Abstract
The potential for antibodies to act as “magic bullets” for treatment of human disease was recognized a century ago, but its full realization has began to occur only during the last decade. A key to their current success is the ability to make libraries of antibodies/B cells, isolate a single species, and engineer it to be safe, efficacious and of high quality. Despite this progress, major challenges to the effective prevention, diagnosis and treatment of a vast majority of diseases remain. Limited success in the development of effective vaccines against diseases such as AIDS and cancer reflects our incomplete understanding of how antibodies are generated and function. Only a miniscule number of antibodies are characterized out of the universe of antibodies generated by the immune system. Knowledge of antibodyomes—the complete sets of antibodies—could help solve these and other challenges.
Introduction
It has been observed since ancient times that humans who recovered from some diseases were resistant (immune) to subsequent infection; a fundamental concept emerged from these observations that one could acquire protection from such diseases by contracting a variant that was already attenuate.Citation1,Citation2 The first well documented successful vaccine, which was against smallpox, was based on such a variant, and had several essential features, including (1) attenuation of the immunogen pathogenicity; (2) virus replication that resulted in persistency of immunogen exposure; (3) conservation of structures shared by the immunogen and the infecting virus; (4) antigenicity, e.g., binding to B cell receptors; and (5) endogenous adjuvants that contributed to its overall immunogenicity, i.e., ability to elicit immune responses. During the last two centuries, these properties were optimized for a number of vaccines that are now successfully used for prevention of 27 diseases, all caused by microbes.
Vaccination has been used successfully for protection from some diseases, but not therapy of an already established disease. It took a century after the invention of the first well documented vaccine, and a paradigm change in science, for a new concept to emerge that immunized animals contain active substances (anti-body) that could be isolated and used for treatment or prevention of disease. This serum therapy was successfully used against diphtheria and other infectious diseases, and garnered the first Nobel prize in physiology or medicine to von Behring. Another century of work and a second paradigm change was required to isolate the active component (antibody) and improve its efficacy.Citation3
The invention of hybridoma technology for immortalization of repertoires (“libraries”) of murine B cellsCitation4 and phage displayCitation5 for generation of combinatorial antibody libraries from miceCitation6,Citation7 and humans,Citation8,Citation9 from which specific monoclonal antibodies (mAbs) can be isolated by panning or screening with an antigen, and the discovery of molecular biology techniques to improve antibody properties have resolved a number of challenges in the development of clinically useful antibody therapeutics.
mAbs are currently used successfully for treatment of a number of diseases, although these are mostly cancer and immune disorders.Citation3,Citation10–Citation16 Only one anti-infective mAb, palivizumab, is currently approved for marketing. Development of vaccine immunogens against some diseases such as AIDS has been a challenge,Citation17,Citation18 although vaccines against some viruses such as papilomavirus have been highly successful.Citation19 The reason for why the vast majority of diseases, notably AIDS and most cancers, cannot be prevented by vaccination, why some individuals are protected and others are not, and how the antigen/host interactions determine its immunogenicity is not well understood. What are the key features that determine success or failure? How can current obstacles be overcome? How can recent advances in technology, including high-throughput sequencing, be used to design better therapeutics and vaccines? How can individualized treatment be made more effective? Will be there a new paradigm change that could lead to conceptually new treatments? To help explore these and other questions, I present an overview of selected recent advances in the development of antibody-based therapeutics and vaccines, and discuss current studies of large sets of antibodies, ideally the complete set, i.e., the antibodyome.
Antibody-Based Therapeutics-Successes and Challenges
Twenty-four mAbs are currently approved by the USA Food and Drug Administration (FDA) for clinical use; most are for therapy of cancer and immune disorders and only one (palivizumab) is indicated for prophylaxis of an infectious disease. Several other antibodies are approved for use in the European Union (catumaxomab) and other countries (nimotuzumab). The number of mAbs entering clinical studies per year has increased significantly from a few in the late 1980s to 34 in 2006.Citation16 The success of antibody-based therapeutics is mostly due to the use of concepts and methodologies developed during the second paradigm change decades ago that resulted in dramatic improvement of three key features in candidate therapeutic antibodies required for FDA approval, i.e., safety, efficacy and quality.
Safety.
Therapeutic antibodies are relatively safe due primarily to their high specificity. Toxicities can result from the antibody effector function, including antibody dependent cell-mediated cytotoxicity (ADCC) and complement dependent cytotoxicity (CDC) after binding to antigen on tissues other than those intended. An example of this is the trastuzumab-associated cardiotoxicity that is potentiated when the antibody is used concurrently or sequentially with an anthracycline.Citation20 Binding to an intended target can also lead to undesirable side effects, especially by immunomodulatory antibodies that could be either suppressory or stimulatory. In a notable case, induction of a systemic inflammatory response characterized by a rapid induction of pro-inflammatory cytokines within 90 minutes of administration of a single dose of the stimulatory anti-CD28 mAb TGN1412 occurred in all six volunteers, leading to critical illness in 12 to 16 hours.Citation21 In another example, administration of suppressory anti-TNFα drugs, including infliximab, certolizumab pegol and adalimumab, can lead to infectious complications.Citation22
Importantly, safety concerns can lead to the withdrawal of the mAb from the market. The psoriasis drug efalizumab was recently withdrawn because of a potential risk of patients developing progressive multifocal leukoencephalopathy (PML), which is a rare, serious, progressive neurologic disease caused by the JC virus (JCV). More than 80% of the general population is infected with JCV. Why the virus becomes activated and causes disease only in minority of the treated patients is unknown, although typically PML occurs in people whose immune systems have been severely weakened. Thus, choosing the most appropriate animal model for toxicity testing is very important and species cross-reactivity should be included when identifying new candidate mAb therapeutics. If such a model doesn’t exist transgenic animals expressing the human antigen and surrogate antibody that is cross-reactive with the human homologous antigen in relevant animals can be used.Citation23
The administration of mAbs can result in adverse acute infusion reactions where cytokine release plays a pivotal role, but other not fully explained mechanisms such as complement activation could be involved; such reactions were reported for many mAbs including infliximab, rituximab, cetuximab, alemtuzumab, trastuzumab and panitumumab.Citation24 Infusional side effects for rituximab can result from release of cellular contents from lysed malignant B cells.Citation25 Administration of mAbs can also lead to hypersensitivity reactions, including anaphylactic shock and serum sickness.Citation22 Pre-existing IgEs that cross-react with therapeutic mAbs can increase the number and severity of such reactions, which can occur even with the first infusion of mAb. A notable example of this occurred with administration of cetuximab.Citation24 Hypersensitivity is often associated with immunogenicity.
Immunogenicity.
Immunogenicity of antibodies and other biologics can be a significant safety and efficacy issue.Citation22,Citation26–Citation31 Murine mAbs were used initially as candidate therapeutics in the 1980s, but their high immunogenicity resulted in high titers of human anti-mouse antibodies (HAMAs), and related toxicities and low potency. Development of the less immunogenic chimeric mAbs, which contain human Fc fragments, and humanized mAbs, which contain mouse complementarity determining regions (CDRs) grafted into human antibody framework, was critical for the clinical success of the products. Fully human antibodies exhibit low immunogenicity on average, and are currently the favored type of antibody in development, although most of the therapeutic antibodies approved for clinical use are still chimeric and humanized mAbs.
Immunogenicity can be influenced by factors related to antibody structure, composition, posttranslational modifications, impurities, heterogeneity, aggregate formation, degradation, formulation, storage conditions, as well as antigen properties, the patient’s immune system and disease status, concomitant medications, dose, route, time and frequency of administration especially when administered as multiple doses over prolonged periods.Citation30 Even fully human antibodies can elicit human anti-human antibodies (HAHAs). In one of the most studied cases of anti-TNFα mAbs, treatment with the fully human adalimumab resulted in HAHAs that varied from <1% to up to 87% for different cohorts of patients, protocols, disease and methods of measurement.Citation32
A likely mechanism for the immunogenicity of fully human mAbs involves the unique antibody sequences that confer antigen binding and specificity, but may appear foreign. Human therapeutic proteins can also break immune tolerance and aggregation can be a major determinant of antibody elicitation.Citation30 Aggregation can result in repetitive structures that may not require T cell help.Citation33 Another possible, but underappreciated, cause of immunogenicity is the antibody allotropy, i.e., the presence of T-cell epitopes within the amino-acid sequence of the antibody.Citation34 Although it was recently found that the IGHG1 polymorphism does not appear to play a major role in the elicitation of antibodies against infliximab, further analysis is required to determine whether G1 allotypes play a role for the immunogenicity of humanized or fully human mAbs.Citation35 Antibody immunogenicity may also affect efficacy through either the pharmacokinetic or neutralizing effects of the antibody responses that are dependent on a number of factors, including the affinity, specificity and concentration of the induced antibodies.Citation31 Because immunogenicity is an important factor in both safety and efficacy, significant efforts to predict and reduce immunogenicity of therapeutic mAbs are ongoing.Citation26–Citation29
Individual immune responses to antibody therapeutics vary widely. A key, and largely unanswered, question is what determines these variations. Despite extensive laboratory and clinical studies that were instrumental in delineating general concepts about critical factors involved in immunogenicity, it is impossible to predict the extent to which a novel therapeutic protein will be immunogenic in human patients. Little is known about the individual antibodies composing the polyclonal response to therapeutic antibodies. The germline antibody repertoire at any given time could be a major determinant of individual differences, and so knowledge of large portions of antibodies generated by the human immune system, preferably the complete set, i.e., the antibodyome, could ultimately help to predict individual immune responses to antibody therapeutics and other biologics.
Efficacy.
Some mAbs have revolutionized treatment of several diseases, e.g., rituximab for non-Hodgkin lymphoma, particularly in combination with chemotherapy.Citation25 Gemtuzumab ozogamicin and alemtuzumab also play an important role in the therapy of hematological malignancies.Citation36 Another example is trastuzumab as adjuvant systemic therapy for human epidermal growth factor receptor type 2 (HER2)-positive breast cancer.Citation37 Results from six trials randomizing more than 14,000 women with HER2-positive early breast cancer to trastuzumab versus non-trastuzumab-based adjuvant chemotherapy demonstrate that the addition of trastuzumab reduces recurrence by approximately 50% and improves overall survival by 30%.Citation38
On average, the efficacy of mAb therapeutics is not high and there is substantial individual variability. Although trastuzumab has clearly revolutionized the treatment of HER2 positive patients, half of the patients still have non-responding tumors, and disease progression occurs within a year in the majority of cases.Citation39 For patients with disease progression, combination with small molecules could be useful, e.g., the addition of the dual tyrosine kinase inhibitor of epidermal growth factor receptor (EGFR) and HER2 lapatinib to capecitabine was shown to provide superior efficacy for women with HER2-positive, advanced breast cancer progressing after treatment with anthracycline-, taxane- and trastuzumab-based therapy.Citation40 Current data do not support the use of trastuzumab for more than one year; the appropriate length of treatment, optimum timing and administration schedule are not known.Citation37 Like other mAbs, trastuzumab does not appear to efficiently cross the blood brain barrier, and it is unclear if the current practice of local therapy of the central nervous system and continued trastuzumab is optimal.Citation39
Anti-angiogenic therapies that target the vascular endothelial growth factor (VEGF), e.g., bevacizumab, and the VEGF receptor (VEGFR) are effective adjuncts for treatment of solid tumors, and are commonly administered in combination with cytotoxic chemotherapy. However, at least half of patients fail to respond to anti-angiogenic treatment of gliomas, and the response duration is modest and variable.Citation41 The use of bevacizumab plus paclitaxel as a first-line treatment of patients with metastatic breast cancer doubled median progression-free survival (PFS; 11.8 months vs. 5.9 months; hazard ratio = 0.60; p < 0.001) compared with paclitaxel alone; however, a statistically significant improvement in overall survival was not provided by the addition of bevacizumab, although a post hoc analysis demonstrated a significant increase in one-year survival for the combination arm.Citation42 The anti-EGFR mAbs cetuximab and panitumumab, either as single agents or in combination with chemotherapy, have demonstrated clinical activity against metastatic colorectal cancer, but appear to benefit only select patients with predictive markers of efficacy, including EGFR overexpression, development of skin rash, and the absence of a K-ras mutation.Citation43 In general, as single agents or in combination, therapeutic mAbs have produced only modest clinical responses in solid tumors.Citation44 There are no mAbs approved for treatment of a number of tumors, e.g., prostate cancer.Citation45
Mechanisms of low efficacy.
The mechanisms underlying the relatively low efficacy of some mAbs and the high variability of responses to treatment are not well known, but are likely to involve multiple factors. Pre-existing resistance or development of resistance is a fundamental problem. Various mechanisms including mutations, activation of multidrug transporters, and overexpression or activation of signaling proteins are operating as exemplified for EGFR-targeted therapies.Citation46 Another major problem is poor penetration into tissues, e.g., solid tumors. A related issue for full-size mAbs is poor or absent binding to regions on the surface of some molecules, i.e., existence of “steric barriers,” e.g., on the HIV envelope glycoprotein (Env).Citation47
Efficacy increase—current approaches.
New approaches are being developed to increase mAb efficacy, including enhanced effector functions, improved half-life; increased tumor and tissue accessibility, and greater stability; the methods used involve both protein- and glyco-engineering, and results to date are encouraging.Citation48,Citation49 mAbs that do not engage the innate immune system’s effector functions are being developed when binding is sufficient.Citation50 Multi-targeted antibodies are being developed and tested in clinical trials, e.g., an antibody targeting HER2/neu and CD3 with preferential binding to activating Fcγ type I/III-receptors, resulting in the formation of tri-cell complexes between tumor cells, T cells and accessory cells.Citation51 Bispecific and multispecific mAbs are currently being developed to a number of targets.
A promising direction is the modulation of immune responses by mAbs targeting regulators of T cell immune responses. The cytotoxic T lymphocyte antigen 4 (CTLA-4) present on activated T cells is an inhibitory regulator of such responses. Human antibodies that abrogate the function of CTLA-4 have been tested in the clinic and found to have clinical activity against melanoma.Citation52,Citation53 It appears that CTLA-4 blockade also enhanced NY-ESO-1 antigen-specific B cell and T cell immune responses in patients with durable objective clinical responses and stable disease suggesting immunotherapeutic designs that combine NY-ESO-1 vaccination with CTLA-4 blockade.Citation53 Therapeutic mAbs that mimic the natural ligand, e.g., the tumor necrosis factor-related apoptosis inducing ligand (TRAIL), have also been developed.Citation54
Currently second and third generation mAbs against already validated targets, e.g., HER2, CD20 and TNFα, are in clinical studies or already approved. Various approaches have been used to discover novel, relevant targets, but progress has been slow. Modifications of the standard panning procedures have been reported, including enhanced selection of cross-reactive antibodies by sequential antigen panningCitation55 and competitive antigen panning for focused selection of antibodies targeting a specific protein domain or subunit.Citation56,Citation57 To ensure better tissue penetration and hidden epitope access, a variety of small engineered antibody domains (about 10-fold smaller than IgG) are being developed.Citation3,Citation58 Knowledge of antibodyomes could be used for generation of semisynthetic libraries for selection of high-affinity binders of small size and minimal immunogenicity ().
Efficacy increase—fundamental challenges and antibodyomes.
A major lesson from the current state of antibody-based therapeutics is that gradual improvement in the properties of existing antibodies and identification of novel antibodies and novel targets is likely to continue in the foreseeable future.Citation3 A fundamental challenge has been to increase dramatically the efficacy of therapeutic antibodies and to apply them to many more diseases. Other major challenges are the development of effective personalized antibody-based therapeutics, and prediction of toxicity or potentially low efficacy in vivo. Knowledge of human antibodyomes undoubtedly could contribute to finding possible solutions to such challenges. New approaches based on high throughput screening and novel knowledge about antibodies already generated by the immune system against potential targets, e.g., in cancer patients, are also needed to significantly expand the number of potentially useful targets.
Quality.
A specific fundamental feature that distinguishes mAb and other biologics from small molecule drugs is their heterogeneity. Heterogeneity of mAbs is due to modifications such as incomplete disulfide bond formation, glycosylation, N-terminal pyroglutamine cyclization, C-terminal lysine processing, deamidation, isomerization, oxidation, amidation of the C-terminal amino acid and modification of the N-terminal amino acids by maleuric acid, as well as noncovalent associations with other molecules, conformational diversity and aggregation.Citation59 Tens of thousands of variants with the same sequence may co-exist (Kathleen Clouse, personal communication).
Development of high quality mAb therapeutics with minimal heterogeneity and contamination is essential for their safety and approval by FDA. Process development for production of a therapeutic antibody is a very complex operation involving recombinant DNA technologies, verification of a strong expression system, gene amplification, characterization of a stable host cell expression system, optimization and design of the mammalian cell culture fermentation system and development of an efficient recovery process resulting in high yields and product quality.Citation60 Titers in the range of 5–10 g/L or even higher, cell densities of more than 20 million cells/ml, and specific productivity of over 20 pg/cell/day (even up to 100 pg/cell per day) have been achieved.Citation61
Genetic delivery of therapeutic mAbs by in vivo production offers a new direction to increase antibody quality and reduce cost; three approaches can be used for the stable long-term expression and secretion of therapeutic antibodies in vivo: (1) direct in vivo administration of integrating vectors carrying a mAb gene, (2) grafting of ex vivo genetically modified autologous cells, and (3) implantation of an encapsulated antibody producing heterologous or autologous cells.Citation62 Knowledge of individual antibody repertoires could help predict potential problems and increase safety and efficacy of such long-term in vivo production of antibodies.
What is next?
Do we expect another series of breakthroughs in the near future to enable development of antibody-based therapeutics with dramatically improved efficacy against diseases? Are the currently used methodologies, and antibodies developed based on these methodologies, reaching their limit? Is it possible to produce conceptually new antibodies able to resolve other long-standing problems, including efficient oral delivery, penetration into solid tumors and low cost of production, that are major drawbacks of antibodies compared to small molecules? Is there value in increasing complexity by making multifunctional antibody based drugs, including nanoparticle conjugates with antibodies in various formats, that could result in novel therapeutics with unique and useful properties? An “omic” approach to antibodies, i.e., exploration of the antibodyome, could help answer some of these questions.
Antibodies Elicited by Vaccination
Vaccine safety and efficacy.
Preventive vaccines are typically administered to healthy children and adults both on small and large scales, which poses somewhat more stringent and specific safety challenges compared with antibody therapeutics. In addition, vaccine side effects could be more difficult to neutralize because of continuing immune responses compared with those due to administration of a therapeutic that could be stopped. In contrast to therapeutic antibodies, the recent history of vaccinology is not marked with significant successes, especially against challenging infectious agents, although hints of modest efficacy were recently reported for an investigational HIV vaccine regimen.Citation63
A notable exception is vaccines against HPV.Citation19 Gardasil and Cervarix were approved by FDA in 2006 and 2009, respectively, and both vaccines are also approved in numerous other countries. Cervarix provides a significant degree of cross-protection against HPV types 31, 33 and 45, as well as the two most common types 16 and 18, which raises the potential overall effectiveness of HPV vaccination from about 70% to 81–86%. It was specifically designed with a novel adjuvant, AS04, to deliver high and sustained levels of antibodies. It is generally well-tolerated, with pain, redness and swelling at the injection site, fatigue, fever, aching, headache, itching and rash or gastrointestinal disturbances being the most common symptoms after vaccination.
Influenza vaccines have been successfully used for more than half a century, but the products must be matched to the virus every year because of the high influenza variability. Progress in the development of an influenza vaccine that elicits antibodies against highly conserved structures has been slow, although there are promising recent findings.Citation64–Citation67 Influenza vaccines are generally safe, but large scale administration, e.g., with the H1N1 vaccine, still raises safety concerns. The swine flu vaccine administered in 1976 induced antibodies against host gangliosides, likely through molecular mimicry,Citation68 that could potentially lead to Guillain-Barre Syndrome. Whether the vaccine caused the disease, and how to protect against this possibility in the future, is debated. Knowledge of human antibodyomes could help in the design of novel vaccines eliciting potent broadly neutralizing antibodies (bnAbs) and prediction of possible side effects.
Therapeutic vaccines.
Attempts to develop therapeutic vaccines, especially against chronic diseases such as cancer, hold promise but have not yet been greatly successful. For example, therapeutic vaccines for lymphomas delivered disappointing results from Phase 3 clinical trials, although there are some indications of clinical efficacy. Several strategies are being developed to improve results, including optimization of antigen delivery and presentation, as well as enhancement of anti-tumor T cell function.Citation69 Another example is therapeutic vaccination against hypertension, which shows some efficacy but is much less effective compared with existing drugs; better immunogens or adjuvants are required so that potent antibodies with high titers are elicited.Citation70
Various approaches are being taken to develop effective therapeutic vaccines, including optimized antigen design, targeting dendritic cells in vivo, various combinations with therapeutics, adoptive T cell transfer and stem cell transplantation.Citation71,Citation72 Advancement in understanding of the structural basis for immunogenicity and immunodominance is needed to enable design of better immunogens.Citation73 Most current vaccine antigens are essentially the native macromolecules of pathogens that are adapted to evade, not induce, immunity. Armed with detailed structural information, one could engineer optimized antigens with immunodominant epitopes by design. To be efficacious, cancer vaccines must break immune tolerance to self antigens, which is a difficult task. A recent example of a vaccine immunogen designed to overcome this problem is the site-specific substitution of a single tyrosine residue of mouse TNFα by an unnatural amino acid (p-nitrophenilalanine), resulting in high-titer antibodies against multiple epitopes that were cross-reactive with wild type TNFα and protected mice against lipopolysaccharide-induced death.Citation74,Citation75 It was suggested that this approach could also be used for elicitation of antibodies targeting predefined specific epitopes,Citation74 but this remains a challenge. Knowledge of antibodyomes, especially germline ones, and how antibodies are elicited following B cell receptor (BCR) interactions with immunogens could help in the design of epitope-specific vaccines.
Guiding the immune system for enhanced elicitation of antibodies with known properties.
Currently approved vaccines were designed without knowledge of the specific sequences and epitopes of the antibodies to be elicited, but, in some cases, the sequences of potent bnAbs are known. For example, several relatively potent bnAbs against HIV-1, including b12, 2G12, 2F5 and 4E10, have been extensively characterized; however, numerous attempts to elicit these antibodies or similar antibodies targeting their epitopes have failed. I have hypothesized that HIV-1 and possibly other microbes have evolved a strategy to reduce or eliminate the immunogenicity of highly conserved functionally important structures containing the epitopes of bnAbs by using “holes,” i.e., absence of germline antibodies that can bind or maintain an efficient immune response to such epitopes, in the human germline BCR repertoire, which is tens of orders of magnitude smaller than all possible antibodies.
In support of this hypothesis, my colleagues and I have found that germline-like putative predecessors of b12, 2G12 and 2F5, including their bivalent formats, did not bind to any of the Envs tested, although the corresponding mature antibodies did.Citation76 Therefore, naïve B cells expressing germline predecessors of bnAbs may not bind structures containing their epitopes and initiate immune responses leading to their elicitation. I have hypothesized that such cells could be activated by using immunogens that may be different than the Env but capable of binding to the germline antibodies, i.e., to serve as primary immunogens. Combining of such primary immunogens with Env-based immunogens could lead to elicitation of bnAbs. This conceptually novel approach could be used not only for elicitation of bnAbs against HIV-1 but also against other diseases including cancer.
All known bnAbs against HIV-1 are highly divergent from the closest corresponding germline antibodies in contrast to bnAbs against SARS CoV,Citation77 Nipah and Hendra viruses (henipaviruses),Citation78 which cause acute infections. Thus, the maturation pathways of HIV-1-specific bnAbs could be much more complex than those of bnAbs against SARS CoV, henipaviruss and likely those for other microbes causing acute infections. Therefore, I have also hypothesized that identifying antibodies that are intermediates in the pathways to maturation could help design conceptually novel vaccine immunogens.Citation76,Citation79,Citation80 Such primary immunogens and one or more additional immunogens could help guide the immune system through the long complex pathways to maturation (). We identified several possible b12 intermediate antibodiesCitation80 and an antigen not related to the Env that binds to some of these antibodies and a germline-like b12 (Xiao, et al. unpublished data). This antigen could serve as primary immunogen and together with Envs could be used in novel candidate vaccines based on two or more immunogens; this approach is general and could be used in other cases with known mAbs (). Knowledge of antibodyomes undoubtedly can help elucidate complex maturation pathways and identify intermediate antibodies needed for the development of such vaccines.
Antibodyomes
Why antibodyomes?
About a decade ago the concept of the “immunome” was introduced as “…the totality of rearranged antibody and antigen receptor genes present in all living humans…”,Citation81 mostly for practical purposes, e.g., collection of known antibody sequences scattered at various laboratories. Later, the term immunome was generalized to denote all immune system related molecules, especially those involved in an immune response. As noted above, knowledge of the complete set of antibodies in an individual (antibodyome) and the antibody binding partners could help in the response to challenges in the development of therapeutics and vaccines. Sequencing of human genomes and the use of other-omic approaches have already demonstrated their utility. Recent advances in large scale sequencing now make exploration of the challenging antibodyome feasible.
Size and dynamics of antibodyomes—challenges.
The theoretical limit of the antibodyome size is the total number of B cells, which is about 10Citation10–10Citation11 for an adult human, although its diversity, i.e., the number of B cells expressing different antibodies could be significantly smaller. There are two additional major challenges—time dependence and tissue distribution. In contrast to the genome, which is relatively constant, the composition of the antibodyome changes relatively quickly with time; every day about 2% of the mature B cells and 30% of the immature ones are replaced by newly produced B cells that can express antibodies with different sequences. The tissue distribution poses an additional problem—only about 2% of the B cells are in the blood, and they traffic between various compartments. The size and turnover rate of the antibodyome varies with B cells subsets, age and health status. The existence of thousands of antibody variants with the same sequence further increases complexity.
Vast number of unknown antibodies.
The human antibodyome is large, but the immune system can generate only an extremely small portion of all antibodies. The theoretical upper limit for all possible antibodies could be as large as 20200∼10260. This number is unimaginably large, larger than the number of different antibody molecules that can fill the whole known universe. Even if we assume that only CDRs are involved in the diversification, i.e., amino acid residues at about 30 positions could be mutated to any of the 20 residues, then the number is still very large (∼1039); if we further assume that the mutations lead to only four of the most frequently used residues, then the number of possible different antibodies, 1018, is still 1013 times larger than the total number of antibody sequences (∼105) in the publicly available databases. We have limited knowledge of the possible number of different human antibodies still preserving the Ig fold at normal physiological conditions, and cannot even estimate what proportion is used for generation of functional antibodies. It is clear that our knowledge of the antibody sequence space and properties is miniscule compared to the unknown human antibodies.
Feasibility of exploring antibodyomes.
The task to sequence and characterize, at least in terms of binding, a number of antibodies that is many orders of magnitude higher than currently known is daunting, but may not be as impossible. If the speed of sequencing continues to increase with the same rate as in the past decade, one could speculate that in the near future we could explore personal antibodyomes as we are now beginning to study personal genomes. Importantly, antibody sequences are relatively short—about 400–450 bp for one variable domain. This nicely coincides with the read length for one of the most advanced high throughput sequencing methods (454 sequencing technology). We have recently used 454 sequencing and obtained about half a million sequences of V domains in a week after beginning the preparation of the DNA, although the analysis is still ongoing (Chen, et al. unpublished). In addition, all antibodies originate from germline antibodies, thus allowing evaluation of sequence quality (the sequences of all germline V, D and Jgenes are known) to a certain extent. For many purposes, knowledge of the complete sets of antibodies expressed by various subsets of B cells and those elicited in response to an antigen will be sufficient. The number of antibodies in such cases, even if measured in dynamics, could be significantly smaller than the entire antibodyome.
Antibodyomes of zebrafish and humans.
Recently, major efforts based on single cell sorting and PCR amplification of the antibody genes, followed by their cloning and sequencing, resulted in accumulation of new potentially useful information for a relatively large number of IgGs.Citation82,Citation83 (Haynes B, et al. unpublished). The heavy chain antibodyome of zebrafish, which has ∼3 × 105 antibody-producing B cells, was sequenced by using the 454 sequencing technology.Citation84 It was found quite unexpectedly that some fish were highly correlated (correlation coefficients up to 0.75) in their VDJ repertoires. Another unexpected finding was that many unique sequences were shared between individual fish (254 sequences between two fish and two—between five fish), suggesting converging evolution and the possibility of estimating evolutionary dynamics. Very recently, large scale (454) sequencing was used to explore human IgM diversity.Citation85 The total diversity of an antibody library was found to be at least 3.5 × 1010. We have sequenced a human IgG antibody library made from about 106 B cells and found that about 70% of the total number of V domain sequences are different (∼2 × 105 VHs and ∼6 × 103 VLs). (Chen, et al. unpublished data). The number of possible different antibodies assuming stochastic pairing is ∼109 which exceeds the number of B cells used. Therefore the diversity is good and additional improvement of selected antibodies by chain shuffling can be useful as it turned out to be the case. (Zhu, et al. unpublished data). These and other results illustrate the wealth of information, some quite unexpected but potentially useful, that could be obtained by exploring antibodyomes. We and other researchers are currently sequencing millions of human antibodies.
Several years ago, we initiated a project to sequence and characterize large number of antibodies in healthy and HIV-1-infected individuals (the HIV-1 antibodyome). We have used standard and 454 sequencing to analyze a large number of antibody heavy and light chains from HIV-1-infected and uninfected adults, and cord blood from newborn infants. (Chen, et al. unpublished). The results suggested new possibilities for identification of putative neutralizing antibodies and characterization of maturation pathways. We believe that knowledge of antibodyomes could help to better understand responses to infection and immunization, and contribute to the development of efficacious AIDS vaccine as well as to the development of therapeutic antibodies.
Possible implications.
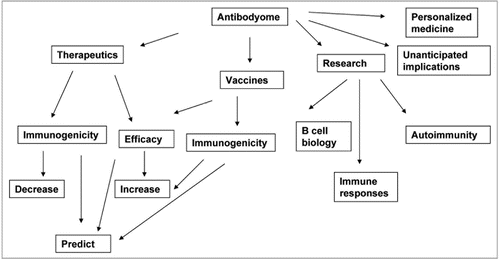
Antibodyomics is in its infancy. Implications for research, diagnosis, prevention and treatment of diseases are enormous, and include deeper understanding of B cell biology and immune diseases, new diagnostic methods based on individual antibodyomes, prediction of individual immune responses to immunization, infection, and therapeutics, and design of novel protein therapeutics (). Most implications cannot be predicted due to the very nature of an “omic” science.
Conclusions and Perspectives
The recent successes of mAbs for prophylaxis and therapy of some diseases raised hopes for their more effective and much wider use in the future. The history of vaccinology and recent successes against infectious agents also provide some grounds for optimism, but there are still many diseases for which there are no effective therapeutics and vaccines. New fundamental insights in the workings of the immune system are needed. Sequencing and characterization of antibodyomes could provide a wealth of knowledge that in combination with advances in other fields of science and technology could help in the development of better therapeutics and vaccines. Hopefully, it would be possible to accomplish the formidable task of exploring personal human antibodyomes during the next decade or two, and a new era of individualized “magic bullets” could be opened.
Glossary
| Antibodyome | = | the complete set of antibodies existing in an organism at any given time |
| Antibodyomics | = | studies of antibodies in aggregate, as a whole |
Figures and Tables
Figure 1 Generation of libraries of engineered antibody domains for selection of small-size high-affinity binders of minimal immunogenicity by grafting or by gene synthesis using information for human CDRs from antibodyome explorations.
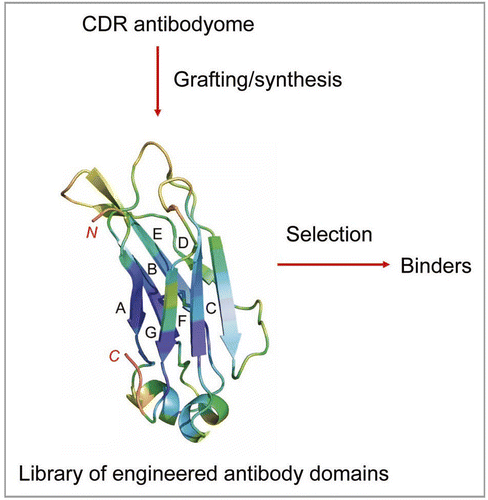
Figure 2 Enhanced elicitation of known antibodies by using primary and secondary immunogens. B cells expressing germline BCRs are activated by binding to a primary immunogen. The maturation pathway through intermediate antibody(ies) to the mature antibody is denoted by black lines. The intermediate BCR(s) cross-reacts with a secondary immunogen(s) leading to further diversification until the mature antibody is elicited. An example with eight mutations is shown where (1) denotes the space of all possible mutants for one mutation (about 200 positions × 20 residues = 4 × 103 possible mutants); (2) (denoted by –) corresponds to x i.e., about 16 × 106 etc., schematically showing the exponentially increasing number of possible mutants and complexity of possible maturation pathways.
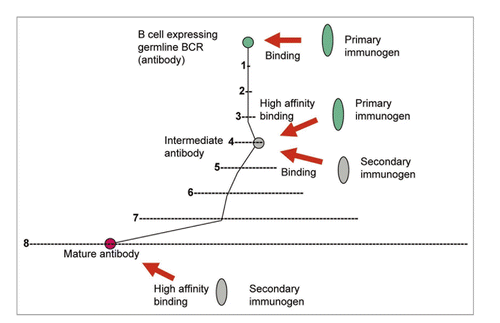
Figure 3 Possible implications of antibodyome exploration.
Acknowledgements
I would like to thank members of the group Protein Interactions especially Weizao Chen, Xiadong Xiao, Emily Streaker and John Owens for discussions, experiments and help. I also thank my collaborators on the HIV antibodyome project especially Barton Haynes for his vision and discussions as well as Robert Blumenthal, Chris Broder, Hana Golding and Ruth Ruprecht for stimulating discussions. I am especially indebted to Janice Reichert for helpful comments and suggestions. This study was supported by the NIH NCI CCR intramural program, Gates Foundation, the NIH intramural AIDS program (IATAP) and the NIH intramural biodefense program.
References
- Paul WE. Fundamental Immunology 2003; New York Lippencott Williams & Wilkins,
- Lombard M, Pastoret PP, Moulin AM. A brief history of vaccines and vaccination. Rev Sci Tech 2007; 26:29 - 48
- Dimitrov DS, Marks JD. Therapeutic antibodies: Current state and future trendsīs a paradigm change coming soon?. Meth Mol Biol 2009; 525:1 - 27
- Kohler G, Milstein C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 1975; 256:495 - 497
- Smith GP. Filamentous fusion phage: novel expression vectors that display cloned antigens on the virion surface. Science 1985; 228:1315 - 1317
- Ward ES, Gussow D, Griffiths AD, Jones PT, Winter G. Binding activities of a repertoire of single immunoglobulin variable domains secreted from Escherichia coli. Nature 1989; 341:544 - 546
- Huse WD, Sastry L, Iverson SA, Kang AS, ting-Mees M, Burton DR, et al. Generation of a large combinatorial library of the immunoglobulin repertoire in phage lambda. Science 1989; 246:1275 - 1281
- Burton DR, Barbas CF, Persson MA, Koenig S, Chanock RM, Lerner RA. A large array of human monoclonal antibodies to type 1 human immunodeficiency virus from combinatorial libraries of asymptomatic seropositive individuals. Proc Natl Acad Sci USA 1991; 88:10134 - 10137
- Marks JD, Hoogenboom HR, Bonnert TP, McCafferty J, Griffiths AD, Winter G. By-passing immunization. Human antibodies from V-gene libraries displayed on phage. J Mol Biol 1991; 222:581 - 597
- Schrama D, Reisfeld RA, Becker JC. Antibody targeted drugs as cancer therapeutics. Nat Rev Drug Discov 2006; 5:147 - 159
- Casadevall A, Dadachova E, Pirofski LA. Passive antibody therapy for infectious diseases. Nat Rev Microbiol 2004; 2:695 - 703
- Waldmann TA. Immunotherapy: past, present and future. Nat Med 2003; 9:269 - 277
- Carter PJ. Potent antibody therapeutics by design. Nat Rev Immunol 2006; 6:343 - 357
- Weiner LM, Dhodapkar MV, Ferrone S. Monoclonal antibodies for cancer immunotherapy. Lancet 2009; 373:1033 - 1040
- Ashkenazi A. Directing cancer cells to self-destruct with pro-apoptotic receptor agonists. Nat Rev Drug Discov 2008; 7:1001 - 1012
- Reichert JM. Monoclonal antibodies as innovative therapeutics. Curr Pharm Biotechnol 2008; 9:423 - 430
- Burton DR. Antibodies, viruses and vaccines. Nat Rev Immunol 2002; 2:706 - 713
- Montefiori D, Sattentau Q, Flores J, Esparza J, Mascola J. Antibody-based HIV-1 vaccines: recent developments and future directions. PLoS Med 2007; 4:348
- Lowy DR, Schiller JT. Prophylactic human papillomavirus vaccines. J Clin Invest 2006; 116:1167 - 1173
- Ewer SM, Ewer MS. Cardiotoxicity profile of trastuzumab. Drug Saf 2008; 31:459 - 467
- Suntharalingam G, Perry MR, Ward S, Brett SJ, Castello-Cortes A, Brunner MD, et al. Cytokine storm in a phase 1 trial of the anti-CD28 monoclonal antibody TGN1412. N Engl J Med 2006; 355:1018 - 1028
- Descotes J, Gouraud A. Clinical immunotoxicity of therapeutic proteins. Expert Opin Drug Metab Toxicol 2008; 4:1537 - 1549
- Dixit R, Coats S. Preclinical efficacy and safety models for mAbs: the challenge of developing effective model systems. IDrugs 2009; 12:103 - 108
- Chung CH. Managing premedications and the risk for reactions to infusional monoclonal antibody therapy. Oncologist 2008; 13:725 - 732
- Winter MC, Hancock BW. Ten years of rituximab in NHL. Expert Opin Drug Saf 2009; 8:223 - 235
- De Groot AS, McMurry J, Moise L. Prediction of immunogenicity: in silico paradigms, ex vivo and in vivo correlates. Curr Opin Pharmacol 2008; 8:620 - 626
- Stas P, Lasters I. Strategies for preclinical immunogenicity assessment of protein therapeutics. IDrugs 2009; 12:169 - 173
- Baker MP, Jones TD. Identification and removal of immunogenicity in therapeutic proteins. Curr Opin Drug Discov Devel 2007; 10:219 - 227
- Onda M. Reducing the immunogenicity of protein therapeutics. Curr Drug Targets 2009; 10:131 - 139
- Schellekens H. How to predict and prevent the immunogenicity of therapeutic proteins. Biotechnol Annu Rev 2008; 14:191 - 202
- Pendley C, Schantz A, Wagner C. Immunogenicity of therapeutic monoclonal antibodies. Curr Opin Mol Ther 2003; 5:172 - 179
- Emi AN, de Carvalho JF, rtur Almeida SC, Bonfa E. Immunogenicity of Anti-TNF-alpha Agents in Autoimmune Diseases. Clin Rev Allergy Immunol 2009;
- Hangartner L, Zinkernagel RM, Hengartner H. Antiviral antibody responses: the two extremes of a wide spectrum. Nat Rev Immunol 2006; 6:231 - 243
- Jefferis R, Lefranc MP. Human immunoglobulin allotypes: Possible implications for immunogenicity. mAbs 2009; 1:332 - 338
- Magdelaine-Beuzelin C, Vermeire S, Goodall M, Baert F, Noman M, Assche GV, et al. IgG1 heavy chain-coding gene polymorphism (G1m allotypes) and development of antibodies-to-infliximab. Pharmacogenet Genomics 2009; 19:383 - 387
- Castillo J, Winer E, Quesenberry P. Newer monoclonal antibodies for hematological malignancies. Exp Hematol 2008; 36:755 - 768
- Mariani G, Fasolo A, De BE, Gianni L. Trastuzumab as adjuvant systemic therapy for HER2-positive breast cancer. Nat Clin Pract Oncol 2009; 6:93 - 104
- Bedard PL, Piccart-Gebhart MJ. Current paradigms for the use of HER2-targeted therapy in early-stage breast cancer. Clin Breast Cancer 2008; 8:157 - 165
- Hall PS, Cameron DA. Current perspective—trastuzumab. Eur J Cancer 2009; 45:12 - 18
- Cameron D, Casey M, Press M, Lindquist D, Pienkowski T, Romieu CG, et al. A phase III randomized comparison of lapatinib plus capecitabine versus capecitabine alone in women with advanced breast cancer that has progressed on trastuzumab: updated efficacy and biomarker analyses. Breast Cancer Res Treat 2008; 112:533 - 543
- Norden AD, Drappatz J, Wen PY. Novel anti-angiogenic therapies for malignant gliomas. Lancet Neurol 2008; 7:1152 - 1160
- Sachdev JC, Jahanzeb M. Evolution of bevacizumab-based therapy in the management of breast cancer. Clin Breast Cancer 2008; 8:402 - 410
- Patel DK. Clinical use of anti-epidermal growth factor receptor monoclonal antibodies in metastatic colorectal cancer. Pharmacotherapy 2008; 28:31 - 41
- Tassev DV, Cheung NK. Monoclonal antibody therapies for solid tumors. Expert Opin Biol Ther 2009; 9:341 - 353
- Jakobovits A. Monoclonal antibody therapy for prostate cancer. Handb Exp Pharmacol 2008; 237 - 256
- Hopper-Borge EA, Nasto RE, Ratushny V, Weiner LM, Golemis EA, Astsaturov I. Mechanisms of tumor resistance to EGFR-targeted therapies. Expert Opin Ther Targets 2009; 13:339 - 362
- Chen W, Zhu Z, Feng Y, Dimitrov DS. Human domain antibodies to conserved sterically restricted regions on gp120 as exceptionally potent cross-reactive HIV-1 neutralizers. Proc Natl Acad Sci USA 2008; 105:17121 - 17126
- Presta LG. Molecular engineering and design of therapeutic antibodies. Curr Opin Immunol 2008; 20:460 - 470
- Jefferis R. Glycosylation as a strategy to improve antibody-based therapeutics. Nat Rev Drug Discov 2009; 8:226 - 234
- Labrijn AF, Aalberse RC, Schuurman J. When binding is enough: nonactivating antibody formats. Curr Opin Immunol 2008; 20:479 - 485
- Kiewe P, Thiel E. Ertumaxomab: a trifunctional antibody for breast cancer treatment. Expert Opin Investig Drugs 2008; 17:1553 - 1558
- Weber J. Ipilimumab: controversies in its development, utility and autoimmune adverse events. Cancer Immunol Immunother 2009; 58:823 - 830
- Yuan J, Gnjatic S, Li H, Powel S, Gallardo HF, Ritter E, et al. CTLA-4 blockade enhances polyfunctional NY-ESO-1 specific T cell responses in metastatic melanoma patients with clinical benefit. Proc Natl Acad Sci USA 2008; 105:20410 - 20415
- Bellail AC, Qi L, Mulligan P, Chhabra V, Hao C. TRAIL agonists on clinical trials for cancer therapy: the promises and the challenges. Rev Recent Clin Trials 2009; 4:34 - 41
- Zhang MY, Shu Y, Phogat S, Xiao X, Cham F, Bouma P, et al. Broadly cross-reactive HIV neutralizing human monoclonal antibody Fab selected by sequential antigen panning of a phage display library. J Immunol Methods 2003; 283:17 - 25
- Zhang MY, Choudhry V, Sidorov IA, Tenev V, Vu BK, Choudhary A, et al. Selection of a novel gp41-specific HIV-1 neutralizing human antibody by competitive antigen panning. J Immunol Methods 2006; 317:21 - 30
- Zhang MY, Dimitrov DS. Novel approaches for identification of broadly cross-reactive HIV-1 neutralizing human monoclonal antibodies and improvement of their potency. Curr Pharm Des 2007; 13:203 - 212
- Xiao X, Feng Y, Vu BK, Ishima R, Dimitrov DS. A large library based on a novel (CH2) scaffold: identification of HIV-1 inhibitors. Biochem Biophys Res Commun 2009; 387:387 - 392
- Liu H, Gaza-Bulseco G, Faldu D, Chumsae C, Sun J. Heterogeneity of monoclonal antibodies. J Pharm Sci 2008; 97:2426 - 2447
- Birch JR, Racher AJ. Antibody production. Adv Drug Deliv Rev 2006; 58:671 - 685
- Zhou JX, Tressel T, Yang X, Seewoester T. Implementation of advanced technologies in commercial monoclonal antibody production. Biotechnol J 2008; 3:1185 - 1200
- Samaranayake H, Wirth T, Schenkwein D, Raty JK, Yla-Herttuala S. Challenges in monoclonal antibody-based therapies. Ann Med 2009; 41:322 - 331
- Rerks-Ngarm S, Pitisuttithum P, Nitayaphan S, Kaewkungwal J, Chiu J, Paris R, et al. Vaccination with ALVAC and AIDSVAX to prevent HIV-1 infection in Thailand. N Engl J Med 2009; 361:2209 - 2220
- Sui J, Hwang WC, Perez S, Wei G, Aird D, Chen LM, et al. Structural and functional bases for broad-spectrum neutralization of avian and human influenza A viruses. Nat Struct Mol Biol 2009; 16:265 - 273
- DiMenna LJ, Ertl HC. Pandemic influenza vaccines. Curr Top Microbiol Immunol 2009; 333:291 - 321
- Ekiert DC, Bhabha G, Elsliger MA, Friesen RH, Jongeneelen M, Throsby M, et al. Antibody recognition of a highly conserved influenza virus epitope. Science 2009; 324:246 - 251
- Kubota-Koketsu R, Mizuta H, Oshita M, Ideno S, Yunoki M, Kuhara M, et al. Broad neutralizing human monoclonal antibodies against influenza virus from vaccinated healthy donors. Biochem Biophys Res Commun 2009; 387:180 - 185
- Nachamkin I, Shadomy SV, Moran AP, Cox N, Fitzgerald C, Ung H, et al. Anti-ganglioside antibody induction by swine (A/NJ/1976/H1N1) and other influenza vaccines: insights into vaccine-associated Guillain-Barre syndrome. J Infect Dis 2008; 198:226 - 233
- Houot R, Levy R. Vaccines for lymphomas: idiotype vaccines and beyond. Blood Rev 2009; 23:137 - 142
- Brown MJ. Therapeutic potential of vaccines in the management of hypertension. Drugs 2008; 68:2557 - 2560
- Chen X, Chang CH, Goldenberg DM. Novel strategies for improved cancer vaccines. Expert Rev Vaccines 2009; 8:567 - 576
- Baxevanis CN, Perez SA, Papamichail M. Combinatorial treatments including vaccines, chemotherapy and monoclonal antibodies for cancer therapy. Cancer Immunol Immunother 2009; 58:317 - 324
- Dormitzer PR, Ulmer JB, Rappuoli R. Structure-based antigen design: a strategy for next generation vaccines. Trends Biotechnol 2008; 26:659 - 667
- Grunewald J, Tsao ML, Perera R, Dong L, Niessen F, Wen BG, et al. Immunochemical termination of self-tolerance. Proc Natl Acad Sci USA 2008; 105:11276 - 11280
- Grunewald J, Hunt GS, Dong L, Niessen F, Wen BG, Tsao ML, et al. Mechanistic studies of the immunochemical termination of self-tolerance with unnatural amino acids. Proc Natl Acad Sci USA 2009; 106:4337 - 4342
- Xiao X, Chen W, Feng Y, Zhu Z, Prabakaran P, Wang Y, et al. Germline-like predecessors of broadly neutralizing antibodies lack measurable binding to HIV-1 envelope glycoproteins: Implications for evasion of immune responses and design of vaccine immunogens. Biochem Biophys Res Commun 2009; 390:404 - 409
- Zhu Z, Chakraborti S, He Y, Roberts A, Sheahan T, Xiao X, et al. Potent cross-reactive neutralization of SARS coronavirus isolates by human monoclonal antibodies. Proc Natl Acad Sci USA 2007; 104:12123 - 12128
- Zhu Z, Dimitrov AS, Bossart KN, Crameri G, Bishop KA, Choudhry V, et al. Potent neutralization of Hendra and Nipah viruses by human monoclonal antibodies. J Virol 2006; 80:891 - 899
- Chen W, Zhu Z, Zhang M, Macagno A, Prabakaran P, Owens J, et al. All known cross reactive HIV-1 neutralizing antibodies are highly divergent from germline and their elicitation may require prolonged periods of time. AIDS Res Hum Retroviruses 2008; 24:11 - 12
- Xiao X, Chen W, Feng Y, Dimitrov DS. Maturation pathways of cross-reactive HIV-1 neutralizing antibodies. Viruses 2009; 1:802 - 817
- Pederson T. The immunome. Mol Immunol 1999; 36:1127 - 1128
- Scheid JF, Mouquet H, Feldhahn N, Seaman MS, Velinzon K, Pietzsch J, et al. Broad diversity of neutralizing antibodies isolated from memory B cells in HIV-infected individuals. Nature 2009; 458:636 - 640
- Wardemann H, Yurasov S, Schaefer A, Young JW, Meffre E, Nussenzweig MC. Predominant autoantibody production by early human B cell precursors. Science 2003; 301:1374 - 1377
- Weinstein JA, Jiang N, White RA III, Fisher DS, Quake SR. High-throughput sequencing of the zebrafish antibody repertoire. Science 2009; 324:807 - 810
- Glanville J, Zhai W, Berka J, Telman D, Huerta G, Mehta GR, et al. Precise determination of the diversity of a combinatorial antibody library gives insight into the human immunoglobulin repertoire. Proc Natl Acad Sci USA 2009; 106:20216 - 20221
Internet links:
- http://aapredbook.aappublications.org/news/vaccstatus.shtml Status of licensure and recommendations for new vaccines
- href="http://www.immunize.org/journalarticles/toi_poten.asp Potential new vaccines
- http://www.fda.gov/CBER/vaccine/licvacc.htm Vaccines licensed for immunization and distribution in the USA
- http://www.accessdata.fda.gov/scripts/cder/drugsatfda/index.cfmfuseaction=Search.Search_Drug_Name Information for any FDA licensed drug including therapeutic antibodies
- http://www.clinicaltrials.gov A registry of federally and privately supported clinical trials conducted in the USA and around the world
- http://nobelprize.org/nobel_prizes/medicine/laureates/1901/behring-lecture.html Nobel Prize lecture of Emil von Behring