Abstract
Monoclonal antibodies represent an attractive therapeutic tool as they are highly specific for their targets, convey effector functions and enjoy robust manufacturing procedures. Humanization of murine monoclonal antibodies has vastly improved their in vivo tolerability. Humanization, the replacement of mouse constant regions and V framework regions for human sequences, results in a significantly less immunogenic product. However, some humanized and even fully human sequence-derived antibody molecules still carry immunological risk. To more fully understand the immunologic potential of humanized and human antibodies, we analyzed CD4+ helper T cell epitopes in a set of eight humanized antibodies. The antibodies studied represented a number of different VH and VL family members carrying unique CDR regions. In spite of these differences, CD4+ T cell epitopes were found only in CDR-sequence containing regions. We were able to incorporate up to two amino acid modifications in a single epitope that reduced the immunogenic potential while retaining full biologic function. We propose that immunogenicity will always be present in some antibody molecules due to the nature of the antigen-specific combining sites. A consequence of this result is modifications to reduce immunogenicity will be centered on the affinity-determining regions. Modifications to CDR regions can be designed that reduce the immunogenic potential while maintaining the bioactivity of the antibody molecule.
Introduction
Monoclonal antibodies (mAbs) represent effective therapeutic agents as they demonstrate significant specificity for their targets and confer effector functions such as receptor-ligand blockade, target cell cytotoxicity and receptor antagonism. However, the use of mAbs in clinical settings has been complicated by a number of technical challenges including the demonstration of immunogenic responses. Immunogenic responses to antibody therapeutics can impact both safety and pharmacokinetic properties which can impact utility and efficacy of the drugs. A heightened awareness of these issues has developed as the field matures and the clinical consequences of immune responses to therapeutics have been reported and detailed. Understanding, controlling and engineering around potential immunogenicity is therefore of interest to the industry.
The use of antibodies as therapeutics has a long history. Prior to the development of mAb technologies, antisera from hyperimmunized animals were used to treat infectious diseases such as botulism and diphtheria. Diphtheria antitoxin, the antigen-specific IgG fraction isolated from the serum of diphtheria immunized horses, is still in use today.Citation1 It is a life-saving therapeutic but is well known to cause significant immunological issues in patients,Citation2 and is administered in a controlled setting where antihistamine is available for immediate application if needed. It is now obvious that injecting a person with a mixture of horse serum-derived proteins could cause immune reactions. It was less apparent that injecting patients with purified mouse-derived antibodies, i.e., murine mAbs, could also cause immune reactions.Citation3 An appreciation of the consequences of an immune response to murine antibodies has lead to the development of engineered antibody constructs that carry a lower risk of immune reactions.Citation4 Engineering of antibodies by sequentially replacing mouse sequence-derived amino acids for human sequences has in fact significantly reduced immunogenicity of this class of therapeutics.Citation5 Chimeric antibodies were the first engineered improvement where the murine constant regions were replaced by human constant regions. The next development was the humanization process. Humanization results in an antibody where only the complementarity determining regions (CDRs) of the variable (V) regions are of mouse-sequence origin. The current state of the art is fully human amino acid sequence derived antibody region therapeutics where antigen specificity has been selected either in vivo by the use of genetically modified mice or by antibody engineering processes combined with screening.Citation6–Citation9 Fully human and humanized antibodies carry a lower risk for inducing immune responses in humans than mouse or chimeric antibodies.Citation5
It is fairly easy to characterize immune reactions such as immediate and delayed hypersensitivity responses. Antibody “inhibitor” responses that impact the efficacy of a protein therapeutic, such as those that develop in hemophilia patients treated with Factor VIII are also easy to characterize due to the severe clinical consequence of the immune response.Citation10–Citation12 The measurement of antibody responses directed at antibody therapeutics is more difficult to assess. These immune responses can be regarded as anti-idiotype responses to the therapeutic when they are directed at the combining site, or as “binding antibodies” when they do not. While the development of hypersensitivity reactions is always of concern, the most often described consequence of the development of an immune response is neutralization of the therapeutic (anti-idiotype response) and loss of efficacy due to modified pharmacokinetics (anti-idiotype and binding antibodies; “anti-drug antibodies”). Low levels of transiently expressed anti-drug antibodies are not of significant concern as they rarely impact clinical outcomes.Citation13 However, the development of high titer, high affinity anti-drug antibodies that interfere with the activity of the antibody therapeutic and in some cases associate with adverse effects are of concern and are the focus of this discussion.Citation13–Citation15
Causes of Immunogenicity
There are many identified and purported causes of immunogenicity for antibody molecules.Citation16–Citation18 In order to minimize immunogenicity, it is recommended that as many of these factors are controlled as possible. Extrinsic factors such as aggregates and adjuvant-like contaminants are well known to cause issues and have been largely resolved by improvements in manufacturing and formulating practices.Citation19,Citation20 Extrinsic factors such as the co-medication of the patient, and the patient’s immunological status can also have profound effects on the reported immunogenicity of protein and antibody therapeutics.Citation10,Citation21 More subtle extrinsic factors that are difficult to eradicate are exemplified by the cytokine release syndrome induced upon administration of therapeutics such as anti-CD3.Citation22 Interestingly, the immune response to a chimeric hamster-mouse anti-CD3 antibody in a mouse model of experimental autoimmune encephalomyelitis (EAE) can be reduced by controlling the extent of cytokine release with cyclosporine A.Citation23 This result suggests that cytokine release is supporting a humoral immune response to the therapeutic by perhaps “short-circuiting” the adaptive helper T cell mediated response. Suppression of cytokine release may also partially explain the observation that co-medication with methotrexate reduces immune responses to antibodies and other protein therapeutics.Citation24–Citation26
Intrinsic factors also influence the immunogenicity of antibodies. Antibodies directed at cell surface markers are deemed to have a higher risk of immunogenicity than antibodies against soluble factors. The reasons for this are not completely understood but may be due to antigen internalization and subsequent processing and presentation by target cells.Citation27 Another intrinsic factor is the presence of carbohydrate side chains attached to the antibody via glycosylation sites conferred by the amino acid sequence of the light chain constant region, the heavy chain constant region or the V region itself.Citation28,Citation29 The presence of a galactose-alpha-1,3-galactose sugar within a carbohydrate structure on the Fab fragment of cetuximab was found to associate with severe anaphylactic reactions to the antibody. IgE antibodies specific for the galactose-alpha-1,3-galactose sugars were pre-existing in most of the donors, an uncontrollable extrinsic factor that contributed to the outcome.Citation30 Notably, when cetuximab was manufactured in a cell line that could not add galactose-alpha-1,3-galactose to the antibody carbohydrate (Chinese hamster ovary derived manufacturing cell line), the resultant product was much less immunogenic.Citation30 Other post-translational modifications to the antibody sequence may confer immunogenicity as well, such as glycation, deamidation and oxidation of amino acid side chains.Citation31,Citation32 Finally, the presence of CD4+ T helper cell epitopes have been described as correlating with immune responses to antibodies and other protein therapeutics.Citation16,Citation17,Citation33-Citation36
The presence of IgG subclass antibodies during immune responses is typically associated with helper T cell activity.Citation37 CD4+ T helper cells secrete cytokines that promote differentiation and isotype class switching by antigen-specific B cells. CD4+ T cells recognize linear sequential peptide fragments derived from the protein immunogen presented in the context of the donors’ HLA class II molecules. The activation, differentiation and migration of antigen-specific CD4+ T cells usually takes a few days after antigen exposure.Citation38,Citation39 This delay gives rise to the canonical “adaptive” antibody response profile characterized by the early temporal expression of IgM by antigen-specific B cells, with a subsequent isotype switching and affinity maturation event occurring once CD4+ T cells are activated. The presence of high affinity, IgG isotype, anti-idiotype antibodies therefore indicates the activity of antibody-specific CD4+ T helper cells. Antibody V region specific CD4+ T helper responses have been well characterized in both mouse and human models,Citation40–Citation43 and are the aim of vaccines that utilize lymphoma idiotype proteins as tumor-specific antigens.Citation44,Citation45 It has not been possible to demonstrate CD4+ T cell specific for isologous constant regions.Citation46 This result is likely due to the imprint of tolerance to the highly expressed constant region proteins. The exception is the ability to generate CD4+ T cell hybridomas to mouse constant region allotypes that represent novel immunoglobulin sequences in mouse strains that do not carry the identical allotype sequence.Citation47 Antibody V regions, by contrast, contain CDRs that are unique to each B cell and are usually present in vanishingly small quantities. This is especially true of the somatically mutated CDR3 regions. The germline portions of V regions have not been demonstrated to induce immune responses in mouse models.Citation40 Tolerance to novel V region sequences, as exemplified by somatically mutated CDR3 regions in particular, is incomplete or not present and V region specific CD4+ T cells are identifiable in the periphery.
CD4+ T cell responses include helper type responses that promote differentiation and activation of B cells and CD8+ cytotoxic cells (Th1, Th2 and T follicular helper cells), regulatory responses that suppress B cell and T cell (Tregs) responses, and inflammatory Th17-type responses. Signaling strength of the T cell receptor epitope has some impact on the subsequent differentiation of CD4+ T cells,Citation48,Citation49 but the biggest effect is provided by the microenvironment where the antigen-specific CD4+ T cell encounters antigen.Citation49,Citation50 The microenvironment is impacted by the presence and type of adjuvant used as well as the route of antigen administration. The differentiation of CD4+ T cells to effector cells has been shown to be somewhat reversible in that the presence of certain cytokines can skew differentiation in cells that have already established a particular lineage.Citation50,Citation51 This result is of interest as patient populations differ by their immunological status; for example the chronic inflammatory status of autoimmune patients may have an impact on the overall immune responses to administered therapeutics.
Antibody “Immunogenicity” may Reflect Homeostasis
It seems counterintuitive that fully human sequence derived antibodies would cause immune responses in humans. Immunoglobulins occur at mg/ml quantities in serum and initiate expression very early in ontogeny, when tolerance to self proteins is imprinted on the nascent immune system.Citation52 CD4+ T cells are tolerized or deleted in the thymus during development, and are anergized and deleted in the periphery upon contact with inappropriately expressed antigen.Citation53,Citation54 Multiple differential subsets of regulatory T cells control inappropriate responses to antigen in the periphery.Citation55,Citation56 B cells are tolerized by deletional and anergistic mechanisms during development, but can also rescue themselves by a process of receptor editing.Citation57 Receptor editing is the developmental process where B cells can rearrange their V region segments a second time, thereby altering specificity to avoid deletion.Citation58,Citation59 However, in spite of these mechanisms, human serum from non-diseased normal donors contains detectable levels of anti-idiotype antibody to a wide variety of autoantibodies.Citation60–Citation65 Anti-idiotype antibodies are akin to anti-drug antibodies against therapeutic mAbs, that is, they are antibodies with specificity for the unique V region of other immunoglobulin molecules. The presence of autoantibodies indicates that the tolerance system is not perfect and in fact, poly-reactivity to autoantigens re-emerges during the somatic hypermutation that takes place during an immune response.Citation66 Anti-idiotype antibodies specific for the newly arising potentially problematic poly-reactive antibodies may represent an additional mechanism for assuring tolerance to self proteins. Indeed, for certain autoimmune diseases, these IgG anti-idiotype antibodies may be protective as the absence of an anti-idiotypic response correlates with the presence of disease in Type I diabetes.Citation62 The presence of anti-idiotype antibodies is one proposed mechanism for the efficacy of IVIg in so many different autoimmune diseases.Citation64,Citation65 Therefore, mounting an antibody-mediated immune response directed at the combining sites of other antibodies likely is a normal, non-pathogenic event.Citation67,Citation68
If antibody-specific CD4+ T helper cells can be activated in normal donors, even under steady-state conditions that lead to non-pathogenic, anti-autoimmune antibodies, then it should be no surprise that administering large quantities of antibody carrying a single specificity could induce anti-idiotype neutralizing IgG responses in some patients. This effect would be especially pronounced if the antibody happens to carry along a CD4+ helper T cell epitope presentable by the patients’ HLA class II molecules in its V region. These types of immune responses to therapeutic antibodies are of most concern as they tend not to diminish with time and can impact efficacy and associate with pharmacokinetic and safety issues.Citation69,Citation70 It is only when autoantibodies, or anti-therapeutic antibody responses, induce clinical sequelae that we pay attention.
Fully Human Antibody Therapeutics can be Immunogenic
Humanized antibodies contain murine-sequence derived CDR regions that have been engrafted, along with any necessary framework back-mutations, into human sequence-derived V regions. Fully human sequence derived antibodies have no murine sequence, and are largely produced via two sources: phage display technologies and transgenic mice. The first fully human sequence-derived antibody to be approved for therapeutic use was adalimumab (Humira), a fully human IgG1 antibody specific for TNFalpha that was selected via phage display of human VH and VL sequences.Citation71 Recently, fully human sequence antibodies isolated from mice carrying genetic modifications such that the murine immunoglobulin genes were disabled and replaced with functional human immunoglobulin loci have been approved for therapeutic use.Citation9,Citation72 These antibodies undergo affinity maturation in vivo and therefore may represent a more naturally occurring set of sequences. The first of these, panitumumab (Vectibix), received approval for marketing in the US in 2006.Citation73 Interestingly, even fully human sequence derived antibodies can induce marked immune responses. Adalimumab has been described as inducing neutralizing responses in a subset of patients that varies depending on the disease and the therapy (5–89%).Citation74–Citation76 Anti-drug antibody has been shown to correlate with a lack of efficacy in some adalimumab treated patients, meeting the generalized criteria for a significant immune response. Another fully human antibody that can induce a marked immune response in human patients is golimumab (Simponi), a fully human anti-TNF antibody derived from genetically modified mice.Citation77 In rheumatoid arthritis patients co-medicated with methotrexate, a total of 16% of patients displayed anti-drug antibodies at follow up.Citation78 The presence of anti-golimumab antibodies correlated with reduced trough levels of circulating antibody, a worrisome clinical event that may impact efficacy. On the other hand, panitumumab displays very low levels of anti-drug antibodies, in the range of 3–4%.Citation79–Citation81 The use of a Biacore type assay that can identify anti-drug antibodies with low affinities showed a 4% incidence.Citation80 No clinical consequences have been reported due to the generation of anti-panitumumab antibodies, and anti-panitumumab antibodies only marginally affect pharmacokinetics.Citation81 Within the subclass of currently approved fully human antibodies, therefore, the percentage of negligible, tolerable and marked immune responses is not clearly different from humanized antibodies ().Citation5 This should not be surprising given the human immune system’s demonstrated ability to mount anti-idiotype antibodies. It does suggest that fully human antibodies may have reached the limit of our ability to select for reduced immunogenicity antibodies without more directed engineering.
Engineering Antibodies with a Reduced Potential for Inducing Anti-Drug Antibodies
Many antibody therapeutics do not induce an immune response in human subjects. While it is difficult to compare products directly, it is clear that some antibody therapeutics do not induce measurable, clinically relevant immune responses. Therefore, it should be possible to engineer antibodies that carry a reduced intrinsic potential for inducing responses. Identifying and modifying CD4+ T cell epitopes present in antibody idiotypes may lead to the creation of antibody variants with a reduced immunogenic potential. There are many methods currently in use to identify CD4+ T cell epitopes in the amino acid sequence of proteins. Most of these methods are algorithms constructed based on HLA class II binding motifs that have been defined within identified peptide epitopes. As predictive methods tend to over-predict the number of functional epitopes in a given sequence, identified peptides must be subsequently tested for activity in vitro.
We chose to search for CD4+ T helper cell epitopes in the sequence of immunoglobulin V regions using a human cell based assay. This empirical method was selected due to the utility of the assay in identifying CD4+ T cell epitopes in other proteinsCitation82,Citation83 with a very good specificity and sensitivity.Citation84 This method identifies CD4+ T cell epitope responses within a test population of community donors. Proliferative responses to each peptide are compiled for approximately 100 donors, and peptides that induce proliferative responses in a statistically significant number of donors are identified. Community donors are presumed to not carry immunological memory to therapeutic proteins, therefore this assay identifies CD4+ T cell epitopes likely to induce immune responses upon initial contact with the protein.
Since VH and VL regions are small, they comprise a total of approximately 70 15-mer peptides overlapping by 12 amino acids. Prescreening for putative CD4+ T cell epitopes by readily available online predictive methods identified potential epitope sequences for subsequent testing scattered throughout the V regions. It was therefore expeditious to simply test all of the peptides at once.
Modification of CD4+ T cell epitopes can also follow algorithm-based methods. Again, we chose to select epitope peptide variants based on functional binding assays of our proposed variant antibodies, and then chose to test the variant peptides in a separate iteration of the cell based assay, as described for other protein therapeutics.Citation82
Results
Humanization can reduce immunogenicity by eliminating CD4+ epitopes in V region frameworks.
Humanization of antibody V regions has greatly reduced their immunogenic potential in vivo. Whereas approximately 40% of chimeric antibodies containing human sequence-derived constant regions and murine-sequence derived V regions induce marked anti-drug antibody responses in vivo, only 9% of humanized and antibodies containing human constant regions and human sequence derived VH and VL framework regions do so.Citation5 When V region peptides derived from the chimeric antibody cetuximab were tested for CD4+ T cell epitopes in vitro, a number of peptides induced prominent responses within our tested population (), consistent with the hypothesis that the presence of CD4+ T cell epitopes correlates with immunogenic potential. However, when the humanized equivalent V region was tested as peptides, there was an absence of prominent CD4+ T cell responses. The humanized version retained the exact mouse sequence-derived CDR sequences; only the framework regions were substituted. This in vitro example shows that the process of humanizing murine V regions can have a substantial impact on the immunogenic potential of an antibody. The responses to the peptides were segregated into either framework-sequence only peptides or CDR-containing peptides where at least one amino acid in the peptide is contributed by a Kabat defined CDR region. Further analysis showed that cetuximab amino acid-sequence framework peptides were among those inducing prominent proliferative responses (). Responses to CDR sequence-containing peptides were also eliminated after humanization. The CDR containing sequences in cetuximab that induce the high responses contain contributions from murine frameworks, and when the similar but not identical sequences from the human frameworks used in the humanization were substituted, the overall response rate to the peptides was significantly reduced. CD4+ T cell proliferative responses to murine framework regions that are similar but not identical to human framework regions show that mouse antibody V regions are encountered by the human immune system as simply a protein immunogen, and that any tolerance induction that has occurred to human framework regions accords no special status to the mouse V regions.
CD4+ T cell epitopes occur only in CDR-containing regions of humanized antibody V regions.
V regions of antibodies are composed of two (VL-JL) or three (VH-DH-JH) genomic segments. Gene segments are rearranged imprecisely, and additional trimming and nucleotide addition can occur at the joints. Imprecision in the joining mechanism is one contributor to the amino acid diversity of antibody CDR regions.Citation85,Citation86 In addition, somatic hypermutation during affinity maturation introduces amino acid variations. The purpose of all this variation is to select for high affinity, antigen-specific antibodies. On the other hand, the creation of novel amino acid sequences by these mechanisms can lead to immune responses as the immune system can not establish tolerance to every single new amino acid sequence randomly generated in this fashion. So once again, the creation of immunogenic epitopes within CDR regions is expected to occur with some frequency.
If our hypothesis is correct, CD4+ T cell epitopes should occur only in CDR region-containing peptides. CDR regions as CD4+ T cell epitopes is not a novel concept to individuals working on idiotype vaccines for lymphomas.Citation87–Citation89 However, the idea that a therapeutic antibody may carry CD4+ T cell epitopes in the CDR regions has not been fully appreciated in the engineering of antibody therapeutics. To formally test our hypothesis and to determine the overall prevalence of CD4+ T cell epitopes in V regions, we assessed the CD4+ T cell epitope content of eight humanized and fully human therapeutic mAbs. Each set of immunoglobulin VH and VL region peptides was tested in vitro as 15-mer peptides in a human donor set of roughly 100 individuals per antibody. Two separate immunoglobulin VH and VL peptide sets were often analyzed together using peripheral blood cells from the same donor set. The datasets, once collected, were separated into germline-derived framework or CDR sequence-containing subsets as described above (). We found a complete absence of prominent CD4+ T cell responses to the framework-derived peptides, consistent with previously published observations.Citation40,Citation89 The data shown represents 188 framework peptides from three different VH subfamilies (VH1, VH3 and VH4) and is compiled responses from a total of 793 donors. The absence of CD4+ T cell proliferative responses by the tested donors demonstrates the power of the human immune response to establish tolerance to framework regions from these VH and VL subfamily members. The prominent CD4+ T cell responses identified in the eight antibodies tested all contained CDR amino acid sequence contribution. The average response rate for framework peptides was lower than the response rate for the CDR region-containing peptides (). The presence of CD4+ T cell epitopes only in the CDR region-containing peptides supports our hypothesis. Interestingly, even though the average response was increased for all CDR region-containing peptides, only a small subset induced prominent responses. For these eight tested antibodies, 2.5% (9 out of 364 tested peptides) of the peptides induced significant responses, with an average stimulation index of greater than 1.5 and average percent responses of greater than 10%. CDR sequence-containing epitope peptides were found in all three CDR regions of both VH and VL segments; no one CDR position was found to be more immunogenic among the eight antibodies tested. Responses to these epitope peptides tended to be associated with either numerous HLA class II alleles within our donor sets, or with widely expressed specific HLA DRB1 or HLA DQB1 class II alleles. The low number of CD4+ T cell epitopes found is consistent with the generally low immunogenicity of therapeutic antibodies, and also shows that CDR regions are not de facto immunogenic. That most CDR regions are not immunogenic suggests that immunogenic regions can be modified to remove this unwanted property; however, as the CDR regions confer affinity and specificity, the modification of these regions to reduce potential immunogenicity might be problematic.
Modification of CD4+ T cell epitope regions in antibody CDRs: is it possible?
Most fully human and humanized antibodies are not notably immunogenic in their approved applications. When an antibody does induce significant levels of clinically relevant anti-drug antibodies, a likely culprit is a CD4+ T helper cell epitope. As we have shown, CD4+ T cell epitopes in antibody V regions seem to occur only in CDR containing peptides. As most CDR regions are not overtly immunogenic, it should be possible to modify the epitope regions to reduce their immunogenic potential. The modification of CD4+ T cell epitopes within the V region of antibody molecules to produce a variant with the lowered immunological potential is termed “deimmunizing;” however, antibody CDR regions confer both affinity and specificity for antigen. Finally, modifications to amino acid sequences could potentially introduce de novo CD4+ T cell epitopes. To investigate whether deimmunization will be generally possible, we have characterized modified V region epitope peptides for three different antibodies in order to assess the potential for making an epitope response worse, better, or for having no impact on the activity of the epitope at all.
A total of five epitope sequences from the eight antibodies shown in were tested in the following way: the amino acid sequence of the epitope peptide was modified based on modeling and alanine scanning data (one epitope sequence) or based on guided selection of variants that were shown a priori to not impact affinity based on an empirical antigen-binding assay (four epitope sequences). A total of 202 peptide variants were tested in the in vitro proliferation assay. Note that changes in the epitope regions were designed to obviate impact on affinity only; no attempt was made to predict the critical HLA or T cell receptor (TCR) contact residues of the peptide epitope. This was due to the absence of any previously characterized HLA binding motifs in the epitope peptides and the subsequent lack of any guidance on TCR contacts. The variant peptides incorporated one or two amino acid changes, and were tested parametrically with at least two replicates of the unmodified parent. Proliferation and average stimulation index data was standardized to the parent epitope peptide responses for each set of variant peptides. Parent peptide response rates were averaged and set at 100%. The average response rate for the entire initial screening dataset for each antibody was defined as 0% in the standardized data. A standardized score for a variant should be as low as possible, as this actually represents the background response rate (typically 2-3%) within the screening dataset. Since the data for all of the variant peptides were now standardized, we compiled the information for the 202 peptides (). What was immediately apparent is that the vast majority of variant peptides had little effect on the average response rate (A) or the average stimulation index (B) of the epitope. The average response rate for all the variants was 70 ± 76% of the unmodified parent peptide, and the average stimulation index was 77 ± 92% of the parent. There were some individual variants that reduced the response, but there were also changes that significantly increased the response. Since both single and double amino acid modified peptide variants were tested, the data were separated based on the number of changes per peptide (). In , all variant peptides with a single modification are shown (n = 65). shows the response rates for all peptides containing two amino acid modifications (n = 131). The small arrows indicate the approximate location of the mean values for each dataset. For the singly modified peptides the average response rate of 94 ± 78% was even closer to the responses of the unmodified parent peptide control, showing that putting a single amino acid modification into an epitope sequence was unlikely to impact the immunogenicity. However, the average response rate for the doubly modified peptides dropped to 57 ± 74%. We were especially surprised to see that for the double mutations, there were more variants with 10% or less of the control response rate (27% of all tested variants peptides) than there were for the single mutants (14%) without a concomitant increase in the numbers of “worse” epitopes: only 6% of all the doubly modified peptide variants displayed response rates over 200% of control as compared to 14% of the singly modified peptides. This suggests that two changes in a CDR are actually safer than making a single change.
For four of the modified epitope regions, changes were made to the sequence based on data showing that the particular point mutation had a minimal effect on affinity and bioactivity. Even so, when the double amino acid mutants were incorporated into the CDRs of full length antibodies only about 40% of the mutants retained full bioactivity. While the variants targeting the combining sites of these antibodies were selected for a lack of impact on affinity, it is of interest to note that incorporation of two random mutations into a protein sequence are predicted to result in an active protein in 41% of all cases. This number is derived from the demonstration that a single mutation in a protein sequence results in an inactive protein 36% of the time.Citation90
Discussion
Taken together, these results suggest that modification of CDR regions to eliminate or reduce the immunogenic activity of CD4+ T cell epitopes is possible. We were successful in the absence of any consideration for the HLA or TCR binding contacts within these epitope peptides. In the best of all circumstances, with two amino acid changes within a single CDR guided by selection of modifications that do not impact binding, a total of approximately 11% of all variants should be effective deimmunizing changes. We arrived at this conclusion by assuming that 40% of the effective double mutants within an epitope will retain full bioactivity, and 27% of all double variant peptides tested will display 10% or less of the parent epitope response rate.
Fully human antibodies derived from transgenic mice are the current apex of therapeutic antibody development. However, as antibodies can be subject to biological regulation by the presence of anti-idiotype antibodies, the presence of a strong CD4+ T cell epitope in fully human antibody CDR regions will always be of concern. This is especially true where extrinsic factors are difficult to control. We have shown that novel CD4+ T cell epitopes within antibodies are found in the CDRs of V regions and therefore represent an engineering challenge to modify. The localization of epitopes to CDR regions also indicates that any human or humanized antibody may have a CD4+ T cell epitope. With careful guidance and a sustained effort, deimmunized fully active antibody variants can be created.
We look forward to the day when the identification and modification of CD4+ T cell epitopes in antibody molecules can be deduced by either specific or general guidelines. The current methods of trial and error are time consuming and add significantly to the cost of creation of lead antibody candidates.
The final step for antibody engineering will be to conclusively demonstrate in humans that the removal of CD4+ T cell epitopes reduces the immunogenicity of a particular antibody. This data will likely come from trials that provide information that can be realistically benchmarked to historical data. While there is scientific information to support the deimmunization of antibodies from mouse models, the sine qua non will be information from clinical trials.
Materials and Methods
Peptides.
All peptides were manufactured as consecutive 15-mers overlapping by 12 amino acids describing the amino acid sequence of both the VH and the VL regions. Peptides were manufactured by Mimotopes (Melborne, Australia). Peptides were pin synthesized, cleaved and lyophilized. Upon arrival, the peptides were resuspended in DMSO at approximately 2 mg/ml. After resuspension the peptide stocks were kept at −80°C.
CD4+ T cell proliferation assays.
Peripheral blood mononuclear cell (PBMC) samples were drawn from community donors at the Stanford Blood Center (Palo Alto, CA). The CD4+ T cell proliferation assay was performed as described.Citation84,Citation91 Briefly, dendritic cells were differentiated from human peripheral blood monocytes by culture in AIM V media (Invitrogen, Carlsbad, CA) plus GM-CSF and IL-4 for 5 days. Dendritic cells were activated by the addition of IL-1 and TNFα, and then collected for use on day 7. Autologous CD4+ T cells were isolated by negative selection from thawed PBMC samples (Stem Cell Technologies), and then co-cultured with irradiated dendritic cells and peptides (5 ug/ml) in AIM V media for 5 days. CD4+ T cells were used at 2 × 105 cells per well, with 2 × 104 dendritic cells as antigen-presenting cells. Proliferation was assessed on day 6. A donor was considered a responder for a specific peptide if the average proliferation of the experimental wells was 2.95 times higher than the control DMSO only wells. This cut-off value has been validated to return the highest sensitivity of positive versus false positive responses.Citation84 Validation of the cut off value was assessed by performing CD4+ T cell proliferation assays using peptides derived from a number of well-characterized protein antigens such as staphylokinase and HPV E6 and E7 and comparing the results to published T cell epitope mapping results. The positive control for the CD4+ T cell assay was tetanus toxoid included in 2-4 replicate wells at 1.25 ug/ml. The tetanus toxoid positive control must reach a stimulation index of at least 3.0 for the peptide proliferation results to be included in the database. All assay wells contained 0.25% DMSO, including the tetanus toxoid positive controls and the no-peptide negative controls.
Abbreviations
| IgG | = | immunoglobulin G |
| CDR | = | complementarity determining region |
| HLA | = | human leukocyte antigen |
| VH | = | immunoglobulin heavy chain variable region |
| VL | = | immunoglobulin light chain variable region |
Figures and Tables
Figure 1 CD4+ T cell responses to peptides derived from cetuximab (black squares) and humanized cetuximab (open circles) V regions. 15-mer peptides overlapping by 12 amino acids describing the VH and VL regions of cetuximab were tested for their ability to induce proliferative responses with CD4+ Tcells and dendritic cells from 87 community donors. peptides derived from humanized cetuximab were tested with 106 community donors.
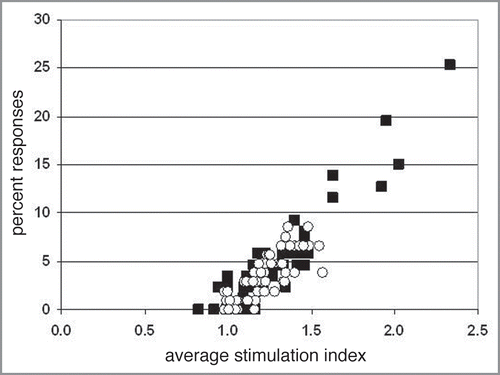
Figure 2 CD4+ T cell responses to framework and CDR-sequence-containing peptides derived from cetuximab (A) and humanized cetuximab (B) V regions. (A) 15-mer peptides overlapping by 12 amino acids describing the VH and VL regions of cetuximab were tested for their ability to induce proliferative responses with CD4+ cells and dendritic cells from 87 community donors. (B) peptides derived from humanized cetuximab were tested with 106 community donors. Framework sequence peptides are depicted by the open circles. CDR sequence-containing peptides are depicted by the black squares.
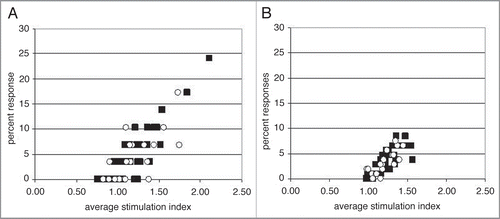
Figure 3 CD4+ T cell epitope responses to eight humanized and fully human antibodies. (A) Framework (circle; n = 188) and CDR region-containing (square; n = 364) peptide responses. (B) Average ± standard deviation for all framework and CDR region containing peptides tested.
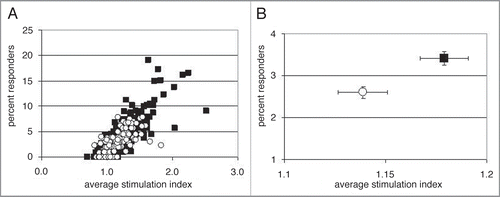
Figure 4 Reduction and exacerbation of in vitro immunogenicity by epitope amino acid sequence modifications. All proliferative responses rates to epitope variants were standardized to the unmodified parent epitope peptide response. (A) Distribution of standardized response rates of the variant peptides. (B) Distribution of standardized average stimulation index of the variant peptides.
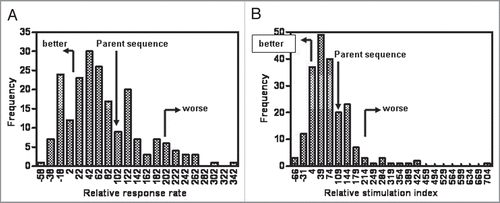
Figure 5 Two amino acid modifications in epitope sequences result in more potential deimmunized peptides. (A) Distribution of response rates to single point mutations. (B) Distribution of response rates to double mutations. (C) Box and whiskers plot standardized to the unmodified parent epitope peptide response. Arrow indicates the approximate location of the median.
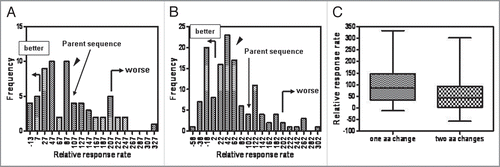
Table 1 Immune responses to fully human antibodies
References
- Wagner KS, Stickings P, White JM, Neal S, Crowcroft NS, Sesardic D, et al. A review of the international issues surrounding the availability and demand for diphtheria antitoxin for therapeutic use. Vaccine 2009; 28:14 - 20
- Silverstein AM. Clemens Freiherr von Pirquet: explaining immune complex disease in 1906. Nat Immunol 2000; 1:453 - 455
- Sgro C. Side-effects of a monoclonal antibody, muromonab CD3/orthoclone OKT3: bibliographic review. Toxicology 1995; 105:23 - 29
- Tsurushita N, Hinton PR, Kumar S. Design of humanized antibodies: from anti-Tac to Zenapax. Methods 2005; 36:69 - 83
- Hwang WY, Foote J. Immunogenicity of engineered antibodies. Methods 2005; 36:3 - 10
- Lonberg N. Fully human antibodies from transgenic mouse and phage display platforms. Curr Opin Immunol 2008; 20:450 - 459
- Lonberg N. Human antibodies from transgenic animals. Nat Biotechnol 2005; 23:1117 - 1125
- Hoogenboom HR. Selecting and screening recombinant antibody libraries. Nat Biotechnol 2005; 23:1105 - 1116
- Green LL. Antibody engineering via genetic engineering of the mouse: XenoMouse strains are a vehicle for the facile generation of therapeutic human monoclonal antibodies. J Immunol Methods 1999; 231:11 - 23
- Oldenburg J, Schroder J, Brackmann HH, Muller-Reible C, Schwaab R, Tuddenham E. Environmental and genetic factors influencing inhibitor development. Semin Hematol 2004; 41:82 - 88
- Lusher JM. Hemophilia treatment. Factor VIII inhibitors with recombinant products: prospective clinical trials. Haematologica 2000; 85:2 - 5
- Rup B. Immunogenicity and immune tolerance coagulation Factors VIII and IX. Dev Biol (Basel) 2003; 112:55 - 59
- Ritter G, Cohen LS, Williams C Jr, Richards EC, Old LJ, Welt S. Serological analysis of human anti-human antibody responses in colon cancer patients treated with repeated doses of humanized monoclonal antibody A33. Cancer Res 2001; 61:6851 - 6859
- Welt S, Ritter G, Williams C Jr, Cohen LS, Jungbluth A, Richards EA, et al. Preliminary report of a phase I study of combination chemotherapy and humanized A33 antibody immunotherapy in patients with advanced colorectal cancer. Clin Cancer Res 2003; 9:1347 - 1353
- Rosenberg AS. Immunogenicity of biological therapeutics: a hierarchy of concerns. Dev Biol (Basel) 2003; 112:15 - 21
- de Groot AS, McMurry J, Moise L. Prediction of immunogenicity: in silico paradigms, ex vivo and in vivo correlates. Curr Opin Pharmacol 2008; 8:620 - 626
- Schellekens H. How to predict and prevent the immunogenicity of therapeutic proteins. Biotechnol Annu Rev 2008; 14:191 - 202
- Holgate RG, Baker MP. Circumventing immunogenicity in the development of therapeutic antibodies. IDrugs 2009; 12:233 - 237
- Shire SJ. Formulation and manufacturability of biologics. Curr Opin Biotechnol 2009; 20:708 - 714
- Rosenberg AS. Effects of protein aggregates: an immunologic perspective. Aaps J 2006; 8:501 - 507
- Hendrickson JE, Desmarets M, Deshpande SS, Chadwick TE, Hillyer CD, Roback JD, et al. Recipient inflammation affects the frequency and magnitude of immunization to transfused red blood cells. Transfusion 2006; 46:1526 - 1536
- Chatenoud L, Ferran C, Legendre C, Thouard I, Merite S, Reuter A, et al. In vivo cell activation following OKT3 administration. Systemic cytokine release and modulation by corticosteroids. Transplantation 1990; 49:697 - 702
- Belmar NA, Lombardo JR, Chao DT, Li O, Ma X, Pong-Afar M, et al. Dissociation of efficacy and cytokine release mediated by an Fc-modified anti-CD3 mAb in a chronic experimental autoimmune encephalomyelitis model. J Neuroimmunol 2009; 212:65 - 73
- Joseph A, Munroe K, Housman M, Garman R, Richards S. Immune tolerance induction to enzyme-replacement therapy by co-administration of short-term, low-dose methotrexate in a murine Pompe disease model. Clin Exp Immunol 2008; 152:138 - 146
- Garman RD, Munroe K, Richards SM. Methotrexate reduces antibody responses to recombinant human alpha-galactosidase A therapy in a mouse model of Fabry disease. Clin Exp Immunol 2004; 137:496 - 502
- Antoni C, Kalden JR. Combination therapy of the chimeric monoclonal anti-tumor necrosis factor alpha antibody (infliximab) with methotrexate in patients with rheumatoid arthritis. Clin Exp Rheumatol 1999; 17:73 - 77
- Gogolak P, Rethi B, Hajas G, Rajnavolgyi E. Targeting dendritic cells for priming cellular immune responses. J Mol Recognit 2003; 16:299 - 317
- Arnold JN, Wormald MR, Sim RB, Rudd PM, Dwek RA. The impact of glycosylation on the biological function and structure of human immunoglobulins. Annu Rev Immunol 2007; 25:21 - 50
- Sheeley DM, Merrill BM, Taylor LC. Characterization of monoclonal antibody glycosylation: comparison of expression systems and identification of terminal alpha-linked galactose. Anal Biochem 1997; 247:102 - 110
- Chung CH, Mirakhur B, Chan E, Le QT, Berlin J, Morse M, et al. Cetuximab-induced anaphylaxis and IgE specific for galactose-alpha-1,3-galactose. N Engl J Med 2008; 358:1109 - 1117
- Eggleton P, Haigh R, Winyard PG. Consequence of neo-antigenicity of the ‘altered self’. Rheumatology 2008; 47:567 - 571
- Doyle HA, Gee RJ, Mamula MJ. Altered immunogenicity of isoaspartate containing proteins. Autoimmunity 2007; 40:131 - 137
- Baker MP, Jones TD. Identification and removal of immunogenicity in therapeutic proteins. Curr Opin Drug Discov Devel 2007; 10:219 - 227
- Chirino AJ, Ary ML, Marshall SA. Minimizing the immunogenicity of protein therapeutics. Drug Discov Today 2004; 9:82 - 90
- Pendley C, Schantz A, Wagner C. Immunogenicity of therapeutic monoclonal antibodies. Curr Opin Mol Ther 2003; 5:172 - 179
- Tangri S, LiCalsi C, Sidney J, Sette A. Rationally engineered proteins or antibodies with absent or reduced immunogenicity. Curr Med Chem 2002; 9:2191 - 2199
- Vinuesa CG, Tangye SG, Moser B, Mackay CR. Follicular B helper T cells in antibody responses and autoimmunity. Nat Rev Immunol 2005; 5:853 - 865
- Miller MJ, Safrina O, Parker I, Cahalan MD. Imaging the single cell dynamics of CD4+ T cell activation by dendritic cells in lymph nodes. J Exp Med 2004; 200:847 - 856
- Jenkins MK, Khoruts A, Ingulli E, Mueller DL, McSorley SJ, Reinhardt RL, et al. In vivo activation of antigen-specific cd4 t cells. Annu Rev Immunol 2001; 19:23 - 45
- Bogen B, Ruffini P. Review: to what extent are T cells tolerant to immunoglobulin variable regions?. Scand J Immunol 2009; 70:526 - 530
- Ostad M, Andersson M, Gruber A, Sundblad A. Expansion of immunoglobulin autoreactive T-helper cells in multiple myeloma. Blood 2008; 111:2725 - 2732
- Munthe LA, Os A, Zangani M, Bogen B. MHC-restricted Ig V region-driven T-B lymphocyte collaboration: B cell receptor ligation facilitates switch to IgG production. J Immunol 2004; 172:7476 - 7484
- Munthe LA, Corthay A, Os A, Zangani M, Bogen B. Systemic autoimmune disease caused by autoreactive B cells that receive chronic help from Ig V region-specific T cells. J Immunol 2005; 175:2391 - 2400
- Houot R, Levy R. Vaccines for lymphomas: idiotype vaccines and beyond. Blood Rev 2009; 23:137 - 142
- Kwak LW, Campbell MJ, Czerwinski DK, Hart S, Miller RA, Levy R. Induction of immune responses in patients with B-cell lymphoma against the surface-immunoglobulin idiotype expressed by their tumors. N Engl J Med 1992; 327:1209 - 1215
- Eyerman MC, Zhang X, Wysocki LJ. T cell recognition and tolerance of antibody diversity. J Immunol 1996; 157:1037 - 1046
- Guo W, Smith D, Guth A, Aviszus K, Wysocki LJ. T cell tolerance to germline-encoded antibody sequences in a lupus-prone mouse. J Immunol 2005; 175:2184 - 2190
- Boyton RJ, Altmann DM. Is selection for TCR affinity a factor in cytokine polarization?. Trends Immunol 2002; 23:526 - 529
- McHeyzer-Williams LJ, Pelletier N, Mark L, Fazilleau N, McHeyzer-Williams MG. Follicular helper T cells as cognate regulators of B cell immunity. Curr Opin Immunol 2009; 21:266 - 273
- Zhou L, Chong MM, Littman DR. Plasticity of CD4+ T cell lineage differentiation. Immunity 2009; 30:646 - 655
- Locksley RM. Nine lives: plasticity among T helper cell subsets. J Exp Med 2009; 206:1643 - 1646
- Schroeder HW Jr, Mortari F, Shiokawa S, Kirkham PM, Elgavish RA, Bertrand FE 3rd. Developmental regulation of the human antibody repertoire. Ann N Y Acad Sci 1995; 764:242 - 260
- Klein L, Hinterberger M, Wirnsberger G, Kyewski B. Antigen presentation in the thymus for positive selection and central tolerance induction. Nat Rev Immunol 2009; 9:833 - 844
- Dembic Z, Weiss S, Bogen B. Thymic selection of immunoglobulin idiotype specific T-cells. Thymus 1994; 22:141 - 152
- Parish IA, Heath WR. Too dangerous to ignore: self-tolerance and the control of ignorant autoreactive T cells. Immunol Cell Biol 2008; 86:146 - 152
- Vignali DA, Collison LW, Workman CJ. How regulatory T cells work. Nat Rev Immunol 2008; 8:523 - 532
- Ferry H, Leung JC, Lewis G, Nijnik A, Silver K, Lambe T, et al. B-cell tolerance. Transplantation 2006; 81:308 - 315
- Hertz M, Nemazee D. Receptor editing and commitment in B lymphocytes. Curr Opin Immunol 1998; 10:208 - 213
- Nussenzweig MC. Immune receptor editing: revise and select. Cell 1998; 95:875 - 878
- Gilles JG, Vanzieleghem B, Saint-Remy JM. Factor VIII Inhibitors. Natural autoantibodies and anti-idiotypes. Semin Thromb Hemost 2000; 26:151 - 155
- Pan ZJ, Anderson CJ, Stafford HA. Anti-idiotypic antibodies prevent the serologic detection of antiribosomal P autoantibodies in healthy adults. J Clin Invest 1998; 102:215 - 222
- Oak S, Gilliam LK, Landin-Olsson M, Torn C, Kockum I, Pennington CR, et al. The lack of anti-idiotypic antibodies, not the presence of the corresponding autoantibodies to glutamate decarboxylase, defines type 1 diabetes. Proc Natl Acad Sci USA 2008; 105:5471 - 5476
- Kenderov A, Minkova V, Mihailova D, Giltiay N, Kyurkchiev S, Kehayov I, et al. Lupus-specific kidney deposits of HSP90 are associated with altered IgG idiotypic interactions of anti-HSP90 autoantibodies. Clin Exp Immunol 2002; 129:169 - 176
- Blank M, Anafi L, Zandman-Goddard G, Krause I, Goldman S, Shalev E, et al. The efficacy of specific IVIG anti-idiotypic antibodies in antiphospholipid syndrome (APS): trophoblast invasiveness and APS animal model. Int Immunol 2007; 19:857 - 865
- Emmi L, Chiarini F. The role of intravenous immunoglobulin therapy in autoimmune and inflammatory disorders. Neurol Sci 2002; 23:1 - 8
- Wardemann H, Nussenzweig MC. B-cell self-tolerance in humans. Adv Immunol 2007; 95:83 - 110
- Jerne NK. Towards a network theory of the immune system. Ann Immunol 1974; 125:373 - 389
- Hebert J, Bernier D, Boutin Y, Jobin M, Mourad W. Generation of anti-idiotypic and anti-anti-idiotypic monoclonal antibodies in the same fusion. Support of Jerne’s Network Theory. J Immunol 1990; 144:4256 - 4261
- Bartelds GM, Wolbink GJ, Stapel S, Aarden L, Lems WF, Dijkmans BA, et al. High levels of human anti-human antibodies to adalimumab in a patient not responding to adalimumab treatment. Ann Rheum Dis 2006; 65:1249 - 1250
- Wagner CL, Schantz A, Barnathan E, Olson A, Mascelli MA, Ford J, et al. Consequences of immunogenicity to the therapeutic monoclonal antibodies ReoPro and Remicade. Dev Biol 2003; 112:37 - 53
- Mahler SM, Marquis CP, Brown G, Roberts A, Hoogenboom HR. Cloning and expression of human V-genes derived from phage display libraries as fully assembled human anti-TNFalpha monoclonal antibodies. Immunotechnology 1997; 3:31 - 43
- Lonberg N, Taylor LD, Harding FA, Trounstine M, Higgins KM, Schramm SR, et al. Antigen-specific human antibodies from mice comprising four distinct genetic modifications. Nature 1994; 368:856 - 859
- Jakobovits A, Amado RG, Yang X, Roskos L, Schwab G. From XenoMouse technology to panitumumab, the first fully human antibody product from transgenic mice. Nat Biotechnol 2007; 25:1134 - 1143
- Radstake TR, Svenson M, Eijsbouts AM, van den Hoogen FH, Enevold C, van Riel PL, et al. Formation of antibodies against infliximab and adalimumab strongly correlates with functional drug levels and clinical responses in rheumatoid arthritis. Ann Rheum Dis 2008;
- Bender NK, Heilig CE, Droll B, Wohlgemuth J, Armbruster FP, Heilig B. Immunogenicity, efficacy and adverse events of adalimumab in RA patients. Rheumatol Int 2007; 27:269 - 274
- West RL, Zelinkova Z, Wolbink GJ, Kuipers EJ, Stokkers PC, van der Woude CJ. Immunogenicity negatively influences the outcome of adalimumab treatment in Crohn’s disease. Aliment Pharmacol Ther 2008; 28:1122 - 1126
- Inman RD, Davis JC Jr, Heijde D, Diekman L, Sieper J, Kim SI, et al. Efficacy and safety of golimumab in patients with ankylosing spondylitis: results of a randomized, double-blind, placebo-controlled, phase III trial. Arthritis Rheum 2008; 58:3402 - 3412
- Kay J, Matteson EL, Dasgupta B, Nash P, Durez P, Hall S, et al. Golimumab in patients with active rheumatoid arthritis despite treatment with methotrexate: a randomized, double-blind, placebo-controlled, dose-ranging study. Arthritis Rheum 2008; 58:964 - 975
- van Cutsem E, Siena S, Humblet Y, Canon JL, Maurel J, Bajetta E, et al. An open-label, single-arm study assessing safety and efficacy of panitumumab in patients with metastatic colorectal cancer refractory to standard chemotherapy. Ann Oncol 2008; 19:92 - 98
- Lofgren JA, Dhandapani S, Pennucci JJ, Abbott CM, Mytych DT, Kaliyaperumal A, et al. Comparing ELISA and surface plasmon resonance for assessing clinical immunogenicity of panitumumab. J Immunol 2007; 178:7467 - 7472
- Muro K, Yoshino T, Doi T, Shirao K, Takiuchi H, Hamamoto Y, et al. A phase 2 clinical trial of panitumumab monotherapy in Japanese patients with metastatic colorectal cancer. Jpn J Clin Oncol 2009; 39:321 - 326
- Harding FA, Liu AD, Stickler M, Razo OJ, Chin R, Faravashi N, et al. A beta-lactamase with reduced immunogenicity for the targeted delivery of chemotherapeutics using antibody-directed enzyme prodrug therapy. Mol Cancer Ther 2005; 4:1791 - 1800
- Stickler M, Valdes AM, Gebel W, Razo OJ, Faravashi N, Chin R, et al. The HLA-DR2 haplotype is associated with an increased proliferative response to the immunodominant CD4+ T-cell epitope in human interferon-beta. Genes Immun 2004; 5:1 - 7
- Stickler M, Chin R, Faravashi N, Gebel W, Razo OJ, Rochanayon N, et al. Human population-based identification of CD4(+) T-cell peptide epitope determinants. J Immunol Methods 2003; 281:95 - 108
- Jung D, Giallourakis C, Mostoslavsky R, Alt FW. Mechanism and control of V(D)J recombination at the immunoglobulin heavy chain locus. Annu Rev Immunol 2006; 24:541 - 570
- Chowdhury D, Sen R. Regulation of immunoglobulin heavy-chain gene rearrangements. Immunol Rev 2004; 200:182 - 196
- Wen YJ, Lim SH. Different properties of T-cell epitopes within complementarity-determining regions 1 and 2 of idiotypic VH in B-lymphoma. Scand J Immunol 1999; 50:296 - 301
- Campbell MJ, Carroll W, Kon S, Thielemans K, Rothbard JB, Levy S, et al. Idiotype vaccination against murine B cell lymphoma. Humoral and cellular responses elicited by tumor-derived immunoglobulin M and its molecular subunits. J Immunol 1987; 139:2825 - 2833
- Baskar S, Kobrin CB, Kwak LW. Autologous lymphoma vaccines induce human T cell responses against multiple, unique epitopes. J Clin Invest 2004; 113:1498 - 1510
- Guo HH, Choe J, Loeb LA. Protein tolerance to random amino acid change. Proc Natl Acad Sci USA 2004; 101:9205 - 9210
- Stickler MM, Estell DA, Harding FA. CD4+ T-cell epitope determination using unexposed human donor peripheral blood mononuclear cells. J Immunother 2000; 23:654 - 660