Abstract
The cytokine-inducible transcription factors signal transducer and activator of transcription 5A and 5B (STAT5A and STAT5B) are important for the proper development of multicellular eukaryotes. Disturbed signaling cascades evoking uncontrolled expression of STAT5 target genes are associated with cancer and immunological failure. Here, we summarize how STAT5 acetylation is integrated into posttranslational modification networks within cells. Moreover, we focus on how inhibitors of deacetylases and tyrosine kinases can correct leukemogenic signaling nodes involving STAT5. Such small molecules can be exploited in the fight against neoplastic diseases and immunological disorders.
Cytokines and Growth Hormones Activate STAT5A and STAT5B
Seven STATs, STAT1, 2, 3, 4, 5A, 5B, and 6, have been found in mammalian cells.Citation1-Citation3 STAT5A and STAT5B are expressed from two different genes. These are located in tandem on human chromosome 17q11.2, and probably arose from gene duplication. The two proteins have approximately 90 kDa and share 94% amino acid sequence identity, with STAT5A being seven amino acids longer than STAT5B.Citation4,Citation5 STAT5A and STAT5B regulate erythropoiesis, lymphopoiesis, and the maintenance of the hematopoietic stem cell population.Citation2,Citation6-Citation8
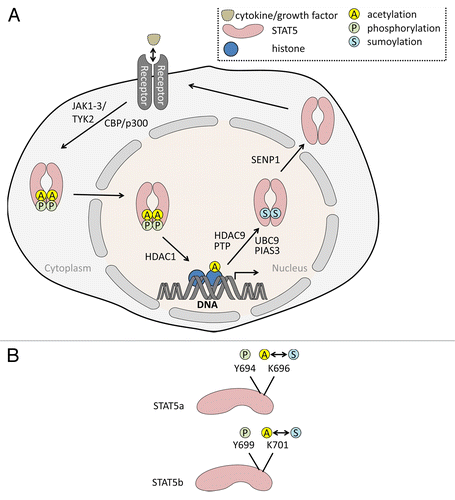
The mechanism of STAT activation has been well elaborated. Binding of ligands (cytokines or growth factors) to cognate receptors activates the associated Janus kinases, JAK1–3 and TYK2 (). These phosphorylate tyrosine residues of the cytosolic receptor subunit.Citation3,Citation9 Via their Src homology-2 (SH2) domains cytosolic STAT5 molecules bind to the phospho-tyrosine residues in the receptors and also become phosphorylated by Janus kinases. Phosphorylation occurs at Y694 (STAT5A) or Y699 (STAT5B) and is crucial for the stable association of STAT5 dimers through their SH2 domains binding phosphorylated tyrosine residues. Activated STAT5 dimers translocate into the nucleus and bind mainly to palindromic interferon gamma activated sequences (GAS) with the consensus TTCNNNGAA.Citation10,Citation11 N-terminal interactions between STAT5 molecules on DNA permit tetramerization,Citation11,Citation12 being critically relevant for cytokine and immune responses.Citation13 Disturbances in the signaling cascades induced through STAT5 are often associated with leukemogenesis and other cancers.Citation5,Citation14,Citation15
Figure 1. (A) STAT5 acetylation-sumoylation-phosphorylation switch. Binding of cytokines to receptors leads to the phosphorylation of STAT5 (P) at tyrosine and serine residues. For example, the interleukins IL-2 and IL-7 induce Janus tyrosine kinases phosphorylating STAT5 at tyrosine residues. The related HATs CBP and p300 catalyze acetylation of STAT5. Phosphorylated STAT5 dimers enter the nucleus and induce STAT5 gene expression promoting cell survival and proliferation. Acetylation (A) of STAT5 rivals sumoylation (S) and thereby allows STAT5 signaling. We postulate that the sumoylation of STAT5 occurs subsequent to a state in which STAT5 is phosphorylated and acetylated, in order to allow gene expression. HDAC9 has been shown to deacetylate STAT5; PTP, phosphatase catalyzing dephosphorylation of STAT5. UBCH9 and PIAS3 belong to the cellular sumoylation machinery (E2/E3 SUMO conjugase/ligase) and transfer SUMO2 to STAT5. SENP1 removes the sumoylation mark and subsequently allows a re-entry of STAT5 into signaling emanating from ligated receptors. The model is based on the works by Ma and colleagues, Van Nguyen and colleagues, and Beier and colleagues (see text for further details). (B) Tyrosine and lysine residues of STAT5 undergoing synergistic and antagonistic posttranslational modifications. The figure shows tyrosine and lysine moieties in STAT5A and STAT5B regulated by phosphorylation, acetylation, and sumoylation. Acetylation and sumoylation of one lysine moiety are mutually exclusive.
STAT5A and STAT5B have overlapping and redundant functions. While both STAT5 isoforms can be activated by the same set of cytokines, some cytokines preferentially activate either STAT5A or STAT5B. For example, prolactin (PRL; stimulates milk production by mammary glands, lactation) predominantly activates STAT5A and growth hormone rather induces STAT5B.Citation16 Moreover, distinct roles of the STAT5 isoforms increasingly become appreciated.Citation17
Acetylation Determines Gene Expression and Signaling
Eukaryotic gene expression is a highly ordered and rapidly adapting process. Dysregulation of genes can lead to cell death or cell cycle disturbances, loss of growth control, and hence ultimately to diseases such as cancer and autoimmunity.Citation18,Citation19 Posttranslational modifications of proteins belonging to the chromatin (the complex consisting of DNA and mostly positively charged histones) and of proteins targeted to other cellular loci affect cellular transcriptomes and proteomes. Acetylation of histones, representing ~20% of the cellular protein mass, and of non-histone proteins is appreciated as an outstanding rheostat for balanced gene expression ensuring homeostasis.Citation20-Citation23 Lysine residues are acetylated by histone acetyltransferases (HATs) using acetyl-CoA as donor for the acetyl group bound as a thioester. Histone deacetylases (HDACs) and Sirtuins catalyze the removal of acetylation marks.Citation19-Citation25
Tumors often have dysregulated acetylation levels. Therefore, histone deacetylase inhibitors (HDACi), small molecules that can regulate acetylation in vivo, are promising candidates for cancer therapy. HDACi fall into structurally diverse classes and block the activity of HDACs by different mechanisms.Citation26 While derivatives of hydroxamic acids attack a Zinc ion (Zn2+) in the catalytic center of HDACs, the fatty acids and benzamides bind near the foot pocket of the catalytic site.Citation27,Citation28 These HDACi do not affect the activity of class III HDACs (Sirtuins), which use a different mechanism of catalysis involving NAD+ instead of Zn2+.Citation25 HDACi are epigenetic drugs considered as candidates for the treatment of cancer, autoimmunity, and neurodegenerative diseases.Citation19-Citation25 Most ongoing clinical studies use HDACi inactivating class I, II, and IV HDACs.Citation20-Citation22
Target genes of STAT5 and STAT5 itself are also regulated by acetylation/deacetylation, and this is one focus of our article. Furthermore, we review how HDACi correct leukemogenic signatures associated with STAT5 activity.
STAT5 is Controlled by Acetylation
Direct effects
A physiological situation causing STAT5 acetylation was identified by Ma and colleagues in 2010. They treated breast cancer-derived cells with the peptide hormone PRL and found an acetylation-dependent PRL receptor (PRLR) dimerization. The HAT CREB-binding protein (CBP) acetylates multiple lysine sites randomly distributed along the cytoplasmic loop of PRLR.Citation29 CBP mainly resides in the nucleus and a redirection of CBP from the nucleus to the cytoplasm has been found in cells treated with PRL, interferons (IFNs), and HDACi.Citation30,Citation31 These mechanisms need further investigation. It is e.g., unclear if posttranslational modifications of CBP are required for its cytosolic accumulation. Furthermore, cytokines may redirect existing CBP from the nucleus to the cytoplasm or the newly synthesized CBP may stop entering the nuclear compartment to accumulate in the cytosol.
PRL-activated PRLR signals to STAT5B, which also becomes acetylated by CBP and undergoes enhanced PRL-induced dimerization dependent on the acetylation of lysine residue K694.Citation29 Furthermore, IL-7 signaling leads to STAT5A acetylation at lysine 696, indicating that acetylation-dependent STAT5 dimerization is also observed in other cytokine signaling pathways.Citation32 Whether STAT5 acetylation is a general mechanism for STAT5 function has to be determined by future studies.
Unphosphorylated STAT5 (U-STAT5) can also form dimers,Citation33,Citation34 and this is also found for other STATs in resting cells (e.g., STAT1–STAT1, STAT1–STAT2, STAT1–STAT3, and STAT1–STAT5 dimers).Citation30,Citation31,Citation35-Citation38 It remains to be clarified if acetylation also determines such interactions between STAT proteins.
Indirect effects
Transcriptional regulation is orchestrated by transcription factor complexes bound to the promoter or enhancer regions on DNA. These complexes recruit histone-modifying enzymes that can either activate or repress transcription by chromatin remodeling.Citation39 In recent years it became clear that both, HATs and HDACs are associated with repression as well as with transcriptional activation.Citation1,Citation21,Citation24,Citation25,Citation40
STAT5 is integrated into the cellular acetylation/deacetylation equilibrium. It was reported that recruitment of the corepressor silencing mediator of retinoid and thyroid transcription (SMRT) to STAT5 target promoter regions can repress gene expression in response to interleukin-3 (IL-3) in murine 32D cells. SMRT was found to interact with both, STAT5A and STAT5B, as well as with STAT3. Repressive effects of this interaction were found for STAT5A, STAT5B, and STAT4, but not for STAT1 and STAT6 in transient transfection reporter assays.Citation41 Binding of SMRT to STAT5 occurs via the STAT5 coiled coil domain and the hyperactive STAT5 point mutant H299R,Citation42 fails to recruit SMRT.Citation41 Due to the fact that the HDACi TSA can activate target gene transcription again, it was concluded that HDACs confer this process inhibiting STAT5-dependent transcription.Citation41
Several studies though report that HDACs promote STAT5’s transcriptional activity. Rascle and colleagues treated IL-2 and IL-3 stimulated murine T and pro-B cell lines with the HDACi TSA.Citation43 Surprisingly, most of the STAT5 target genes were repressed by TSA, indicating that histone deacetylase activity is required for STAT5 dependent gene activation. This study further shows that the DNA binding of STAT5 is not perturbed by TSA treatment, indicating that STAT5 dimerization, phosphorylation and activation remain intact after HDACi treatment.Citation43
Similarly, Sebastián and colleagues found that in macrophages TSA does not abrogate tyrosine phosphorylation of STAT5A/STAT5B on Y694/699 induced by the cytokine granulocyte-monocyte colony stimulating factor (GM-CSF).Citation44 Both, studies showed that 20–200 nM TSA disturbs the recruitment of the basal transcription machinery including mRNA polymerase II to STAT5 target genes.Citation43,Citation44 A study assessing the effects of IFN-β on STAT1/STAT2/IRF9 (ISGF3) also found that TSA blocks the interaction of polymerase II with ISGF3 target genes in human kidney-derived 293T cells.Citation45
Interestingly, the STAT5/HDAC1 interaction seems to be cell type-specific, since it cannot be detected in macrophages.Citation44 This might explain the different findings for the function of HDACs in STAT5 target gene expression induced by IL-3, since different cell lines were used in these studies.Citation41,Citation43 Likewise, different cytokines could variably induce processes that are positively or negatively controlled by HDACs and HDACi. Furthermore, one should consider that most cytokines act very rapidly, HDACi require far longer to alter the cellular transcriptome. This could well explain different data on whether HDAC inhibition alters STAT phosphorylation or not.Citation1,Citation40 It is plausible that the effects of HDACi on STAT phosphorylation, which were measured, after hours, cannot be directly related to the effects on STAT phosphorylation measured after minutes.
Data collected for the transcription factor inhibitory domain-1 (ID-1), being a main target of STATs, also argue in favor of HDACs being required for its expression.Citation46 Analysis on the regulation of ID-1 revealed that STAT5 recruits HDAC1 to the ID-1 promoter. Here, the transcription factor C/EBPβ has to be deacetylated by HDAC1 to allow transcription of ID-1.Citation47
Taken together these findings suggest that recruitment of HDACs by STAT5 can lead to different molecular modifications. HDACs can deacetylate histones in the promoter region of STAT5 target genes, co-factors like C/EBPβ, or STAT5 itself.Citation29,Citation41,Citation43,Citation44,Citation47,Citation48
A study testing oncolytic viruses also shed light on the control of STAT5 signaling in cells with drug-induced hyperacetylation. Such viruses can be used for tumor therapy as they preferentially target and eliminate fast growing tumor cells. The host immune response restricts the efficacy of such tumor therapy via the induction of IFNs.Citation49 The subsequent activation of STAT1 and STAT2 and their anti-viral target genes eliminate the oncolytic viruses and induce anti-viral resistance. HDACi evoke acetylation of STAT1 and chromatin and thereby block the activation of the anti-viral host response.Citation1 The HDACi valproic acid (VPA),Citation50 and other HDACi can inhibit STAT5 phosphorylation and expression of the transcription factor T-BET promoting IFNγ production.Citation51 Accordingly, efficacy of oncolytic herpes simplex virus (oHSV) infection in an orthotopic glioblastoma mouse model was augmented without an enhanced risk for HSV encephalitis.Citation51 At the cellular level, VPA attenuated the recruitment and activation of natural killer (NK) cells and macrophages into tumor-bearing brains post-oHSV infection. VPA also impairs the activation of pro-inflammatory gene expression of NK cells, e.g., of granzyme B and perforin. These data suggest that VPA antagonizes the initial phase of the inflammatory immune response against therapeutic approaches with oHSV efficacy.Citation51
Data supporting that HDACs are required for STAT5 dependent gene activation contrast findings demonstrating that STAT5 dimerization is dependent on acetylation.Citation29,Citation32 It is unclear why the same target genes of STAT5 can be activated or repressed upon TSA treatment. One possibility might be the recruitment of cell specific co-factors that mediate different outcomes after STAT5 activation and HDACi treatment.
STAT5 Acetylation and B and T Cell Development
STAT5A and STAT5B are key regulators of hematopoietic development.Citation2,Citation6-Citation8 They are the downstream effectors of several cytokine receptors like EPO-R, IL-2R and IL-7R, and they modulate cell proliferation, apoptosis, and differentiation. STAT5 is expressed in several hematopoietic lineages and is indispensable for erythroid and lymphocyte development and maturation.Citation6,Citation52,Citation53 Mice lacking Stat5A/Stat5B are perinatal lethal, highly anemic, and show elevated levels of apoptosis in erythroid progenitor cells.Citation54,Citation55 Complete STAT5 inactivation impairs the development of T and B lymphocytes, while the myeloid cell population is not or barely effected. Interestingly, IL-7R−/− mice show a similar phenotype, which indicates a key role of STAT5 in IL-7 signaling and lymphocyte development. Furthermore, STAT5A and STAT5B regulate the quiescence of hematopoietic stem cells.Citation7 STAT5 is regulated through IL-2 and IL-7 signaling during T and B cell development and plays an instructive and permissive role during lymphocyte maturation. On the one hand STAT5 regulates the expression of developmental genes like early B cell factor (EBF) and on the other hand activated STAT5 is critical for cell survival and proliferation.Citation53,Citation56,Citation57 The proliferative and anti-apoptotic function of STAT5 during lymphocyte development, implicates STAT5 in the pathogenesis of human B cell precursor acute lymphoblastic leukemia (B-ALL).Citation53 Hence, understanding how STAT5 shapes cellular development and maturation can provide important insights into the role of STAT5 in tumorigenesis.
The JAK-STAT response has to be tightly controlled to allow proper lymphocyte development. One well-described mechanism to regulate the strength of IL-7 signaling is the modulation by negative feedback mechanisms. IL-7R signaling has to be shut down during lymphocyte development in order to maintain homeostasis and to prevent hyperproliferation.Citation58 One mediator of this downregulation is the suppressor of cytokine signaling (SOCS) family proteins mediate this by inhibiting phosphorylated JAKs, thus blocking the phosphorylation of STAT5.Citation59 Furthermore, Henriques and coworkers demonstrated that IL-7 signaling is also regulated by receptor degradation. IL-7R is rapidly internalized and degraded upon IL-7 signaling.Citation60
Of note, SOCS1 also integrates STAT5A, the tumor suppressor p53, and responses regulated by the DNA damage induced kinases ataxia telangiectasia mutated/ataxia telangiectasia mutated related (ATM/ATR).Citation61 Via such mechanisms, SOCS1 may act as a tumor suppressor maintaining genomic stability.Citation62 Truly, posttranslational modifications of STAT5 are likely to fine-tune such elaborated regulatory circuits. Furthermore, the expression of SOCS1 and SOCS3 is suppressed by HDAC8 in leukemic cells carrying the oncogenic kinases BCR-ABL or JAK2VF. The HDACi TSA and sodium butyrate can augment the levels of these SOCS proteins and suppress phosphorylation of JAK2 and STAT5.Citation63 An HDAC-dependent repression of SOCS1 and SOCS3 has also been reported in colon cancer-derived cells.Citation64 It is plausible that such mechanism, in addition to the activation of phosphatases in HDACi-treated cells,Citation30 are responsible for the frequently observed inhibition of STAT phosphorylation in cells incubated with HDACi.Citation1,Citation40,Citation65
A recent study revealed dynamic post-translational modifications of STAT5 as a highly specific mechanism to adjust transcriptional activity of STAT5. Van Nguyen and colleagues presented a model in which sumoylation and acetylation antagonistically regulate STAT5 phosphorylation and transcriptional activity (). Sumoylation is mediated through an activating enzyme (E1), a SUMO-conjugase UBC9 (E2), and SUMO-ligases (E3), and this process can be reverted through SUMO-specific proteases (SENPs). This dynamic modification can alter the localization, interaction and activity of proteins.Citation66 Van Nguyen and colleagues showed that sumoylation of STAT5 in lymphoid cells leads to an inactivation of STAT5 and a block in early T and B cell development similar to the defect observed in STAT5 deficient lymphocytes.Citation6,Citation52,Citation53 In the absence of SENP1 sumoylated STAT5 accumulates in the cells and leads to an inactivation of STAT5 activity during lymphocyte development. Sumoylation of STAT5A occurs mainly on Lysine 696, which is also the major target for acetylation (; K701 in STAT5B). Interestingly, sumoylation of STAT5 prevents subsequent acetylation;Citation32 acetylation is essential for STAT5 dimerization and transcriptional activity.Citation29 This effect of SENP1 is specific for lymphocytes and cannot be observed in myeloid or erythroid cells.Citation32 The reason for this is still unknown. Interestingly, SENP1−/− mice have defective erythropoiesis caused by decreased levels of hypoxia-inducible factor-1α (HIF-1α) and a subsequent downregulation of erythropoietin (EPO) production. The EPO/STAT5 signaling pathway itself though appears unaffected by SENP1 deficiency,Citation67 again indicating specificity. One possible explanation for these observations relies on the fact that STAT5 has different functions in various cell types. STAT5 can have instructive and permissive roles. For example, STAT5 has been show to regulate cell survival during early lymphopoiesis rather than inducing maturation steps.Citation53,Citation68-Citation70 Consistent with these findings, van Nguyen and colleagues observed a downregulation of the apoptosis inhibitor BCL2 in SENP1−/− lymphocytes.Citation32
The regulation of other STAT5 target genes is also dependent on STAT5 acetylation. Beier and colleagues,Citation71 demonstrated that HDAC9 deficiency, but not a lack of HDAC6 or situin-1 (SIRT1), leads to the stabilization of acetylated STAT5 and the activation of its target genes in regulatory T cells (). Taken together these data clearly indicate an important role of STAT5 acetylation during lymphocyte development and maturation.
Modulation of STAT5 Signaling with Epigenetic Drugs
Chronic myeloid, acute myeloid, and lymphatic leukemia
Constitutively phosphorylated STAT5 can be found in leukemia-derived cell lines and in primary samples of acute myeloid leukemia (AML), chronic myeloid leukemia (CML), and acute lymphoid leukemia (ALL).Citation72 Inhibition of STAT5 appears as a potent strategy to target leukemia,Citation73,Citation74 warranting further investigations on the molecular mechanisms of oncogenic STAT5 activation.
Leukemic cells frequently express a mutant of the class III receptor tyrosine kinase (RTK) FMS-like tyrosine kinase 3 (FLT3-ITD), carrying an internal tandem duplication mutation in the juxtamembrane region,Citation75 or the leukemia fusion protein BCR-ABL (translocation t[9;22][q34;q11]).Citation76 FLT3-ITD is mainly found in AML and BCR-ABL typically occurs in CML. As both leukemogenic proteins accelerate proliferation and suppress apoptosis as well as differentiation, they represent promising therapeutic targets.Citation77-Citation79 Expression of BCR-ABL or FLT3-ITD stimulates STAT5 phosphorylation and its translocation into the nuclear compartment (). Remarkably, FLT3-ITD activates STAT5 mainly at the endoplasmic reticulum (ER) and specifically FLT3-ITD anchored at the ER is a transforming oncogene.Citation80,Citation81 Remarkably, activation of STAT5 by these two oncogenes has overlapping and distinct features. For example, the nuclear translocation of phosphorylated STAT5 activated by BCR-ABL, but not of STAT5 activated by FLT3-ITD, is controlled by SRC family kinases.Citation82
Figure 2. (A) Oncogenic STAT5 activation through FLT3-ITD. FLT3-ITD is a constitutively active, oncogenic driver mutant of FLT3. FLT3-ITD is frequently found in AML and ALL (acute myeloid/lymphoid leukemia). In contrast to FLT3, FLT3-ITD mainly locates to the endoplasmic reticulum (ER), from where it propels STAT5 signaling. Reactive oxygen species (ROS) are involved in the aberrant signaling of FLT3-ITD to STAT5. Resulting gene expression patterns linked to STAT5 promote uncontrolled cell proliferation and apoptosis resistance. It is unknown whether FLT3-ITD-dependent STAT5 activation is linked to the acetylation (A) or sumoylation (S) of STAT5. HSP90 stabilizes FLT3-ITD but is dispensable for the stability of FLT3. For (A) FLT3-ITD is provided as an example for an oncogenic kinase activating STAT5; similar findings were e.g., made for BCR-ABL. (B) HDACi antagonize STAT5 and FLT3-ITD. Histone deacetylase inhibitors (HDACi) eliminate FLT3-ITD and STAT5 via different, partially overlapping molecular mechanisms in AML cells. HDACi induce expression of the UBE2L6 gene encoding the E2 ubiquitin ligase UBCH8. In conjunction with SIAH1 and SIAH2 UBCH8 promotes the poly-ubiquitinylation and proteasomal degradation of FLT3-ITD. Acetylation events may play a role, e.g., HSP90 acetylation occurs in cells treated with HDACi that can block HDAC6. Acetylation of SIAH1, SIAH2, and UBCH8 has so far never been reported. Caspases are activated by HDACi in leukemic cells and these can cleave STAT5. The apoptotic cleavage of STAT5 impairs STAT5-dependent gene expression and this accelerates cellular demise.
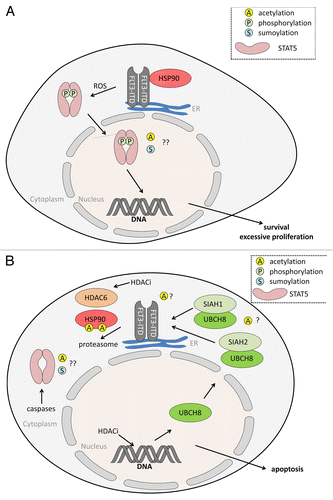
There is an intensive search for factors responsible for the oncogenic nature of FLT3-ITD. A pro-tumorigenic role of reactive oxygen species (ROS) in AML and CML has been reported. In FLT3-ITD-positive AMLs, ROS inactivate the tyrosine phosphatase DEP1 to promote FLT3-ITD phosphorylation,Citation83 and in CML ROS promote chemoresistant tumor reservoirs.Citation84 Activation of STAT5 by FLT3-ITD at the ER is linked to H2O2 production mediated by the flavocytochrome b558 subunit p22phox/CYBA.Citation85
Interestingly, HDACi are able to impair the pro-tumorigenic activation of STAT5 by leukemogenic factors such as FLT3-ITD or BCR-ABL. For example, HDACi can increase the in vitro and in vivo activities of combinations between HDACi (SAHA, MS-275) and the dual BCR-ABL/Aurora kinase inhibitor KW-2449 against BCR-ABL expressing human CML and ALL cells. This even applies to cells sensitive or resistant to imatinib (BCR-ABL T315I and E255K mutants).Citation86 Furthermore, this study included the analysis of mice bearing IM-resistant ALL xenografts (BV173 cells carrying BCR-ABL with an E255K mutation), in which a prolonged survival of double treated mice was observed. At the molecular level, such treatment inactivated BCR-ABL and its downstream signaling to STAT5 and other signaling molecules. Damage to leukemic cells was also evoked by the generation of ROS and DNA damage evidenced by γH2AX staining;Citation86 γH2AX is a stress-induced serine phosphorylated form of histone H2AX.Citation87 Of note, increased lethality of KW-2449 plus HDACi was specific for primary CD34-positive leukemic stem cells from patients with CML and spared normal CD34-positive stem cells from healthy donors.Citation86 While this work clearly demonstrates that attenuating BCR-ABL by various strategies induces leukemia cell apoptosis linked to the inactivation of STAT5 phosphorylation, remaining questions are whether STAT5 became acetylated by HDACi under the conditions chosen. Likewise, BCR-ABL may become acetylated and inactivated by HDACi. Moreover, it remains to be shown, e.g., by RNAi or precise chemical inhibition, whether inactivation of STAT5 is causally linked to pro-apoptotic effects caused by HDACi. Such knowledge can be of high clinical value as STAT5 mediates the maintenance of BCR-ABL-positive leukemic cells,Citation88 and promotes their chemoresistance.Citation89,Citation90
HDACi also reduce the activity of oncoproteins through modulating their interaction with the chaperone HSP90 (). This heat shock protein supports the proper folding and stability of proteins including FLT3-ITD and BCR-ABL.Citation78,Citation91 Since acetylation/deacetylation cycles control the functions of HSP90, its inhibition offers a strategy for pharmacological intervention strategies including HDACi.Citation92,Citation93 Indeed, it was reported that both, pan-HDACi as well as the specific elimination of HDAC6 by RNAi propelled hyperacetylation to inhibit the ATP binding necessary for chaperoning functions of HSP90.Citation94 The possibility to modulate acetylation of cancer-relevant proteins by inactivation of HDAC6 has prompted an intensive search on such agents.Citation95 Using a compound library screen agents with a central naphthoquinone structure were identified as selective inhibitors of HDAC6. In FLT3-ITD-positive MV4-11 cells such compounds diminish mutant FLT3, activation of its downstream target STAT5, and phosphorylation of the mitogen activated protein (MAP) kinases extracellular signal-regulated kinase-1/-2 (ERK1 and ERK2).Citation96
Based on the findings that HDACi deplete oncoproteins via inhibition of HDAC6 and the subsequent acetylation of HSP90, combinations between drugs targeting HDACs and HSP90 were tested against leukemic cells. The acetylated HSP90 showed increased binding to the HSP90 inhibitor 17-allylamino-demothoxy geldanamycin (17-AAG, a drug blocking HSP90s ATP-binding pocket). Moreover, HDAC6 became degraded by the proteasome in 17-AAG treated cells and this in turn enhanced HSP90 hyperacetylation. Cotreatment with 17-AAG and siRNA to HDAC6 or applying the HDACi tubacin had a superior effect on BCR-ABL positive cells than treatment with the single agents. This drug combination also attenuated the viability of primary AML and CML samples.Citation94 Beneficial effects of 17-AAG plus the HDACi SAHA and butyrate were also found in another study.Citation97 This work shows that such combinations are active against BCR-ABL-positive primary and permanent leukemic cells, independent of whether BCR-ABL is sensitive or resistant to its specific inhibitor imatinib mesylate (K562 or LAMA84 cells, respectively). It was found that 17-AAG/HDACi cotreatment evoked a BCL2 and caspase dependent cell death linked to mitochondrial injury, an increased binding of BCR-ABL to HSP70, and inactivation of ERK1 and ERK2. Apoptosis seen in K562 cells exposed to 17-AAG and SAHA was correspondingly associated with reduced DNA binding of STAT5 and attenuated expression of MCL1 and BCL-XL, which are positively regulated target genes of STAT5.Citation97
These data suggest that HDAC6-dependent deacetylation of HSP90 is a main target of HDACi. However, tubacin—a compound initially believed to be specific for HDAC6—turned out to target other HDACs as well.Citation26 Thus, additional HDACs may control the acetylation status of HSP90 and some existing data have to be reassessed. Moreover, it should also be noted that STAT5 itself can be a client protein of HSP90 and this may also contribute to anti-neoplastic effects of HDACi. In human erythroleukemia cells positive for the JAK2 V617F mutant, which is frequently found in myelofibrosis, exposure to 17-AAG depleted STAT5.Citation98
Indeed, HSP90 acetylation has been observed with HDACi not affecting HDAC6. The HDACi MS-275 is active against the AML cell lines MOLM13 and MV4–11 cells carrying FLT3-ITD hetero- and homozygously.Citation99 In the micromolar range MS-275 promotes the ubiquitin dependent proteasomal degradation of FLT3-ITD in leukemic cells (). Accordingly, STAT5 was inactivated upon exposure to MS-275. Although acetylation of STAT5 was not tested in this work, acetylation of HSP90 was detected and appears correlated with the proteolysis of FLT3-ITD.Citation99 Given that MS-275 specifically targets HDAC1, -2, and -3,Citation26 the induction of HSP90 acetylation is in contrast to data showing that HSP90 is a bona fide substrate for HDAC6.Citation94,Citation100,Citation101 Similar results were collected with other HDACi not targeting HDAC6. We characterized valproic acid (VPA) as an HDACi and we also found that this compound preferentially blocks the class I HDACs HDAC1–3.Citation27,Citation50,Citation102,Citation103 Further studies revealed that VPA selectively inhibits the four class I HDACs.Citation26 Although ineffective against HDAC6, VPA/imatinib combinations significantly increased acetylation of HSP90 in imatinib resistant CML cells.Citation104
Further lines of evidence argue that other HDACs control HSP90 and that mechanisms beyond HSP90 inhibition regulate oncoprotein levels. For example, effects seen with a genetic approach targeting HDAC6 achieved much weaker effects than application of HDACi.Citation94,Citation101 A further conundrum is the observation that on the one hand direct HSP90 inhibition with 17-AAG attenuates the stability of HDAC6 while on the other LBH-589 potently causes the acetylation of HSP90 and though does not affect the levels of HDAC6.Citation94
We recently found a mechanism by which HDACi/TKi combinations target aberrant FLT3-ITD and STAT5 hyperactivation. Targeting FLT3-ITD genetically by RNA interference (RNAi) or pharmacologically with three structurally different TKi (AC220/compound-102/PKC412), we noted that FLT3-ITD antagonized pro-apoptotic effects of panbinostat (LBH589).Citation105 Accordingly, application of the HDACi LBH589 with TKi synergistically induced cell death of FLT3-ITD-positive AML cell cultures. At the molecular level this tied in with a mainly proteasomal degradation of FLT3-ITD and the processing of STAT5 by caspase-3.Citation105 Of note, FLT3-ITD-negative leukemic cells as well as normal peripheral blood mononuclear cells (PBMCs) were largely resistant to pro-apoptotic effects of such drug combinations. TKi/HDACi combinations might hence be a promising strategy for the treatment of FLT3-ITD-positive AMLs.Citation105
Further studies have to be conducted to decipher the exact molecular mechanism by which HDACi/TKi combinations kill ITD-positive leukemic cells. In cells exposed to HDACi there is an increased expression of enzymes of the ubiquitin-proteasome-system (UPS).Citation24,Citation78,Citation102,Citation106-Citation108 These increased levels of E2 ubiquitin conjugases and/or E3 ubiquitin ligases may accelerate proteasomal degradation independent of HSP90 (). Additionally, it is possible that such drug regimen target the localization of ITD, the enzymes promoting its proteasomal degradation, unidentified negative regulators of ITD degradation, or other pathways. For example, SOCS2 has recently been shown to accelerate the proteasomal degradation of ligand stimulated FLT3 and of constitutively phosphorylated FLT3-ITD in leukemic cells. Both, phosphorylation of STAT5 and ERK1/2 signaling decreased and the proliferation of FLT3-ITD-positive cells were slowed down upon overexpression of SOCS2.Citation109 As the SOCS1/3 genes are induced in cancer cells plated with HDACi,Citation63,Citation64 an interesting question is whether the SOCS2 gene is also suppressed by HDACs and how this may affect cell fate.
Moreover, it is unknown which HDAC(s) are responsible for the synergistic effects of HDACi/TKi combinatorial treatment. Such knowledge may point to a very specific strategy against AMLs positive for ITD. For example, an increased proteasomal degradation of FLT3-ITD occurs in MV4–11 cells treated with LBH589,Citation106 and also with LBH589/TKi regimen.Citation105 The underlying mechanism is an induction of the E2 ubiquitin conjugase UBCH8 (UBE2L6) and the recognition of FLT3-ITD by the E3 ubiquitin ligases SIAH1 and SIAH2.Citation106 It is tempting to speculate that the reduction of FLT3-ITD phosphorylation which stabilizes the protein and leads to its translocation to the cell surface,Citation105 is antagonized by the accelerated HDACi-mediated proteasomal degradation. Of note, the UBCH8-SIAH1/SIAH2 module enhancing proteasomal degradation by poly-ubiquitinylation targets many other oncologically relevant factors, too.Citation78 Examples are the leukemia fusion proteins AML1-ETO and PML-RARα.Citation78,Citation107 Precisely deciphering such mechanisms might be a critical step toward personalized therapy.
Chronic eosinophilic leukemia and mastocytosis
Additional leukemogenic proteins activate STAT5 and its target genes. Examples are the leukemia fusion proteins FIP1-like-1–platelet-derived growth factor receptor α (FIP1L1-PDGFRA; mRNA processing factor-receptor tyrosine kinase (RTK)-fusion) and E-20 six (leukemia virus, E26; ETS) variant gene 6–platelet-derived growth factor receptor β (ETV6-PDGFRB aka TEL[translocation ets leukemia]-PDGFRB; transcription factor-RTK fusion).Citation110,Citation111 In addition to STAT5, both fusion proteins also induce STAT1 and STAT3 as well as nuclear factor-kappa B (NFκB) in human CD34-positive hematopoietic progenitor and stem cells.Citation110 ETV6-PDGFRB stems from the translocation t(5;12)(q33;p13),Citation110,Citation112 and FIP1L1-PDGFRA arises from an approximately 800 kb internal deletion in chromosome 4q12.Citation113-Citation115 Expression of these fusion proteins is linked to chronic eosinophilic leukemia (CEL) characterized by transformation of eosinophilic precursors into leukemic cells and clonal hypereosinophilia.Citation113,Citation114 FIP1L1-PDGFRA causes up to 60% of CEL cases and correlates with a more aggressive disease phenotype than that seen in patients with hypereosinophilia devoid of FIP1L1-PDGFRA expression.Citation111,Citation113 In permanent and primary CEL cells, FIP1L1-PDGFRA activates STAT5 by a still unknown pathway. This mechanism seems not to involve JAK2,Citation111 which classically phosphorylates STAT5.Citation3,Citation5,Citation116 JAK2 though mediates the activation of STAT3 and NFκB, PI3/AKT kinases, and the expression c-MYC and survivin promoting the survival of CEL cells,Citation111 and JAK1 is another important inducer of STAT5 signaling.Citation117
Similar to FLT3-ITD, FIP1L1-PDGFRA, but not its corresponding mRNA, is reduced in EOL-1 cells incubated with HDACi (apicidin or butyrate). These cells endogenously express FIP1L1-PDGFRA and undergo differentiation upon inhibition of HDACs.Citation118 Also p-STAT5 became attenuated when the cells were exposed to apicidin or butyrate. Nevertheless, the pan-HDACi TSA,Citation26 failed to antagonize FIP1L1-PDGFRA and p-STAT5.Citation118 Although these variable findings for different HDACi seem unexpected, HDAC2 is a further protein, which becomes degraded when cells are exposed to the less potent HDACi VPA and butyrate (effective in the mM range), but not when they are treated with TSA (a pan-HDACi effective in the nanomaolar range).Citation50,Citation102,Citation103 An explanation for these data is that in conjunction with the HDACi-inducible E2 ubiquitin conjugase UBCH8 the E3 ubiquitin ligase RLIM promotes poly-ubiquitinylation of HDAC2 for its proteasomal degradation. However, TSA enhances the proteasomal degradation of RLIM and thereby antagonizes the effect caused by the induction of UBCH8.Citation78,Citation102 Further experiments need to be conducted to clarify how FIP1L1-PDGFRA is degraded in vivo. Interestingly, in murine model cell systems and in primary patient material FIP1L1-PDGFRA and ETV6-PDGFRB are less ubiquitinylated and more stable than their wild-type counterparts. The E3 ubiquitin ligase CBL contributes to this process and accelerating the turnover of ETV6-PDGFRB reduced STAT5 activation and cell proliferation.Citation119 It remains to be investigated if acetylation affects CBL and its interactions with other components of the ubiquitinylation machinery.
The TKi imatinib is not only a highly appreciated drug against CML,Citation77 but equally shows promising activity against CEL in cultured cells and in patients.Citation113,Citation120 Drug combinations consisting of PDGFR inhibitors and HDACi may be promising for the treatment of diseases caused by aberrant PDGF signaling.Citation121,Citation122 Moreover, combining such epigenetic regulators with agents antagonizing Janus and TYK2 kinases,Citation3,Citation9 might block oncogenic signaling involving STAT5.
Expression of FIP1L1/PDGFRA is also linked to systemic mastocytosis (SM),Citation120 which is not to be confused with the mastocytosis driven by mutations in the cell surface receptor c-KIT (CD117).Citation75,Citation115 Mast cell hyperproliferation and accumulation in organs as well as allergic responses are tightly linked to dysregulated JAK-STAT signaling pathways. Especially STAT5 causally mediates the proliferation, survival, and mediator release by mast cells activated through ligation of immunoglobulin E receptors, stem cell factor (SCF), and IL-3.Citation123 The ligand for c-KIT is stem cell factor,Citation124 and mutants of c-KIT and PDGFRA also drive the development of gastrointestinal stromal tumors (GIST).Citation125,Citation126 Remarkably, mutations occur almost exclusively at amino acid 816 within the kinase domain of c-KIT (KIT-Asp816, KITD816V mutant), and this event causes transcriptional activation of STAT5.Citation127 Studies in mice as well as the analysis of human mastocytosis samples thoroughly elaborated that particularly KITD816V-driven STAT5 signaling is causally linked to the growth and survival of neoplastic mast cells.Citation128
Several strategies have been suggested against tumors caused by mutant c-KIT. These are especially the TKi imatinib, sunitinib, dasatinib, and PKC412.Citation125 Due to secondary mutations that confer drug resistance co-targeting of transforming pathways is needed.Citation125 An interruption of HDAC activity has also been suggested as a strategy against several myeloid neoplasms linked to inherent and acquired resistance of c-KIT and excessive STAT5 activation.Citation129
A study analyzing murine and canine malignant mast cell lines with mutant c-KIT status, normal canine mast cells found that the broad-range HDACi AR-42 halts cancer cell proliferation and tumorigenic signaling involving STAT5 (in addition to STAT3 and the AKT kinase). An elegant feature of this work is the use of spontaneously occurring primary canine malignant mast cells. The results collected by Lin and colleagues argue in favor of HDACi-based therapeutic approaches against malignant mast cells.Citation130 AR-42 caused biological effects typically seen with a pan-HDACi, hyperacetylation of histones H3, H4, of tubulin-α, and induction of the p21 gene. Furthermore, this HDACi abrogated transcription of the KIT gene and a loss of HSP90-KIT interactions independent of the HSP90 acetylation status.Citation130 Enhanced proteasomal degradation in HDACi-treated leukemic cells in the absence of HSP90 acetylation was also reported,Citation107 and HSP90 hyperacetylation in HDAC6 knockout mice does not impair survival.Citation131
HDACi may as well be particularly effective against KIT-driven GIST. Mühlenberg and colleagues found that a larger panel of pan- and class-specific HDACi (the hydroxamates TSA, SAHA, LBH-589, and the monocarboxylates VPA and sodium butyrate),Citation26 attenuate oncogenic KIT and its downstream signaling pathways preventing apoptotic cell death. It was again seen that HDACi reduced KIT mRNA expression and that HSP90 functions stabilizing oncogenic KIT were lost. A further notable aspect of this study is the reported strong synergism of imatinib/SAHA and LBH589 combinations.Citation126 While Lin and colleagues found no evidence for HSP90 acetylation in mast cell tumors, Mühlenberg and colleagues detected an HDACi-induced HSP90 acetylation in GIST cells. Therefore, we assume that while HSP90 acetylation somehow marks the effective inhibition of HDACs in certain cell types, this acetylation event is not linked to anti-cancer effects or the loss of oncoprotein stability evoked by HDACi. The unexpected finding that HDACi resistant HL-60 AML cell clones and BCR-ABL-independent imatinib-resistant K562 cell clones show constitutive HSP90 hyperacetylation,Citation132,Citation133 may even suggest that acetylation of this chaperone is an adaption or a side effect antagonizing beneficial drug effects.
Conclusion
Both, pro- and anti-apoptotic functions of STAT5 might be controlled through acetylation. This may involve direct effects on STAT5 as well as the crosstalk of STAT5 with other transcription factors. Whether negative regulators of the JAK-STAT pathway, phosphatases, SOCS proteins, and protein inhibitors of activated STATs can be modulated by TKi and HDACi is another open question. Whether the basal acetylation of various STATs is biologically relevant and whether this posttranslational modification depends on particular conditions equally remains to be clarified. There may also be cell type- and tissue-dependent effects and consequences. Although there are many reports now on how acetylation influences protein structure and function, it is enigmatic and speculative whether acetylation has to be a permanent modification to remain a certain protein activity. Alternatively, acetylation may drive protein complex formation and subsequent modifications, and then becomes biologically dispensable. Alternatively, acetylation of STATs may be a very dynamic process facilitated by the rather large number of HATs and HDACs. Future research should decipher whether STAT acetylation is altered in primary tumors at various stages. Such analyses will reveal putative associations of STAT acetylation of carcinogenesis. The fact that STAT5 acetylation is critical for distinct mechanisms and can be target by HDACi makes this a promising way to target elevated STAT5 signaling in malignant cells.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
Work done in our groups is supported by the Deutsche Krebshilfe (110908 and 110125), the Wilhelm-Sander-Stiftung (2010.078.2), the Bundesministerium für Bildung und Forschung/Center for Sepsis Control and Care, and the Deutsche Forschungsgemeinschaft/Graduiertenkolleg RTG1705.
References
- Wieczorek M, Ginter T, Brand P, Heinzel T, Krämer OH. Acetylation modulates the STAT signaling code. Cytokine Growth Factor Rev 2012; 23:293 - 305; http://dx.doi.org/10.1016/j.cytogfr.2012.06.005; PMID: 22795479
- O’Shea JJ, Holland SM, Staudt LM. JAKs and STATs in immunity, immunodeficiency, and cancer. N Engl J Med 2013; 368:161 - 70; http://dx.doi.org/10.1056/NEJMra1202117; PMID: 23301733
- Haan C, Kreis S, Margue C, Behrmann I. Jaks and cytokine receptors--an intimate relationship. Biochem Pharmacol 2006; 72:1538 - 46; http://dx.doi.org/10.1016/j.bcp.2006.04.013; PMID: 16750817
- Krämer O, Moriggl R. Acetylation and sumoylation control STAT5 activation antagonistically. JAK-STAT 2012; 1:203 - 7; http://dx.doi.org/10.4161/jkst.21232
- Ferbeyre G, Moriggl R. The role of Stat5 transcription factors as tumor suppressors or oncogenes. Biochim Biophys Acta 2011; 1815:104 - 14; PMID: 20969928
- Yao Z, Cui Y, Watford WT, Bream JH, Yamaoka K, Hissong BD, Li D, Durum SK, Jiang Q, Bhandoola A, et al. Stat5a/b are essential for normal lymphoid development and differentiation. Proc Natl Acad Sci U S A 2006; 103:1000 - 5; http://dx.doi.org/10.1073/pnas.0507350103; PMID: 16418296
- Wang Z, Li G, Tse W, Bunting KD. Conditional deletion of STAT5 in adult mouse hematopoietic stem cells causes loss of quiescence and permits efficient nonablative stem cell replacement. Blood 2009; 113:4856 - 65; http://dx.doi.org/10.1182/blood-2008-09-181107; PMID: 19258595
- Schepers H, Wierenga ATJ, Vellenga E, Schuringa JJ. STAT5-mediated self-renewal of normal hematopoietic and leukemic stem cells. JAK-STAT 2011; 1:13 - 22; http://dx.doi.org/10.4161/jkst.19316
- Haan C, Behrmann I, Haan S. Perspectives for the use of structural information and chemical genetics to develop inhibitors of Janus kinases. J Cell Mol Med 2010; 14:504 - 27; PMID: 20132407
- Horvath CM, Wen Z, Darnell JE Jr.. A STAT protein domain that determines DNA sequence recognition suggests a novel DNA-binding domain. Genes Dev 1995; 9:984 - 94; http://dx.doi.org/10.1101/gad.9.8.984; PMID: 7774815
- Soldaini E, John S, Moro S, Bollenbacher J, Schindler U, Leonard WJ. DNA binding site selection of dimeric and tetrameric Stat5 proteins reveals a large repertoire of divergent tetrameric Stat5a binding sites. Mol Cell Biol 2000; 20:389 - 401; http://dx.doi.org/10.1128/MCB.20.1.389-401.2000; PMID: 10594041
- John S, Vinkemeier U, Soldaini E, Darnell JE Jr., Leonard WJ. The significance of tetramerization in promoter recruitment by Stat5. Mol Cell Biol 1999; 19:1910 - 8; PMID: 10022878
- Lin JX, Li P, Liu D, Jin HT, He J, Ata Ur Rasheed M, Rochman Y, Wang L, Cui K, Liu C, et al. Critical Role of STAT5 transcription factor tetramerization for cytokine responses and normal immune function. Immunity 2012; 36:586 - 99; http://dx.doi.org/10.1016/j.immuni.2012.02.017; PMID: 22520852
- Moriggl R, Sexl V, Kenner L, Duntsch C, Stangl K, Gingras S, Hoffmeyer A, Bauer A, Piekorz R, Wang D, et al. Stat5 tetramer formation is associated with leukemogenesis. Cancer Cell 2005; 7:87 - 99; http://dx.doi.org/10.1016/j.ccr.2004.12.010; PMID: 15652752
- Nelson EA, Sharma SV, Settleman J, Frank DA. A chemical biology approach to developing STAT inhibitors: molecular strategies for accelerating clinical translation. Oncotarget 2011; 2:518 - 24; PMID: 21680956
- Schindler C, Plumlee C. Inteferons pen the JAK-STAT pathway. Semin Cell Dev Biol 2008; 19:311 - 8; http://dx.doi.org/10.1016/j.semcdb.2008.08.010; PMID: 18765289
- Tang JZ, Zuo ZH, Kong XJ, Steiner M, Yin Z, Perry JK, Zhu T, Liu DX, Lobie PE. Signal transducer and activator of transcription (STAT)-5A and STAT5B differentially regulate human mammary carcinoma cell behavior. Endocrinology 2010; 151:43 - 55; http://dx.doi.org/10.1210/en.2009-0651; PMID: 19966185
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011; 144:646 - 74; http://dx.doi.org/10.1016/j.cell.2011.02.013; PMID: 21376230
- Dieker J, Muller S. Epigenetic histone code and autoimmunity. Clin Rev Allergy Immunol 2010; 39:78 - 84; http://dx.doi.org/10.1007/s12016-009-8173-7; PMID: 19662539
- Müller S, Krämer OH. Inhibitors of HDACs--effective drugs against cancer?. Curr Cancer Drug Targets 2010; 10:210 - 28; http://dx.doi.org/10.2174/156800910791054149; PMID: 20201785
- Schneider G, Krämer OH, Fritsche P, Schüler S, Schmid RM, Saur D. Targeting histone deacetylases in pancreatic ductal adenocarcinoma. J Cell Mol Med 2010; 14:6A 1255 - 63; http://dx.doi.org/10.1111/j.1582-4934.2009.00974.x; PMID: 19929947
- Grant S, Dai Y. Histone deacetylase inhibitors and rational combination therapies. Adv Cancer Res 2012; 116:199 - 237; PMID: 23088872
- Botrugno OA, Santoro F, Minucci S. Histone deacetylase inhibitors as a new weapon in the arsenal of differentiation therapies of cancer. Cancer Lett 2009; 280:134 - 44; http://dx.doi.org/10.1016/j.canlet.2009.02.027; PMID: 19345000
- Buchwald M, Krämer OH, Heinzel T. HDACi--targets beyond chromatin. Cancer Lett 2009; 280:160 - 7; http://dx.doi.org/10.1016/j.canlet.2009.02.028; PMID: 19342155
- Spange S, Wagner T, Heinzel T, Krämer OH. Acetylation of non-histone proteins modulates cellular signalling at multiple levels. Int J Biochem Cell Biol 2009; 41:185 - 98; http://dx.doi.org/10.1016/j.biocel.2008.08.027; PMID: 18804549
- Bradner JE, Mak R, Tanguturi SK, Mazitschek R, Haggarty SJ, Ross K, Chang CY, Bosco J, West N, Morse E, et al. Chemical genetic strategy identifies histone deacetylase 1 (HDAC1) and HDAC2 as therapeutic targets in sickle cell disease. Proc Natl Acad Sci U S A 2010; 107:12617 - 22; http://dx.doi.org/10.1073/pnas.1006774107; PMID: 20616024
- Krämer OH. HDAC2: a critical factor in health and disease. Trends Pharmacol Sci 2009; 30:647 - 55; http://dx.doi.org/10.1016/j.tips.2009.09.007; PMID: 19892411
- Bressi JC, Jennings AJ, Skene R, Wu Y, Melkus R, De Jong R, O’Connell S, Grimshaw CE, Navre M, Gangloff AR. Exploration of the HDAC2 foot pocket: Synthesis and SAR of substituted N-(2-aminophenyl)benzamides. Bioorg Med Chem Lett 2010; 20:3142 - 5; http://dx.doi.org/10.1016/j.bmcl.2010.03.091; PMID: 20392638
- Ma L, Gao JS, Guan Y, Shi X, Zhang H, Ayrapetov MK, Zhang Z, Xu L, Hyun YM, Kim M, et al. Acetylation modulates prolactin receptor dimerization. Proc Natl Acad Sci U S A 2010; 107:19314 - 9; http://dx.doi.org/10.1073/pnas.1010253107; PMID: 20962278
- Krämer OH, Knauer SK, Greiner G, Jandt E, Reichardt S, Gührs KH, Stauber RH, Böhmer FD, Heinzel T. A phosphorylation-acetylation switch regulates STAT1 signaling. Genes Dev 2009; 23:223 - 35; http://dx.doi.org/10.1101/gad.479209; PMID: 19171783
- Tang X, Gao JS, Guan YJ, McLane KE, Yuan ZL, Ramratnam B, Chin YE. Acetylation-dependent signal transduction for type I interferon receptor. Cell 2007; 131:93 - 105; http://dx.doi.org/10.1016/j.cell.2007.07.034; PMID: 17923090
- Van Nguyen T, Angkasekwinai P, Dou H, Lin FM, Lu LS, Cheng J, Chin YE, Dong C, Yeh ET. SUMO-specific protease 1 is critical for early lymphoid development through regulation of STAT5 activation. Mol Cell 2012; 45:210 - 21; http://dx.doi.org/10.1016/j.molcel.2011.12.026; PMID: 22284677
- Neculai D, Neculai AM, Verrier S, Straub K, Klumpp K, Pfitzner E, Becker S. Structure of the unphosphorylated STAT5a dimer. J Biol Chem 2005; 280:40782 - 7; http://dx.doi.org/10.1074/jbc.M507682200; PMID: 16192273
- Bernadó P, Pérez Y, Blobel J, Fernández-Recio J, Svergun DI, Pons M. Structural characterization of unphosphorylated STAT5a oligomerization equilibrium in solution by small-angle X-ray scattering. Protein Sci 2009; 18:716 - 26; PMID: 19309697
- Braunstein J, Brutsaert S, Olson R, Schindler C. STATs dimerize in the absence of phosphorylation. J Biol Chem 2003; 278:34133 - 40; http://dx.doi.org/10.1074/jbc.M304531200; PMID: 12832402
- Ndubuisi MI, Guo GG, Fried VA, Etlinger JD, Sehgal PB. Cellular physiology of STAT3: Where’s the cytoplasmic monomer?. J Biol Chem 1999; 274:25499 - 509; http://dx.doi.org/10.1074/jbc.274.36.25499; PMID: 10464281
- Kretzschmar AK, Dinger MC, Henze C, Brocke-Heidrich K, Horn F. Analysis of Stat3 (signal transducer and activator of transcription 3) dimerization by fluorescence resonance energy transfer in living cells. Biochem J 2004; 377:289 - 97; http://dx.doi.org/10.1042/BJ20030708; PMID: 12974672
- Stancato LF, David M, Carter-Su C, Larner AC, Pratt WB. Preassociation of STAT1 with STAT2 and STAT3 in separate signalling complexes prior to cytokine stimulation. J Biol Chem 1996; 271:4134 - 7; http://dx.doi.org/10.1074/jbc.271.8.4134; PMID: 8626752
- Narlikar GJ, Fan HY, Kingston RE. Cooperation between complexes that regulate chromatin structure and transcription. Cell 2002; 108:475 - 87; http://dx.doi.org/10.1016/S0092-8674(02)00654-2; PMID: 11909519
- Krämer OH, Heinzel T. Phosphorylation-acetylation switch in the regulation of STAT1 signaling. Mol Cell Endocrinol 2010; 315:40 - 8; http://dx.doi.org/10.1016/j.mce.2009.10.007; PMID: 19879327
- Nakajima H, Brindle PK, Handa M, Ihle JN. Functional interaction of STAT5 and nuclear receptor co-repressor SMRT: implications in negative regulation of STAT5-dependent transcription. EMBO J 2001; 20:6836 - 44; http://dx.doi.org/10.1093/emboj/20.23.6836; PMID: 11726519
- Onishi M, Nosaka T, Misawa K, Mui AL, Gorman D, McMahon M, Miyajima A, Kitamura T. Identification and characterization of a constitutively active STAT5 mutant that promotes cell proliferation. Mol Cell Biol 1998; 18:3871 - 9; PMID: 9632771
- Rascle A, Johnston JA, Amati B. Deacetylase activity is required for recruitment of the basal transcription machinery and transactivation by STAT5. Mol Cell Biol 2003; 23:4162 - 73; http://dx.doi.org/10.1128/MCB.23.12.4162-4173.2003; PMID: 12773560
- Sebastián C, Serra M, Yeramian A, Serrat N, Lloberas J, Celada A. Deacetylase activity is required for STAT5-dependent GM-CSF functional activity in macrophages and differentiation to dendritic cells. J Immunol 2008; 180:5898 - 906; PMID: 18424709
- Sakamoto S, Potla R, Larner AC. Histone deacetylase activity is required to recruit RNA polymerase II to the promoters of selected interferon-stimulated early response genes. J Biol Chem 2004; 279:40362 - 7; http://dx.doi.org/10.1074/jbc.M406400200; PMID: 15194680
- Wood AD, Chen E, Donaldson IJ, Hattangadi S, Burke KA, Dawson MA, Miranda-Saavedra D, Lodish HF, Green AR, Göttgens B. ID1 promotes expansion and survival of primary erythroid cells and is a target of JAK2V617F-STAT5 signaling. Blood 2009; 114:1820 - 30; http://dx.doi.org/10.1182/blood-2009-02-206573; PMID: 19571317
- Xu M, Nie L, Kim SH, Sun XH. STAT5-induced Id-1 transcription involves recruitment of HDAC1 and deacetylation of C/EBPbeta. EMBO J 2003; 22:893 - 904; http://dx.doi.org/10.1093/emboj/cdg094; PMID: 12574125
- Nguyên TL, Abdelbary H, Arguello M, Breitbach C, Leveille S, Diallo JS, Yasmeen A, Bismar TA, Kirn D, Falls T, et al. Chemical targeting of the innate antiviral response by histone deacetylase inhibitors renders refractory cancers sensitive to viral oncolysis. Proc Natl Acad Sci U S A 2008; 105:14981 - 6; http://dx.doi.org/10.1073/pnas.0803988105; PMID: 18815361
- Nguyen TL, Wilson MG, Hiscott J. Oncolytic viruses and histone deacetylase inhibitors--a multi-pronged strategy to target tumor cells. Cytokine Growth Factor Rev 2010; 21:153 - 9; http://dx.doi.org/10.1016/j.cytogfr.2010.03.002; PMID: 20395162
- Göttlicher M, Minucci S, Zhu P, Krämer OH, Schimpf A, Giavara S, Sleeman JP, Lo Coco F, Nervi C, Pelicci PG, et al. Valproic acid defines a novel class of HDAC inhibitors inducing differentiation of transformed cells. EMBO J 2001; 20:6969 - 78; http://dx.doi.org/10.1093/emboj/20.24.6969; PMID: 11742974
- Alvarez-Breckenridge CA, Yu J, Price R, Wei M, Wang Y, Nowicki MO, Ha YP, Bergin S, Hwang C, Fernandez SA, et al. The histone deacetylase inhibitor valproic acid lessens NK cell action against oncolytic virus-infected glioblastoma cells by inhibition of STAT5/T-BET signaling and generation of gamma interferon. J Virol 2012; 86:4566 - 77; http://dx.doi.org/10.1128/JVI.05545-11; PMID: 22318143
- Hoelbl A, Kovacic B, Kerenyi MA, Simma O, Warsch W, Cui Y, Beug H, Hennighausen L, Moriggl R, Sexl V. Clarifying the role of Stat5 in lymphoid development and Abelson-induced transformation. Blood 2006; 107:4898 - 906; http://dx.doi.org/10.1182/blood-2005-09-3596; PMID: 16493008
- Malin S, McManus S, Cobaleda C, Novatchkova M, Delogu A, Bouillet P, Strasser A, Busslinger M. Role of STAT5 in controlling cell survival and immunoglobulin gene recombination during pro-B cell development. Nat Immunol 2010; 11:171 - 9; http://dx.doi.org/10.1038/ni.1827; PMID: 19946273
- Socolovsky M, Fallon AE, Wang S, Brugnara C, Lodish HF. Fetal anemia and apoptosis of red cell progenitors in Stat5a-/-5b-/- mice: a direct role for Stat5 in Bcl-X(L) induction. Cell 1999; 98:181 - 91; http://dx.doi.org/10.1016/S0092-8674(00)81013-2; PMID: 10428030
- Socolovsky M, Nam H, Fleming MD, Haase VH, Brugnara C, Lodish HF. Ineffective erythropoiesis in Stat5a(-/-)5b(-/-) mice due to decreased survival of early erythroblasts. Blood 2001; 98:3261 - 73; http://dx.doi.org/10.1182/blood.V98.12.3261; PMID: 11719363
- Kikuchi K, Lai AY, Hsu CL, Kondo M. IL-7 receptor signaling is necessary for stage transition in adult B cell development through up-regulation of EBF. J Exp Med 2005; 201:1197 - 203; http://dx.doi.org/10.1084/jem.20050158; PMID: 15837809
- Nosaka T, Kawashima T, Misawa K, Ikuta K, Mui AL, Kitamura T. STAT5 as a molecular regulator of proliferation, differentiation and apoptosis in hematopoietic cells. EMBO J 1999; 18:4754 - 65; http://dx.doi.org/10.1093/emboj/18.17.4754; PMID: 10469654
- Marshall AJ, Fleming HE, Wu GE, Paige CJ. Modulation of the IL-7 dose-response threshold during pro-B cell differentiation is dependent on pre-B cell receptor expression. J Immunol 1998; 161:6038 - 45; PMID: 9834086
- Ram PA, Waxman DJ. SOCS/CIS protein inhibition of growth hormone-stimulated STAT5 signaling by multiple mechanisms. J Biol Chem 1999; 274:35553 - 61; http://dx.doi.org/10.1074/jbc.274.50.35553; PMID: 10585430
- Henriques CM, Rino J, Nibbs RJ, Graham GJ, Barata JT. IL-7 induces rapid clathrin-mediated internalization and JAK3-dependent degradation of IL-7Ralpha in T cells. Blood 2010; 115:3269 - 77; http://dx.doi.org/10.1182/blood-2009-10-246876; PMID: 20190194
- Calabrese V, Mallette FA, Deschênes-Simard X, Ramanathan S, Gagnon J, Moores A, Ilangumaran S, Ferbeyre G. SOCS1 links cytokine signaling to p53 and senescence. Mol Cell 2009; 36:754 - 67; http://dx.doi.org/10.1016/j.molcel.2009.09.044; PMID: 20005840
- Mallette FA, Calabrese V, Ilangumaran S, Ferbeyre G. SOCS1, a novel interaction partner of p53 controlling oncogene-induced senescence. Aging (Albany NY) 2010; 2:445 - 52; PMID: 20622265
- Gao SM, Chen CQ, Wang LY, Hong LL, Wu JB, Dong PH, Yu FJ. Histone deacetylases inhibitor sodium butyrate inhibits JAK2/STAT signaling through upregulation of SOCS1 and SOCS3 mediated by HDAC8 inhibition in myeloproliferative neoplasms. Exp Hematol 2013; 41:261 - , e4; http://dx.doi.org/10.1016/j.exphem.2012.10.012; PMID: 23111066
- Xiong H, Du W, Zhang YJ, Hong J, Su WY, Tang JT, Wang YC, Lu R, Fang JY. Trichostatin A, a histone deacetylase inhibitor, suppresses JAK2/STAT3 signaling via inducing the promoter-associated histone acetylation of SOCS1 and SOCS3 in human colorectal cancer cells. Mol Carcinog 2012; 51:174 - 84; http://dx.doi.org/10.1002/mc.20777; PMID: 21520296
- Klampfer L. Signal transducers and activators of transcription (STATs): Novel targets of chemopreventive and chemotherapeutic drugs. Curr Cancer Drug Targets 2006; 6:107 - 21; http://dx.doi.org/10.2174/156800906776056491; PMID: 16529541
- Geiss-Friedlander R, Melchior F. Concepts in sumoylation: a decade on. Nat Rev Mol Cell Biol 2007; 8:947 - 56; http://dx.doi.org/10.1038/nrm2293; PMID: 18000527
- Cheng J, Kang X, Zhang S, Yeh ET. SUMO-specific protease 1 is essential for stabilization of HIF1alpha during hypoxia. Cell 2007; 131:584 - 95; http://dx.doi.org/10.1016/j.cell.2007.08.045; PMID: 17981124
- Kosan C, Saba I, Godmann M, Herold S, Herkert B, Eilers M, Möröy T. Transcription factor miz-1 is required to regulate interleukin-7 receptor signaling at early commitment stages of B cell differentiation. Immunity 2010; 33:917 - 28; http://dx.doi.org/10.1016/j.immuni.2010.11.028; PMID: 21167753
- Saba I, Kosan C, Vassen L, Möröy T. IL-7R-dependent survival and differentiation of early T-lineage progenitors is regulated by the BTB/POZ domain transcription factor Miz-1. Blood 2011; 117:3370 - 81; http://dx.doi.org/10.1182/blood-2010-09-310680; PMID: 21258009
- Kondo M, Akashi K, Domen J, Sugamura K, Weissman IL. Bcl-2 rescues T lymphopoiesis, but not B or NK cell development, in common gamma chain-deficient mice. Immunity 1997; 7:155 - 62; http://dx.doi.org/10.1016/S1074-7613(00)80518-X; PMID: 9252128
- Beier UH, Wang L, Hancock WW. Combination of isoform-selective histone/protein deacetylase inhibitors improves Foxp3+ T-regulatory cell function. Cell Cycle 2012; 11:3351 - 2; http://dx.doi.org/10.4161/cc.21876; PMID: 22918251
- Friedbichler K, Kerenyi MA, Kovacic B, Li G, Hoelbl A, Yahiaoui S, Sexl V, Müllner EW, Fajmann S, Cerny-Reiterer S, et al. Stat5a serine 725 and 779 phosphorylation is a prerequisite for hematopoietic transformation. Blood 2010; 116:1548 - 58; http://dx.doi.org/10.1182/blood-2009-12-258913; PMID: 20508164
- Nelson EA, Walker SR, Xiang M, Weisberg E, Bar-Natan M, Barrett R, Liu S, Kharbanda S, Christie AL, Nicolais M, et al. The STAT5 Inhibitor Pimozide Displays Efficacy in Models of Acute Myelogenous Leukemia Driven by FLT3 Mutations. Genes Cancer 2012; 3:503 - 11; http://dx.doi.org/10.1177/1947601912466555; PMID: 23264850
- Page BD, Khoury H, Laister RC, Fletcher S, Vellozo M, Manzoli A, Yue P, Turkson J, Minden MD, Gunning PT. Small molecule STAT5-SH2 domain inhibitors exhibit potent antileukemia activity. J Med Chem 2012; 55:1047 - 55; http://dx.doi.org/10.1021/jm200720n; PMID: 22148584
- Masson K, Rönnstrand L. Oncogenic signaling from the hematopoietic growth factor receptors c-Kit and Flt3. Cell Signal 2009; 21:1717 - 26; http://dx.doi.org/10.1016/j.cellsig.2009.06.002; PMID: 19540337
- Hochhaus A, La Rosée P, Müller MC, Ernst T, Cross NC. Impact of BCR-ABL mutations on patients with chronic myeloid leukemia. Cell Cycle 2011; 10:250 - 60; http://dx.doi.org/10.4161/cc.10.2.14537; PMID: 21220945
- Cross NC, White HE, Müller MC, Saglio G, Hochhaus A. Standardized definitions of molecular response in chronic myeloid leukemia. Leukemia 2012; 26:2172 - 5; http://dx.doi.org/10.1038/leu.2012.104; PMID: 22504141
- Krämer OH, Stauber RH, Bug G, Hartkamp J, Knauer SK. SIAH proteins: critical roles in leukemogenesis. Leukemia 2013; 27:792 - 802; http://dx.doi.org/10.1038/leu.2012.284; PMID: 23038274
- Scott E, Hexner E, Perl A, Carroll M. Targeted signal transduction therapies in myeloid malignancies. Curr Oncol Rep 2010; 12:358 - 65; http://dx.doi.org/10.1007/s11912-010-0126-z; PMID: 20809224
- Schmidt-Arras D, Böhmer SA, Koch S, Müller JP, Blei L, Cornils H, Bauer R, Korasikha S, Thiede C, Böhmer FD. Anchoring of FLT3 in the endoplasmic reticulum alters signaling quality. Blood 2009; 113:3568 - 76; http://dx.doi.org/10.1182/blood-2007-10-121426; PMID: 19204327
- Choudhary C, Olsen JV, Brandts C, Cox J, Reddy PN, Böhmer FD, Gerke V, Schmidt-Arras DE, Berdel WE, Müller-Tidow C, et al. Mislocalized activation of oncogenic RTKs switches downstream signaling outcomes. Mol Cell 2009; 36:326 - 39; http://dx.doi.org/10.1016/j.molcel.2009.09.019; PMID: 19854140
- Chatain N, Ziegler P, Fahrenkamp D, Jost E, Moriggl R, Schmitz-Van de Leur H, Müller-Newen G. Src family kinases mediate cytoplasmic retention of activated STAT5 in BCR-ABL-positive cells. Oncogene 2013; 32:3587 - 97; http://dx.doi.org/10.1038/onc.2012.369; PMID: 22926520
- Godfrey R, Arora D, Bauer R, Stopp S, Müller JP, Heinrich T, Böhmer SA, Dagnell M, Schnetzke U, Scholl S, et al. Cell transformation by FLT3 ITD in acute myeloid leukemia involves oxidative inactivation of the tumor suppressor protein-tyrosine phosphatase DEP-1/ PTPRJ. Blood 2012; 119:4499 - 511; http://dx.doi.org/10.1182/blood-2011-02-336446; PMID: 22438257
- Nieborowska-Skorska M, Kopinski PK, Ray R, Hoser G, Ngaba D, Flis S, Cramer K, Reddy MM, Koptyra M, Penserga T, et al. Rac2-MRC-cIII-generated ROS cause genomic instability in chronic myeloid leukemia stem cells and primitive progenitors. Blood 2012; 119:4253 - 63; http://dx.doi.org/10.1182/blood-2011-10-385658; PMID: 22411871
- Woolley JF, Naughton R, Stanicka J, Gough DR, Bhatt L, Dickinson BC, Chang CJ, Cotter TG. H2O2 production downstream of FLT3 is mediated by p22phox in the endoplasmic reticulum and is required for STAT5 signalling. PLoS One 2012; 7:e34050; http://dx.doi.org/10.1371/journal.pone.0034050; PMID: 22807997
- Nguyen T, Dai Y, Attkisson E, Kramer L, Jordan N, Nguyen N, Kolluri N, Muschen M, Grant S. HDAC inhibitors potentiate the activity of the BCR/ABL kinase inhibitor KW-2449 in imatinib-sensitive or -resistant BCR/ABL+ leukemia cells in vitro and in vivo. Clin Cancer Res 2011; 17:3219 - 32; http://dx.doi.org/10.1158/1078-0432.CCR-11-0234; PMID: 21474579
- Roos WP, Kaina B. DNA damage-induced cell death: from specific DNA lesions to the DNA damage response and apoptosis. Cancer Lett 2013; 332:237 - 48; http://dx.doi.org/10.1016/j.canlet.2012.01.007; PMID: 22261329
- Hoelbl A, Schuster C, Kovacic B, Zhu B, Wickre M, Hoelzl MA, Fajmann S, Grebien F, Warsch W, Stengl G, et al. Stat5 is indispensable for the maintenance of bcr/abl-positive leukaemia. EMBO Mol Med 2010; 2:98 - 110; http://dx.doi.org/10.1002/emmm.201000062; PMID: 20201032
- Warsch W, Kollmann K, Eckelhart E, Fajmann S, Cerny-Reiterer S, Hölbl A, Gleixner KV, Dworzak M, Mayerhofer M, Hoermann G, et al. High STAT5 levels mediate imatinib resistance and indicate disease progression in chronic myeloid leukemia. Blood 2011; 117:3409 - 20; http://dx.doi.org/10.1182/blood-2009-10-248211; PMID: 21220747
- Li G, Miskimen KL, Wang Z, Xie XY, Tse W, Gouilleux F, Moriggl R, Bunting KD. Effective targeting of STAT5-mediated survival in myeloproliferative neoplasms using ABT-737 combined with rapamycin. Leukemia 2010; 24:1397 - 405; http://dx.doi.org/10.1038/leu.2010.131; PMID: 20535152
- Neckers L, Workman P. Hsp90 molecular chaperone inhibitors: are we there yet?. Clin Cancer Res 2012; 18:64 - 76; http://dx.doi.org/10.1158/1078-0432.CCR-11-1000; PMID: 22215907
- Mollapour M, Neckers L. Post-translational modifications of Hsp90 and their contributions to chaperone regulation. Biochim Biophys Acta 2012; 1823:648 - 55; http://dx.doi.org/10.1016/j.bbamcr.2011.07.018; PMID: 21856339
- Jackson SE. Hsp90: structure and function. Top Curr Chem 2013; 328:155 - 240; http://dx.doi.org/10.1007/128_2012_356; PMID: 22955504
- Rao R, Fiskus W, Yang Y, Lee P, Joshi R, Fernandez P, Mandawat A, Atadja P, Bradner JE, Bhalla K. HDAC6 inhibition enhances 17-AAG--mediated abrogation of hsp90 chaperone function in human leukemia cells. Blood 2008; 112:1886 - 93; http://dx.doi.org/10.1182/blood-2008-03-143644; PMID: 18591380
- Dallavalle S, Pisano C, Zunino F. Development and therapeutic impact of HDAC6-selective inhibitors. Biochem Pharmacol 2012; 84:756 - 65; http://dx.doi.org/10.1016/j.bcp.2012.06.014; PMID: 22728920
- Inks ES, Josey BJ, Jesinkey SR, Chou CJ. A novel class of small molecule inhibitors of HDAC6. ACS Chem Biol 2012; 7:331 - 9; http://dx.doi.org/10.1021/cb200134p; PMID: 22047054
- Rahmani M, Reese E, Dai Y, Bauer C, Kramer LB, Huang M, Jove R, Dent P, Grant S. Cotreatment with suberanoylanilide hydroxamic acid and 17-allylamino 17-demethoxygeldanamycin synergistically induces apoptosis in Bcr-Abl+ Cells sensitive and resistant to STI571 (imatinib mesylate) in association with down-regulation of Bcr-Abl, abrogation of signal transducer and activator of transcription 5 activity, and Bax conformational change. Mol Pharmacol 2005; 67:1166 - 76; http://dx.doi.org/10.1124/mol.104.007831; PMID: 15625278
- Bareng J, Jilani I, Gorre M, Kantarjian H, Giles F, Hannah A, Albitar M. A potential role for HSP90 inhibitors in the treatment of JAK2 mutant-positive diseases as demonstrated using quantitative flow cytometry. Leuk Lymphoma 2007; 48:2189 - 95; http://dx.doi.org/10.1080/10428190701607576; PMID: 17926180
- Nishioka C, Ikezoe T, Yang J, Takeuchi S, Koeffler HP, Yokoyama A. MS-275, a novel histone deacetylase inhibitor with selectivity against HDAC1, induces degradation of FLT3 via inhibition of chaperone function of heat shock protein 90 in AML cells. Leuk Res 2008; 32:1382 - 92; http://dx.doi.org/10.1016/j.leukres.2008.02.018; PMID: 18394702
- Boyault C, Sadoul K, Pabion M, Khochbin S. HDAC6, at the crossroads between cytoskeleton and cell signaling by acetylation and ubiquitination. Oncogene 2007; 26:5468 - 76; http://dx.doi.org/10.1038/sj.onc.1210614; PMID: 17694087
- Bali P, Pranpat M, Bradner J, Balasis M, Fiskus W, Guo F, Rocha K, Kumaraswamy S, Boyapalle S, Atadja P, et al. Inhibition of histone deacetylase 6 acetylates and disrupts the chaperone function of heat shock protein 90: a novel basis for antileukemia activity of histone deacetylase inhibitors. J Biol Chem 2005; 280:26729 - 34; http://dx.doi.org/10.1074/jbc.C500186200; PMID: 15937340
- Krämer OH, Zhu P, Ostendorff HP, Golebiewski M, Tiefenbach J, Peters MA, Brill B, Groner B, Bach I, Heinzel T, et al. The histone deacetylase inhibitor valproic acid selectively induces proteasomal degradation of HDAC2. EMBO J 2003; 22:3411 - 20; http://dx.doi.org/10.1093/emboj/cdg315; PMID: 12840003
- Brandl A, Wagner T, Uhlig KM, Knauer SK, Stauber RH, Melchior F, Schneider G, Heinzel T, Krämer OH. Dynamically regulated sumoylation of HDAC2 controls p53 deacetylation and restricts apoptosis following genotoxic stress. J Mol Cell Biol 2012; 4:284 - 93; http://dx.doi.org/10.1093/jmcb/mjs013; PMID: 22493095
- Buchi F, Pastorelli R, Ferrari G, Spinelli E, Gozzini A, Sassolini F, Bosi A, Tombaccini D, Santini V. Acetylome and phosphoproteome modifications in imatinib resistant chronic myeloid leukaemia cells treated with valproic acid. Leuk Res 2011; 35:921 - 31; http://dx.doi.org/10.1016/j.leukres.2011.01.033; PMID: 21382639
- Pietschmann K, Bolck HA, Buchwald M, Spielberg S, Polzer H, Spiekermann K, Bug G, Heinzel T, Böhmer FD, Krämer OH. Breakdown of the FLT3-ITD/STAT5 axis and synergistic apoptosis induction by the histone deacetylase inhibitor panobinostat and FLT3-specific inhibitors. Mol Cancer Ther 2012; 11:2373 - 83; http://dx.doi.org/10.1158/1535-7163.MCT-12-0129; PMID: 22942377
- Buchwald M, Pietschmann K, Müller JP, Böhmer FD, Heinzel T, Krämer OH. Ubiquitin conjugase UBCH8 targets active FMS-like tyrosine kinase 3 for proteasomal degradation. Leukemia 2010; 24:1412 - 21; http://dx.doi.org/10.1038/leu.2010.114; PMID: 20508617
- Krämer OH, Müller S, Buchwald M, Reichardt S, Heinzel T. Mechanism for ubiquitylation of the leukemia fusion proteins AML1-ETO and PML-RARalpha. FASEB J 2008; 22:1369 - 79; http://dx.doi.org/10.1096/fj.06-8050com; PMID: 18073335
- Pietschmann K, Buchwald M, Müller S, Knauer SK, Kögl M, Heinzel T, Krämer OH. Differential regulation of PML-RARα stability by the ubiquitin ligases SIAH1/SIAH2 and TRIAD1. Int J Biochem Cell Biol 2012; 44:132 - 8; http://dx.doi.org/10.1016/j.biocel.2011.10.008; PMID: 22037423
- Kazi JU, Rönnstrand L. Suppressor of cytokine signaling 2 (SOCS2) associates with FLT3 and negatively regulates downstream signaling. Mol Oncol 2013; 7:693 - 703; http://dx.doi.org/10.1016/j.molonc.2013.02.020; PMID: 23548639
- Montano-Almendras CP, Essaghir A, Schoemans H, Varis I, Noël LA, Velghe AI, Latinne D, Knoops L, Demoulin JB. ETV6-PDGFRB and FIP1L1-PDGFRA stimulate human hematopoietic progenitor cell proliferation and differentiation into eosinophils: the role of nuclear factor-κB. Haematologica 2012; 97:1064 - 72; http://dx.doi.org/10.3324/haematol.2011.047530; PMID: 22271894
- Li B, Zhang G, Li C, He D, Li X, Zhang C, Tang F, Deng X, Lu J, Tang Y, et al. Identification of JAK2 as a mediator of FIP1L1-PDGFRA-induced eosinophil growth and function in CEL. PLoS One 2012; 7:e34912; http://dx.doi.org/10.1371/journal.pone.0034912; PMID: 22523564
- Lierman E, Cools J. TV6 and PDGFRB: a license to fuse. Haematologica 2007; 92:145 - 7; http://dx.doi.org/10.3324/haematol.11187; PMID: 17296561
- Cools J. FIP1L1-PDGFR alpha, a therapeutic target for the treatment of chronic eosinophilic leukemia. Verh K Acad Geneeskd Belg 2005; 67:169 - 76; PMID: 16089297
- Montgomery ND, Dunphy CH, Mooberry M, Laramore A, Foster MC, Park SI, Fedoriw YD. Diagnostic complexities of eosinophilia. Arch Pathol Lab Med 2013; 137:259 - 69; http://dx.doi.org/10.5858/arpa.2011-0597-RA; PMID: 23368869
- Metcalfe DD. Mast cells and mastocytosis. Blood 2008; 112:946 - 56; http://dx.doi.org/10.1182/blood-2007-11-078097; PMID: 18684881
- Furth PA, Nakles RE, Millman S, Diaz-Cruz ES, Cabrera MC. Signal transducer and activator of transcription 5 as a key signaling pathway in normal mammary gland developmental biology and breast cancer. Breast Cancer Res 2011; 13:220; http://dx.doi.org/10.1186/bcr2921; PMID: 22018398
- Haan C, Rolvering C, Raulf F, Kapp M, Drückes P, Thoma G, Behrmann I, Zerwes HG. Jak1 has a dominant role over Jak3 in signal transduction through γc-containing cytokine receptors. Chem Biol 2011; 18:314 - 23; http://dx.doi.org/10.1016/j.chembiol.2011.01.012; PMID: 21439476
- Kaneko M, Ishihara K, Takahashi A, Hong J, Hirasawa N, Zee O, Ohuchi K. Mechanism for the differentiation of EoL-1 cells into eosinophils by histone deacetylase inhibitors. Int Arch Allergy Immunol 2007; 143:Suppl 1 28 - 32; http://dx.doi.org/10.1159/000101401; PMID: 17541273
- Toffalini F, Kallin A, Vandenberghe P, Pierre P, Michaux L, Cools J, Demoulin JB. The fusion proteins TEL-PDGFRbeta and FIP1L1-PDGFRalpha escape ubiquitination and degradation. Haematologica 2009; 94:1085 - 93; http://dx.doi.org/10.3324/haematol.2008.001149; PMID: 19644140
- Yamada Y, Cancelas JA. FIP1L1/PDGFR alpha-associated systemic mastocytosis. Int Arch Allergy Immunol 2010; 152:Suppl 1 101 - 5; http://dx.doi.org/10.1159/000312134; PMID: 20523072
- Mahboobi S, Dove S, Sellmer A, Winkler M, Eichhorn E, Pongratz H, Ciossek T, Baer T, Maier T, Beckers T. Design of chimeric histone deacetylase- and tyrosine kinase-inhibitors: a series of imatinib hybrides as potent inhibitors of wild-type and mutant BCR-ABL, PDGF-Rbeta, and histone deacetylases. J Med Chem 2009; 52:2265 - 79; http://dx.doi.org/10.1021/jm800988r; PMID: 19301902
- Uecker A, Sicker M, Beckers T, Mahboobi S, Hägerstrand D, Ostman A, Böhmer FD. Chimeric tyrosine kinase-HDAC inhibitors as antiproliferative agents. Anticancer Drugs 2010; 21:759 - 65; http://dx.doi.org/10.1097/CAD.0b013e32833ccf25; PMID: 20613486
- Morales JK, Falanga YT, Depcrynski A, Fernando J, Ryan JJ. Mast cell homeostasis and the JAK-STAT pathway. Genes Immun 2010; 11:599 - 608; http://dx.doi.org/10.1038/gene.2010.35; PMID: 20535135
- Drube S, Schmitz F, Göpfert C, Weber F, Kamradt T. C-Kit controls IL-1β-induced effector functions in HMC-cells. Eur J Pharmacol 2012; 675:57 - 62; http://dx.doi.org/10.1016/j.ejphar.2011.11.035; PMID: 22173128
- Ashman LK, Griffith R. Therapeutic targeting of c-KIT in cancer. Expert Opin Investig Drugs 2013; 22:103 - 15; http://dx.doi.org/10.1517/13543784.2013.740010; PMID: 23127174
- Mühlenberg T, Zhang Y, Wagner AJ, Grabellus F, Bradner J, Taeger G, Lang H, Taguchi T, Schuler M, Fletcher JA, et al. Inhibitors of deacetylases suppress oncogenic KIT signaling, acetylate HSP90, and induce apoptosis in gastrointestinal stromal tumors. Cancer Res 2009; 69:6941 - 50; http://dx.doi.org/10.1158/0008-5472.CAN-08-4004; PMID: 19706776
- Chaix A, Lopez S, Voisset E, Gros L, Dubreuil P, De Sepulveda P. Mechanisms of STAT protein activation by oncogenic KIT mutants in neoplastic mast cells. J Biol Chem 2011; 286:5956 - 66; http://dx.doi.org/10.1074/jbc.M110.182642; PMID: 21135090
- Harir N, Boudot C, Friedbichler K, Sonneck K, Kondo R, Martin-Lannerée S, Kenner L, Kerenyi M, Yahiaoui S, Gouilleux-Gruart V, et al. Oncogenic Kit controls neoplastic mast cell growth through a Stat5/PI3-kinase signaling cascade. Blood 2008; 112:2463 - 73; http://dx.doi.org/10.1182/blood-2007-09-115477; PMID: 18579792
- Mithraprabhu S, Grigoriadis G, Khong T, Spencer A. Deactylase inhibition in myeloproliferative neoplasms. Invest New Drugs 2010; 28:Suppl 1 S50 - 7; http://dx.doi.org/10.1007/s10637-010-9590-4; PMID: 21127942
- Lin TY, Fenger J, Murahari S, Bear MD, Kulp SK, Wang D, Chen CS, Kisseberth WC, London CA. AR-42, a novel HDAC inhibitor, exhibits biologic activity against malignant mast cell lines via down-regulation of constitutively activated Kit. Blood 2010; 115:4217 - 25; http://dx.doi.org/10.1182/blood-2009-07-231985; PMID: 20233974
- Zhang Y, Kwon S, Yamaguchi T, Cubizolles F, Rousseaux S, Kneissel M, Cao C, Li N, Cheng HL, Chua K, et al. Mice lacking histone deacetylase 6 have hyperacetylated tubulin but are viable and develop normally. Mol Cell Biol 2008; 28:1688 - 701; http://dx.doi.org/10.1128/MCB.01154-06; PMID: 18180281
- Fiskus W, Rao R, Fernandez P, Herger B, Yang Y, Chen J, Kolhe R, Mandawat A, Wang Y, Joshi R, et al. Molecular and biologic characterization and drug sensitivity of pan-histone deacetylase inhibitor-resistant acute myeloid leukemia cells. Blood 2008; 112:2896 - 905; http://dx.doi.org/10.1182/blood-2007-10-116319; PMID: 18660379
- Lee SM, Bae JH, Kim MJ, Lee HS, Lee MK, Chung BS, Kim DW, Kang CD, Kim SH. Bcr-Abl-independent imatinib-resistant K562 cells show aberrant protein acetylation and increased sensitivity to histone deacetylase inhibitors. J Pharmacol Exp Ther 2007; 322:1084 - 92; http://dx.doi.org/10.1124/jpet.107.124461; PMID: 17569822