Abstract
Pulmonary artery hypertension (PAH) is a proliferative disorder associated with enhanced pulmonary artery smooth muscle cell proliferation and suppressed apoptosis. The sustainability of this phenotype requires the activation of pro-survival transcription factor like the signal transducers and activators of transcription-3 (STAT3). Using multidisciplinary and translational approaches, we and others have demonstrated that STAT3 activation in both human and experimental models of PAH accounts for the modulation of the expression of several proteins already known as implicated in PAH pathogenesis, as well as for signal transduction to other transcription factors. Furthermore, recent data demonstrated that STAT3 could be therapeutically targeted in different animal models and some molecules are actually in clinical trials for cancer or PAH treatment.
Introduction
Pulmonary arterial hypertension (PAH) is a disease affecting the pulmonary vasculature, characterized by vasoconstricted and remodeled pulmonary arteries,Citation1 increasing pulmonary vascular resistance (PVR) and pulmonary pressure. This is associated with a primary, compensatory right ventricular hypertrophy which rapidly will turn into right heart failure.Citation2 Current approved therapies, despite improving quality of life remain insufficient to reverse PAH and improve survival; mortality rates are still unacceptably high (≈10% within 1 year, ≈35% within 3 yearsCitation3,Citation4).
It is well recognize that the pathogenesis of PAH has fundamental similarities with cancer as pulmonary artery smooth muscle cells (PASMCs) adopt a pro-proliferative, pro-survival, invasive phenotypeCitation5-Citation7 (). Also, as in cancer, there is an environment of persistent inflammation.Citation8 In some patients, there is a major genetic predisposition in the form of heterozygous mutations in BMPR-II (bone morphogenetic type II receptor) leading to an impaired function of SMAD (mothers against decapentaplegic homolog) pathwayCitation9-Citation11 and an increased p38/MAPK (mitogen-activated protein kinase) activation.Citation12,Citation13 Mutations have also been detected in the ALK1 (activin-like kinase 1) geneCitation14 and polymorphisms have been found in the sequence coding for the serotonin transporter 5-HTTCitation15,Citation16 and the gene KCNA5 coding for the potassium channel Kv1.5,Citation17 leading to consider genomic instability as a part of PAH development. Plexiform lesions are complex vascular structures observed in idiopathic forms of PAH, and resemble neo-plastic disorders with an abnormal and “quasi malignant” endothelial cell growth.Citation6,Citation18 Oncogenic pathways, like p21 and p27, and the lack of tumor suppressor, like p53 have been implicated in PAH etiology as well.Citation19-Citation23 The metabolic switch from glucose oxidation to glycolysis seen in cancer has also been described in PAH,Citation24,Citation25 with for origin the inhibition of the mitochondrial gate-keeping enzyme the pyruvate dehydrogenase (PDH). This is associated with mitochondrial suppression,Citation25-Citation27 hyperpolarization of the mitochondrial membrane potential (ΔΨm) and the inhibition of mitochondrial reactive oxygen species (ROS) generation, both being implicated in apoptosis suppression,Citation26,Citation28-Citation30 another important feature that PAH shares with cancer.
Table 1. Cancer hallmarks shared with PAH
The signal transducers and activators of transcription (STAT) protein family regulates diverse cellular processes including growth and survival, and is frequently deregulated in cancer and several other disorders. The family is composed of 7 isoforms (STAT1–4, 5A, 5B and 6) that are activated, phosphorylated in response to cytokines, growth factors or agonists. The role of STAT3 in PAH has been suggested in 2007Citation31 and strengthened in the last couple of years, even leading to the conclusion that STAT3 activation might be an early event in PAH etiology, at the origin of several signaling cascades and that its role is critical in the sustainability of the pathologic phenotype. In this review, we will make a statement of the current knowledge regarding STAT3 implication in PAH as well as an overview of the speculated additional role that it could play and that might be the subject of future studies.
STAT3 Upstream Signals
STAT3 is a cytoplasmic latent transcription factor activated by phosphorylation on its tyrosine 705 residue (PY705) (allowing nuclear translocation and DNA binding of STAT3 after dimerizationCitation35) in response to cytokines such as interleukin-6 (IL-6),Citation36 growth factors such as platelet-derived growth factor (PDGF)Citation36 and agonists such as endothelin-1 (ET1) and angiotensin II (AngII).Citation37 The secretion of these factors is deregulated in PAH, their levels are increased in the serum of PAH patients.Citation38-Citation40 Following pulmonary artery endothelial cells (PAECs) injury occurring in the early stages of the disease, and alteration of their function as a barrier, PASMCs are in direct contact with these factors, thus enhancing pathways of growth, proliferation and resistance to apoptosis. A 2.8-, 3.2-, 2.5-, 2.8- and 1.9-fold increase in PY705-STAT3 has been described in healthy-PASMCs treated for 48 h with ET1, AngII, PDGF, IL-6 and TNF respectivelyCitation33 suggesting the important role of STAT3 in these processes (). A second phosphorylation event occurs on the serine 727 residue (PS-727) of the STAT3 C-terminal activation domain. This phosphorylation seems to be necessary for maximal gene expression, since its mutation prevents STAT3 transcriptional functions;Citation41 however, the exact role of this site of phosphorylation is still unclear.
Figure 1. Spectrum of STAT3 implication in PAH. STAT3 is activated in response to cytokines, growth factors and agonists that are dysregulated in PAH, signal transduction trough their receptors (tyrosine kinase, G-protein-coupled, immunoglobin like or integrin), and involvement of SHP2 and Src. STAT3 activates a broad range of transcription factors and proteins, all implicated in the management of proliferation and resistance to apoptosis that lead to the development of PAH. By downregulating miR-204, STAT3 abolishes SHP2 repression, enhances Src activation and finally sustains its own activation. Several STAT3 inhibitors (in red) might be beneficial for the treatment of PAH. Some of them are in early phase clinical trial for the treatment of cancer. This might as well facilitate their utilization for PAH treatment as data on their tolerability and efficiency will be available soon. DHEA is actually undergoing a phase 3 clinical trials for chronic obstructive pulmonary diseases and pulmonary hypertension.
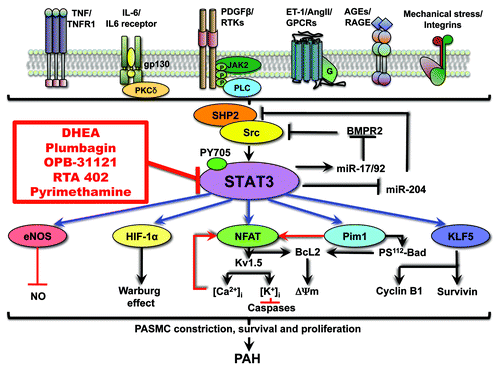
Src and JAKs are among the proteins the most frequently involved in the transduction of the signal between the fixation of the agonist on the receptor and the phosphorylation of STAT3.Citation42,Citation43 Some studies have mentioned the possible implication of JAKs proteins in PAH early in the decade, determining an increase in JAKs mRNA levels in rats with hypoxia induced-PAHCitation44 or through the beneficial effect of the JAK2 inhibitor AG490 in reversing PAECs proliferation rates.Citation31 However, using either microarray or western blot, we failed to determine that JAK2 is upregulated/activated in PAH-PASMC compared with healthy PASMC.Citation30 At the opposite, we found an accurate 2-fold increase in both c-Src and Src-related protein SHP2 in PAH-PASMC compare with healthy PASMC (). These results have been reproduced in the hypoxia and monocrotaline experimental models of PAH as well.Citation30 Recently, JAK2 has been found by microarray study to be expressed more highly in limited cutaneous systemic sclerosis (lcSSc) patients, especially when associated with PAH, than in controls and in idiopathic PAH where JAK2 levels were not affected.Citation45 This suggests that JAK2 activation more likely occur in autoimmune disease-associated PAH. However, state of phosphorylation of JAK2 has not been measured in this study, poorly allowing any conclusion on whether or not JAK2 is activated. Nonetheless, both Src and JAK2 need to be considered with interest, since their relationship is complex and might be compensatory, the inhibition of one enhancing the activation of the second as seen in both cancerCitation46,Citation47 and VSMC diseases,Citation48 leading to the development of mechanism of resistance to the treatment.
Typically, growth factors like PDGF bind on receptor tyrosine kinases (RTK). These receptors are activated by ligand fixation and undergo dimerization, which juxtaposes the cytoplasmic tyrosine kinase domains,Citation49 facilitates autophosphorylation on specific tyrosine residues, conformational changes and stabilization of the active kinase.Citation50 Enzymes and adaptor/scaffolding proteins recognize phosphotyrosine residues, typically through Src homology-2 [SH2, such as phospholipase C-γ (PLC-γ), Janus associated kinases JAKs, v-Src sarcoma (Schmidt-Ruppin A-2) viral oncogene homolog (Src), Src homology 2 domain-containing phosphatase 2 (Shp2) and growth factor receptor-bound protein 2 (Grb2)] or phosphotyrosine-binding [PTB, such as insulin-receptor substrate-1/2 (IRS-1/2) or c-Jun N-terminal kinase-interacting protein (JIP)] domains, are phosphorylated and activated leading to the initiation of signal transduction pathways, STAT3 included. Interleukins acts through IL-receptors that are composed of two transmembrane glycoproteins chains, required for the generation of high-affinity IL-binding sites and signal-transduction. Signal transduction induces tyrosine phosphorylation, primarily of JAK2 and TYK2 (another member of the JAK family), which, in turn, phosphorylate and activate STATs. Other signal transduction molecules, including Src,Citation51,Citation52 SHP2 and suppressor of cytokine signaling (SOCS) are also recruited, associated or not to JAK.Citation53-Citation55 ET1 and AngII exert their action through G-protein coupled receptors (GPCR), 7 transmembrane domain receptors that interact with a heterodimeric G-protein after ligand stimulation. G-proteins consist in a GDP/GTP-binding α-subunit and a βγ-subunit complex.Citation56,Citation57 Both Gα and Gβγ subunits can modulate downstream effectors activity: activation or inhibition of adenylate cyclase, activation of phospholipase C (PLC), activation of the small G protein RhoA GTPase, activation of the mitogen-activated protein kinase 1 (ERK). A JAK/Src-dependent STAT3 activation have been also reported after ET1 or AngII-dependent GPCR activation.Citation58-Citation61 This can be due to direct signal transduction from the GPCR as well as a reciprocal cross-communication between RTKs and GPCRs, since numerous GPCRs have been shown to usurp the signaling machinery of RTKs and that signal transduction by RTKs is mediated, at least in part by transactivation of GPCR.Citation62-Citation68
TNF receptors are as well able to constitutively form a complex with endogenous c-Src and JAK2 and to use them to engage signaling cascades, activate transcription factors, and alter gene expression.Citation69
Recently, in human carotid artery smooth muscle cells (CASMC), activation of the STAT3/Pim1/NFATc axis has been associated with signal transduction trough the receptor for advanced glycation end products (RAGE) after sensitization by the Nε-carboxymethyl lysine (CML).Citation70 As RAGE has been previously implicated in PAH pathogenesis, especially by enhancing PAH-PASMCs migration,Citation71,Citation72 it is possible that STAT3 might also be activated by fixation of AGEs, or other ligands, on RAGECitation70,Citation73 ().
Hemodynamic forces that are increased by pressure overload, including shear stress and cyclic strain, have been recognized as important modulators of vascular cell morphology and function and are also upstream activators of STAT3. STAT3 activation has been determined in thoracic aorta embryonic A7r5A SMCs under cyclic strain (10% average strain at 60 cycles/min for various time periods).Citation74 This mechanism is independent of JAK2 but Src-dependent for STAT3 tyrosine phosphorylation and p30/MAPK/ERK-dependent for STAT3 Serine 727 phosphorylation.Citation74
STAT3 Plays Its Role of Signaling Hub in PAH
The role of STAT3 in PAH, particularly with regard to the sustainability of the proproliferative and anti-apoptotic phenotype, has been studied more in deep and several downstream STAT3 targets have been identified. The expression of Pim1 (provirus integration site for Moloney murine leukemia virus), a proto-oncogene encoding a serine/threonine protein kinaseCitation75 has been described as a key downstream signal of STAT3 activation in PAH. Overexpression of Pim1 increases PASMCs proliferation within the remodeled PAs by promoting (1) bad phosphorylation/inhibition and mitochondrial hyperpolarizationCitation33 and (2) enhancing NFATc2 (nuclear factor of activated T-cells) activity. NFAT is a transcription factor that has been previously showed to account for the downregulation of K+ channels like Kv1.5, leading to cell depolarization, increased calcium levels and cell proliferation.Citation26,Citation34,Citation76 On the other hand NFATc2 activation also upregulates Bcl-2 leading to mitochondrial hyperpolarization and apoptosis resistance.Citation26,Citation34,Citation76 Interestingly, several STAT binding sites have been identified within the promoter regionsCitation77 of NFATc1, NFATc2 and NFATc3 (the three major NFAT isoforms implicated in PAHCitation26) and Pim1. Chromatin immunoprecipitation associated with PCR (ChIP/PCR) experiments confirmed the interaction of STAT3 with the promoters of NFATc and Pim1 suggesting that STAT3 acts in an ingenious manner by increasing the expression of both the effecter NFATc and its activator Pim1.
Survivin, a member of the inhibitor of apoptosis family of proteins, has been described as upregulated in PAH and playing a major role in PAH pathogenesis.Citation5 STAT3 activation has been associated with cancerCitation78,Citation79 and systemic vascular remodelingCitation70 and showed to cause an upregulation of Survivin mRNA levels. This has been shown in PAH recently, where STAT3 inhibition in PAH-PASMCs was associated with decreased Survivin mRNA levels. Survivin is another example of STAT3 downstream target, increasing further the role and impact of STAT3 activation in PAH. More recently it has been determined that this STAT3-dependent expression of Survivin is not direct but through activation of the transcription factor Krüppel-like factor 5 (KLF5).Citation33 Once activated, this zinc-finger-type transcription factor promotes the upregulation of the cyclin B1 and triggers the expression of Survivin, and then contributes to PASMC proliferation and survival. Again, this suggests that STAT3 is a key mediator of PAH pathology as it activates a broad range of transcription factors and proteins, all implicated in the management of proliferation, and resistance to apoptosis that lead to the development of PAH.
Migration, invasion and motility of PASMCs in PAH have not been extensively studied. It is well characterized that PDGF is a potent inducer of PASMCs migration,Citation80-Citation82 at least by activation of the ERK/MAPK axis. PASMCs stimulated by BMP2 or S100A4 have also increased level of migration due to activation of BMPR2 and RAGE receptors.Citation71,Citation72 Several studies have demonstrated that hypoxia stimulates PASMCs migration,Citation83-Citation85 in a manner that is not dependent of Hif-1α. Finally, it is also suggested that PASMCs migration is regulated by sphingosine-1 via ROCK activation.Citation86 STAT3 has been described to be implicated in SMCs invasion.Citation87,Citation88 Indeed, cellular invasiveness has been determined as dependent on the balance between two opposing forces: the proinvasive oncogenes Src-STAT3 and the anti-invasive tumor suppressors p53-PTEN. STAT3 is a required downstream effector of Src to induce podosome structures and related invasive phenotypes. STAT3 promotes Src phenotypes through the suppression of p53 and the p53-inducible protein caldesmon, a known podosome antagonist. In contrast, enhanced p53 attenuates STAT3 function and Src-induced podosome formation by upregulating the tumor suppressor phosphatase PTEN.Citation87
PKCδ seems to also have a critical role in smooth muscle cells motilityCitation89 as PKCδ-deficient SMCs showed an abnormal cytoskeleton structure and decreased migration in response to mechanical stress (60 cycles/min and 5, 15 or 20% elongation). The protein kinase C delta (PKCδ) has been found increased in expression in PAs of chronic hypoxia-induced PAH rats.Citation90 Among the 13 PKC members, PKCδ is the only PKC member that is a substrate for protein tyrosine kinases. For example, growth factors and Src kinases have been shown to induce tyrosine phosphorylation of PKC.Citation91 Moreover, PKCδ is able to interact with the SH2 domain and part of the adjacent C-terminal transactivation domain of STAT3 to significantly enhance the interaction of STAT3 and the IL-6 receptor subunit glycoprotein (gp) 130, which is the initial step for STAT3 activation by IL-6. These findings suggest strongly that PKCδ is implicated at several levels in the transduction of the signal upstream to STAT3.Citation92
Finally, in patients with pulmonary arterial hypertension, reduced bioavailability of nitric oxide (NO) has been observed and associated with reduced pulmonary levels of the endothelium NO synthase (eNOS).Citation93 Interestingly, a PKCδ/STAT3 axis has been implicated in the downregulation of eNOS expression and subsequent decrease in NO generation.Citation94 Indeed, PKCδ activation following ET-1 stimulation in ovine fetal PAECs has been associated with STAT3 phosphorylation. This, increased the binding of STAT3 on the eNOS promoter resulting in inhibition of eNOS expression.Citation94 Thus, PKCδ/STAT3 pathway might be at least in part responsible for the downregulation of the NO signaling in PAH.
Reciprocal Regulations between STATs and miRNA in PAH
Active interactions between STATs and miRNAs have been described in pathways driving immune cells maturation and cancer, with the presence of reciprocal regulations between STATs and miRNA.Citation95 The inappropriate STAT3 activation seen in PAH has been linked as well with miRNA expression. miR-204, which is located within the intron 6 of the human transient receptor potential melastatin 3 (TRPM3), has been showed negatively regulated, like TRPM3, by STAT3 binding on the promoter.Citation30 miR-204 has been previously described as tumor suppressor in cancerCitation96 as its downregulation induces PDGF-dependent proliferation.Citation97 The identified direct target of miR-204 in PAH is SHP2, a cytoplasmic tyrosine phosphatase implicated in Src activation.Citation98,Citation99 Thus, by downregulating miR-204, STAT3 abolishes SHP2 repression, enhances Src activation and finally sustains its own activation (). The understanding of this feedback mechanism is of great therapeutic interest since it could explain that targeting STAT3 signaling upstream to Src activation would only have a short impact, as the loop is still active. In PAH, therapeutic interventions restoring miR-204 expression should be an interesting strategy equivalent with Src and STAT3 inhibitors.
Brock and colleagues demonstrated that in a model of HPAECs stimulated with IL-6, STAT3 activation results in STAT3 binding on the promoter region of the miR-17/92 gene (C13orf25), enhancing transcription of the gene and expression of the miRNA and BMPR2 downregulation.Citation100 Indeed, by computational analysis, several miRNAs encoded by the cluster miR-17/92 have been identified to directly target BMPR2. Src is also an interacting partner of the BMPR2 C terminus domain. BMPR2 mutation resulting in a truncated BMPR-II C-terminal domain is able to increase c-Src phosphorylation in response to BMP-2.Citation101 This suggests that the effect of STAT3 on BMPR2 and subsequently on Src might be another mechanism built to maintain its own activation ().
STAT3 and Associated Therapies (Anti-Cancer and DHEA)
There is evidence that dehydroepiandrosterone (DHEA) is an efficient STAT3 inhibitor in PAH (), confirming previous findings in the liver.Citation102 The exact molecular mechanism by which DHEA decreases STAT3 activation remains to be established. Nonetheless, we have evidence that DHEA effects in vascular tissue is likely mediated through a plasma membrane receptor coupled to G protein.Citation76 Other mechanisms cannot be ruled out, for example, DHEA has been shown to increase PPAR-γ mRNA levels.Citation103 PPAR-γ could be implicated in the DHEA-dependent STAT3 inhibition as activation of the peroxisome proliferator-activated receptor gamma (PPAR-γ), a member of the nuclear receptor family which is downregulated in PAH,Citation104 have an inhibitory effect on STAT3.Citation105-Citation107 A direct physical protein-protein interaction has been shown between PPAR-γ and activated STAT3, resulting in decreased transcriptional activity of STAT3. PPAR-γ agonist ciglitazone treatments in glioblastoma cell lines showed a decreased expression of phosphorylated form of STAT3 associated with an increased expression of STAT3 inhibitors like the suppressor of cytokine signaling (SOCS) 3 and the protein inhibitor of activated STAT3 (PIAS3).Citation108 Considering that PPAR-γ agonists used in the treatment of PAH patients (rosiglitazone and pioglitazone) could be associated with adverse cardiovascular events,Citation109 DHEA might be a smart alternative way to increase PPAR-γ and decrease STAT3 activation in PAH. DHEA is actually undergoing a phase 3 clinical trials for chronic obstructive pulmonary diseases and pulmonary hypertension.Citation110
Plumbagin is a natural vegetal organic compound known to block STAT3 in cancer cells. This molecule has been tested in PAH animal models, and has been shown to reverses experimental PAH through Src/STAT3/NFAT inhibitionCitation111 (). Nonetheless, little is known about human tolerance of the molecule and side effects, making its translation to clinical use unlikely.
Several STAT3 inhibitors are in early phase clinical trials for the treatment of diverse malignancies. STA-21 has completed phase I/II clinical trials for the treatment of psoriasis,Citation60 but no results are publicly available yet. OPB-31121 () is currently in phase I in patients with advanced solid tumors.Citation59 Pyrimethamine is in phase I/II trials for the treatment of chronic lymphocytic leukemia and small lymphocytic leukemia.Citation61 RTA 402 is an antioxidant inflammation modulator able to modulate NFκB, STAT3 and Nrf2 [nuclear factor (erythroid-derived 2)-like 2]. A phase I trial has been completed in 34 patients with solid tumors and lymphoid malignancies. RTA 402 is well tolerated up to 900 mg/day with prolonged exposure up to 12 mo. Data indicate appropriate modulation of the targets NFκB, STAT3 and Nrf2 and suggest clinical efficiency, including a partial response observed in a patient with thyroid cancer, and prolonged disease stabilization in nine patients, six of which lasted for 6–12 mo (3 melanoma, 2 renal cell, 1 medullary thyroid). The recruitment of patients with solid tumors and lymphoid malignancies for the phase II trials has begun.Citation62
STAT3 Implication in Endothelial Cells, Right Ventricle and Inflammation
Although these results are very promising for PAH, the impact of these drugs on PAEC, RV and inflammatory response are crucial. Indeed, while triggering apoptosis in PASMCs may improve PAH, triggering apoptosis of remaining and unaltered PAECs might at the opposite worsen it. Another important factor to consider in PAH therapy is RV health since survival in PAH seems to be determined by the condition of the RV better than the degree of the afterload increase.Citation112 Thus, PAH therapies, if not beneficial for the RV, must at least be not detrimental.
Right ventricle
Several evidences have shown that STAT3 plays a critical role in cardiac hypertrophy in response to stimuli like increased afterloads, infarct, ischemia and neurohormones. First of all, a transgenic conditional STAT3 overexpression in mice cardiomyocytes is sufficient to provoke a spontaneous ventricular hypertrophy,Citation113 suggesting that STAT3 is critical for cardiomyocyte compensatory response. This response is followed by increased transcription of antioxidant proteins like SOD-2, and induction of anti-apoptotic and cyto-protective proteins like Bcl-xL and Hsp70.Citation114-Citation117 Cardiomyocytes appear to be more resistant to H2O2 induced-apoptosis when STAT3 expression is high.Citation118 STAT3 is involved as well in response to mechanical stress.Citation119,Citation120 In a feline model of heart under pressure, STAT3 have been showed activated at 48 h in a manner at least in part dependent of Src activation.Citation121 Src/STAT3 activation has not been particularly measured in RV hypertrophy but could be a mechanism implicated in the protective response of RV by compensation. The question of possible adverse effects of STAT3 inhibition is intriguing. Nonetheless, we believe that as soon as STAT3 inhibition is effective in the decrease of pulmonary pressure and resistance, the mechanisms of RV compensation would not be necessary anymore, STAT3 inhibition in RV being hence without consequences.
Endothelial cells
Increased and sustained STAT3 phosphorylation has also been identified in the endothelium in plexiform lesions of idiopathic lung tissues as well as in PAECs isolated from idiopathic PAH human lungs, suggesting the implication of STAT3 in the proliferative and survival phenotype of PAEC within plexogenic lesions.Citation31 Later, the downregulation of the caveolin-1 protein (known to initiate the coupling between integrins and the Ras-ERK pathway to promote cell cycle progression)Citation122,Citation123 has been identified as a mechanism promoting STAT3 activation. In PAH-PAECs and PAECs exposed to monocrotaline pyrrole, STAT3 is located in cytoplasmic vesicles like endosomes that might be involved in the transport of STAT3 to the nucleus. Caveolin-1 overexpression has been shown to increase the targeting of STAT3 to lysosomes, thus inhibiting STAT3 transcriptional activity. A marked cytoplasmic sequestration of activated PY-STAT3 has been also found in PAECs in the rat model of monocrotaline-induced PAH as well as in human plexiform lesions.Citation122,Citation123 Therefore, STAT3 inhibition might be beneficial as well to reverse plexiform lesion. It might be of particular interest to further study this aspect in the Hypoxia/Sugen rat modelCitation110 or the overexpressing Mts1/S100A4Citation124 mice model, that both develop lesions that somewhat resemble human plexogenic arteriopathies.
However, STAT3 is not only implicated in promoting proliferation or survival, but also in enhancing angiogenesis. Another important therapeutic strategy for PAH consists in the inducting the re-endothelization of vessels (endothelial barrier recovery) and promoting the formation of new vessels by mobilizing endothelial progenitor cells. It is believed that endothelial progenitor cells (EPCs) constitute one aspect of the endothelium repair process by providing beneficial endothelial-derived factors, like HGF or FGH that mobilize and recruit Lin-c-kit+Sca-1+CD34+ progenitor cells from bone marrow into the injured organ. STAT3, along with ERK/MAPK and PI3K/Akt, is one of the predominant pathways activated in response to these factors,Citation125 suggesting that STAT3 inhibition might counteract re-endothelization processes and worsen the disease. Importantly, PAECs isolated from idiopathic patients, despite persistent STAT3 activation, were found less able to produce organized networks in tube formation assays than control cells in response to angiogenic factors.Citation31 This demonstrates that STAT3 is not the only player involved in the appropriate recruitment and migration of PAECs during the angiogenic response. Angiogenesis is a complex mechanism involving several other molecules; therefore STAT3 inhibition might not be that detrimental in the overall angiogenic function and it is critical to continue to increase the knowledge in this area.
Inflammation
Growing evidence demonstrate that inflammation plays a key role in triggering and maintaining pulmonary vascular remodeling in PAH. Indeed, different cell types such as chemokines, cytokines and antibodies play a role in the pathogenesis of PAH. The inflammatory milieu in PAH is composed of resident and recruited macrophages, dendritic cells, T and B cells and mast cells.Citation126 B lymphocytes are probably the critical actors of inflammation by transforming into plasma cell auto-antibody, antigen presentation, cytokine production, differentiation of effectors T-cells and collaboration with DC.Citation127 While regulatory lymphocyte CD4+ CD25 (Treg) activity is usually decreased in autoimmune disorder, increased circulating Treg and decreased CD8+ cytotoxic T cells have been reported in idiopathic PAH,Citation128 controlling T and B cells activity and DC function. DCs are antigen-presenting cells responsible for the initiation of the inflammatory response. DCs have the ability to differentiate into other cell types including ECs, which might be critical for PAH pathogenesis. An increased number of DCs has been found in pulmonary vascular lesion in human and MCT-induced PAH.Citation129 Immature dendritic cells have beenCitation129 found in pulmonary vascular remodeling, suggesting their involvement in PAH immunopathology.
Many studies in cancer support that STAT3 inhibition decreases immunosuppression in tumor microenvironment. Indeed, STAT3 activation inhibits the production of Th1-type cytokines such as IL-12 and IFN-γ, mediators critical for potent anti-tumor immune responses and that are also decreased in PAH. Futhermore, STAT3 regulates the release of IL-10, VEGF and IL-6, factors that prevent DCs maturation into antigen-presenting cellsCitation130-Citation133 and thus alter the inflammation response. Thus the presence of activated STAT3 in PAH could explain the presence of immature DC in the pulmonary vasculature in PAH. Thus STAT3 impairs both adaptive and innate immune responses against tumorCitation134 and its inhibition might have a beneficial impact on restoring normal inflammation processes in the lungs.
However, we cannot rule out the possibility that STAT3 inhibition may decrease T-cells activation, especially via the downregulation of the Pim-1/NFAT axis, which is shown to be implicated in PAH and inflammatory cells.Citation26,Citation28,Citation135 Further studies would be needed to establish the role of STAT3 inhibition in inflammation processes in PAH and to determine if the overall effect of this therapy is beneficial.
Speculations
STAT3 implication in mitochondria/oxygen sensing
A recent study described the potential implication of STAT3 in the mitochondrial dependent energy metabolism.Citation136 Indeed, the mitochondrial protein GRIM-19 (gene associated with retinoid interferon-induced mortality), a component of the electron transport chain (ETC) complex I, is known to interact with STAT3 and inhibit its transcriptional activity.Citation137-Citation140 This interaction is associated with a mitochondrial STAT3 localization and seems to be critical for ETC function and energy production by glucose oxydation.Citation136 This phenomenon is independent of either STAT3 binding to DNA, phosphorylation on the residue tyrosine 705 or dimerization, but is dependent on phosphorylation on the residue serine 727.
Another study also demonstrated that a transgenic STAT3 constitutive activation in primary mouse embryonic fibroblasts enhances a HIF-1α-dependent increase in glycolysis and a HIF-1α-independent reduction in mitochondrial respiration.Citation141 In fact, H. Yu and co-authors have previously shown that a constitutive STAT3 activation (downstream of Src activation) is required for HIF-1α mRNA expression in response to hypoxia or growth factors in cancer, STAT3 acting directly at the level of HIF-1α promoter to enhance transcription.Citation142 Thus, STAT3 might play an important role as a master metabolic regulator in STAT3-dependent human cancer cell lines, and in PAH.
Possible role in NFκB activation
Moreover, in several type of tumor, the constitutive activation of STAT3 is associated with an increased expression of STAT3 that accumulates in a non-phosphorylated form U-STAT3. U-STAT3 has been demonstrated as able to link a non-phosphorylated form of NFκB (nuclear factor kappa-light-chain-enhancer of activated B cells), U-NFκB to form a complex that accumulates in the nucleus and activates a selection of gene.Citation143-Citation145 Other studies using tyrosine 705 mutants showed in keeping results as these mutant can associate with NFκB and induce the expression of genes such as mras or met, which are likely to play an important role in cell transformation by STAT3.Citation146-Citation148 These findings increase the spectrum of the functions that STAT3 might have in PAH, as STAT3 can induce different transcriptional programs depending on which sites are phosphorylated.
STAT5 and organelle integrity
There is evidence that PAH pathogenesis is associated with dysfunction in organelle function and trafficking. Golgi dysfunctions have been first described in human idiopathic PAH and the HIV-macaque model of pulmonary hypertension, with an increase and cytoplasmic dispersal of the Golgi matrix protein/tether giantin and p115.Citation149 ER stress has been also found with activation of the ER unfolded protein response, activation of the ER shape regulator Nogo, a tubular and bundled ERCitation150 and disrupting the ER-mitochondrial unit.Citation27,Citation151 Interestingly, non-phosphorylated STAT5 has been recently reported unexpectedly associated with the Golgi and ER with the non-genomic function to preserve the structural and the functional integrity of these organelles. Indeed, STAT5 siRNA on endothelial cell result in cystis dilatation of the ER with accumulation of Nogo at the boundaries, dilatation and fragmentation of the Golgi, distortion of the nucleus and mitochondrial fragmentation. These data strongly suggest the involvement of STAT5-related ER/Golgi/mitochondrial dysfunctions in the disease pathogenesis.Citation152
Conclusion
PAH is a lethal disease for which treatments are limited. Over the past two years, STAT3 has been demonstrated as a key mediator of PAH pathology and thus has been revealed as a new therapeutic target. Pharmaceutical agents, such as DHEA and other STAT3 inhibitors like plumbagin, are able to reverse experimental PAH by restoring most of the molecular abnormalities seen in PAH-PASMCs: they decrease the expression/activation of Pim1, Survivin and NFAT, and upregulate BMPR2 and miR-204 (). All these downstream effects contribute to decrease PAH-PASMC proliferation and survival, making STAT3 an interesting therapeutic target for PAH treatment. Targeting STAT3 has the potential to not only inhibit cell proliferation, survival and motility but also immune escape and altered immunologic environment and we believe that STAT3 inhibition will result in positive outcome in PAH patients. Several STAT3 inhibitors have been designed for the cancer and are currently in clinical trials for cancer. Since they seem to be safe and well tolerated, their translation to bedside might be facilitated for PAH clinic. STAT3 inhibition opens hence new clinical avenues for PAH.
| Abbreviations: | ||
| ALK1 | = | activin-like kinase 1 |
| Ang-II | = | angiotensin II |
| BMPR2 | = | bone morphogenetic type II receptor |
| CASMC | = | carotid artery smooth muscle cells |
| ChIP/PCR | = | chromatin immunoprecipitation associated with PCR |
| CML | = | Nε-carboxymethyl lysine |
| DHEA | = | dehydroepiandrosterone |
| DC | = | dendritic cell |
| ER | = | endoplasmic reticulum |
| EPC | = | endothelial progenitor cells |
| ET-1 | = | endothelin-1 |
| ETC | = | electron transport chain |
| eNOS | = | endothelium nitric oxide synthase |
| GPCR | = | G-protein coupled receptor |
| GRB2 | = | growth factor receptor-bound protein 2 |
| HIF-1α | = | hypoxia inducible factor 1α |
| IL-6 | = | interleukin-6 |
| INFγ | = | interferon γ |
| IRS-1/2 | = | insulin-receptor substrate-1/2 |
| JIP | = | c-Jun amino-terminal kinase-interacting protein |
| KLF5 | = | Krüppel-like factor 5 |
| MAPK | = | mitogen-activated protein kinase |
| NFAT | = | buclear factor of activated T-cells |
| NFκB | = | nuclear factor kappa-light-chain-enhancer of activated B cells PIAS3 |
| Nrf2 | = | nuclear factor (erythroid-derived 2)-like 2 |
| NO | = | nitric oxide |
| PAEC | = | pulmonary artery endothelial cells |
| PAH | = | pulmonary arterial hypertension |
| PASMC | = | pulmonary artery smooth muscle cells |
| PDGF | = | platelet-derived growth factor |
| PDH | = | pyruvate dehydrogenase |
| PIM1 | = | provirus integration site for Moloney murine leukemia virus |
| PKCδ | = | protein kinase Cδ |
| PLC | = | phospholipase C |
| PPARγ | = | peroxisome proliferator-activated receptor gamma |
| PTB | = | phosphotyrosine-binding |
| PVR | = | pulmonary vascular resistance |
| RAGE | = | receptor for advanced glycation end products |
| ROS | = | reactive oxygen species |
| RTK | = | receptor tyrosine kinase |
| SH2 | = | Src homology 2 domain |
| SHC | = | Src homology 2 domain-containing |
| SHP2 | = | Src homology 2 domain-containing phosphatase 2 |
| SMAD | = | mothers against decapentaplegic homolog |
| SOCS | = | suppressor of cytokine signaling |
| SRC | = | v-Src sarcoma (Schmidt-Ruppin A-2) viral oncogene homolog |
| STAT | = | signal transducer and activator of transcription |
| TRPM3 | = | transient receptor potential melastatin 3 |
| TNFα | = | tumor necrosis factor α |
| ΔΨm | = | mitochondrial membrane potential |
Acknowledgments
R.P. is supported by an Alberta Innovates—Health Solutions post-doctoral fellowship. J.M. received Canadian Institutes of Health Research and Canadian Thoracic Society graduate scholarships. This work was also supported by Canada Research Chairs (CRC) and by Canadian Institutes of Health Research (CIHR) to S.B.
References
- Archer S, Rich S. Primary pulmonary hypertension: a vascular biology and translational research “Work in progress”. Circulation 2000; 102:2781 - 91; http://dx.doi.org/10.1161/01.CIR.102.22.2781; PMID: 11094047
- Ahmad S. Pulmonary hypertension and right heart failure. Chest 1995; 108:1773; http://dx.doi.org/10.1378/chest.108.6.1773; PMID: 7497812
- Humbert M, Sitbon O, Chaouat A, Bertocchi M, Habib G, Gressin V, et al. Survival in patients with idiopathic, familial, and anorexigen-associated pulmonary arterial hypertension in the modern management era. Circulation 2010; 122:156 - 63; http://dx.doi.org/10.1161/CIRCULATIONAHA.109.911818; PMID: 20585011
- Benza RL, Miller DP, Gomberg-Maitland M, Frantz RP, Foreman AJ, Coffey CS, et al. Predicting survival in pulmonary arterial hypertension: insights from the Registry to Evaluate Early and Long-Term Pulmonary Arterial Hypertension Disease Management (REVEAL). Circulation 2010; 122:164 - 72; http://dx.doi.org/10.1161/CIRCULATIONAHA.109.898122; PMID: 20585012
- McMurtry MS, Archer SL, Altieri DC, Bonnet S, Haromy A, Harry G, et al. Gene therapy targeting survivin selectively induces pulmonary vascular apoptosis and reverses pulmonary arterial hypertension. J Clin Invest 2005; 115:1479 - 91; http://dx.doi.org/10.1172/JCI23203; PMID: 15931388
- Voelkel NF, Cool C, Lee SD, Wright L, Geraci MW, Tuder RM. Primary pulmonary hypertension between inflammation and cancer. Chest 1998; 114:Suppl 225S - 30S; http://dx.doi.org/10.1378/chest.114.3_Supplement.225S; PMID: 9741573
- Paulin R, Courboulin A, Barrier M, Bonnet S. From oncoproteins/tumor suppressors to microRNAs, the newest therapeutic targets for pulmonary arterial hypertension. J Mol Med (Berl). 2011.
- Dorfmüller P, Perros F, Balabanian K, Humbert M. Inflammation in pulmonary arterial hypertension. Eur Respir J 2003; 22:358 - 63; http://dx.doi.org/10.1183/09031936.03.00038903; PMID: 12952274
- Zhang S, Fantozzi I, Tigno DD, Yi ES, Platoshyn O, Thistlethwaite PA, et al. Bone morphogenetic proteins induce apoptosis in human pulmonary vascular smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 2003; 285:L740 - 54; PMID: 12740218
- Nohe A, Hassel S, Ehrlich M, Neubauer F, Sebald W, Henis YI, et al. The mode of bone morphogenetic protein (BMP) receptor oligomerization determines different BMP-2 signaling pathways. J Biol Chem 2002; 277:5330 - 8; http://dx.doi.org/10.1074/jbc.M102750200; PMID: 11714695
- Deng Z, Morse JH, Slager SL, Cuervo N, Moore KJ, Venetos G, et al. Familial primary pulmonary hypertension (gene PPH1) is caused by mutations in the bone morphogenetic protein receptor-II gene. Am J Hum Genet 2000; 67:737 - 44; http://dx.doi.org/10.1086/303059; PMID: 10903931
- Yang X, Long L, Southwood M, Rudarakanchana N, Upton PD, Jeffery TK, et al. Dysfunctional Smad signaling contributes to abnormal smooth muscle cell proliferation in familial pulmonary arterial hypertension. Circ Res 2005; 96:1053 - 63; http://dx.doi.org/10.1161/01.RES.0000166926.54293.68; PMID: 15845886
- Moreno-Vinasco L, Gomberg-Maitland M, Maitland ML, Desai AA, Singleton PA, Sammani S, et al. Genomic assessment of a multikinase inhibitor, sorafenib, in a rodent model of pulmonary hypertension. Physiol Genomics 2008; 33:278 - 91; http://dx.doi.org/10.1152/physiolgenomics.00169.2007; PMID: 18303084
- Girerd B, Montani D, Coulet F, Sztrymf B, Yaici A, Jaïs X, et al. Clinical outcomes of pulmonary arterial hypertension in patients carrying an ACVRL1 (ALK1) mutation. Am J Respir Crit Care Med 2010; 181:851 - 61; http://dx.doi.org/10.1164/rccm.200908-1284OC; PMID: 20056902
- Eddahibi S, Chaouat A, Morrell N, Fadel E, Fuhrman C, Bugnet AS, et al. Polymorphism of the serotonin transporter gene and pulmonary hypertension in chronic obstructive pulmonary disease. Circulation 2003; 108:1839 - 44; http://dx.doi.org/10.1161/01.CIR.0000091409.53101.E8; PMID: 14530202
- Eddahibi S, Humbert M, Fadel E, Raffestin B, Darmon M, Capron F, et al. Serotonin transporter overexpression is responsible for pulmonary artery smooth muscle hyperplasia in primary pulmonary hypertension. J Clin Invest 2001; 108:1141 - 50; PMID: 11602621
- Remillard CV, Tigno DD, Platoshyn O, Burg ED, Brevnova EE, Conger D, et al. Function of Kv1.5 channels and genetic variations of KCNA5 in patients with idiopathic pulmonary arterial hypertension. Am J Physiol Cell Physiol 2007; 292:C1837 - 53; http://dx.doi.org/10.1152/ajpcell.00405.2006; PMID: 17267549
- Lee SD, Shroyer KR, Markham NE, Cool CD, Voelkel NF, Tuder RM. Monoclonal endothelial cell proliferation is present in primary but not secondary pulmonary hypertension. J Clin Invest 1998; 101:927 - 34; http://dx.doi.org/10.1172/JCI1910; PMID: 9486960
- Mizuno S, Bogaard HJ, Kraskauskas D, Alhussaini A, Gomez-Arroyo J, Voelkel NF, et al. p53 gene deficiency promotes hypoxia-induced pulmonary hypertension and vascular remodeling in mice. Am J Physiol Lung Cell Mol Physiol 2011; 300:L753 - 61; http://dx.doi.org/10.1152/ajplung.00286.2010; PMID: 21335523
- Chen SJ, Wang YB, Chen O, Zhu XB, Ma Y. [Effect of p21 gene transfection mediated by replication deficient adenovirus on the pulmonary hypertensive rat model]. Zhonghua Er Ke Za Zhi 2008; 46:139 - 42; PMID: 19099691
- Fouty BW, Grimison B, Fagan KA, Le Cras TD, Harral JW, Hoedt-Miller M, et al. p27(Kip1) is important in modulating pulmonary artery smooth muscle cell proliferation. Am J Respir Cell Mol Biol 2001; 25:652 - 8; PMID: 11713109
- Yu L, Quinn DA, Garg HG, Hales CA. Cyclin-dependent kinase inhibitor p27Kip1, but not p21WAF1/Cip1, is required for inhibition of hypoxia-induced pulmonary hypertension and remodeling by heparin in mice. Circ Res 2005; 97:937 - 45; http://dx.doi.org/10.1161/01.RES.0000188211.83193.1a; PMID: 16195480
- Ravi Y, Selvendiran K, Meduru S, Citro L, Naidu S, Khan M, et al. Dysregulation of PTEN in Cardiopulmonary Vascular Remodeling Induced by Pulmonary Hypertension. Cell Biochem Biophys 2011; In press http://dx.doi.org/10.1007/s12013-011-9332-z; PMID: 22205501
- Koppenol WH, Bounds PL, Dang CV. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat Rev Cancer 2011; 11:325 - 37; http://dx.doi.org/10.1038/nrc3038; PMID: 21508971
- Sutendra G, Bonnet S, Rochefort G, Haromy A, Folmes KD, Lopaschuk GD, et al. Fatty acid oxidation and malonyl-CoA decarboxylase in the vascular remodeling of pulmonary hypertension. Sci Transl Med 2010; 2:44ra58; http://dx.doi.org/10.1126/scitranslmed.3001327; PMID: 20702857
- Bonnet S, Rochefort G, Sutendra G, Archer SL, Haromy A, Webster L, et al. The nuclear factor of activated T cells in pulmonary arterial hypertension can be therapeutically targeted. Proc Natl Acad Sci U S A 2007; 104:11418 - 23; http://dx.doi.org/10.1073/pnas.0610467104; PMID: 17596340
- Sutendra G, Dromparis P, Wright P, Bonnet S, Haromy A, Hao Z, et al. The role of Nogo and the mitochondria-endoplasmic reticulum unit in pulmonary hypertension. Sci Transl Med 2011; 3:88ra55; http://dx.doi.org/10.1126/scitranslmed.3002194; PMID: 21697531
- Paulin R, Courboulin A, Meloche J, Mainguy V, Dumas de la Roque E, Saksouk N, et al. Signal transducers and activators of transcription-3/pim1 axis plays a critical role in the pathogenesis of human pulmonary arterial hypertension. Circulation 2011; 123:1205 - 15; http://dx.doi.org/10.1161/CIRCULATIONAHA.110.963314; PMID: 21382889
- Paulin R, Meloche J, Jacob MH, Bisserier M, Courboulin A, Bonnet S. Dehydroepiandrosterone inhibits the Src/STAT3 constitutive activation in pulmonary arterial hypertension. Am J Physiol Heart Circ Physiol 2011; 301:H1798 - 809; http://dx.doi.org/10.1152/ajpheart.00654.2011; PMID: 21890685
- Courboulin A, Paulin R, Giguère NJ, Saksouk N, Perreault T, Meloche J, et al. Role for miR-204 in human pulmonary arterial hypertension. J Exp Med 2011; 208:535 - 48; http://dx.doi.org/10.1084/jem.20101812; PMID: 21321078
- Masri FA, Xu W, Comhair SA, Asosingh K, Koo M, Vasanji A, et al. Hyperproliferative apoptosis-resistant endothelial cells in idiopathic pulmonary arterial hypertension. Am J Physiol Lung Cell Mol Physiol 2007; 293:L548 - 54; http://dx.doi.org/10.1152/ajplung.00428.2006; PMID: 17526595
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011; 144:646 - 74; http://dx.doi.org/10.1016/j.cell.2011.02.013; PMID: 21376230
- Courboulin A, Tremblay VL, Barrier M, Meloche J, Jacob MH, Chapolard M, et al. Krüppel-like factor 5 contributes to pulmonary artery smooth muscle proliferation and resistance to apoptosis in human pulmonary arterial hypertension. Respir Res 2011; 12:128; http://dx.doi.org/10.1186/1465-9921-12-128; PMID: 21951574
- Bonnet S, Archer SL, Allalunis-Turner J, Haromy A, Beaulieu C, Thompson R, et al. A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell 2007; 11:37 - 51; http://dx.doi.org/10.1016/j.ccr.2006.10.020; PMID: 17222789
- Bromberg JF, Horvath CM, Besser D, Lathem WW, Darnell JE Jr.. Stat3 activation is required for cellular transformation by v-src. Mol Cell Biol 1998; 18:2553 - 8; PMID: 9566875
- Darnell JE Jr.. STATs and gene regulation. Science 1997; 277:1630 - 5; http://dx.doi.org/10.1126/science.277.5332.1630; PMID: 9287210
- Banes-Berceli AK, Ketsawatsomkron P, Ogbi S, Patel B, Pollock DM, Marrero MB. Angiotensin II and endothelin-1 augment the vascular complications of diabetes via JAK2 activation. Am J Physiol Heart Circ Physiol 2007; 293:H1291 - 9; http://dx.doi.org/10.1152/ajpheart.00181.2007; PMID: 17526654
- Csiszar A, Labinskyy N, Olson S, Pinto JT, Gupte S, Wu JM, et al. Resveratrol prevents monocrotaline-induced pulmonary hypertension in rats. Hypertension 2009; 54:668 - 75; http://dx.doi.org/10.1161/HYPERTENSIONAHA.109.133397; PMID: 19597040
- Schermuly RT, Dony E, Ghofrani HA, Pullamsetti S, Savai R, Roth M, et al. Reversal of experimental pulmonary hypertension by PDGF inhibition. J Clin Invest 2005; 115:2811 - 21; http://dx.doi.org/10.1172/JCI24838; PMID: 16200212
- Frasch HF, Marshall C, Marshall BE. Endothelin-1 is elevated in monocrotaline pulmonary hypertension. Am J Physiol 1999; 276:L304 - 10; PMID: 9950893
- Wen Z, Zhong Z, Darnell JE Jr.. Maximal activation of transcription by Stat1 and Stat3 requires both tyrosine and serine phosphorylation. Cell 1995; 82:241 - 50; http://dx.doi.org/10.1016/0092-8674(95)90311-9; PMID: 7543024
- Banes AK, Shaw SM, Tawfik A, Patel BP, Ogbi S, Fulton D, et al. Activation of the JAK/STAT pathway in vascular smooth muscle by serotonin. Am J Physiol Cell Physiol 2005; 288:C805 - 12; http://dx.doi.org/10.1152/ajpcell.00385.2004; PMID: 15601754
- Potula HS, Wang D, Quyen DV, Singh NK, Kundumani-Sridharan V, Karpurapu M, et al. Src-dependent STAT-3-mediated expression of monocyte chemoattractant protein-1 is required for 15(S)-hydroxyeicosatetraenoic acid-induced vascular smooth muscle cell migration. J Biol Chem 2009; 284:31142 - 55; http://dx.doi.org/10.1074/jbc.M109.012526; PMID: 19736311
- Wang GS, Qian GS, Mao BL, Cai WQ, Chen WZ, Chen Y. [Changes of interleukin-6 and Janus kinases in rats with hypoxic pulmonary hypertension]. Zhonghua Jie He He Hu Xi Za Zhi 2003; 26:664 - 7; PMID: 14703438
- Christmann RB, Hayes E, Pendergrass S, Padilla C, Farina G, Affandi AJ, et al. Interferon and alternative activation of monocyte/macrophages in systemic sclerosis-associated pulmonary arterial hypertension. Arthritis Rheum 2011; 63:1718 - 28; http://dx.doi.org/10.1002/art.30318; PMID: 21425123
- Byers LA, Sen B, Saigal B, Diao L, Wang J, Nanjundan M, et al. Reciprocal regulation of c-Src and STAT3 in non-small cell lung cancer. Clin Cancer Res 2009; 15:6852 - 61; http://dx.doi.org/10.1158/1078-0432.CCR-09-0767; PMID: 19861436
- Sen B, Peng S, Woods DM, Wistuba I, Bell D, El-Naggar AK, et al. STAT5A-mediated SOCS2 expression regulates Jak2 and STAT3 activity following c-Src inhibition in head and neck squamous carcinoma. Clin Cancer Res 2012; 18:127 - 39; http://dx.doi.org/10.1158/1078-0432.CCR-11-1889; PMID: 22090359
- Singh NK, Wang D, Kundumani-Sridharan V, Van Quyen D, Niu J, Rao GN. 15-Lipoxygenase-1-enhanced Src-Janus kinase 2-signal transducer and activator of transcription 3 stimulation and monocyte chemoattractant protein-1 expression require redox-sensitive activation of epidermal growth factor receptor in vascular wall remodeling. J Biol Chem 2011; 286:22478 - 88; http://dx.doi.org/10.1074/jbc.M111.225060; PMID: 21536676
- Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell 2000; 103:211 - 25; http://dx.doi.org/10.1016/S0092-8674(00)00114-8; PMID: 11057895
- Hubbard SR. Juxtamembrane autoinhibition in receptor tyrosine kinases. Nat Rev Mol Cell Biol 2004; 5:464 - 71; http://dx.doi.org/10.1038/nrm1399; PMID: 15173825
- Reddy EP, Korapati A, Chaturvedi P, Rane S. IL-3 signaling and the role of Src kinases, JAKs and STATs: a covert liaison unveiled. Oncogene 2000; 19:2532 - 47; http://dx.doi.org/10.1038/sj.onc.1203594; PMID: 10851052
- Hofmeister R, Khaled AR, Benbernou N, Rajnavolgyi E, Muegge K, Durum SK. Interleukin-7: physiological roles and mechanisms of action. Cytokine Growth Factor Rev 1999; 10:41 - 60; http://dx.doi.org/10.1016/S1359-6101(98)00025-2; PMID: 10379911
- Takahashi-Tezuka M, Hibi M, Fujitani Y, Fukada T, Yamaguchi T, Hirano T. Tec tyrosine kinase links the cytokine receptors to PI-3 kinase probably through JAK. Oncogene 1997; 14:2273 - 82; http://dx.doi.org/10.1038/sj.onc.1201071; PMID: 9178903
- Xia K, Mukhopadhyay NK, Inhorn RC, Barber DL, Rose PE, Lee RS, et al. The cytokine-activated tyrosine kinase JAK2 activates Raf-1 in a p21ras-dependent manner. Proc Natl Acad Sci U S A 1996; 93:11681 - 6; http://dx.doi.org/10.1073/pnas.93.21.11681; PMID: 8876196
- Yetter A, Uddin S, Krolewski JJ, Jiao H, Yi T, Platanias LC. Association of the interferon-dependent tyrosine kinase Tyk-2 with the hematopoietic cell phosphatase. J Biol Chem 1995; 270:18179 - 82; http://dx.doi.org/10.1074/jbc.270.31.18179; PMID: 7629131
- Cabrera-Vera TM, Vanhauwe J, Thomas TO, Medkova M, Preininger A, Mazzoni MR, et al. Insights into G protein structure, function, and regulation. Endocr Rev 2003; 24:765 - 81; http://dx.doi.org/10.1210/er.2000-0026; PMID: 14671004
- Milligan G, Stoddart LA, Brown AJ. G protein-coupled receptors for free fatty acids. Cell Signal 2006; 18:1360 - 5; http://dx.doi.org/10.1016/j.cellsig.2006.03.011; PMID: 16716567
- Corre I, Baumann H, Hermouet S. Regulation by Gi2 proteins of v-fms-induced proliferation and transformation via Src-kinase and STAT3. Oncogene 1999; 18:6335 - 42; http://dx.doi.org/10.1038/sj.onc.1203010; PMID: 10597233
- Ram PT, Horvath CM, Iyengar R. Stat3-mediated transformation of NIH-3T3 cells by the constitutively active Q205L Galphao protein. Science 2000; 287:142 - 4; http://dx.doi.org/10.1126/science.287.5450.142; PMID: 10615050
- Liang H, Venema VJ, Wang X, Ju H, Venema RC, Marrero MB. Regulation of angiotensin II-induced phosphorylation of STAT3 in vascular smooth muscle cells. J Biol Chem 1999; 274:19846 - 51; http://dx.doi.org/10.1074/jbc.274.28.19846; PMID: 10391929
- Marrero MB, Schieffer B, Paxton WG, Heerdt L, Berk BC, Delafontaine P, et al. Direct stimulation of Jak/STAT pathway by the angiotensin II AT1 receptor. Nature 1995; 375:247 - 50; http://dx.doi.org/10.1038/375247a0; PMID: 7746328
- Daub H, Weiss FU, Wallasch C, Ullrich A. Role of transactivation of the EGF receptor in signalling by G-protein-coupled receptors. Nature 1996; 379:557 - 60; http://dx.doi.org/10.1038/379557a0; PMID: 8596637
- Shah BH, Catt KJ. GPCR-mediated transactivation of RTKs in the CNS: mechanisms and consequences. Trends Neurosci 2004; 27:48 - 53; http://dx.doi.org/10.1016/j.tins.2003.11.003; PMID: 14698610
- Wetzker R, Böhmer FD. Transactivation joins multiple tracks to the ERK/MAPK cascade. Nat Rev Mol Cell Biol 2003; 4:651 - 7; http://dx.doi.org/10.1038/nrm1173; PMID: 12923527
- Akekawatchai C, Holland JD, Kochetkova M, Wallace JC, McColl SR. Transactivation of CXCR4 by the insulin-like growth factor-1 receptor (IGF-1R) in human MDA-MB-231 breast cancer epithelial cells. J Biol Chem 2005; 280:39701 - 8; http://dx.doi.org/10.1074/jbc.M509829200; PMID: 16172123
- Delcourt N, Thouvenot E, Chanrion B, Galéotti N, Jouin P, Bockaert J, et al. PACAP type I receptor transactivation is essential for IGF-1 receptor signalling and antiapoptotic activity in neurons. EMBO J 2007; 26:1542 - 51; http://dx.doi.org/10.1038/sj.emboj.7601608; PMID: 17332755
- Hobson JP, Rosenfeldt HM, Barak LS, Olivera A, Poulton S, Caron MG, et al. Role of the sphingosine-1-phosphate receptor EDG-1 in PDGF-induced cell motility. Science 2001; 291:1800 - 3; http://dx.doi.org/10.1126/science.1057559; PMID: 11230698
- Mira E, Lacalle RA, González MA, Gómez-Moutón C, Abad JL, Bernad A, et al. A role for chemokine receptor transactivation in growth factor signaling. EMBO Rep 2001; 2:151 - 6; http://dx.doi.org/10.1093/embo-reports/kve027; PMID: 11258708
- Pincheira R, Castro AF, Ozes ON, Idumalla PS, Donner DB. Type 1 TNF receptor forms a complex with and uses Jak2 and c-Src to selectively engage signaling pathways that regulate transcription factor activity. J Immunol 2008; 181:1288 - 98; PMID: 18606683
- Meloche J, Paulin R, Courboulin A, Lambert C, Barrier M, Bonnet P, et al. RAGE-dependent activation of the oncoprotein Pim1 plays a critical role in systemic vascular remodeling processes. Arterioscler Thromb Vasc Biol 2011; 31:2114 - 24; http://dx.doi.org/10.1161/ATVBAHA.111.230573; PMID: 21680901
- Lawrie A, Spiekerkoetter E, Martinez EC, Ambartsumian N, Sheward WJ, MacLean MR, et al. Interdependent serotonin transporter and receptor pathways regulate S100A4/Mts1, a gene associated with pulmonary vascular disease. Circ Res 2005; 97:227 - 35; http://dx.doi.org/10.1161/01.RES.0000176025.57706.1e; PMID: 16002749
- Spiekerkoetter E, Guignabert C, de Jesus Perez V, Alastalo TP, Powers JM, Wang L, et al. S100A4 and bone morphogenetic protein-2 codependently induce vascular smooth muscle cell migration via phospho-extracellular signal-regulated kinase and chloride intracellular channel 4. Circ Res 2009; 105:639 - 47, 13, 647; http://dx.doi.org/10.1161/CIRCRESAHA.109.205120; PMID: 19713532
- Sakaguchi T, Yan SF, Yan SD, Belov D, Rong LL, Sousa M, et al. Central role of RAGE-dependent neointimal expansion in arterial restenosis. J Clin Invest 2003; 111:959 - 72; PMID: 12671045
- Kakisis JD, Pradhan S, Cordova A, Liapis CD, Sumpio BE. The role of STAT-3 in the mediation of smooth muscle cell response to cyclic strain. Int J Biochem Cell Biol 2005; 37:1396 - 406; http://dx.doi.org/10.1016/j.biocel.2005.01.009; PMID: 15833272
- Padma R, Nagarajan L. The human PIM-1 gene product is a protein serine kinase. Cancer Res 1991; 51:2486 - 9; PMID: 1826633
- Bonnet S, Paulin R, Sutendra G, Dromparis P, Roy M, Watson KO, et al. Dehydroepiandrosterone reverses systemic vascular remodeling through the inhibition of the Akt/GSK3-beta/NFAT axis. Circulation 2009; 120:1231 - 40; http://dx.doi.org/10.1161/CIRCULATIONAHA.109.848911; PMID: 19752325
- Chen X, Xu H, Yuan P, Fang F, Huss M, Vega VB, et al. Integration of external signaling pathways with the core transcriptional network in embryonic stem cells. Cell 2008; 133:1106 - 17; http://dx.doi.org/10.1016/j.cell.2008.04.043; PMID: 18555785
- Li J, Piao YF, Jiang Z, Chen L, Sun HB. Silencing of signal transducer and activator of transcription 3 expression by RNA interference suppresses growth of human hepatocellular carcinoma in tumor-bearing nude mice. World J Gastroenterol 2009; 15:2602 - 8; http://dx.doi.org/10.3748/wjg.15.2602; PMID: 19496189
- Glienke W, Maute L, Wicht J, Bergmann L. Curcumin inhibits constitutive STAT3 phosphorylation in human pancreatic cancer cell lines and downregulation of survivin/BIRC5 gene expression. Cancer Invest 2010; 28:166 - 71; http://dx.doi.org/10.3109/07357900903287006; PMID: 20121547
- Yabkowitz R, Mansfield PJ, Ryan US, Suchard SJ. Thrombospondin mediates migration and potentiates platelet-derived growth factor-dependent migration of calf pulmonary artery smooth muscle cells. J Cell Physiol 1993; 157:24 - 32; http://dx.doi.org/10.1002/jcp.1041570104; PMID: 8408239
- Yamboliev IA, Gerthoffer WT. Modulatory role of ERK MAPK-caldesmon pathway in PDGF-stimulated migration of cultured pulmonary artery SMCs. Am J Physiol Cell Physiol 2001; 280:C1680 - 8; PMID: 11350764
- Leung WC, Lawrie A, Demaries S, Massaeli H, Burry A, Yablonsky S, et al. Apolipoprotein D and platelet-derived growth factor-BB synergism mediates vascular smooth muscle cell migration. Circ Res 2004; 95:179 - 86; http://dx.doi.org/10.1161/01.RES.0000135482.74178.14; PMID: 15192024
- Sarkar J, Gou D, Turaka P, Viktorova E, Ramchandran R, Raj JU. MicroRNA-21 plays a role in hypoxia-mediated pulmonary artery smooth muscle cell proliferation and migration. Am J Physiol Lung Cell Mol Physiol 2010; 299:L861 - 71; http://dx.doi.org/10.1152/ajplung.00201.2010; PMID: 20693317
- Negash S, Narasimhan SR, Zhou W, Liu J, Wei FL, Tian J, et al. Role of cGMP-dependent protein kinase in regulation of pulmonary vascular smooth muscle cell adhesion and migration: effect of hypoxia. Am J Physiol Heart Circ Physiol 2009; 297:H304 - 12; http://dx.doi.org/10.1152/ajpheart.00077.2008; PMID: 19411288
- Osada-Oka M, Ikeda T, Akiba S, Sato T. Hypoxia stimulates the autocrine regulation of migration of vascular smooth muscle cells via HIF-1alpha-dependent expression of thrombospondin-1. J Cell Biochem 2008; 104:1918 - 26; http://dx.doi.org/10.1002/jcb.21759; PMID: 18384112
- Harvey KA, Welch Z, Sliva D, Siddiqui RA. Role of Rho kinase in sphingosine 1-phosphate-mediated endothelial and smooth muscle cell migration and differentiation. Mol Cell Biochem 2010; 342:7 - 19; http://dx.doi.org/10.1007/s11010-010-0461-2; PMID: 20401628
- Mukhopadhyay UK, Mooney P, Jia L, Eves R, Raptis L, Mak AS. Doubles game: Src-Stat3 versus p53-PTEN in cellular migration and invasion. Mol Cell Biol 2010; 30:4980 - 95; http://dx.doi.org/10.1128/MCB.00004-10; PMID: 20733006
- Gao H, Priebe W, Glod J, Banerjee D. Activation of signal transducers and activators of transcription 3 and focal adhesion kinase by stromal cell-derived factor 1 is required for migration of human mesenchymal stem cells in response to tumor cell-conditioned medium. Stem Cells 2009; 27:857 - 65; http://dx.doi.org/10.1002/stem.23; PMID: 19350687
- Li C, Wernig F, Leitges M, Hu Y, Xu Q. Mechanical stress-activated PKCdelta regulates smooth muscle cell migration. FASEB J 2003; 17:2106 - 8; PMID: 12958154
- Shi Y, Wang C, Han S, Pang B, Zhang N, Wang J, et al. Determination of PKC isoform-specific protein expression in pulmonary arteries of rats with chronic hypoxia-induced pulmonary hypertension. Med Sci Monit 2012; 18:BR69 - 75; PMID: 22293869
- Kronfeld I, Kazimirsky G, Lorenzo PS, Garfield SH, Blumberg PM, Brodie C. Phosphorylation of protein kinase Cdelta on distinct tyrosine residues regulates specific cellular functions. J Biol Chem 2000; 275:35491 - 8; http://dx.doi.org/10.1074/jbc.M005991200; PMID: 10945993
- Novotny-Diermayr V, Zhang T, Gu L, Cao X. Protein kinase C delta associates with the interleukin-6 receptor subunit glycoprotein (gp) 130 via Stat3 and enhances Stat3-gp130 interaction. J Biol Chem 2002; 277:49134 - 42; http://dx.doi.org/10.1074/jbc.M206727200; PMID: 12361954
- Giaid A, Saleh D. Reduced expression of endothelial nitric oxide synthase in the lungs of patients with pulmonary hypertension. N Engl J Med 1995; 333:214 - 21; http://dx.doi.org/10.1056/NEJM199507273330403; PMID: 7540722
- Sud N, Black SM. Endothelin-1 impairs nitric oxide signaling in endothelial cells through a protein kinase Cdelta-dependent activation of STAT3 and decreased endothelial nitric oxide synthase expression. DNA Cell Biol 2009; 28:543 - 53; http://dx.doi.org/10.1089/dna.2009.0865; PMID: 19754268
- Kohanbash G, Okada H. MicroRNAs and STAT interplay. Semin Cancer Biol 2012; 22:70 - 5; http://dx.doi.org/10.1016/j.semcancer.2011.12.010; PMID: 22210182
- Wu W, Lin Z, Zhuang Z, Liang X. Expression profile of mammalian microRNAs in endometrioid adenocarcinoma. Eur J Cancer Prev 2009; 18:50 - 5; http://dx.doi.org/10.1097/CEJ.0b013e328305a07a; PMID: 19077565
- Wang FE, Zhang C, Maminishkis A, Dong L, Zhi C, Li R, et al. MicroRNA-204/211 alters epithelial physiology. FASEB J 2010; 24:1552 - 71; http://dx.doi.org/10.1096/fj.08-125856; PMID: 20056717
- Senis YA, Tomlinson MG, Ellison S, Mazharian A, Lim J, Zhao Y, et al. The tyrosine phosphatase CD148 is an essential positive regulator of platelet activation and thrombosis. Blood 2009; 113:4942 - 54; http://dx.doi.org/10.1182/blood-2008-08-174318; PMID: 19246339
- Wu JH, Goswami R, Cai X, Exum ST, Huang X, Zhang L, et al. Regulation of the platelet-derived growth factor receptor-beta by G protein-coupled receptor kinase-5 in vascular smooth muscle cells involves the phosphatase Shp2. J Biol Chem 2006; 281:37758 - 72; http://dx.doi.org/10.1074/jbc.M605756200; PMID: 17018529
- Brock M, Trenkmann M, Gay RE, Michel BA, Gay S, Fischler M, et al. Interleukin-6 modulates the expression of the bone morphogenic protein receptor type II through a novel STAT3-microRNA cluster 17/92 pathway. Circ Res 2009; 104:1184 - 91; http://dx.doi.org/10.1161/CIRCRESAHA.109.197491; PMID: 19390056
- Rhee ST, Buchman SR. Colocalization of c-Src (pp60src) and bone morphogenetic protein 2/4 expression during mandibular distraction osteogenesis: in vivo evidence of their role within an integrin-mediated mechanotransduction pathway. Ann Plast Surg 2005; 55:207 - 15; http://dx.doi.org/10.1097/01.sap.0000164576.10754.aa; PMID: 16034255
- Kopplow K, Wayss K, Enzmann H, Mayer D. Dehydroepiandrosterone causes hyperplasia and impairs regeneration in rat liver. Int J Oncol 2005; 27:1551 - 8; PMID: 16273211
- Poczatková H, Bogdanová K, Uherková L, Cervenková K, Riegrová D, Rypka M, et al. Dehydroepiandrosterone effects on the mRNA levels of peroxisome proliferator-activated receptors and their coactivators in human hepatoma HepG2 cells. Gen Physiol Biophys 2007; 26:268 - 74; PMID: 18281744
- Hansmann G, Wagner RA, Schellong S, Perez VA, Urashima T, Wang L, et al. Pulmonary arterial hypertension is linked to insulin resistance and reversed by peroxisome proliferator-activated receptor-gamma activation. Circulation 2007; 115:1275 - 84; PMID: 17339547
- Ji JD, Kim HJ, Rho YH, Choi SJ, Lee YH, Cheon HJ, et al. Inhibition of IL-10-induced STAT3 activation by 15-deoxy-Delta12,14-prostaglandin J2. Rheumatology (Oxford) 2005; 44:983 - 8; http://dx.doi.org/10.1093/rheumatology/keh657; PMID: 15840591
- Kim HJ, Rho YH, Choi SJ, Lee YH, Cheon H, Um JW, et al. 15-Deoxy-delta12,14-PGJ2 inhibits IL-6-induced Stat3 phosphorylation in lymphocytes. Exp Mol Med 2005; 37:179 - 85; PMID: 16000871
- Wang LH, Yang XY, Zhang X, Huang J, Hou J, Li J, et al. Transcriptional inactivation of STAT3 by PPARgamma suppresses IL-6-responsive multiple myeloma cells. Immunity 2004; 20:205 - 18; http://dx.doi.org/10.1016/S1074-7613(04)00030-5; PMID: 14975242
- Ehrmann J, Strakova N, Vrzalikova K, Hezova R, Kolar Z. Expression of STATs and their inhibitors SOCS and PIAS in brain tumors. In vitro and in vivo study. Neoplasma 2008; 55:482 - 7; PMID: 18999875
- Juurlink DN, Gomes T, Lipscombe LL, Austin PC, Hux JE, Mamdani MM. Adverse cardiovascular events during treatment with pioglitazone and rosiglitazone: population based cohort study. BMJ 2009; 339:b2942; http://dx.doi.org/10.1136/bmj.b2942; PMID: 19690342
- Abe K, Toba M, Alzoubi A, Ito M, Fagan KA, Cool CD, et al. Formation of plexiform lesions in experimental severe pulmonary arterial hypertension. Circulation 2010; 121:2747 - 54; http://dx.doi.org/10.1161/CIRCULATIONAHA.109.927681; PMID: 20547927
- Courboulin A, Barrier M, Perreault T, Bonnet P, Tremblay VL, Paulin R, et al. Plumbagin reverses proliferation and resistance to apoptosis in experimental PAH. Eur Respir J 2012; 40:618 - 29; http://dx.doi.org/10.1183/09031936.00084211; PMID: 22496325
- Mauritz GJ, Kind T, Marcus JT, Bogaard HJ, van de Veerdonk M, Postmus PE, et al. Progressive changes in right ventricular geometric shortening and long-term survival in pulmonary arterial hypertension. Chest 2012; 141:935 - 43; http://dx.doi.org/10.1378/chest.10-3277; PMID: 21960697
- Kunisada K, Negoro S, Tone E, Funamoto M, Osugi T, Yamada S, et al. Signal transducer and activator of transcription 3 in the heart transduces not only a hypertrophic signal but a protective signal against doxorubicin-induced cardiomyopathy. Proc Natl Acad Sci U S A 2000; 97:315 - 9; http://dx.doi.org/10.1073/pnas.97.1.315; PMID: 10618415
- Negoro S, Kunisada K, Fujio Y, Funamoto M, Darville MI, Eizirik DL, et al. Activation of signal transducer and activator of transcription 3 protects cardiomyocytes from hypoxia/reoxygenation-induced oxidative stress through the upregulation of manganese superoxide dismutase. Circulation 2001; 104:979 - 81; http://dx.doi.org/10.1161/hc3401.095947; PMID: 11524388
- Stephanou A, Latchman DS. Transcriptional regulation of the heat shock protein genes by STAT family transcription factors. Gene Expr 1999; 7:311 - 9; PMID: 10440232
- Yamauchi-Takihara K, Kishimoto T. A novel role for STAT3 in cardiac remodeling. Trends Cardiovasc Med 2000; 10:298 - 303; http://dx.doi.org/10.1016/S1050-1738(01)00066-4; PMID: 11343970
- Hilfiker-Kleiner D, Hilfiker A, Fuchs M, Kaminski K, Schaefer A, Schieffer B, et al. Signal transducer and activator of transcription 3 is required for myocardial capillary growth, control of interstitial matrix deposition, and heart protection from ischemic injury. Circ Res 2004; 95:187 - 95; http://dx.doi.org/10.1161/01.RES.0000134921.50377.61; PMID: 15192020
- Lu Y, Zhou J, Xu C, Lin H, Xiao J, Wang Z, et al. JAK/STAT and PI3K/AKT pathways form a mutual transactivation loop and afford resistance to oxidative stress-induced apoptosis in cardiomyocytes. Cell Physiol Biochem 2008; 21:305 - 14; http://dx.doi.org/10.1159/000129389; PMID: 18441519
- Haas M, Askari A, Xie Z. Involvement of Src and epidermal growth factor receptor in the signal-transducing function of Na+/K+-ATPase. J Biol Chem 2000; 275:27832 - 7; PMID: 10874030
- Takeishi Y, Huang Q, Abe J, Glassman M, Che W, Lee JD, et al. Src and multiple MAP kinase activation in cardiac hypertrophy and congestive heart failure under chronic pressure-overload: comparison with acute mechanical stretch. J Mol Cell Cardiol 2001; 33:1637 - 48; http://dx.doi.org/10.1006/jmcc.2001.1427; PMID: 11549343
- Willey CD, Palanisamy AP, Johnston RK, Mani SK, Shiraishi H, Tuxworth WJ, et al. STAT3 activation in pressure-overloaded feline myocardium: role for integrins and the tyrosine kinase BMX. Int J Biol Sci 2008; 4:184 - 99; http://dx.doi.org/10.7150/ijbs.4.184; PMID: 18612371
- Huang J, Kaminski PM, Edwards JG, Yeh A, Wolin MS, Frishman WH, et al. Pyrrolidine dithiocarbamate restores endothelial cell membrane integrity and attenuates monocrotaline-induced pulmonary artery hypertension. Am J Physiol Lung Cell Mol Physiol 2008; 294:L1250 - 9; http://dx.doi.org/10.1152/ajplung.00069.2007; PMID: 18390833
- Huang J, Wolk JH, Gewitz MH, Mathew R. Progressive endothelial cell damage in an inflammatory model of pulmonary hypertension. Exp Lung Res 2010; 36:57 - 66; http://dx.doi.org/10.3109/01902140903104793; PMID: 20128682
- Greenway S, van Suylen RJ, Du Marchie Sarvaas G, Kwan E, Ambartsumian N, Lukanidin E, et al. S100A4/Mts1 produces murine pulmonary artery changes resembling plexogenic arteriopathy and is increased in human plexogenic arteriopathy. Am J Pathol 2004; 164:253 - 62; http://dx.doi.org/10.1016/S0002-9440(10)63115-X; PMID: 14695338
- Katsha AM, Ohkouchi S, Xin H, Kanehira M, Sun R, Nukiwa T, et al. Paracrine factors of multipotent stromal cells ameliorate lung injury in an elastase-induced emphysema model. Mol Ther 2011; 19:196 - 203; http://dx.doi.org/10.1038/mt.2010.192; PMID: 20842104
- Tuder RM, Groves B, Badesch DB, Voelkel NF. Exuberant endothelial cell growth and elements of inflammation are present in plexiform lesions of pulmonary hypertension. Am J Pathol 1994; 144:275 - 85; PMID: 7508683
- Ulrich S, Taraseviciene-Stewart L, Huber LC, Speich R, Voelkel N. Peripheral blood B lymphocytes derived from patients with idiopathic pulmonary arterial hypertension express a different RNA pattern compared with healthy controls: a cross sectional study. Respir Res 2008; 9:20; http://dx.doi.org/10.1186/1465-9921-9-20; PMID: 18269757
- Ulrich S, Nicolls MR, Taraseviciene L, Speich R, Voelkel N. Increased regulatory and decreased CD8+ cytotoxic T cells in the blood of patients with idiopathic pulmonary arterial hypertension. Respiration 2008; 75:272 - 80; http://dx.doi.org/10.1159/000111548; PMID: 18025812
- Perros F, Dorfmüller P, Souza R, Durand-Gasselin I, Mussot S, Mazmanian M, et al. Dendritic cell recruitment in lesions of human and experimental pulmonary hypertension. Eur Respir J 2007; 29:462 - 8; http://dx.doi.org/10.1183/09031936.00094706; PMID: 17107989
- Gabrilovich D. Mechanisms and functional significance of tumour-induced dendritic-cell defects. Nat Rev Immunol 2004; 4:941 - 52; http://dx.doi.org/10.1038/nri1498; PMID: 15573129
- Gabrilovich DI, Chen HL, Girgis KR, Cunningham HT, Meny GM, Nadaf S, et al. Production of vascular endothelial growth factor by human tumors inhibits the functional maturation of dendritic cells. Nat Med 1996; 2:1096 - 103; http://dx.doi.org/10.1038/nm1096-1096; PMID: 8837607
- Ohm JE, Gabrilovich DI, Sempowski GD, Kisseleva E, Parman KS, Nadaf S, et al. VEGF inhibits T-cell development and may contribute to tumor-induced immune suppression. Blood 2003; 101:4878 - 86; http://dx.doi.org/10.1182/blood-2002-07-1956; PMID: 12586633
- Visinoni F. [Milestone bring forth new idea on pathological laboratory]. Zhonghua Bing Li Xue Za Zhi 2004; 33:283 - 4; PMID: 15256132
- Lee H, Pal SK, Reckamp K, Figlin RA, Yu H. STAT3: a target to enhance antitumor immune response. Curr Top Microbiol Immunol 2011; 344:41 - 59; http://dx.doi.org/10.1007/82_2010_51; PMID: 20517723
- Macian F. NFAT proteins: key regulators of T-cell development and function. Nat Rev Immunol 2005; 5:472 - 84; http://dx.doi.org/10.1038/nri1632; PMID: 15928679
- Wegrzyn J, Potla R, Chwae YJ, Sepuri NB, Zhang Q, Koeck T, et al. Function of mitochondrial Stat3 in cellular respiration. Science 2009; 323:793 - 7; http://dx.doi.org/10.1126/science.1164551; PMID: 19131594
- Huang G, Lu H, Hao A, Ng DC, Ponniah S, Guo K, et al. GRIM-19, a cell death regulatory protein, is essential for assembly and function of mitochondrial complex I. Mol Cell Biol 2004; 24:8447 - 56; http://dx.doi.org/10.1128/MCB.24.19.8447-8456.2004; PMID: 15367666
- Lu H, Cao X. GRIM-19 is essential for maintenance of mitochondrial membrane potential. Mol Biol Cell 2008; 19:1893 - 902; http://dx.doi.org/10.1091/mbc.E07-07-0683; PMID: 18287540
- Lufei C, Ma J, Huang G, Zhang T, Novotny-Diermayr V, Ong CT, et al. GRIM-19, a death-regulatory gene product, suppresses Stat3 activity via functional interaction. EMBO J 2003; 22:1325 - 35; http://dx.doi.org/10.1093/emboj/cdg135; PMID: 12628925
- Zhang J, Yang J, Roy SK, Tininini S, Hu J, Bromberg JF, et al. The cell death regulator GRIM-19 is an inhibitor of signal transducer and activator of transcription 3. Proc Natl Acad Sci U S A 2003; 100:9342 - 7; http://dx.doi.org/10.1073/pnas.1633516100; PMID: 12867595
- Demaria M, Misale S, Giorgi C, Miano V, Camporeale A, Campisi J, et al. STAT3 can serve as a hit in the process of malignant transformation of primary cells. Cell Death Differ 2012; 19:1390 - 7; http://dx.doi.org/10.1038/cdd.2012.20; PMID: 22402588
- Niu G, Briggs J, Deng J, Ma Y, Lee H, Kortylewski M, et al. Signal transducer and activator of transcription 3 is required for hypoxia-inducible factor-1alpha RNA expression in both tumor cells and tumor-associated myeloid cells. Mol Cancer Res 2008; 6:1099 - 105; http://dx.doi.org/10.1158/1541-7786.MCR-07-2177; PMID: 18644974
- Hagihara K, Nishikawa T, Sugamata Y, Song J, Isobe T, Taga T, et al. Essential role of STAT3 in cytokine-driven NF-kappaB-mediated serum amyloid A gene expression. Genes Cells 2005; 10:1051 - 63; http://dx.doi.org/10.1111/j.1365-2443.2005.00900.x; PMID: 16236134
- Yu Z, Zhang W, Kone BC. Signal transducers and activators of transcription 3 (STAT3) inhibits transcription of the inducible nitric oxide synthase gene by interacting with nuclear factor kappaB. Biochem J 2002; 367:97 - 105; http://dx.doi.org/10.1042/BJ20020588; PMID: 12057007
- Yoshida Y, Kumar A, Koyama Y, Peng H, Arman A, Boch JA, et al. Interleukin 1 activates STAT3/nuclear factor-kappaB cross-talk via a unique TRAF6- and p65-dependent mechanism. J Biol Chem 2004; 279:1768 - 76; http://dx.doi.org/10.1074/jbc.M311498200; PMID: 14593105
- Yang J, Chatterjee-Kishore M, Staugaitis SM, Nguyen H, Schlessinger K, Levy DE, et al. Novel roles of unphosphorylated STAT3 in oncogenesis and transcriptional regulation. Cancer Res 2005; 65:939 - 47; PMID: 15705894
- Yang J, Liao X, Agarwal MK, Barnes L, Auron PE, Stark GR. Unphosphorylated STAT3 accumulates in response to IL-6 and activates transcription by binding to NFkappaB. Genes Dev 2007; 21:1396 - 408; http://dx.doi.org/10.1101/gad.1553707; PMID: 17510282
- Lee H, Herrmann A, Deng JH, Kujawski M, Niu G, Li Z, et al. Persistently activated Stat3 maintains constitutive NF-kappaB activity in tumors. Cancer Cell 2009; 15:283 - 93; http://dx.doi.org/10.1016/j.ccr.2009.02.015; PMID: 19345327
- Sehgal PB, Mukhopadhyay S, Patel K, Xu F, Almodóvar S, Tuder RM, et al. Golgi dysfunction is a common feature in idiopathic human pulmonary hypertension and vascular lesions in SHIV-nef-infected macaques. Am J Physiol Lung Cell Mol Physiol 2009; 297:L729 - 37; http://dx.doi.org/10.1152/ajplung.00087.2009; PMID: 19648286
- Voeltz GK, Prinz WA, Shibata Y, Rist JM, Rapoport TA. A class of membrane proteins shaping the tubular endoplasmic reticulum. Cell 2006; 124:573 - 86; http://dx.doi.org/10.1016/j.cell.2005.11.047; PMID: 16469703
- Li W, Dunmore BJ, Morrell NW. Bone morphogenetic protein type II receptor mutations causing protein misfolding in heritable pulmonary arterial hypertension. Proc Am Thorac Soc 2010; 7:395 - 8; http://dx.doi.org/10.1513/pats.201002-024AW; PMID: 21030519
- Lee JE, Yang YM, Liang FX, Gough DJ, Levy DE, Sehgal PB. Nongenomic STAT5-dependent effects on Golgi apparatus and endoplasmic reticulum structure and function. Am J Physiol Cell Physiol 2012; 302:C804 - 20; http://dx.doi.org/10.1152/ajpcell.00379.2011; PMID: 22159083