by exposure of the membrane to X-ray film after development using the CDP Star detection system (Sigma, C0712).
Measurement of nitrite production
Nitrite production was measured by Griess Assay. Briefly, 100 μl of supernatant was collected from each well of a 6-well plate in triplicate. Samples and sodium nitrite standards were incubated in the dark for 10 min with 50 μl 1% sulphanilamide in 5% orthophosphoric acid and then for a further 10 min with 50 μl 0.1% naphthylenediamine dihydrochloride before their absorbance was measured at 562 nm. Nitrite concentrations were estimated by comparing the absorbance of the unknown samples with those of standards.
STAT3 reporter assay
INS-1E cells were seeded at a density of 105 cells/well into 24-well plates. Twenty-four hours later cells were transfected with a STAT3-responsive dual firefly/Renilla luciferase reporter construct (Cignal reporter, Qiagen, CCS-90028L), using Attractene transfection reagent according to the manufacturer’s instructions (Qiagen, 301005). Following a 4-h incubation period, cells were treated with cytokines for a further 18 h and the STAT3 activity established using the dual-luciferase reporter assay system (Promega, E1910).
Statistics
Results are expressed as mean values ± SEM unless otherwise stated and statistical significance between pairs of experimental groups was assessed by Student’s t-test.
Additional material
Download Zip (249.7 KB)Acknowledgments
We wish to acknowledge, with thanks, the Oxford Centre for Islet Transplantation and the Worcestershire Clinical Research Unit for the provision of human islets. We thank Haima Raman for technical assistance funded by the Nuffield Foundation Bursary scheme. We also thank Dr Hannah Welters and Dr Sarah Richardson for helpful discussions during the preparation of this manuscript.
The work was supported by funding from the European Union’s Seventh Framework Programme PEVNET (FP7/2007-2013) under grant agreement number 261441.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Material
Supplemental materials may be found here:
http://www.landesbioscience.com/journals/islets/article/24249
References
- Mandrup-Poulsen T. The role of interleukin-1 in the pathogenesis of IDDM. Diabetologia 1996; 39:1005 - 29; http://dx.doi.org/10.1007/BF00400649; PMID: 8877284
- Eizirik DL, Mandrup-Poulsen T. A choice of death--the signal-transduction of immune-mediated beta-cell apoptosis. Diabetologia 2001; 44:2115 - 33; http://dx.doi.org/10.1007/s001250100021; PMID: 11793013
- Rapoport MJ, Mor A, Vardi P, Ramot Y, Winker R, Hindi A, et al. Decreased secretion of Th2 cytokines precedes Up-regulated and delayed secretion of Th1 cytokines in activated peripheral blood mononuclear cells from patients with insulin-dependent diabetes mellitus. J Autoimmun 1998; 11:635 - 42; http://dx.doi.org/10.1006/jaut.1998.0240; PMID: 9878085
- Berman MA, Sandborg CI, Wang Z, Imfeld KL, Zaldivar F Jr., Dadufalza V, et al. Decreased IL-4 production in new onset type I insulin-dependent diabetes mellitus. J Immunol 1996; 157:4690 - 6; PMID: 8906850
- Marselli L, Dotta F, Piro S, Santangelo C, Masini M, Lupi R, et al. Th2 cytokines have a partial, direct protective effect on the function and survival of isolated human islets exposed to combined proinflammatory and Th1 cytokines. J Clin Endocrinol Metab 2001; 86:4974 - 8; http://dx.doi.org/10.1210/jc.86.10.4974; PMID: 11600573
- Kaminski A, Kaminski ER, Morgan NG. Pre-incubation with interleukin-4 mediates a direct protective effect against the loss of pancreatic beta-cell viability induced by proinflammatory cytokines. Clin Exp Immunol 2007; 148:583 - 8; http://dx.doi.org/10.1111/j.1365-2249.2007.03375.x; PMID: 17403060
- Souza KL, Gurgul-Convey E, Elsner M, Lenzen S. Interaction between pro-inflammatory and anti-inflammatory cytokines in insulin-producing cells. J Endocrinol 2008; 197:139 - 50; http://dx.doi.org/10.1677/JOE-07-0638; PMID: 18372240
- Pickup JC, Crook MA. Is type II diabetes mellitus a disease of the innate immune system?. Diabetologia 1998; 41:1241 - 8; http://dx.doi.org/10.1007/s001250051058; PMID: 9794114
- Donath MY, Shoelson SE. Type 2 diabetes as an inflammatory disease. Nat Rev Immunol 2011; 11:98 - 107; http://dx.doi.org/10.1038/nri2925; PMID: 21233852
- Campbell IL, Cutri A, Wilson A, Harrison LC. Evidence for IL-6 production by and effects on the pancreatic beta-cell. J Immunol 1989; 143:1188 - 91; PMID: 2501390
- Donath MY, Böni-Schnetzler M, Ellingsgaard H, Halban PA, Ehses JA. Cytokine production by islets in health and diabetes: cellular origin, regulation and function. Trends Endocrinol Metab 2010; 21:261 - 7; http://dx.doi.org/10.1016/j.tem.2009.12.010; PMID: 20096598
- Olsson A, Johansson U, Korsgren O, Frisk G. Inflammatory gene expression in Coxsackievirus B-4-infected human islets of Langerhans. Biochem Biophys Res Commun 2005; 330:571 - 6; http://dx.doi.org/10.1016/j.bbrc.2005.03.016; PMID: 15796921
- Richardson SJ, Willcox A, Bone AJ, Foulis AK, Morgan NG. The prevalence of enteroviral capsid protein vp1 immunostaining in pancreatic islets in human type 1 diabetes. Diabetologia 2009; 52:1143 - 51; http://dx.doi.org/10.1007/s00125-009-1276-0; PMID: 19266182
- Richardson SJ, Willcox A, Bone AJ, Morgan NG, Foulis AK. Immunopathology of the human pancreas in type-I diabetes. Semin Immunopathol 2011; 33:9 - 21; http://dx.doi.org/10.1007/s00281-010-0205-0; PMID: 20424842
- Roy B, Bhattacharjee A, Xu B, Ford D, Maizel AL, Cathcart MK. IL-13 signal transduction in human monocytes: phosphorylation of receptor components, association with Jaks, and phosphorylation/activation of Stats. J Leukoc Biol 2002; 72:580 - 9; PMID: 12223527
- Matikainen S, Sareneva T, Ronni T, Lehtonen A, Koskinen PJ, Julkunen I. Interferon-alpha activates multiple STAT proteins and upregulates proliferation-associated IL-2Ralpha, c-myc, and pim-1 genes in human T cells. Blood 1999; 93:1980 - 91; PMID: 10068671
- Kaminski A, Welters HJ, Kaminski ER, Morgan NG. Human and rodent pancreatic beta-cells express IL-4 receptors and IL-4 protects against beta-cell apoptosis by activation of the PI3K and JAK/STAT pathways. Biosci Rep 2010; 30:169 - 75; http://dx.doi.org/10.1042/BSR20090021; PMID: 19531027
- Lee TS, Chau LY. Heme oxygenase-1 mediates the anti-inflammatory effect of interleukin-10 in mice. Nat Med 2002; 8:240 - 6; http://dx.doi.org/10.1038/nm0302-240; PMID: 11875494
- Ellingsgaard H, Ehses JA, Hammar EB, Van Lommel L, Quintens R, Martens G, et al. Interleukin-6 regulates pancreatic alpha-cell mass expansion. Proc Natl Acad Sci U S A 2008; 105:13163 - 8; http://dx.doi.org/10.1073/pnas.0801059105; PMID: 18719127
- Villanueva EC, Myers MG Jr.. Leptin receptor signaling and the regulation of mammalian physiology. Int J Obes (Lond) 2008; 32:Suppl 7 S8 - 12; http://dx.doi.org/10.1038/ijo.2008.232; PMID: 19136996
- Neilson LM, Zhu J, Xie J, Malabarba MG, Sakamoto K, Wagner KU, et al. Coactivation of janus tyrosine kinase (Jak)1 positively modulates prolactin-Jak2 signaling in breast cancer: recruitment of ERK and signal transducer and activator of transcription (Stat)3 and enhancement of Akt and Stat5a/b pathways. Mol Endocrinol 2007; 21:2218 - 32; http://dx.doi.org/10.1210/me.2007-0173; PMID: 17550976
- Kelly-Welch AE, Hanson EM, Boothby MR, Keegan AD. Interleukin-4 and interleukin-13 signaling connections maps. Science 2003; 300:1527 - 8; http://dx.doi.org/10.1126/science.1085458; PMID: 12791978
- Hershey GK. IL-13 receptors and signaling pathways: an evolving web. J Allergy Clin Immunol 2003; 111:677 - 90, quiz 691; http://dx.doi.org/10.1067/mai.2003.1333; PMID: 12704343
- Heinrich PC, Behrmann I, Haan S, Hermanns HM, Müller-Newen G, Schaper F. Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem J 2003; 374:1 - 20; http://dx.doi.org/10.1042/BJ20030407; PMID: 12773095
- Andrews RP, Ericksen MB, Cunningham CM, Daines MO, Hershey GK. Analysis of the life cycle of stat6. Continuous cycling of STAT6 is required for IL-4 signaling. J Biol Chem 2002; 277:36563 - 9; http://dx.doi.org/10.1074/jbc.M200986200; PMID: 12121972
- Carey GB, Semenova E, Qi X, Keegan AD. IL-4 protects the B-cell lymphoma cell line CH31 from anti-IgM-induced growth arrest and apoptosis: contribution of the PI-3 kinase/AKT pathway. Cell Res 2007; 17:942 - 55; http://dx.doi.org/10.1038/sj.cr.2007.90; PMID: 17968425
- Yang L, Wang L, Lin HK, Kan PY, Xie S, Tsai MY, et al. Interleukin-6 differentially regulates androgen receptor transactivation via PI3K-Akt, STAT3, and MAPK, three distinct signal pathways in prostate cancer cells. Biochem Biophys Res Commun 2003; 305:462 - 9; http://dx.doi.org/10.1016/S0006-291X(03)00792-7; PMID: 12763015
- Lust JA, Donovan KA, Kline MP, Greipp PR, Kyle RA, Maihle NJ. Isolation of an mRNA encoding a soluble form of the human interleukin-6 receptor. Cytokine 1992; 4:96 - 100; http://dx.doi.org/10.1016/1043-4666(92)90043-Q; PMID: 1633265
- Nishimura Y, Nitto T, Inoue T, Node K. IL-13 attenuates vascular tube formation via JAK2-STAT6 pathway. Circ J 2008; 72:469 - 75; http://dx.doi.org/10.1253/circj.72.469; PMID: 18296848
- Pedranzini L, Dechow T, Berishaj M, Comenzo R, Zhou P, Azare J, et al. Pyridone 6, a pan-Janus-activated kinase inhibitor, induces growth inhibition of multiple myeloma cells. Cancer Res 2006; 66:9714 - 21; http://dx.doi.org/10.1158/0008-5472.CAN-05-4280; PMID: 17018630
- Donath MY, Böni-Schnetzler M, Ellingsgaard H, Ehses JA. Islet inflammation impairs the pancreatic beta-cell in type 2 diabetes. Physiology (Bethesda) 2009; 24:325 - 31; http://dx.doi.org/10.1152/physiol.00032.2009; PMID: 19996363
- Zaccone P, Phillips J, Conget I, Gomis R, Haskins K, Minty A, et al. Interleukin-13 prevents autoimmune diabetes in NOD mice. Diabetes 1999; 48:1522 - 8; http://dx.doi.org/10.2337/diabetes.48.8.1522; PMID: 10426368
- Pennline KJ, Roque-Gaffney E, Monahan M. Recombinant human IL-10 prevents the onset of diabetes in the nonobese diabetic mouse. Clin Immunol Immunopathol 1994; 71:169 - 75; http://dx.doi.org/10.1006/clin.1994.1068; PMID: 8181185
- Rapoport MJ, Jaramillo A, Zipris D, Lazarus AH, Serreze DV, Leiter EH, et al. Interleukin 4 reverses T cell proliferative unresponsiveness and prevents the onset of diabetes in nonobese diabetic mice. J Exp Med 1993; 178:87 - 99; http://dx.doi.org/10.1084/jem.178.1.87; PMID: 8315397
- Gaddy DF, Riedel MJ, Bertera S, Kieffer TJ, Robbins PD. dsAAV8-mediated gene transfer and β-cell expression of IL-4 and β-cell growth factors are capable of reversing early-onset diabetes in NOD mice. Gene Ther 2011; 19:791 - 9; http://dx.doi.org/10.1038/gt.2011.181; PMID: 22089495
- Rehman KK, Trucco M, Wang Z, Xiao X, Robbins PD. AAV8-mediated gene transfer of interleukin-4 to endogenous beta-cells prevents the onset of diabetes in NOD mice. Mol Ther 2008; 16:1409 - 16; http://dx.doi.org/10.1038/mt.2008.116; PMID: 18560422
- Scheller J, Chalaris A, Schmidt-Arras D, Rose-John S. The pro- and anti-inflammatory properties of the cytokine interleukin-6. Biochim Biophys Acta 2011; 1813:878 - 88; http://dx.doi.org/10.1016/j.bbamcr.2011.01.034; PMID: 21296109
- Drucker C, Gewiese J, Malchow S, Scheller J, Rose-John S. Impact of interleukin-6 classic- and trans-signaling on liver damage and regeneration. J Autoimmun 2010; 34:29 - 37; http://dx.doi.org/10.1016/j.jaut.2009.08.003; PMID: 19717281
- Doganci A, Eigenbrod T, Krug N, De Sanctis GT, Hausding M, Erpenbeck VJ, et al. The IL-6R alpha chain controls lung CD4+CD25+ Treg development and function during allergic airway inflammation in vivo. J Clin Invest 2005; 115:313 - 25; PMID: 15668741
- Hebenstreit D, Wirnsberger G, Horejs-Hoeck J, Duschl A. Signaling mechanisms, interaction partners, and target genes of STAT6. Cytokine Growth Factor Rev 2006; 17:173 - 88; http://dx.doi.org/10.1016/j.cytogfr.2006.01.004; PMID: 16540365
- Wadt KA, Larsen CM, Andersen HU, Nielsen K, Karlsen AE, Mandrup-Poulsen T. Ciliary neurotrophic factor potentiates the beta-cell inhibitory effect of IL-1beta in rat pancreatic islets associated with increased nitric oxide synthesis and increased expression of inducible nitric oxide synthase. Diabetes 1998; 47:1602 - 8; http://dx.doi.org/10.2337/diabetes.47.10.1602; PMID: 9753298
- Oh YS, Lee YJ, Park EY, Jun HS. Interleukin-6 treatment induces beta-cell apoptosis via STAT-3-mediated nitric oxide production. Diabetes Metab Res Rev 2011; 27:813 - 9; http://dx.doi.org/10.1002/dmrr.1233; PMID: 22069265
- Kolios G, Wright KL, Linehan JD, Robertson DA, Westwick J. Interleukin-13 inhibits nitric oxide production in human colonic mucosa. Hepatogastroenterology 2000; 47:714 - 7; PMID: 10919016
- Bogdan C, Thüring H, Dlaska M, Röllinghoff M, Weiss G. Mechanism of suppression of macrophage nitric oxide release by IL-13: influence of the macrophage population. J Immunol 1997; 159:4506 - 13; PMID: 9379051
- Asfari M, Janjic D, Meda P, Li G, Halban PA, Wollheim CB. Establishment of 2-mercaptoethanol-dependent differentiated insulin-secreting cell lines. Endocrinology 1992; 130:167 - 78; http://dx.doi.org/10.1210/en.130.1.167; PMID: 1370150
- McCluskey JT, Hamid M, Guo-Parke H, McClenaghan NH, Gomis R, Flatt PR. Development and functional characterization of insulin-releasing human pancreatic beta cell lines produced by electrofusion. J Biol Chem 2011; 286:21982 - 92; http://dx.doi.org/10.1074/jbc.M111.226795; PMID: 21515691
- Kissler HJ, Niland JC, Olack B, Ricordi C, Hering BJ, Naji A, et al. Validation of methodologies for quantifying isolated human islets: an Islet Cell Resources study. Clin Transplant 2010; 24:236 - 42; http://dx.doi.org/10.1111/j.1399-0012.2009.01052.x; PMID: 19719726
- Morgan NG, Diakogiannaki E, Russell MA. The incubation and monitoring of cell viability in primary rat islets of Langerhans and pancreatic beta-cell lines. Methods Mol Biol 2009; 560:53 - 64; http://dx.doi.org/10.1007/978-1-59745-448-3_5; PMID: 19504243
- Welters HJ, Oknianska A, Erdmann KS, Ryffel GU, Morgan NG. The protein tyrosine phosphatase-BL, modulates pancreatic beta-cell proliferation by interaction with the Wnt signalling pathway. J Endocrinol 2008; 197:543 - 52; http://dx.doi.org/10.1677/JOE-07-0262; PMID: 18413347

