Abstract
JAK-STAT signaling occurs in virtually every tissue of the body, and so does glucose metabolism. In this review, we summarize the regulation of glucose metabolism in the myocardium and ponder whether JAK-STAT signaling participates in this regulation. Despite a paucity of data directly pertaining to cardiac myocytes, we conclude that JAK-STAT signaling may contribute to the development of insulin resistance in the myocardium in response to various hormones and cytokines.
Myocardial Glucose Metabolism
Overview
Life critically depends on the beating of the heart, an energy-consuming process fueled by hydrolysis of adenosine triphosphate (ATP) to adenosine diphosphate (ADP). In fact, the heart is the organ with the highest specific oxygen consumption, reflecting its intense, mostly aerobic, metabolic activity. The most important substrates for energy production in the normal myocardium are fatty acids, glucose and lactate, in decreasing order of importance. Together, anaerobic and aerobic metabolism of these substrates account for almost all of energy production in the normal adult heart, the respective contribution of each depending on the metabolic and hormonal status.Citation1 Catabolic breakdown of glucose occurs in two stages: glycolysis, an anaerobic, cytoplasmic stage with low ATP yield (2 ATP/glucose), followed by aerobic oxidation of glycolysis-derived pyruvate in the mitochondria. Pyruvate is first converted to acetyl-CoA by the action of pyruvate dehydrogenase complex (PDC), the rate-limiting enzyme for glucose oxidation. Acetyl-CoA then enters the Krebs cycle, wherein it is oxidized to CO2 with production of reducing equivalents, thereafter used in the electron transport chain to produce ATP with a much higher yield (34 ATP/glucose).Citation2
Importance of glucose metabolism for the myocardium
Among the myocardial substrates, glucose accounts for less than 25% of the energy production under normal conditions.Citation3 It should not be surmised from this rather low figure that glucose is entirely dispensable for the heart. Indeed, although isolated perfused hearts can aerobically run for hours on fatty acids only, glucose becomes extremely important during episodes of ischemia and reperfusion.Citation4 There are mostly two reasons for this requirement for glucose during metabolic stress: (1) energy can be obtained from glucose through glycolysis even in situations of hypoxia or ischemia and (2) ATP obtained from glycolysis, although scarce, is important for the maintenance of ionic homeostasis. Indeed ATP production and use is highly compartmentalized in the myocardium, and glycolytic ATP is preferentially used to fuel the sarcolemmal and sarcoplasmic reticulum ion pumps.Citation5,Citation6
Regulation of glucose metabolism
Glucose metabolism in the myocardium is tightly regulated; there are three major steps regulating the rates at which the two stages of glucose breakdown proceed (): (1) Glucose transport from the extracellular space; (2) the phosphofructokinase reaction, which is the first committing step of glycolysis; and (3) the intramitochondrial conversion of pyruvate to acetyl-CoA, which is the first step of pyruvate oxidation.
Figure 1. Principal points of regulation of glucose metabolism in cardiac myocytes. Glucose enters cardiac myocytes by facilitated diffusion through GLUT (mostly GLUT4) transporters and to a minor extent by cotransport with sodium through SGLT1. Glycolysis yields pyruvate, which is converted to acetyl-CoA to undergo mitochondrial oxidation in the Krebs cycle. Principal points of regulation are transmembrane transport, regulated by translocation of GLUT4, the PFK-1 reaction, which is stimulated by F2,6BP, and activity of the pyruvate dehydrogenase complex, regulated by phosphorylation by PDH kinases or dephosphorylation by PDH phosphatases. See text for details. Abbreviations: AMPK, AMP-activated protein kinase; F1,6BP, fructose-1,6-bisphosphate; F2,6BP, fructose-2,6-bisphosphate; F6P, fructose-6-phosphate; GLUT1 or 4, facilitative glucose transporters; Ins, insulin; IRα/β, insulin receptor, subunit α, respectively β; PDC, pyruvate dehydrogenase complex; PDK1-4, pyruvate dehydrogenase kinase 1 to 4; PDPC1-2, pyruvate dehydrogenase phosphatase 1 or 2; PFK-1, 6-phosphofructo-1-kinase; PFK-2, 6-phosphofructo-2-kinase; SGLT1, sodium-glucose cotransporter 1; WISK, wortmannin-sensitive and insulin-stimulated protein kinase.
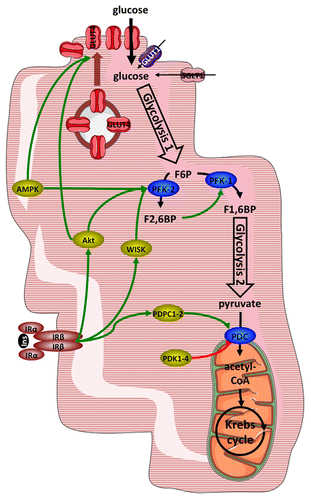
1) Glucose transport occurs mostly by facilitated diffusion through selective transport proteins of the GLUT family. In cardiac myocytes, mostly two isoforms of glucose transporter, GLUT1 and GLUT4, are involved. GLUT1, which predominates during fetal and early postnatal periodCitation7 is located mainly in the sarcolemma under basal conditions.Citation7,Citation8 GLUT4 on the other hand is the main isoform present in fully differentiated cardiac myocyte. GLUT4 is mainly located in intracellular membrane compartments and is translocated to the cell surface in response to various stimuli. As a result, the major determinant of glucose uptake into cardiac myocytes at physiological glucose concentrations is the number of GLUT4 transporters present at the cell surface. However, in addition to facilitated diffusion, cotransport of sodium and glucose by the cotransporter SGLT1 has been recently reported in mouse heart and found to be stimulated in response to insulin and leptin.Citation9
The most important stimuli triggering translocation of GLUT4 in cardiac myocytes are insulin, ischemia and workload.Citation10,Citation11 Signaling in response to insulin and leading to stimulation of glucose transport in short involves recruitment and activating tyrosine phosphorylation of insulin receptor substrates proteins (IRS-1, -2, and -3), activation of phosphoinositide-3-kinases (PI3K), and activating phosphorylation of Akt.Citation12 Ischemia on the other hand increases the AMP to ATP ratio within the cardiac myocytes, leading to activation of the AMP-activated kinase (AMPK) by both allostery and phosphorylation on threonine 172 of the catalytic AMPKα subunit.Citation13 Both activated Akt and AMPK phosphorylate and inactivate the Rab-GTPase AS160; this relieves an inhibition of GLUT4 translocation.Citation14 For a more detailed discussion of the mechanisms controlling GLUT4 translocation in the myocardium, the interested reader is referred to a recent review.Citation15
2) The phosphofructokinase reaction converts fructose-6-phosphate to fructose-1,6-bisphosphate. This is the first committed step in glycolysis, and as such, it determines the rate at which glycolysis proceeds downstream. The activity of 6-phosphofructo-1-kinase (PFK-1) is allosterically regulated in negative feedback loops by ATP and citrate. Importantly, in cardiac myocytes it is strongly activated by a glucose metabolite that is not part of the glycolysis pathway sensu stricto, fructose-2,6-bisphosphate. This metabolite is produced from fructose-6-phosphate by the enzyme 6-phosphofructo-2-kinase 2 (PFK-2; the heart isoenzyme is different in both gene and function from the liver and muscle isoenzymes).Citation16 Similar to glucose transport, activity of the heart PFK-2 is stimulated in response to insulin, ischemia and workload. Insulin signaling activates a wortmannin-sensitive protein kinase (WISK), whose molecular identity remains unsure, but which is distinct from Akt; ischemia, as described above, activates AMPK and workload activates Akt. All three kinases phosphorylate and activate PFK-2, thereby accelerating glycolysis.
3) Activity of the pyruvate dehydrogenase complex (PDC) commits glycolysis-derived pyruvate to intramitochondrial oxidation in the Krebs cycle. The PDC is a multienzyme complex carrying out three successive reactions leading to the biosynthesis of acetyl-CoA. The first and rate-limiting reaction is decarboxylation of pyruvate by the pyruvate dehydrogenase enzyme (PDH-E1). The PDH-E1 can be inhibited by phosphorylation on three specific serine residues on its α subunit by PDH kinase, of which there exists four isoforms (PDK1–4).Citation17 Conversely, two PDH phosphatases (PDPC1 and PDPC2) dephosphorylate and activate PDH-E1; this phosphatase activity is stimulated by insulin.Citation18
JAK-STAT Signaling in the Myocardium
Overview
Many signaling pathways have been found in myocardium, including the Janus kinase (JAK)-signal transducer and activator of transcription (STAT) pathway. The JAK-STAT signaling pathway transduces signals from extracellular ligands such as cytokines, growth factors and hormones to the nucleus to orchestrate the appropriate cellular response.
Four members of the JAK family, (JAK1, JAK2, JAK3, and TYK2) and seven STAT proteins (STAT1, STAT2, STAT3, STAT4, STAT5A, STAT5B, and STAT6) have been identified in mammals.Citation19 Although STAT proteins are structurally related, their activations and effects present a high degree of specificity for the different STAT isoforms. The JAK/STAT pathway can be activated by many cytokines, including all interferons and most interleukins, many growth factors and some hormones.
In non-stimulated cells, JAK proteins are associated with the membrane receptors and are inactive. Inactive STAT proteins are located in the cytosol in a monomeric form. Activation occurs when the cytokine binds to its membrane receptor, the bound-receptor dimerizes and JAK proteins are activated by trans-phosphorylation. Activated JAK proteins can phosphorylate the tyrosine residues located in the intracellular section of the receptor, which become the docking sites for the Src 2 domains (SH2) of the STAT proteins that are recruited. Recruited STAT proteins are thus activated via JAK proteins by phosphorylation on the tyrosine residue situated near the SH2 domain. Once phosphorylated on the tyrosine residue, STAT monomers form hetero- or homo-dimers, which translocate to the nucleus where they can bind to the DNA and induce the transcription of target genes.Citation20 Although this is the classical activation process, other tyrosine kinases are able to activate STAT proteins; for example, growth factor receptors containing an intrinsic tyrosine kinase activity (EGF and PDGF receptors) as well as non-receptor tyrosine kinases (Src, Abl) can directly activate the members of the STAT family.
Activation of the JAK-STAT signaling pathway is a complex process, which can be stopped or negatively regulated by several processes including dephosphorylation, nuclear export and negative regulators such as SOCS (suppressors of cytokine signaling) and PIAS (protein inhibitor of activated STAT).Citation21 For a more detailed discussion of the mechanisms of STAT regulation, the interested reader is referred to a recent review.Citation22
Role of JAK-STAT in the heart
JAK1, JAK2, and TYK2 and all the members of STAT family are expressed in the heart.Citation23,Citation24 Among these proteins STAT3 and STAT1 are the most studied.
Multiple studies have demonstrated a beneficial and protective role of STAT3 in the heart. This role has mainly been pointed out by data from animal experiments. As STAT3 knockout mice result in early embryonic lethality,Citation25 specific cardiac myocyte STAT3 knockout (STAT3-KO) mice have been a useful tool to investigate STAT3 in the heart. The use of these mice and of the pharmacological inhibitor of JAK2 (AG490) demonstrates a protective and anti-apoptotic role of STAT3. This role has mainly been demonstrated in the model of ischemia reperfusion injury. Thus STAT3-KO mice submitted to 1 h ischemia followed by 24 h of reperfusion show a significantly increased infarct size.Citation26 STAT3 is also involved in pro-survival processes such as ischemic pre- and post-conditioning. Ischemic pre- and post-conditioning protocols result in a significant reduction in infarct size and an improvement of cardiac function when compared with non-conditioned hearts. In STAT3-KO mice and in experiments using pharmacological JAK inhibitors, these protective effects are reduced.Citation24,Citation27-Citation29 It is also possible to mimic ischemic conditioning with pharmacological compounds. In this context STAT3 has been shown to play a role in cardioprotection afforded by tumor necrosis factor α, insulin, melatonin, sphingosine-1-phosphate, and high density lipoproteins.Citation30-Citation34 The beneficial effect of STAT3 in the heart is confirmed in experiments using mice overexpressing STAT3 in the myocardium. These mice are less sensitive to ischemia-reperfusion injury and to doxorubicin (a cardiotoxic drug) exposure than wild-type mice.Citation35 For a more detailed discussion on the effects of STAT3 in the heart, the interested reader is referred to a review.Citation36 Similar cardioprotective effect have been described for STAT5 activation.Citation37
In addition of these pro-survival actions, STAT3 is involved in adaptive hypertrophy. Hypertrophy is initially beneficial and contributes to reduce wall stress and oxygen consumption in the overloaded heart; this serves to maintain normal cardiac output. Transgenic mice with myocardium STAT3 overexpression show signs of hypertrophy by 12 weeks of age. The hearts display enlarged left ventricles and enhanced expression of hypertrophic genes (β-myosin heavy chain, atrial natriuretic peptide).Citation35 Members of the IL-6 family (LIF, CT-1 and IL-6) activate the JAK-STAT3 signaling pathway via the activation of the gp130 receptor and have been shown to be potent mediators of cardiac hypertrophy.Citation23,Citation38 gp130 receptor engagement can prevent heart failure through inhibition of apoptosis and induction of compensatory hypertrophy, via STAT3 activation.Citation39
In contrast to the protective role of STAT3, STAT1 has been attributed deleterious actions. Indeed, in cultured cardiomyocytes STAT1 is activated by hypoxia–reoxygenation and enhances apoptosis by activating the pro-apoptotic targets caspase1, Fas, and FasL.Citation40 Inhibition of STAT1 confers protection against hypoxia–reoxygenation. In vivo, STAT1 is activated during ischemia-reperfusion and hearts from STAT1-KO mice submitted to ischemia-reperfusion have a smaller infarct than wild type mice.Citation41 Recently, STAT1 has been shown to reduce autophagy, which participates in post-infarction cardioprotection.Citation41 Interestingly, experiments in cultured fibroblasts and cardiac cells using STAT overexpression showed that the pro-apoptotic action of STAT1 can be counteracted by the relative expression of STAT3.Citation40,Citation42 The balance between STAT1 and STAT3 activation might thus play a role in the determination of cell fate.
Interaction between JAK-STAT Signaling and Myocardial Glucose Metabolism
Participation of JAK-STAT in insulin and AMPK signaling
Given the ubiquitous nature of both JAK-STAT signaling and glucose metabolism, one may wonder about the involvement of the former in the regulation of the latter in the heart. The first question to ponder is whether JAK-STAT signaling is activated in response to insulin or metabolic stress, the two most important stimuli of glucose metabolism in the myocardium. Indeed it has been shown very early that insulin activates JAK2 in all insulin-responsive tissues, including the heart, in vivo in rats;Citation43 this observation was further extended to JAK1 in cultured cells.Citation44 Independently STAT5 was found to be tyrosine-phosphorylated in response to insulin;Citation45 it was later confirmed that STAT5 phosphorylation could occur both independently of and through JAK activation.Citation46,Citation47 STAT3 was also found to be activated in response to insulin in the heart, thus mediating the cardioprotective effects of the hormone.Citation31,Citation48 In an intriguing crosstalk between canonical pathways, JAK2 activated by other hormones such as growth hormone (GH) or leptin can phosphorylate IRS-1 and IRS-2 on tyrosine residues and thereby recruit and activate PI3K.Citation49,Citation50 This however is not sufficient to stimulate glucose metabolism.
Similarly, several studies have reported activation of STAT3, and perhaps STAT1, 5, and 6, in the ischemic myocardium or in cardiac myocytes submitted to simulated ischemia,Citation37,Citation40,Citation51-Citation53 situations that entail AMPK activation. The participation of AMPK in STAT activation, or AMPK activation downstream to STAT activation, have to the best of our knowledge not been reported. We have observed activation of STAT5 in cardiac myocytes in response to the ATP-synthase inhibitor oligomycin concomitantly with strong AMPK activation, but again without proof of causality.Citation54
Participation of JAK-STAT in the regulation of glucose metabolism
Having established that JAK-STAT signaling could be activated in response to stimuli that increase glucose metabolism, we now have to consider whether JAK-STAT signaling actually contributes to the stimulation of glucose metabolism. Regarding this issue the literature is remarkably scarce, and almost nonexistent as to the myocardium. In skeletal myotubes, which are similar to cardiac myocytes in the regulation of glucose metabolism, insulin-stimulated GLUT4 translocation and glucose uptake is not affected by JAK2 silencing, whereas the proliferative effects of insulin are blunted.Citation55 Similarly, pharmacological inhibition of JAK2 with AG490 fails to prevent the stimulatory effect of leptin on glucose transport in myotubes.Citation56 On the other hand leptin increases Na-glucose cotransport in the myocardium by increasing expression of SGLT1,Citation9 which was shown to be driven by JAK2 activity.Citation57
Let us now turn our attention to the converse possibility, which is that JAK-STAT signaling impedes myocardial glucose metabolism instead of stimulating it. Indeed several factors known to activate JAK-STAT signaling in the myocardium have also inhibitory effects on glucose metabolism (). These include angiotensin II (Ang II),Citation58-Citation60 low concentrations of cardiotrophin-1 (CT-1),Citation54,Citation61 GH,Citation50,Citation62 and leukemia inhibitory factor (LIF).Citation63,Citation64 A common effect of these factors is the upregulation of suppressor of cytokine signaling 3 (SOCS3) expression,Citation54,Citation60,Citation64 although to date this has only been shown in non-myocardial tissues for GH.Citation65 SOCS3, in addition to exerting a negative feedback on JAK-STAT signaling in the myocardium,Citation66-Citation68 is able to reduce insulin signaling by preventing autophosphorylation of the insulin receptor,Citation69 reducing interaction of IRS with the IR and with PI3KCitation70 and by promoting proteasomal degradation of IRS.Citation71 In cardiac myocytes overexpression on SOCS3 has indeed been associated with insulin resistance induced by PPARα and PPARδ agonistsCitation72 and by low concentrations of cardiotrophin-1.Citation54 The requirement of JAK-STAT signaling for the upregulation of SOCS3 expression has not to date been firmly established in cardiac myocytes; SOCS3 overexpression has only been tightly associated with STAT3 activation.Citation66,Citation68 In other tissues and cell types SOCS3 transcription is driven by activated STAT3Citation73 and STAT5,Citation74,Citation75 and it seems reasonable to assume that it could be similar in the myocardium. Indeed, in cardiac myocytes exposed to low concentrations of CT-1, pharmacological inhibition of STAT5 activity suppressed SOCS3 overexpression and restored insulin signaling and insulin-stimulated glucose transport.Citation54 Upstream of STATs SOCS3 expression seems to be at least in part dependent on JAK2 activity, as it can be reduced by the JAK2 inhibitor AG490.Citation76
Figure 2. Interference of JAK-STAT signaling with insulin signaling and glucose transport in cardiac myocytes. Top: Normal insulin signaling leading to Akt activation and translocation of GLUT4. Right: activation of JAK-STAT signaling by Ang II, LIF, and CT-1 leading to SOCS3 overexpression and GLUT4 repression. Left: disruption of insulin signaling by SOCS3, with dissociation and degradation of IRS-1. Bottom: sequestration of IRS-1 by JAK2 activated by the ligand-bound AGTR1. See text for details. Abbreviations: Ang II, angiotensin II; AGTR1, angiotensin receptor type 1; CT-1, cardiotrophin-1; gp130, glycoprotein 130; IRS-1, insulin receptor substrate 1; LIF, leukemia inhibitory factor; LIFR, LIF receptor; mTORC2, mammalian target of rapamycin complex 2; PDPK1, phosphoinositide-dependent protein kinase 1; PI3Kα, phosphoinositide 3-kinase α; PIP3, phosphatidylinositol-3,4,5-trisphosphate; pY, phosphotyrosine; SOCS3, suppressor of cytokine signaling 3.
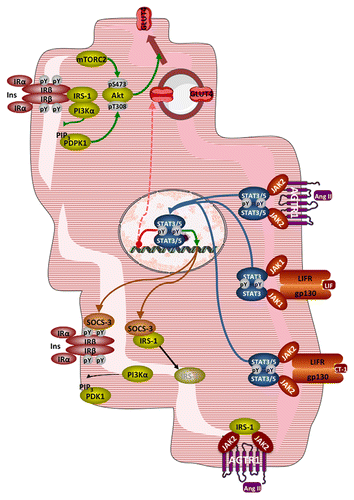
Obviously a reduction in glucose metabolism could also result from diminished expression of the main glucose transporter GLUT4. Both LIF and low concentrations of CT-1 reduce GLUT4 expression in cardiac myocytes.Citation54,Citation64 Whereas there is no evidence for the JAK-STAT axis involvement in this effect of LIF, only “guilt by association”, pharmacological inhibition of STAT5 activity indeed restored GLUT4 expression reduced by a low dose of CT-1.
Other mechanisms, independent of gene expression, may operate to reduce insulin signaling when the JAK-STAT axis is activated. Thus in cardiac myocytes stimulated concomitantly with both Ang II and insulin, the branch of insulin signaling downstream of IRS phosphorylation leading to stimulation of glucose metabolism, i.e. PI3K activation and subsequent Akt recruitment and activation, is reduced.Citation77 This occurs while IRS association with JAK2 is increased, suggesting that JAK2 activation reduces insulin signaling to glucose transport by sequestering IRS.Citation78 In contrast ERK1/2 activation in response to insulin is potentiated by Ang II;Citation77 ERK1/2 activation is known to be detrimental to the stimulation of glucose transport in cardiac myocytes.Citation79
In line with these observations, in myotubes rendered insulin resistant by incubation with ceramides JAK2 silencing restored Akt activation and insulin-stimulated glucose transport.Citation55 Collectively these results suggest that JAK2 may depress the Akt to glucose uptake signaling axis selectively in insulin-resistant states.
A last mechanism by which JAK-STAT signaling activation could reduce glucose metabolism is by driving expression of a PDH kinase; in adipocytes the expression of PDK4 is mediated by STAT5 in response to prolactin.Citation80 Indeed Ang II, which activates STAT5,Citation58 induces PDK4 expression in cardiac myocytes, although JAK-STAT signaling was not investigated in the latter study.Citation81 We observed however in cardiac myocytes exposed to CT-1 and displaying STAT5 phosphorylation a slight reduction of PDH-E1 phosphorylation.Citation54
In conclusion, despite a paucity of data directly pertaining to the myocardium, it appears that JAK-STAT signaling does not significantly participate in the stimulation of glucose metabolism by insulin or metabolic stress in the heart. On the contrary, JAK-STAT signaling most likely mediates the development of insulin resistance induced by various cytokines. Again, the literature is remarkably scarce of results obtained in heart-relevant experimental models; many of the above conclusions are extrapolated from data obtained in skeletal muscle or adipocytes, and therefore should be taken with caution. Whether this shortage of information results from an actual lack of experiments or from abstaining to report negative results remains unknown.
| Abbreviations: | ||
| Acetyl-CoA | = | acetyl-coenzyme A |
| ADP | = | adenosine diphosphate |
| AMP | = | adenosine monophosphate |
| ATP | = | adenosine triphosphate |
| AMPK | = | AMP-activated protein kinase |
| Ang II | = | angiotensin II |
| AGTR1 | = | angiotensin receptor type 1 |
| CT-1 | = | cardiotrophin-1 |
| F1,6BP | = | fructose-1,6-bisphosphate F2,6BP, fructose-2,6-bisphosphate |
| F6P | = | fructose-6-phosphate |
| GLUT1 or 4 | = | facilitative glucose transporters |
| gp130 | = | glycoprotein 130 |
| Ins | = | insulin |
| IRα/β | = | insulin receptor, subunit α/β |
| IRS | = | insulin receptor substrate |
| LIF | = | leukemia inhibitory factor |
| LIFR | = | LIF receptor |
| mTORC2 | = | mammalian target of rapamycin complex 2 |
| PDC | = | pyruvate dehydrogenase complex |
| PDH-E1 | = | pyruvate dehydrogenase enzyme |
| PDK1-4 | = | pyruvate dehydrogenase kinase 1 to 4 |
| PDPK1 | = | Phosphoinositide-dependent protein kinase 1 |
| PDPC1-2 | = | pyruvate dehydrogenase phosphatase 1 or 2 |
| PFK-1 | = | 6-phosphofructo-1-kinase |
| PFK-2 | = | 6-phosphofructo-2-kinase |
| PI3Kα | = | phosphoinositide 3-kinase α |
| PIP3 | = | phosphatidylinositol-3,4,5-trisphosphate |
| SGLT1 | = | sodium-glucose cotransporter 1 |
| SOCS3 | = | suppressor of cytokine signaling 3 |
| WISK | = | wortmannin-sensitive and insulin-stimulated protein kinase |
Disclosure of Potential Conflicts of Interest
There is no conflict of interest to disclose.
Acknowledgments
Research in the authors’ labs is supported by grants from the Swiss National Science Foundation (310030_146537 to C Montessuit), the Fondation Carlos et Elsie de Reuter (to C Montessuit), the Fondation Gustave et Simone Prévot (to C Montessuit and MA Frias) and the Wolfermann Naegli Stiftung and Jubiläumsstiftung (to MA Frias).
References
- Opie LH. The Heart: Physiology, from Cell to Circulation. 3rd ed. Philadelphia, New York: Lippincott-Raven; 1998.
- Stryer L. Biochemistry. 2nd ed. San Francisco: W.H. Freeman and Company; 1981.
- Lopaschuk GD, Gamble J. The 1993 Merck Frosst Award. Acetyl-CoA carboxylase: an important regulator of fatty acid oxidation in the heart. Can J Physiol Pharmacol 1994; 72:1101 - 9; http://dx.doi.org/10.1139/y94-156; PMID: 7882173
- Tamm C, Benzi R, Papageorgiou I, Tardy I, Lerch R. Substrate competition in postischemic myocardium. Effect of substrate availability during reperfusion on metabolic and contractile recovery in isolated rat hearts. Circ Res 1994; 75:1103 - 12; http://dx.doi.org/10.1161/01.RES.75.6.1103; PMID: 7955147
- Dhar-Chowdhury P, Malester B, Rajacic P, Coetzee WA. The regulation of ion channels and transporters by glycolytically derived ATP. Cell Mol Life Sci 2007; 64:3069 - 83; http://dx.doi.org/10.1007/s00018-007-7332-3; PMID: 17882378
- Saks V, Dzeja P, Schlattner U, Vendelin M, Terzic A, Wallimann T. Cardiac system bioenergetics: metabolic basis of the Frank-Starling law. J Physiol 2006; 571:253 - 73; http://dx.doi.org/10.1113/jphysiol.2005.101444; PMID: 16410283
- Santalucía T, Camps M, Castelló A, Muñoz P, Nuel A, Testar X, Palacin M, Zorzano A. Developmental regulation of GLUT-1 (erythroid/Hep G2) and GLUT-4 (muscle/fat) glucose transporter expression in rat heart, skeletal muscle, and brown adipose tissue. Endocrinology 1992; 130:837 - 46; http://dx.doi.org/10.1210/en.130.2.837; PMID: 1370797
- Slot JW, Geuze HJ, Gigengack S, James DE, Lienhard GE. Translocation of the glucose transporter GLUT4 in cardiac myocytes of the rat. Proc Natl Acad Sci U S A 1991; 88:7815 - 9; http://dx.doi.org/10.1073/pnas.88.17.7815; PMID: 1881917
- Banerjee SK, McGaffin KR, Pastor-Soler NM, Ahmad F. SGLT1 is a novel cardiac glucose transporter that is perturbed in disease states. Cardiovasc Res 2009; 84:111 - 8; http://dx.doi.org/10.1093/cvr/cvp190; PMID: 19509029
- Wheeler TJ, Fell RD, Hauck MA. Translocation of two glucose transporters in heart: effects of rotenone, uncouplers, workload, palmitate, insulin and anoxia. Biochim Biophys Acta 1994; 1196:191 - 200; http://dx.doi.org/10.1016/0005-2736(94)00211-8; PMID: 7841183
- Sun D, Nguyen N, DeGrado TR, Schwaiger M, Brosius FC 3rd. Ischemia induces translocation of the insulin-responsive glucose transporter GLUT4 to the plasma membrane of cardiac myocytes. Circulation 1994; 89:793 - 8; http://dx.doi.org/10.1161/01.CIR.89.2.793; PMID: 8313568
- Bertrand L, Horman S, Beauloye C, Vanoverschelde J-L. Insulin signalling in the heart. Cardiovasc Res 2008; 79:238 - 48; http://dx.doi.org/10.1093/cvr/cvn093; PMID: 18390897
- Hardie DG, Ross FA, Hawley SA. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol 2012; 13:251 - 62; http://dx.doi.org/10.1038/nrm3311; PMID: 22436748
- Thong FSL, Bilan PJ, Klip A. The Rab GTPase-activating protein AS160 integrates Akt, protein kinase C, and AMP-activated protein kinase signals regulating GLUT4 traffic. Diabetes 2007; 56:414 - 23; http://dx.doi.org/10.2337/db06-0900; PMID: 17259386
- Montessuit C, Lerch R. Regulation and dysregulation of glucose transport in cardiomyocytes. Biochim Biophys Acta 2013; 1833:848 - 56; http://dx.doi.org/10.1016/j.bbamcr.2012.08.009; PMID: 22967513
- Rider MH, Bertrand L, Vertommen D, Michels PA, Rousseau GG, Hue L. 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase: head-to-head with a bifunctional enzyme that controls glycolysis. Biochem J 2004; 381:561 - 79; http://dx.doi.org/10.1042/BJ20040752; PMID: 15170386
- Bowker-Kinley MM, Davis WI, Wu P, Harris RA, Popov KM. Evidence for existence of tissue-specific regulation of the mammalian pyruvate dehydrogenase complex. Biochem J 1998; 329:191 - 6; PMID: 9405293
- Caruso M, Maitan MA, Bifulco G, Miele C, Vigliotta G, Oriente F, Formisano P, Beguinot F. Activation and mitochondrial translocation of protein kinase Cdelta are necessary for insulin stimulation of pyruvate dehydrogenase complex activity in muscle and liver cells. J Biol Chem 2001; 276:45088 - 97; http://dx.doi.org/10.1074/jbc.M105451200; PMID: 11577086
- Darnell JE Jr.. STATs and gene regulation. Science 1997; 277:1630 - 5; http://dx.doi.org/10.1126/science.277.5332.1630; PMID: 9287210
- Levy DE, Darnell JE Jr.. Stats: transcriptional control and biological impact. Nat Rev Mol Cell Biol 2002; 3:651 - 62; http://dx.doi.org/10.1038/nrm909; PMID: 12209125
- Schindler C, Levy DE, Decker T. JAK-STAT signaling: from interferons to cytokines. J Biol Chem 2007; 282:20059 - 63; http://dx.doi.org/10.1074/jbc.R700016200; PMID: 17502367
- Kiu H, Nicholson SE. Biology and significance of the JAK/STAT signalling pathways. Growth Factors 2012; 30:88 - 106; http://dx.doi.org/10.3109/08977194.2012.660936; PMID: 22339650
- Pan J, Fukuda K, Saito M, Matsuzaki J, Kodama H, Sano M, Takahashi T, Kato T, Ogawa S. Mechanical stretch activates the JAK/STAT pathway in rat cardiomyocytes. Circ Res 1999; 84:1127 - 36; http://dx.doi.org/10.1161/01.RES.84.10.1127; PMID: 10347087
- Xuan YT, Guo Y, Han H, Zhu Y, Bolli R. An essential role of the JAK-STAT pathway in ischemic preconditioning. Proc Natl Acad Sci U S A 2001; 98:9050 - 5; http://dx.doi.org/10.1073/pnas.161283798; PMID: 11481471
- Takeda K, Noguchi K, Shi W, Tanaka T, Matsumoto M, Yoshida N, Kishimoto T, Akira S. Targeted disruption of the mouse Stat3 gene leads to early embryonic lethality. Proc Natl Acad Sci U S A 1997; 94:3801 - 4; http://dx.doi.org/10.1073/pnas.94.8.3801; PMID: 9108058
- Hilfiker-Kleiner D, Hilfiker A, Fuchs M, Kaminski K, Schaefer A, Schieffer B, Hillmer A, Schmiedl A, Ding Z, Podewski E, et al. Signal transducer and activator of transcription 3 is required for myocardial capillary growth, control of interstitial matrix deposition, and heart protection from ischemic injury. Circ Res 2004; 95:187 - 95; http://dx.doi.org/10.1161/01.RES.0000134921.50377.61; PMID: 15192020
- Hattori R, Maulik N, Otani H, Zhu L, Cordis G, Engelman RM, Siddiqui MA, Das DK. Role of STAT3 in ischemic preconditioning. J Mol Cell Cardiol 2001; 33:1929 - 36; http://dx.doi.org/10.1006/jmcc.2001.1456; PMID: 11708838
- Smith RM, Suleman N, Lacerda L, Opie LH, Akira S, Chien KR, Sack MN. Genetic depletion of cardiac myocyte STAT-3 abolishes classical preconditioning. Cardiovasc Res 2004; 63:611 - 6; http://dx.doi.org/10.1016/j.cardiores.2004.06.019; PMID: 15306216
- Boengler K, Buechert A, Heinen Y, Roeskes C, Hilfiker-Kleiner D, Heusch G, Schulz R. Cardioprotection by ischemic postconditioning is lost in aged and STAT3-deficient mice. Circ Res 2008; 102:131 - 5; http://dx.doi.org/10.1161/CIRCRESAHA.107.164699; PMID: 17967780
- Lecour S, Smith RM, Woodward B, Opie LH, Rochette L, Sack MN. Identification of a novel role for sphingolipid signaling in TNF alpha and ischemic preconditioning mediated cardioprotection. J Mol Cell Cardiol 2002; 34:509 - 18; http://dx.doi.org/10.1006/jmcc.2002.1533; PMID: 12056855
- Fuglesteg BN, Suleman N, Tiron C, Kanhema T, Lacerda L, Andreasen TV, Sack MN, Jonassen AK, Mjøs OD, Opie LH, et al. Signal transducer and activator of transcription 3 is involved in the cardioprotective signalling pathway activated by insulin therapy at reperfusion. Basic Res Cardiol 2008; 103:444 - 53; http://dx.doi.org/10.1007/s00395-008-0728-x; PMID: 18500485
- Lamont KT, Somers S, Lacerda L, Opie LH, Lecour S. Is red wine a SAFE sip away from cardioprotection? Mechanisms involved in resveratrol- and melatonin-induced cardioprotection. J Pineal Res 2011; 50:374 - 80; http://dx.doi.org/10.1111/j.1600-079X.2010.00853.x; PMID: 21342247
- Somers SJ, Frias M, Lacerda L, Opie LH, Lecour S. Interplay between SAFE and RISK pathways in sphingosine-1-phosphate-induced cardioprotection. Cardiovasc Drugs Ther 2012; 26:227 - 37; http://dx.doi.org/10.1007/s10557-012-6376-2; PMID: 22392184
- Frias MA, Pedretti S, Hacking D, Somers S, Lacerda L, Opie LH, James RW, Lecour S. HDL protects against ischemia reperfusion injury by preserving mitochondrial integrity. Atherosclerosis 2013; 228:110 - 6; http://dx.doi.org/10.1016/j.atherosclerosis.2013.02.003; PMID: 23497785
- Kunisada K, Negoro S, Tone E, Funamoto M, Osugi T, Yamada S, Okabe M, Kishimoto T, Yamauchi-Takihara K. Signal transducer and activator of transcription 3 in the heart transduces not only a hypertrophic signal but a protective signal against doxorubicin-induced cardiomyopathy. Proc Natl Acad Sci U S A 2000; 97:315 - 9; http://dx.doi.org/10.1073/pnas.97.1.315; PMID: 10618415
- Boengler K, Hilfiker-Kleiner D, Drexler H, Heusch G, Schulz R. The myocardial JAK/STAT pathway: from protection to failure. Pharmacol Ther 2008; 120:172 - 85; http://dx.doi.org/10.1016/j.pharmthera.2008.08.002; PMID: 18786563
- Yamaura G, Turoczi T, Yamamoto F, Siddqui MAQ, Maulik N, Das DK. STAT signaling in ischemic heart: a role of STAT5A in ischemic preconditioning. Am J Physiol Heart Circ Physiol 2003; 285:H476 - 82; PMID: 12860560
- Barry SP, Townsend PA, Latchman DS, Stephanou A. Role of the JAK-STAT pathway in myocardial injury. Trends Mol Med 2007; 13:82 - 9; http://dx.doi.org/10.1016/j.molmed.2006.12.002; PMID: 17194625
- Hirota H, Chen J, Betz UAK, Rajewsky K, Gu Y, Ross JJ Jr., Müller W, Chien KR. Loss of a gp130 cardiac muscle cell survival pathway is a critical event in the onset of heart failure during biomechanical stress. Cell 1999; 97:189 - 98; http://dx.doi.org/10.1016/S0092-8674(00)80729-1; PMID: 10219240
- Stephanou A, Brar BK, Scarabelli TM, Jonassen AK, Yellon DM, Marber MS, Knight RA, Latchman DS. Ischemia-induced STAT-1 expression and activation play a critical role in cardiomyocyte apoptosis. J Biol Chem 2000; 275:10002 - 8; http://dx.doi.org/10.1074/jbc.275.14.10002; PMID: 10744676
- McCormick J, Suleman N, Scarabelli TM, Knight RA, Latchman DS, Stephanou A. STAT1 deficiency in the heart protects against myocardial infarction by enhancing autophagy. J Cell Mol Med 2012; 16:386 - 93; http://dx.doi.org/10.1111/j.1582-4934.2011.01323.x; PMID: 21447043
- Shen Y, Devgan G, Darnell JE Jr., Bromberg JF. Constitutively activated Stat3 protects fibroblasts from serum withdrawal and UV-induced apoptosis and antagonizes the proapoptotic effects of activated Stat1. Proc Natl Acad Sci U S A 2001; 98:1543 - 8; http://dx.doi.org/10.1073/pnas.98.4.1543; PMID: 11171987
- Saad MJA, Carvalho CRO, Thirone ACP, Velloso LA. Insulin induces tyrosine phosphorylation of JAK2 in insulin-sensitive tissues of the intact rat. J Biol Chem 1996; 271:22100 - 4; http://dx.doi.org/10.1074/jbc.271.36.22100; PMID: 8703019
- Gual P, Baron V, Lequoy V, Van Obberghen E. Interaction of Janus kinases JAK-1 and JAK-2 with the insulin receptor and the insulin-like growth factor-1 receptor. Endocrinology 1998; 139:884 - 93; http://dx.doi.org/10.1210/en.139.3.884; PMID: 9492017
- Chen J, Sadowski HB, Kohanski RA, Wang L-H. Stat5 is a physiological substrate of the insulin receptor. Proc Natl Acad Sci U S A 1997; 94:2295 - 300; http://dx.doi.org/10.1073/pnas.94.6.2295; PMID: 9122188
- Sawka-Verhelle D, Tartare-Deckert S, Decaux J-F, Girard J, Van Obberghen E. Stat 5B, activated by insulin in a Jak-independent fashion, plays a role in glucokinase gene transcription. Endocrinology 2000; 141:1977 - 88; http://dx.doi.org/10.1210/en.141.6.1977; PMID: 10830280
- Le MN, Kohanski RA, Wang L-H, Sadowski HB. Dual mechanism of signal transducer and activator of transcription 5 activation by the insulin receptor. Mol Endocrinol 2002; 16:2764 - 79; http://dx.doi.org/10.1210/me.2002-0017; PMID: 12456798
- Coffer PJ, van Puijenbroek A, Burgering BM, Klop-de Jonge M, Koenderman L, Bos JL, Kruijer W. Insulin activates Stat3 independently of p21ras-ERK and PI-3K signal transduction. Oncogene 1997; 15:2529 - 39; http://dx.doi.org/10.1038/sj.onc.1201429; PMID: 9399641
- Kellerer M, Koch M, Metzinger E, Mushack J, Capp E, Häring HU. Leptin activates PI-3 kinase in C2C12 myotubes via janus kinase-2 (JAK-2) and insulin receptor substrate-2 (IRS-2) dependent pathways. Diabetologia 1997; 40:1358 - 62; http://dx.doi.org/10.1007/s001250050832; PMID: 9389430
- Thirone ACP, Carvalho CRO, Saad MJA. Growth hormone stimulates the tyrosine kinase activity of JAK2 and induces tyrosine phosphorylation of insulin receptor substrates and Shc in rat tissues. Endocrinology 1999; 140:55 - 62; http://dx.doi.org/10.1210/en.140.1.55; PMID: 9886807
- Goodman MD, Koch SE, Afzal MR, Butler KL. STAT subtype specificity and ischemic preconditioning in mice: is STAT-3 enough?. Am J Physiol Heart Circ Physiol 2011; 300:H522 - 6; http://dx.doi.org/10.1152/ajpheart.00231.2010; PMID: 21131482
- Negoro S, Kunisada K, Tone E, Funamoto M, Oh H, Kishimoto T, Yamauchi-Takihara K. Activation of JAK/STAT pathway transduces cytoprotective signal in rat acute myocardial infarction. Cardiovasc Res 2000; 47:797 - 805; http://dx.doi.org/10.1016/S0008-6363(00)00138-3; PMID: 10974228
- Omura T, Yoshiyama M, Ishikura F, Kobayashi H, Takeuchi K, Beppu S, Yoshikawa J. Myocardial ischemia activates the JAK-STAT pathway through angiotensin II signaling in in vivo myocardium of rats. J Mol Cell Cardiol 2001; 33:307 - 16; http://dx.doi.org/10.1006/jmcc.2000.1303; PMID: 11162135
- Asrih M, Gardier S, Papageorgiou I, Montessuit C. Dual effect of the heart-targeting cytokine cardiotrophin-1 on glucose transport in cardiomyocytes. J Mol Cell Cardiol 2013; 56:106 - 15; http://dx.doi.org/10.1016/j.yjmcc.2012.12.015; PMID: 23277190
- Thirone ACP, JeBailey L, Bilan PJ, Klip A. Opposite effect of JAK2 on insulin-dependent activation of mitogen-activated protein kinases and Akt in muscle cells: possible target to ameliorate insulin resistance. Diabetes 2006; 55:942 - 51; http://dx.doi.org/10.2337/diabetes.55.04.06.db05-1265; PMID: 16567515
- Bates SH, Gardiner JV, Jones RB, Bloom SR, Bailey CJ. Acute stimulation of glucose uptake by leptin in l6 muscle cells. Horm Metab Res 2002; 34:111 - 5; http://dx.doi.org/10.1055/s-2002-23192; PMID: 11972298
- Hosseinzadeh Z, Bhavsar SK, Shojaiefard M, Saxena A, Merches K, Sopjani M, Alesutan I, Lang F. Stimulation of the glucose carrier SGLT1 by JAK2. Biochem Biophys Res Commun 2011; 408:208 - 13; http://dx.doi.org/10.1016/j.bbrc.2011.03.036; PMID: 21406183
- McWhinney CD, Dostal D, Baker K. Angiotensin II activates Stat5 through Jak2 kinase in cardiac myocytes. J Mol Cell Cardiol 1998; 30:751 - 61; http://dx.doi.org/10.1006/jmcc.1998.0639; PMID: 9602424
- McWhinney CD, Hunt RA, Conrad KM, Dostal DE, Baker KM. The type I angiotensin II receptor couples to Stat1 and Stat3 activation through Jak2 kinase in neonatal rat cardiac myocytes. J Mol Cell Cardiol 1997; 29:2513 - 24; http://dx.doi.org/10.1006/jmcc.1997.0489; PMID: 9299374
- Calegari VC, Alves M, Picardi PK, Inoue RY, Franchini KG, Saad MJA, Velloso LA. Suppressor of cytokine signaling-3 Provides a novel interface in the cross-talk between angiotensin II and insulin signaling systems. Endocrinology 2005; 146:579 - 88; http://dx.doi.org/10.1210/en.2004-0466; PMID: 15514089
- Latchman DS. Cardiotrophin-1 (CT-1): a novel hypertrophic and cardioprotective agent. Int J Exp Pathol 1999; 80:189 - 96; http://dx.doi.org/10.1046/j.1365-2613.1999.00114.x; PMID: 10583628
- Lu C, Schwartzbauer G, Sperling MA, Devaskar SU, Thamotharan S, Robbins PD, McTiernan CF, Liu J-L, Jiang J, Frank SJ, et al. Demonstration of direct effects of growth hormone on neonatal cardiomyocytes. J Biol Chem 2001; 276:22892 - 900; http://dx.doi.org/10.1074/jbc.M011647200; PMID: 11303022
- Kunisada K, Hirota H, Fujio Y, Matsui H, Tani Y, Yamauchi-Takihara K, Kishimoto T. Activation of JAK-STAT and MAP kinases by leukemia inhibitory factor through gp130 in cardiac myocytes. Circulation 1996; 94:2626 - 32; http://dx.doi.org/10.1161/01.CIR.94.10.2626; PMID: 8921810
- Florholmen G, Thoresen GH, Rustan AC, Jensen J, Christensen G, Aas V. Leukaemia inhibitory factor stimulates glucose transport in isolated cardiomyocytes and induces insulin resistance after chronic exposure. Diabetologia 2006; 49:724 - 31; http://dx.doi.org/10.1007/s00125-006-0150-6; PMID: 16489447
- Adams TE, Hansen JA, Starr R, Nicola NA, Hilton DJ, Billestrup N. Growth hormone preferentially induces the rapid, transient expression of SOCS-3, a novel inhibitor of cytokine receptor signaling. J Biol Chem 1998; 273:1285 - 7; http://dx.doi.org/10.1074/jbc.273.3.1285; PMID: 9430658
- Yasukawa H, Hoshijima M, Gu Y, Nakamura T, Pradervand S, Hanada T, Hanakawa Y, Yoshimura A, Ross J Jr., Chien KR. Suppressor of cytokine signaling-3 is a biomechanical stress-inducible gene that suppresses gp130-mediated cardiac myocyte hypertrophy and survival pathways. J Clin Invest 2001; 108:1459 - 67; PMID: 11714737
- Calegari VC, Bezerra RMN, Torsoni MA, Torsoni AS, Franchini KG, Saad MJA, Velloso LA. Suppressor of cytokine signaling 3 is induced by angiotensin II in heart and isolated cardiomyocytes, and participates in desensitization. Endocrinology 2003; 144:4586 - 96; http://dx.doi.org/10.1210/en.2003-0046; PMID: 12960061
- Hamanaka I, Saito Y, Yasukawa H, Kishimoto I, Kuwahara K, Miyamoto Y, Harada M, Ogawa E, Kajiyama N, Takahashi N, et al. Induction of JAB/SOCS-1/SSI-1 and CIS3/SOCS-3/SSI-3 is involved in gp130 resistance in cardiovascular system in rat treated with cardiotrophin-1 in vivo. Circ Res 2001; 88:727 - 32; http://dx.doi.org/10.1161/hh0701.088512; PMID: 11304496
- Senn JJ, Klover PJ, Nowak IA, Zimmers TA, Koniaris LG, Furlanetto RW, Mooney RA. Suppressor of cytokine signaling-3 (SOCS-3), a potential mediator of interleukin-6-dependent insulin resistance in hepatocytes. J Biol Chem 2003; 278:13740 - 6; http://dx.doi.org/10.1074/jbc.M210689200; PMID: 12560330
- Emanuelli B, Peraldi P, Filloux C, Chavey C, Freidinger K, Hilton DJ, Hotamisligil GS, Van Obberghen E. SOCS-3 inhibits insulin signaling and is up-regulated in response to tumor necrosis factor-α in the adipose tissue of obese mice. J Biol Chem 2001; 276:47944 - 9; PMID: 11604392
- Rui L, Yuan M, Frantz D, Shoelson S, White MF. SOCS-1 and SOCS-3 block insulin signaling by ubiquitin-mediated degradation of IRS1 and IRS2. J Biol Chem 2002; 277:42394 - 8; http://dx.doi.org/10.1074/jbc.C200444200; PMID: 12228220
- Asrih M, Lerch R, Papageorgiou I, Pellieux C, Montessuit C. Differential regulation of stimulated glucose transport by free fatty acids and PPARα or -δ agonists in cardiac myocytes. Am J Physiol Endocrinol Metab 2012; 302:E872 - 84; http://dx.doi.org/10.1152/ajpendo.00427.2011; PMID: 22297301
- Auernhammer CJ, Bousquet C, Melmed S. Autoregulation of pituitary corticotroph SOCS-3 expression: characterization of the murine SOCS-3 promoter. Proc Natl Acad Sci U S A 1999; 96:6964 - 9; http://dx.doi.org/10.1073/pnas.96.12.6964; PMID: 10359822
- Davey HW, McLachlan MJ, Wilkins RJ, Hilton DJ, Adams TE. STAT5b mediates the GH-induced expression of SOCS-2 and SOCS-3 mRNA in the liver. Mol Cell Endocrinol 1999; 158:111 - 6; http://dx.doi.org/10.1016/S0303-7207(99)00175-6; PMID: 10630411
- Emanuelli B, Peraldi P, Filloux C, Sawka-Verhelle D, Hilton D, Van Obberghen E. SOCS-3 is an insulin-induced negative regulator of insulin signaling. J Biol Chem 2000; 275:15985 - 91; http://dx.doi.org/10.1074/jbc.275.21.15985; PMID: 10821852
- Fasshauer M, Kralisch S, Klier M, Lossner U, Bluher M, Klein J, Paschke R. Insulin resistance-inducing cytokines differentially regulate SOCS mRNA expression via growth factor- and Jak/Stat-signaling pathways in 3T3-L1 adipocytes. J Endocrinol 2004; 181:129 - 38; http://dx.doi.org/10.1677/joe.0.1810129; PMID: 15072573
- Carvalheira JBC, Calegari VC, Zecchin HG, Nadruz W Jr., Guimarães RB, Ribeiro EB, Franchini KG, Velloso LA, Saad MJA. The cross-talk between angiotensin and insulin differentially affects phosphatidylinositol 3-kinase- and mitogen-activated protein kinase-mediated signaling in rat heart: implications for insulin resistance. Endocrinology 2003; 144:5604 - 14; http://dx.doi.org/10.1210/en.2003-0788; PMID: 12960006
- Velloso LA, Folli F, Sun XJ, White MF, Saad MJA, Kahn CR. Cross-talk between the insulin and angiotensin signaling systems. Proc Natl Acad Sci U S A 1996; 93:12490 - 5; http://dx.doi.org/10.1073/pnas.93.22.12490; PMID: 8901609
- Asrih M, Pellieux C, Papageorgiou I, Lerch R, Montessuit C. Role of ERK1/2 activation in microtubule stabilization and glucose transport in cardiomyocytes. Am J Physiol Endocrinol Metab 2011; 301:E836 - 43; http://dx.doi.org/10.1152/ajpendo.00160.2011; PMID: 21771966
- White UA, Coulter AA, Miles TK, Stephens JM. The STAT5A-mediated induction of pyruvate dehydrogenase kinase 4 expression by prolactin or growth hormone in adipocytes. Diabetes 2007; 56:1623 - 9; http://dx.doi.org/10.2337/db06-1286; PMID: 17360981
- Mori J, Basu R, McLean BA, Das SK, Zhang L, Patel VB, Wagg CS, Kassiri Z, Lopaschuk GD, Oudit GY. Agonist-induced hypertrophy and diastolic dysfunction are associated with selective reduction in glucose oxidation: a metabolic contribution to heart failure with normal ejection fraction. Circ Heart Fail 2012; 5:493 - 503; http://dx.doi.org/10.1161/CIRCHEARTFAILURE.112.966705; PMID: 22705769