Abstract
Most therapeutic monoclonal antibodies (mAbs) licensed for human use or in clinical development are indicated for treatment of patients with cancer and inflammatory/autoimmune disease, and as such are designed to directly interact with the immune system. A major hurdle for the development and early clinical investigation of many of these immunomodulatory mAbs is their inherent risk for adverse immune-mediated drug reactions in humans such as infusion reactions, cytokine storms, immunosuppression and autoimmunity. A thorough understanding of the immunopharmacology of a mAb in humans and animals is required to both anticipate the clinical risk of adverse immunotoxicological events and to select a safe starting dose for first-in-human (FIH) clinical studies. This review summarizes the most common adverse immunotoxicological events occurring in humans with immunomodulatory mAbs and outlines non-clinical strategies to define their immunopharmacology and assess their immunotoxic potential, as well as reduce the risk of immunotoxicity through rational mAb design. Tests to assess the relative risk of mAb candidates for cytokine release syndrome, innate immune system (dendritic cell) activation and immunogenicity in humans are also described. The importance of selecting a relevant and sensitive toxicity species for human safety assessment in which the immunopharmacology of the mAb is similar to that expected in humans is highlighted, as is the importance of understanding the limitations of the species selected for human safety assessment and supplementation of in vivo safety assessment with appropriate in vitro human assays. A tiered approach to assess effects on immune status, immune function and risk of infection and cancer, governed by the mechanism of action and structural features of the mAb, is described. Finally, the use of immunopharmacology and immunotoxicity data in determining a minimum anticipated biologic effect Level (MABEL) and in the selection of safe human starting dose is discussed.
Introduction
Since the major indications for therapeutic monoclonal antibodies (mAbs), defined here as mAbs, fragments thereof and Fc-fusion proteins, are cancer and inflammatory/autoimmune disease,Citation1–Citation8 a large proportion of the products approved for human use () or in clinical development are designed to directly or indirectly modulate one or more aspects of the immune system (humoral, cell-mediated and innate immunity), and therefore have the potential to induce either immune suppression or immune activation. Therapeutic mAbs, including immunomodulatory mAbs, have generally proven to be safe, and in many cases, effective pharmaceuticals. Their toxicity is usually related to exaggerated pharmacology and can, in many cases, be predicted based on an understanding of the intended function of the mAb and the results of appropriate non-clinical studies in pharmacologically-responsive test systems; however the recent well-publicized adverse events observed with an immunomodulatory anti-CD28 superagonist mAb (TGN-1412) in a clinical trial in the United KingdomCitation9 have highlighted the potential toxicity of some therapeutic mAb approaches, as well as the potential pitfalls in interpreting and extrapolating non-clinical findings to the clinical setting. The profound toxic effects observed in healthy volunteers in this trial has emphasized the importance in considering all available biological data, including knowledge of the comparative pharmacological effects in animals and humans, when evaluating the safety of mAbs and in the selection of the starting dose in humans. Such data will be scrutinized more than ever by the regulatory authorities in the years to come.
For immunomodulatory mAbs, a thorough understanding of the relative immunopharmacology of a mAb in humans and animals, i.e., an understanding of comparative immunology, is required to (1) select a pharmacologically-relevant species for toxicology assessment, (2) to understand the limitations of the chosen animal species and whether in vivo safety data should be supplemented with in vitro assays with human cells, (3) to try and predict the immunological response and the risk of adverse immunotoxicological events occurring in humans and (4) to select a safe human starting dose for FIH clinical studies based on the minimum anticipated biological effect level (MABEL).Citation10–Citation13 This review aims to provide a comprehensive overview of potential non-clinical safety assessment strategies and practical considerations in defining the immunopharmacological and immunotoxicological potential of immunomodulatory mAbs, as well as strategies to minimize undesirable immunological effects, using a range of ex vivo, in vitro and in vivo tests.
General Toxicity of mAbs
There are multiple features of mAbs that govern their toxic potential. Their size and specificity, i.e., large protein drugs with high affinity that display highly selective binding to specific antigens or epitopes, reduce the potential for non-mechanism-based toxicity, although toxicity resulting from cross-reactivity with non-target antigens or non-specific binding remains a theoretical possibility. mAbs are proteins comprised of natural amino 3 acids and their metabolism is well-defined (catabolism into constituent amino acids) so they cannot be converted to reactive intermediates or toxic metabolites. Since they are limited by size to the extracellular space and do not interact directly with DNA, mAbs are not directly genotoxic. The primary toxicity of mAbs is due to exaggerated pharmacology related to blocking or enhancing the activities of the target molecule on the target cells or tissues, e.g., immunosuppression or immune activation with immunomodulatory mAbs or effects on wound healing with anti-angiogenic mAbs. Toxicity can also result from binding to target antigen in tissues other than those necessary for therapeutic effect. The skin toxicity (acneiform rash) observed with cetuximab (anti-EGFR; Erbitux)Citation14 and the cardiotoxicity observed with trastuzumab (anti-HER2; Herceptin)Citation15 have been attributed to the expression of the targeted antigens in skin and cardiac muscle respectively. The likelihood of toxicity occurring at non-therapeutic sites is dependent on not only the pharmacological effect on the target but also on the degree of target antigen expression and the role of the target in normal physiologic processes. If the biology and tissue distribution of the target are well-defined, potential target organs of toxicity can often be identified and predicted. In this context the choice of IgG isotype (1, 2 or 4) and the design of the Fc portion of the antibody to minimize or enhance Fc-mediated antibody-dependent cellular cytotoxicity (ADCC) and complement-dependent cytotoxicity (CDC) activity can have major influence on the toxicity to target and non-target tissues. A mAb specific for a target antigen that is expressed on cancer or auto-pathogenic cells but also highly expressed on normal cells and involved in normal cell function, e.g., rituximab (Rituxan), efalizumab (Raptiva), ipilimumab (anti-CTLA-4), adalimumab (Humira), cetuximab, trastuzumab is likely to have more potential toxicity than a mAb against an antigen that is either not expressed in humans, e.g., palivizumab (anti-RSV; Synagis), or that has a restricted tissue expression or function.
Immunopharmacology and Immunotoxicity of mAbs
Immunomodulatory mAbs (and Fc-fusion proteins) indicated for the treatment of inflammatory and autoimmune diseases or to prevent organ transplant rejection are often designed to bind directly to T cells, B cells, granulocytes, antigen-presenting cells (APCs; dendritic cells (DCs), macrophages) or other immune cells and mediators (cytokines, chemokines, growth factors, complement components) in order to deplete them or suppress their function, prevent their homing to lymphoid organs and inflammatory sites or induce anergy.Citation1–Citation5,Citation16,Citation17 Examples include muromonab-CD3 (Orthoclone OKT3), alefacept (Amevive), natalizumab (Tysabri), infliximab (Remicade), adalimumab, etanercept (Enbrel), efalizumab, abatacept (Orencia), eculizumab (Soliris) and rituximab ( and ). The majority of these anti-inflammatory mAbs are of the IgG1 isotype that have been pre-selected for low/no Fc effector function, although several are IgG2 or IgG4 isotypes. Unintended immune suppression as a consequence of immune cell depletion can also result from the administration of some cancer therapeutic mAbs (IgG1 subtype with strong Fc effector function) particularly those for the treatment of leukemias, e.g., rituximab, alemtuzumab.Citation18
In contrast, immunomodulatory mAbs that induce immune activation are primarily indicated for cancer.Citation19–Citation21 These mAbs are designed to break tolerance and augment anti-tumor immune responses by activating T cells, B cells, Natural Killer (NK) cells or DCs through direct agonism of an immune activation receptor, antagonism of an immune inhibitory receptor, or by depleting or inhibiting specific populations of T reg cells. Examples include mAbs directed at targets such as CTLA-4, GITR, OX40 and CD40. There are no immune-activating mAbs of this type that have been approved for marketing at this time, although there are a number in later stage clinical trials. There are also approved products such rituximab and alemtuzumab of the IgG1 isotype, where a primary mode of action is tumor cell cytotoxicity as a consequence of immune activation triggered via Fc-mediated binding such as ADCC and CDC. In ADCC, mAbs interact directly with FcγR (CD16, CD32a)-expressing cells such as NK cells, macrophages, B cells, DCs, neutrophils and eosinophils leading to cellular activation, target cell killing and release of pro-inflammatory cytokines, e.g., TNFα, IFNγ, IL-6. In CDC, mAbs interact with the C1q component of complement, leading to activation of the complement system and release of components (anaphylatoxins and opsonins) that can directly interact with receptors on immune cells (C3aR, C5aR, CR1, CR3) leading to their activation, migration and other effects.
Many of the immunomodulatory effects of mAbs are desirable and intended immunopharmacology that is required for clinical efficacy. However, activation or suppression/depletion of non-target immune cells and mediators, or permanent non-reversible changes to immune target cells/pathways, or any unintended sequelae of the intended pharmacology, e.g., cell and tissue injury, inflammation, ‘cytokine storms,’ tumor lysis syndrome, infection and cancer, autoimmunity, hypersensitivity, would be considered to be or reflect immunotoxicity. These often adverse consequences of immune modulation by mAbs have recently been reviewedCitation22,Citation23 and are discussed further below. Such immunotoxicity can result from exaggerated or prolonged activity of the mAb binding to the desired target antigen on the desired target cells/mediators, modulating a target with pleiotropic immune functions, including those whose modification is not required for therapeutic benefit, or modulating a target that is also expressed on non-immune cells or other immune cells besides those that are the intended therapeutic focus. Some of these immunological safety concerns can be reduced or circumvented by rational mAb design, e.g., through the use of an ‘inert’ IgG isotype with little or no effector function, or by screening mAb candidates for reduced cytokine release, DC activation and immunogenicity potential.
Adverse effects of immunosuppression
Generalized immunosuppression results from chronic administration of anti-inflammatory mAbs that are designed to reduce the activity of T cells and B cells, and often given in conjunction with other immunosuppressive drugs, e.g., methotrexate or steroids. This immunosuppression, if widespread, pronounced and prolonged, can lead to an increased risk of opportunistic bacterial, fungal or parasitic infection, chronic viral infection, e.g., EBV, CMV, or virally-induced cancers, e.g., lymphoma, skin cancer, cancer of the lips, Karposi’s sarcoma, hepatocellular carcinoma, cervical cancer. RA patients treated chronically with anti-TNFα biologics such as infliximab, adalimumab or etanercept are at increased risk for infection with Mycobacterium tuberculosis, Listeria monocytogenes, Salmonella and other facultative intracellular pathogens, opportunistic pathogens such as Pneumocystis carinii, and for certain types of cancer, e.g., lymphomas/carcinomas.Citation24 Frequent infections are also observed in patients treated with alemtuzumabCitation25 and rituximab.Citation26 Chronic treatment of MS patients with the anti-VLA-4 mAb natalizumab as a monotherapyCitation28 or in combination with IFNβCitation27 may increase the risk of progressive multifocal leukoencephalopathy (PML) caused by polyoma JC virus. Natalizumab is designed to inhibit inflammatory T cell migration to the brain, and the increased incidence of PML may be due to reduced homing of virus-clearing T helper and cytotoxic T cells to the brain.Citation29 PML has also recently been observed in a small number of psoriasis patients treated with efalizumab, an anti-CD11a (LFA-1) mAb that also affects lymphocyte recirculation and has been withdrawn from the market, and more recently with rituximab, which depletes B cell subsets.Citation30
mAbs for cancer therapy, e.g., alemtuzumab, rituximab, are often designed to kill leukemia cells via ADCC and CDC. However, the molecules recognized by these mAbs might also be expressed on normal lymphocytes/myeloid cells and other tissue types, and hence undesirable cytopenia and immunosuppression (immunotoxicity) and tissue injury can result.Citation25,Citation26
Adverse effects of immune activation
Some mAbs are designed to activate immune cells such as T cells, NK cells, B cells and DCs. Such activation, particularly if strong and polyclonal (and persistent due to the long half-life of mAbs), could lead not only to the desired activation of cancer-specific immune cells, but also to the undesirable activation of autopathogenic cells and development of autoimmunity observed with alemtuzumab,Citation31 anti-CTLA-4 ipilizumabCitation32 and anti-TNFα biologics in a small number of patients.Citation33 There is also the theoretical possibility that immune-activating mAbs could increase allergic responses, e.g., asthma, urticaria, rhinitis to common environmental and food allergens, although this has not been reported.
Immunomodulatory mAbs may also produce infusion and hypersensitivity reactions. These are generic terms describing a set of related clinical and laboratory findings that can be caused by several immune-mediated mechanisms, including allergic reactions, pseudoallergic reactions, and cytokine release syndrome (CRS).Citation34 True allergic reactions, which are mediated by anti-drug IgE, require prior exposure to the mAb and consequently do not occur on the first infusion, except in rare cases where patients have pre-existing antibodies that cross react with the drug.Citation35 Pseudoallergic reactions (IgE-independent reactions mediated possibly by direct immune cell and complement activation) and CRS both occur primarily on the first infusion of drug, although they can also occur on subsequent administrations. The symptoms of all three types of immunologically-mediated infusion reactions overlap, making it difficult to identify the cause without additional laboratory work. Many approved mAbs carry a black box warning for infusion reactions, particularly CRS, which is associated with systemic increases in cytokines including TNFα, IFNγ and IL-6. CRS as a consequence of mAb infusion may be related to the intended immunopharmacological activity of the mAb and mediated by target-specific binding that either triggers cellular activation, leading to cytokine release, e.g., OKT3 or TGN1412, or, when associated with Fc-mediated effector functions such as ADCC and CDC, leads to cellular lysis and cytokine release, e.g., rituximab, alemtuzumab. CRS can be mild to life-threatening and characterized by fever, chills, nausea, rash, myalgia, flushing, vascular leak, shortness of breath or hypotension.Citation34 As indicated above, many of these clinical symptoms are also seen in association with hypersensitivity or pseudoallergic reactions, making it difficult to distinguish these two reactions without additional data. In some cases, oral drugs such as diphenhydramine, acetaminophen and methylprednisolone are administered prior to the mAb to reduce the effect and symptoms of infusion reactions.
Assessment of Risk of Immunotoxicity in Humans
The risk of immunotoxicity in humans, especially for immunomodulatory mAbs with one or more risk factors, e.g., novel target or mechanism of action (MoA), agonist activity, steep dose-response curve, poorly predictive animal models, needs to be carefully and thoroughly assessed in non-clinical studies in vitro and in vivo. Understanding the risk of immunotoxicity in humans and the ability of the animal model(s) to predict these effects requires defining and comparing the immunopharmacology in human and animal systems by performing in silico, in vitro and in vivo immunopharmacology and toxicology studies. Immunotoxic effects in humans are sometimes predictable or at least not unexpected based on the MoA, e.g., autoimmunity with the immune-activating anti-CTLA-4 or infections with TNFα inhibitors, but in other cases predicting immunotoxicity is difficult, e.g., autoimmune thyroiditis (Graves disease) in MS patients administered Campath.Citation31 It should be noted that some immunotoxicity may be clinically acceptable depending on the risk/benefit ratio to the patient. Some risk of decreased host defense due to mAb-induced immunosuppression may also be deemed acceptable depending on the indication.
General Approach to Safety Testing of mAbs
Non-clinical safety testing programs for mAbs must be rationally-designed with a strong scientific understanding of the product, including its method of manufacture, purity, sequence, structure and class/isotype, pharmacological and immunological effects, the biology of the target and intended clinical use, e.g., indication, patient population and dosing regimen. The conventional development strategies applied to new chemical entities (NCEs; typically small molecular compounds), which are usually clearly-defined and prescriptive, i.e., containing studies to provide data on genetic toxicology, single and repeat dose toxicology, absorption, distribution, metabolism and excretion (ADME), safety pharmacology, carcinogenicity and reproductive toxicology, are generally inappropriate for mAbs, but may be required if novel chemical linkers and chelators or modified amino acids form part of the product. Non-clinical safety studies with mAbs, as with all pharmaceuticals, aim to identify possible causes of toxicity prior to undertaking clinical studies. Such studies should be designed to define safe dosing regimens for clinical trials, including recommending a safe starting dose for FIH studies, identify target organs for toxicity and reversibility of any toxic effect, characterize the pharmacological response and how it relates to any observed toxicity, identify immunological and toxicological safety parameters for clinical monitoring and determine the kinetics of the mAb and how they relate to toxicity and efficacy.Citation36 These studies may comprise in vivo studies in animals, as well as in silico studies and in vitro and ex vivo studies with cells and tissues as discussed in the following sections. illustrates how these tests can be applied throughout the development timeline for a mAb.
Design of a Non-Clinical Safety Assessment Program for an Immunomodulatory mAb
A starting point in the design of a non-clinical safety testing program is to refer to the guidelines recognized or published by the International Conference on Harmonization (ICH) and regulatory bodies such as the European Medicines Agency (EMA) and the US Food and Drug Administration (FDA). Some of these guidelines, although often not specifically designed to address mAb safety, can provide important guidance on relevant test strategies/models and regulatory expectation for mAb safety assessment; guidelines relevant to mAb safety assessment are shown in . ICH guidelines take precedence over regional guidance unless the regional guidance includes topics not addressed in ICH guidance.
The ICHM3 document, for which a step 4 update was recently released,Citation37 provides guidance for the timing, duration and dose levels for non-clinical safety assessment of all pharmaceuticals, but does not provide extensive information on design of the assessment program. The most important points of reference for the conduct of non-clinical safety assessment of mAbs are the ICHS6 guideline relating specifically to biotechnology-derived products,Citation38 including a newly released Addendum (step 2)Citation39 and the FDA Points to Consider (PTC) document.Citation40 Very little guidance on immunotoxicity assessment is provided in ICHS6, the FDA guidance or in ICHM3, which references ICHS8, Immunotoxicity Testing for Human Pharmaceuticals.Citation41 Although the ICHS8 Immunotoxicity guideline41 states that it does not relate to biotechnology-derived products, many of the principles and testing strategies are relevant to mAbs. In addition to ICHS8, there is a prior FDA guidelineCitation42 (Immunotoxicology Evaluation of Investigational New Drugs) that includes a section on drug allergy (specifically excluded in ICHS8), a draft guidance on immunotoxicity testing published by the Japanese Health AuthorityCitation43 and an EMEA guideline that specifically deals with immunogenicity of recombinant proteins, including mAbs.Citation44
In addition to these guidelines, reference to the published data on marketed mAb products such as European Public Assessment Reports (EPARS), Summary Basis of Approvals (SBAs), Summary of Product Characteristics (SPCs), Scientific Discussions and other documents relating to these products published by EMA and FDA, as well as literature reviews and internet searches are also very useful in providing an awareness of regulatory expectations and identifying suitable strategies, test systems, e.g., in vivo and in vivo biological and immunological models, and reagents to aid in program and study design. A firm understanding of the regulatory requirements for mAbs and related products will ensure that the most sensitive and regulatory-compliant test systems are utilized to maximize the chances of gaining regulatory approval for clinical testing or market authorization in the shortest time-frame.
Circumventing Immunotoxicity by Rational mAb Design
The choice of mAb structure, e.g., choice of IgG isotype for natural mAbs, domains for chimeric molecules, or whether a whole mAb or fragment should be used, can influence certain aspects of immunopharmacology and hence these can be rationally-chosen depending on the indication and desired pharmacological activity of the mAb. There are four isotypes of IgG in humans: IgG1, IgG2, IgG3 and IgG4 (). Only IgG1, IgG2 and IgG4 are used therapeutically since these isotypes bind the neonatal Fc receptor (FcRn) and hence have an extended half-life in humans (about 20 days).Citation45,Citation46 IgG3 shows only low affinity binding for FcRn and consequently has a half-life of only 6–8 d hence mAbs are rarely developed on an IgG3 framework. IgG1, IgG2 and IgG4 differ in their binding capacity to activating FcγRs (FcγRIIIA/CD16 and FcγRIIA/CD32A) on immune effector cells, e.g., NK cells, phagocytes, and in their ability to induce ADCC or bind the first C1q component of the classical complement pathway and mediate CDC ().Citation45 The cellular expression and function of FcγRs has recently been reviewed.Citation47 IgG1 (and IgG3) bind all FcγRs and fix complement and thus have the greatest potential for Fc-mediated effector function (). IgG4 and IgG2 on the other hand do not bind or bind weakly to FcγRs and hence have little or no effector function, although IgG2 can bind more strongly to certain allelic forms of FcγRIIA (131H and 131R) and FcγRIIIA (V158) in some individuals. IgG2 has very poor complement fixation activity whereas IgG4 does not fix complement ().Citation45–Citation47 Protein engineering makes it possible to create chimeric molecules that have binding and functional characteristics not observed in nature, or to optimize functional characteristics of domains like the Fc region to increase their binding or effector functions beyond that seen in the parent isotype. It is important to consider these structural modifications when evaluating the risks of such molecules.
When targeting inflammatory diseases, it is undesirable to have mAb-mediated activation of immune cells (NK cells, phagocytes, DCs) and induction of cytokines via FcγR interaction on these cells. Unless cell depletion is a desired pharmacologic effect, mAbs that bind to cellular receptors, e.g., to activate NK or T cells for cancer therapy or to inhibit the function of cells involved in inflammatory (and normal) immune responses must be designed to avoid ADCC/CDC. Avoidance of these effects is usually achieved through the use of the more inert IgG4 or IgG2 mAbs.Citation46 IgG4 has an instability in the hinge region that results in the production of half-antibodies (10–30% of the total) both in vitro and in vivo, as observed with natalizumab.Citation48 These half-antibodies need to be monitored, controlled and characterized because the half-antibodies can exchange their Fab arms with endogenous IgG4 in vivo.Citation48 For these reasons, many companies are less interested in developing IgG4 mAbs for therapeutic use, and are using either IgG2 or IgG1 mAbs that have been pre-selected for no/low Fc effector function activity. Development of IgG2 therapeutics may also have issues because it has the propensity for disulfide (S-S) rearrangement leading to isomer and dimer formation. Indeed, the majority of the currently licensed mAbs for inflammatory disease therapy are IgG1 with low or no effector function (). Other structural changes that can be considered include mutations in the CH2 domain to completely prevent FcγR interactionCitation49 and mAb aglycosylation to completely remove effector function;Citation45 however, immunogenicity of any non-natural mutation or structure needs to be considered.
The use of an IgG4 or IgG2 isotype or use of an antibody containing mutations in the Fc region that minimize receptor binding and effector function would likely reduce the infusion reactions and cytokine release syndromes seen with a number of the licensed mAbs (mainly IgG1). However, preservation (or even optimization) of Fc effector function such as that mediated by IgG1 mAbs may be necessary for efficacy if direct killing of cancer or inflammatory cells via ADCC or CDC is required; in such cases Fc-mediated side effects may be unavoidable. Fragments of mAbs lacking the Fc region should be considered if mAb effector function is not wanted, when inhibiting an immune receptor to avoid receptor cross-linking and activation, or if a short half-life is desirable. For example, a Fab may be a desirable format for agonist activity on an immune-activating receptor (provided that polymerization of the receptor is not needed for signaling to occur), where prolonged immune activation is not desirable, or to increase the chance of reaching the intended target by extravasation and tumor penetration, or when target cell aggregation needs to be avoided, e.g., abciximab (ReoPro) and platelets. In vitro studies should be performed to confirm the expected effector function (+/− ADCC/CDC activity) and biological activity of the chosen IgG isotype or mutated construct.
Assessing Potential Immunotoxicity Concerns of mAbs by Evaluating the Biology and Expression of the Target and the Intended Clinical Population
The immunotoxicity risk analysis for a mAb should begin with a thorough literature review of the immunobiology/MoA of its target that includes an assessment of the potential to unintentionally modulate related immune mechanisms. The cellular and tissue expression and function of the target in normal and diseased humans (where the risk of immunotoxicity could be greater), as well as in the animal species used for toxicology studies should be determined. If expression data are limited, one should consider the use of commercial antibodies to determine the expression of the target by immunohistochemistry (IHC) of a range of frozen human and animal tissues. Consideration should be given to whether the function of the target is well-defined and whether expression is restricted to the target cells or other immune and non-immune cells. The availability of immunopharmacology and safety data either from humans who lack or have reduced levels of the target or who overexpress the target, or from antigen knockout or transgenic mice (if available) should be determined. Human and animal pharmacology and toxicology data generated with mAbs with a similar MoA, e.g., targeting the same/similar immunological pathway, or generated in animals treated with surrogate mAbs against the same target (animal homolog) should be assessed if available. Consideration should also be given to whether there are any potentially unwanted immune effects that pose particular risk to the intended clinical population. It is important at this stage of risk assessment to identify the particular questions to be asked, and to determine whether they might best be investigated in vitro with human/animal cells or in vivo in animals or by some combination of the two. Correlation of an immune effect in vitro and in vivo in animals with the same effect in vitro with human cells might be a strong indicator of predictivity for response in humans.
In Vitro Studies with Immunomodulatory mAbs
A number of in silico and in vitro tests can be performed on mAbs to characterize their immunopharmacological and potential immunotoxicological effects, as well as to reduce the risk of some types of immunotoxicity, such as cytokine storms and immunogenicity/hypersensitivity.
In vitro immunopharmacology studies
The relative specificity of the candidate mAb binding to the immune system in humans and animals should be determined using methods such as flow cytometry, cell-based assays or competitive immunoassays. In addition, the binding of the candidate mAb to human and animal tissues can be determined by IHC in tissue cross reactivity (TCR) studies, although if the target distribution has not been well characterized using other tools, then these may merely identify previously unknown sites of expression of the intended target, rather than identifying sites of off-target binding. The relative affinity and immunopharmacological activity of the candidate mAb for the immune target in humans and animal species used for toxicology studies should be determined using clinically-relevant in vitro/ex vivo assays, e.g., to assess cell depletion, suppression, activation, cytokine production, effects on global immune regulators. The full dose-response curve should be thoroughly characterized in humans and animals in vitro by exploring immunological effects at both the low and high end of the curve using clinically-relevant cell-based assays (if available). Consideration should be given to the shape of the curve(s): is there a bell-shaped curve of activity or a steep concentration:response curve? Is the response in humans and animals comparable? These questions and data are important when considering how many identified risk factors a given biologic may have and how these contribute to calculation of the MABEL for FIH dose selection. Potential unwanted immunological effects should be assessed in these assays, e.g., to demonstrate lack of agonism of an antagonist mAb, lack of cell depletion etc. Consider if (based on the above), full human relevant immunopharmacology can be elicited in the toxicology species and how predictive of human immunotoxicity the toxicology species are likely to be. Are there any immunological effects in humans that might preclude clinical development? Are there any potential immunotoxicities in humans that will not be predicted in animals and need to be assessed in in vitro studies with human cells or in the clinical studies? Also, the number of risk factors and their implications should be given consideration. Are there any Fc-mediated effector functions of the mAb and can these be elicited in animals to a similar extent as in humans? If unknown then further investigation in animals may be required, e.g., ADCC and CDC assays with animal cells.
Assessment of potential for cytokine release.
As mentioned above, therapeutic mAbs and Fc-fusion proteins have the potential to trigger systemic CRS in man, either by cross-linking and clustering of the antigen target on immune cells by the Fab arms, by interaction of the Fc region with Fcgamma receptors (FcγR) on NK cells and neutrophils, or a combination of the two.Citation50–Citation52 Though multiple cytokines may be present, the classic signature of CRS consists of the pro-inflammatory cytokines TNFα, IFNγ and IL-6. The systemic and local presence of these molecules and the associated inflammation and hemodynamic effects damage tissues and organs, and can result in disseminated intravascular coagulation, organ failure and death if left untreated. Analysis of serum cytokines in humans provides a primary means of monitoring the development and resolution of this syndrome, with supportive evidence provided by clinical signs/symptoms, peripheral blood differential cell counts, and flow cytometric analyses; however, the most severe clinical sequelae of CRS do not occur in animal models, in spite of the generation of high systemic levels of cytokines.Citation53 In view of this, in vitro studies with human cells may be of more value in trying to assess the risk of CRS prior to FIH studies.
Although many biotechnology companies with portfolios of immunomodulatory mAbs conduct some kind of in vitro candidate mAb screening to evaluate their potential to induce cytokine release, there is no regulatory requirement for such testing. Among independent testing facilities, universities and companies that do conduct screening cytokine release assays (CRAs), there is no agreement on assay format, validation protocols or appropriate standard procedures and controls. The methods used to screen proteins for pharmacologically-mediated cytokine release overlap those described for pyrogen testing (encompassing both endotoxin and non-endotoxin pyrogens, such as peptidoglycan). Test systems that have been used include diluted and undiluted human whole blood and isolated PBMCs with or without a solid phase.Citation51–Citation54 CRAs typically include one or two positive controls that are known to be associated with a high clinical incidence of CRS, such as an anti-CD3, an anti-CD52 (alemtuzumab) or an CD28 agonistic mAb similar to TGN1412, as well as appropriate negative controls. A variety of assay formats can be used to measure cytokines, but multiplex assays, in which multiple analytes can be measured in a single sample, are most popularly used. FACS analysis that can detect both immunophenotype and intracellular cytokine concentrations has also been described.Citation55
Conducting in vitro CRAs shows due diligence in the assessment of human risk, can provide useful comparative data against known positives, and can be a useful complement to in vivo animal studies. However, it may merely demonstrate what was already expected based on the immunopharmacology and structure of the molecules evaluated. There are a number of molecular characteristics that increase the potential to stimulate cytokine release, some of which are alluded to in the Final Report of the Expert Scientific Group (ESG) on Phase I Clinical Trials and the subsequent EMEA Guideline on Strategies to Identify and Mitigate Risk for FIH Clinical Trials with Investigational Medicinal Products (EMEA/CHMP/SWP/28367/07).Citation56 Molecules that have higher potential to cause clinically relevant cytokine release events include those that bind targets such as Toll-like Receptors expressed on immune cells or other cell types rich in cytokines; bind “master switches“ of the immune system; have Fcγ functionality leading to ADCC or CDC (particularly if the Fcγ portion of the molecule has been engineered to increase binding or activity); are multivalent, permitting cross linking of targets or have multiple binding specificities, permitting simultaneous binding of multiple cell types; cause proliferation and expansion of immune cells; have agonistic activity on targets within a biological amplification cascade; or are expressed in microbial cells (particularly E. coli).
One of the concerns about conducting in vitro CRAs is repeatability and predictivity of results. In vitro assays, in which antibody was incubated with human PBMCs and cytokines measured, were conducted for TGN1412 prior to initial clinical testing. The information about these assays was redacted from the Investigator’s Brochure that was made public following the clinical trial, but was summarized in the Final Report of the ESG as follows: “Co-incubation of PBMC with soluble TGN1412 resulted in polyclonal T cell proliferation and secretion of T cell specific cytokines. The degree of TGN1412-induced proliferation varied among different blood donors, while conventional, co-stimulatory human-specific anti-CD28 antibodies were generally unable to induce substantial cellular proliferation. TGN1412 was, therefore, considered to be unique in its ability to deliver mitogenic signals via CD28 without co-engagement of the T cell receptor.“
The cytokines secreted were not specified in the ESG report, although other data suggests that the predominant cytokine signature observed with CD28-stimulated T cell proliferation is Th2-like (IL-4 and IL-10).Citation57 The National Institute of Biological Standards & Controls subsequently evaluated TGN1412 for its ability to induce pro-inflammatory cytokine release (TNF, IL-6 and IL-8) in an in vitro system using soluble TGN1412 added to isolated human PBMCs or 1:5 diluted whole blood.Citation52 Under these experimental conditions, no cytokine release was observed. In subsequent experiments TGN1412 did stimulate pro-inflammatory cytokine release in either PBMC or 1:5 diluted whole blood test systems if the antibody was immobilized by air drying to plastic or anti-Fc antibody capture, or if the antibody was added in aqueous phase in the presence of endothelial cells.Citation52 Additionally, the dose response curve was bell-shaped. We have shown consistent cytokine release in undiluted human whole blood with both a TGN1412 analogue and alemtuzumab in solution without the need for mAb immobilization (R. Allenspach, unpublished observations).
These points highlight the difficulty of being sure that a negative in vitro cytokine release assay result can be reliably used to make assumptions about risk for patients. Careful consideration of the structure and pharmacology of the test mAb and the intended patient population may guide decisions about which type of assay(s) to conduct and which test systems to be used. Fresh whole blood or isolated PBMCs from healthy volunteers are the most commonly used test systems. Both have the disadvantage of inter-individual variability and in neither case are the immune cell populations present in peripheral blood representative of the total immune populations in the body. Another potential disadvantage is that the target distribution or concentration in cells from healthy volunteers may be different in patients, e.g., leukemic patients. Whole blood from human volunteers is readily available and requires little manipulation, but the percentage of target cells may be relatively low and blood is further diluted during testing. We found that a dilution of the whole blood by more than 10% will by itself lead to a disproportionate reduction of cytokines released. Whole blood contains complement and other soluble mediators that are lacking from isolated PBMC preparations. PBMCs are also readily obtained, although they require further handling than whole blood. Using PBMCs as a test system to evaluate the effects of mAbs directed to immune cell targets concentrates the population of targeted cells and increases the likelihood of detecting a response related to intercellular signaling. Assay formats that include mAb immobilization via Fc targeting may be inappropriate to evaluate antibodies that mediate their effects by Fc binding. Antibodies that cause cytokine release by ADCC may produce relatively minimal reactions in test systems that lack the targeted epitope, e.g., on cancer cells. In such cases it may be useful to conduct assays in systems that include cells that express the intended target.Citation58
In summary, the prevalence and potential severity of immunologically-mediated infusion reactions in patients treated with immunomodulatory mAbs indicates that the potential for CRS, which is likely to occur on the first infusion, be carefully evaluated prior to initiating clinical testing. In addition to measuring cytokines in animal studies, appropriately designed in vitro cytokine release assays can be useful adjuncts to help evaluate this risk. If sponsors believe that there is no risk, and do not conduct assays to evaluate the risk, their rationale should be thoroughly explained to regulatory authorities.
Assessment for potential to activate DCs
DCs are the most potent APCs representing a bridge between the innate and adaptive immune systems. Immunomodulatory mAbs may directly or indirectly interact with the phenotype and function of DCs, thereby affecting both adaptive and innate immune functions. Those effects can either reflect the intended therapeutic MoA or represent side effects with potential safety relevance that can be investigated in vitro with human DCs. The in vitro assessment of mAb-mediated effects on human DCs may provide relevant information about the MoA of a mAb. Since DCs are key players in the generation of anti-drug antibody (ADA) responses (), the assessment may also indicate potential side effects on the phenotype and function of DCs that could impact its immunogenic potential.
DCs effectively internalize antigens and process them for major histocompatibility complex (MHC)-restricted presentation to stimulate naïve T cells, but they require a second co-stimulatory signal for effective activation.Citation59 Maturation of DCs is triggered by so-called danger signals and associated with a decrease in antigen-processing capacity, an upregulation of maturation markers, including co-stimulatory molecules, adhesion molecules, chemokine receptors and MHC molecules, along with secretion of T-cell stimulating and polarizing cytokines and a rearrangement of the cytoskeleton. This enables them to deliver effective co-stimulation. Danger signals, mainly recognized by the families of toll-like receptors (TLR) and Nod-like receptors (NLR), can be delivered by pathogen-associated molecular patterns (PAMPs) of exogenous origin or by endogenous signals of cell damage-associated molecular patterns (DAMPs).Citation60 In addition, maturation is also mediated via CD40L ligation provided by T helper cells, by inflammatory cytokines released from bystander cells of the innate immune system such as NK cells and by antigen-antibody complexes.Citation61
DCs generated from human blood monocytes by cultivation in the presence of IL-4 and granulocyte-macrophage colony stimulating factor (GM-CSF) represent a model system with a high similarity of expression markers and functional features to immature myeloid DCs in the human body. In the presence of maturation stimuli, these cells show the typical upregulation of the co-stimulatory molecules CD80 and CD86, of other maturation markers as CD83 and CD40, of MHC and adhesion molecules. The changes in surface marker expression, e.g., by a mAb can be monitored by flow cytometry. In addition, monocyte-derived DCs may also be triggered to release pro-inflammatory and T cell stimulating cytokines that can be analyzed in the culture supernatant by multiplexed cytokine-detection systems. In vivo modulation of DCs by indirect mechanisms such as mAb-mediated cytokine secretion from other immune cells might also be evaluated in vitro, but would require a relevant cell co-culturing system.
mAbs and other proteins purified from eukaryotic cell lines may contain impurities that function as classical PAMPs and DAMPs such as lipopolysaccharide (LPS), heat shock protein, mobility group box 1 protein and others, and hence have the potential to stimulate DC maturation and immune activation. In addition, the presence of misfolded or partially degraded drug protein associated with the exposure of hydrophobic regions may stimulate DCs. Importantly, aggregated mAb has the risk of DC activation via FcγR engagement by a mechanism similar to that described for antigen-antibody complexesCitation62,Citation63 and might result in effective activation of drug-specific T cells in vivo. Also, formulation excipients such as polysorbate and leachable substances from plastic containers cannot fully be excluded to act as danger signals for DCs.Citation64 These types of risks might be assessed in in vitro DC test systems. Since this assay has the potential to detect a variety of endogenous and exogenous DC stimuli including effects of the mAb, but also components of the formulation, including product- and process-related impurities such as LPS, the influence of irrelevant factors must be excluded or subtracted from the relevant ones. A challenge of the assay is the high inter-individual variability in DC response to stimuli. Further validation of this assay is required to determine if it has utility in detecting immunological effects of mAb formulations that are relevant for human safety assessment.
Predictive immunogenicity testing
The formation of anti-drug antibodies (ADA) to mAbs and other therapeutic proteins could potentially lead to severe immunotoxicological reactions, such as IgE-mediated anaphylactic reactions,Citation35 or immune complex disease, e.g., vasculitis, glomerulonephritis,Citation65 as well as to a loss of clinical exposure and efficacy. ADA raised against therapeutic mAbs have not been shown to cross-react with endogenous antibodies or induce autoimmunity, but some patients treated with the therapeutic proteins pegylated megakaryocyte growth and development factor (PEG-MGDF) and erythropoietin (EPO; Eprex) developed ADA that were cross-reactive to their respective endogenous counterparts, leading to severe thrombocytopenia with PEG-MGDF and pure red-cell aplasia with Eprex.Citation66,Citation67 Hence, it is important to reduce the immunogenicity risk prior to human testing. Minimizing immunogenicity will be especially important for alternative high affinity protein binding scaffolds (antibody mimics) containing modified, non-human sequences that are beginning to enter the clinic. These include domain antibodies (dAbs), fibronectins, minibodies, nanobodies or fusion proteins designed to expand half lives of the drugs or to gain multivalent binding possibilities. As these drugs may differ vastly in their protein sequence from the wild type protein, immunogenicity and cross-reactivity to the endogenous counterpart needs special attention.
Formation of ADA can be induced in at least two different ways. T cell-dependent and -independent pathways have been described for B cell activation. A strong, high affinity IgG response is T cell-dependent and requires involvement of CD4+ T helper cells (TH cells). The immune response is analogous to a response against foreign antigens: naive TH cells are specifically activated by professional APCs, such as DCs, and in turn induce activation of drug-specific B cells (). Depending on circumstances, e.g., dosing schedule or immune status of the patient, breaking of T and B cell tolerance to endogenous proteins such as EPO or MGDF has also been observed. On the other hand, a direct activation of B cells without direct involvement of TH cells has been described for repetitive structures on the surface of viruses or bacteria (PAMPS). Aggregation of the biotherapeutic drugs leads to protein clusters containing repetitive structures that may mimic the surface of pathogens, and therefore can lead to direct activation of B cells and also breakdown of B cell tolerance;Citation68 however, a T cell-independent activation of B cells leads to only low affinity IgM and IgG responses without formation of B cell memory or affinity maturation.
Besides the protein sequence, other factors influence immunogenicity to therapeutic mAbs and proteins. Important factors directly linked to the drug itself are protein modifications such as glycosylation or PEGylation that may alter the likelihood of immunogenicity. It has been shown for example that inclusion of galactose-α1,3-galactose into glycans of mAbs, which may occur as a result of production in a SP2/0 cell system, can lead to IgE-mediated immunogenicity to therapeutic antibodies.Citation43 In contrast, PEGylation has been described to reduce immunogenicity, but not in all cases.Citation66 A further factor influencing immunogenicity is the route of administration. Intramuscular injection appears to be less immunogenic as compared with intravenous or subcutaneous administration of the drug. Here, immuno-surveillance mechanisms seem to play an important role. Injection into tissues with reduced numbers of immune cells, especially professional APCs like DCs, results in a reduced risk for immunogenicity. A simultaneous presence of a danger signal, as described earlier, is mandatory to drive initiation of a T cell-mediated immune response to the biotherapeutic protein. A danger signal may come from necrotic cell death which results from tissue injury after subcutaneous injection. Alternatively, danger signals may come directly from the drug or from formulation ingredients. In addition to these drug-related factors, patient characteristics also influence immunogenicity. One major patient factor is the highly polymorphic human leukocyte antigen (HLA) genes, which are the restriction elements for the T cell receptor. Depending on the HLA genotype of the patient, a different set of peptides derived from the protein drug is presented to T cells that may result in activation of T cells in one patient, but not in another. Other factors influencing immunogenicity include the type of the disease, the health status of the patient or co- and pre-medication. For example, adalimumab induced immunogenicity in only 1% of rheumatoid arthritis (RA) patients when co-administered with methotrexate, whereas without concomitant methotrexate the incidence of an unwanted immune response was 12%.Citation69,Citation70
The demand to improve the safety and efficacy profile of new drugs has led to a heightened consideration of applying emerging technologies, based mainly on in silico and in vitro methods, to attempt to predict immunogenicity. Attempts to develop novel in vivo tools are also under way. Traditional animal models, such as cynomolgus monkeys, rats, rabbits or mice, are of limited value for predicting human immunogenicity, although ranking of potential immunogenicity into low or high risk may be possible. The incidence of immunogenicity observed in these animal models is generally much higher than is observed in humans, not only because the human therapeutic mAbs/proteins appear as foreign in animal models, but also because the immune system and especially the MHC genes differ greatly between different species.
Nevertheless, a comparative immunogenicity evaluation has been demonstrated for interferon-α2b in wild-type mice or mice transgenic for interferon-α2b.Citation68 Interferon-α2b preparations containing aggregates increased the immune response relative to native interferon-α2b preparations in the wild-type mice and in addition, aggregates were able to break the immune tolerance of interferon-α2b transgenic mice. HLA transgenic mice expressing the most common HLA-DR alleles of the Caucasian population are available, but human proteins are still immunogenic in these mice.Citation71–Citation74 Generation of double mAb/HLA transgenic mice takes a long time, often fails and the immune system still differs from the human immune system.Citation75 For example, DC subsets or the phenotype of T regs is different in mice and humans. To overcome these differences, new xenotransplantation mouse models, based on NOD/SCID/γc−/− or Rag2−/−/γc−/− strains have been developed.Citation76,Citation77 These mice lack functional T, B and NK cells and have impaired capability to secrete cytokines. By engrafting human CD34+ positive cord blood stem cells, a human-like immune system evolves in these mice. The drawback of this system is that every mouse that is used for immunogenicity prediction needs to be transplanted, and this of course means that a single mouse represents only a single human individual. Therefore, several mice have to be transplanted to gain significant population coverage. In addition, as already discussed for the HLA transgenic mice, these mice might need to be transgenic for the human mAb/protein as well in order to have a situation comparable to the human system.
In contrast to the in vivo approaches, in silico and in vitro prediction methods specifically focus on the contribution of T cells to ADA formation. The advantage of these methods is that they are human-based and so there is no issue regarding species differences. In addition, these approaches are relatively easy to apply and their short time course fits well into a research and development program of a new protein-based therapeutic agent. In silico tools are either based on in vitro peptide binding dataCitation78–Citation80 or on energy minimization models, which use the crystal structure of HLA molecules to calculate binding affinities.Citation81 All these tools have in common that they predict the binding affinity or binding probability of a defined peptide sequence to a defined HLA allele. In silico tools do not take into account the antigen processing and presentation processes for HLA class II, as these processes are highly complex and not yet predictable. Therefore, the currently available in silico tools do not predict the antigen presentation process as a whole. In addition, even if a sequence is accurately predicted to be presented in the context of HLA class II, this does not mean that T cells will respond to this epitope in vivo as tolerance mechanisms may prevent this. Therefore, these tools tend to be over-predictive. Nevertheless, they are very easy to use and allow a rapid analysis and comparison of different protein sequences. Hence, in silico HLA binding prediction is very useful in guiding protein design processes. In order to validate the in silico prediction, additional binding assays might need to be performed.
Only in vitro identification of HLA class II peptides, which were processed by APCs, will take the antigen uptake, processing and presentation processes into account. In this approach, APCs such as human monocyte-derived DCs, are challenged with the biotherapeutic drug candidates and HLA class II-presented peptides, which are derived from the biotherapeutic protein, are identified by mass spectrometry.Citation82 This approach allows an accurate identification of immunodominant epitopes, but, similar to in silico methods, false positive peptides might be identified as epitopes because tolerance of T cells is not taken into account. To verify peptide sequences identified by in silico or in vitro methods, human T cell activation assays need to be performed. These assays can be based on APCs and T cells derived either from healthy blood donors or from the desired patient population, which can be necessary if an increased immunogenicity risk is expected in that population. In addition, using T cell assays with full length proteins instead of peptides is useful to rank different similar drug candidates relative to each other or relative to similar compounds with known immunogenicity.
Information derived from such assays can feed into a candidate selection process and support selection of candidates with a favorable immunogenicity profile; however, it is difficult to accurately determine the predictivity of these immunogenicity screening tests since multiple mAb candidates with different immunogenicity profiles in these in vivo and in vitro models are seldom allowed to enter long-term human clinical trials to obtain comparative immunogenicity data from humans. Assessment of the currently approved mAbs does show some degree of correlation between in vitro immunogenicity and immunogenicity in humans.Citation83
In general, an immunogenicity risk-based approach should be taken when determining which of the available approaches to predict immunogenicity should be applied to a new mAb candidate.Citation84 Especially for protein-based therapeutics with high risk to develop immunogenicity or when there is a high probability that neutralizing antibody responses will cross-react with the endogenous counterpart of the biotherapeutic, special attention should be paid to immunogenicity assessment in research and development.
In Vivo Studies with Immunomodulatory mAbs—Species Selection and Qualification
Species selection.
Toxicology studies with mAbs should be performed in a pharmacologically-relevant species, i.e., one that both expresses the target antigen recognized by the mAb and evokes a similar pharmacological response following mAb binding as that expected in humans.Citation37–Citation39 For mAbs with strong effector function, e.g., IgG1, it is also important to demonstrate that the mAb exhibits comparable effector function in animals to that predicted in humans. In this way the most sensitive animal model available for predicting human safety is utilized. Cross-reactivity, or lack thereof, can often be predicted by an in silico analysis of sequence and structural homology/identity between the human antigen protein or targeted epitopes and the cognate proteins in conventional species used for toxicology studies. The in silico data can be used to select species (with a high homology to human) for initial mAb binding and pharmacology studies and TCR studies; however it is recognized that high homology in animals does not guarantee mAb binding and pharmacological activity. Toxicology assessment should usually be performed in two relevant species if available, one rodent and one non-rodent. For further guidance relating to species selection see ICHS6,Citation38,Citation39 and recent reviews.Citation12,Citation36 In reality, many mAbs have been tested in only one species (mainly primate) because only one relevant species could be identified. The NHP should be demonstrated to be the most suitable species to ethically justify its use and strategies should be used to minimize primate use.Citation85 The cynomolgus monkey is the preferred NHP species for toxicology studies since it is an Old World monkey of medium size and requires lower amounts of test compound for dosing than the rhesus monkey or baboon, and the cynomolgus monkey has historically been the most common species for toxicology testing, including immunotoxicology and reproductive toxicology, of human mAbs. If binding and relevant pharmacology is seen in NHP and rodents, then studies in both NHP and rodents should be performed. If a mAb has a comparable safety profile in 4-week toxicity studies in NHPs and rodents then it may be that the rodent study could be restricted to 4 weeks duration.Citation39 The duration of dosing in NHPs and rodents may depend on whether neutralizing antibodies are elicited to the human mAb. The presence of neutralizing antibodies might prompt the termination of a study if exposure to the mAb is lost or below the expected clinical exposure in the majority of the monkeys thereby preventing a meaningful toxicological evaluation or there are severe adverse effects, e.g., anaphylaxis, that preclude further dosing. The use of high mAb dose levels, e.g., 100–200 mg/kg, as well as increasing the number of animals within a study, might allow significant mAb exposure for the duration of the study in a greater number of animals. If no binding/pharmacology is observed in any of the commonly used toxicology species, alternative toxicology models such as surrogate mAbs and human transgenic models can be considered.
Species qualification strategies.
In some cases, the recombinant human and animal proteins will be available so that species-specific binding can be simply assessed by ELISA or BIAcore analysis. If the target is expressed on blood cells or other readily sampled cells, then species-specific binding can be determined by flow cytometry where binding of the mAb to cells from a range of species can be assessed.
In species where mAb binding is observed or predicted, clinically-relevant pharmacology, e.g., inhibition of chemotaxis, inhibition or induction of T cell activation or cytokine-mediated effects, can be assessed using a relevant bioassay (if available) and the pharmacological effects compared to those observed with the mAb on human cells. This allows a determination of the comparative pharmacology between humans and the toxicology species to be considered in the selection of a safe starting dose in humans. Some investigators use a 5–10-fold reduction in potency between animals versus humans as a maximum cut-off for species selection, although it could be argued that a mAb with a lower relative potency than this could still be used provided that complete inhibition of the target for the duration of each dosing interval in the study can still be achieved in animals at practical toxicology dose levels, which often can be predicted by PK/PD modeling. In situ hybridization and other techniques can also be used to assess target expression and distribution in tissues to further support species selection. TCR studies by IHC analysis of the mAb on frozen tissues from humans and the chosen animal species might provide confirmatory support for the relevance of a toxicology species by demonstrating a similar tissue binding profile with the mAb on human and animal tissues.
Use of a surrogate mAb.
If no relevant conventional species exists, then alternative toxicology models might be useful to assess safety of a mAb. Since the toxicity of mAbs is often associated with exaggerated pharmacology, the use of a surrogate mAb binding to the homologous target in rodents (or primates) might provide important mechanism-related safety data. Surrogate antibodies have been used successfully to assess the safety of both infliximab (anti-TNFα)Citation86 and efalizumab (anti-CD11a)Citation87 in short-term, chronic and reproductive toxicity studies and efficacy studies. However, since these studies do not use the drug product, differences in mAb binding properties of the surrogate and downstream signaling events in the animals, coupled with likely differences in the function and expression of the rodent homologue compared with its human counterpart, mean that data from these studies should be interpreted with caution.
If the structural homolog of the human target is not present in rodents, then a mAb targeting the pharmacological homolog, i.e., a structurally different molecule with the same function (if available), may prove acceptable to regulatory authorities. The surrogate mAb may be a mouse anti-mouse homolog, as in the case of the cV1Q mouse surrogate mAb of infliximabCitation86 or a rat anti-mouse homolog. ‘Mousification’ of the rat mAb (as in the case of the muM17 surrogate mAb of efalizumab)Citation87 could be considered, depending on the immunogenicity of the rat molecule in mice. The mouse or rat isotype used for the surrogate should be chosen to mimic the half-life/exposure and effector function, e.g., ADCC and CDC activity, expected with the human mAb in humans. When a surrogate mAb is used, studies should be performed to show that the target of the surrogate mAb is expressed in the same cells and tissues in the mouse as the human target is in humans, and that the specificity and pharmacological activity of the human and surrogate mAbs are similar, as described above. Surrogate mAbs should be produced under controlled conditions and be well-characterized as outlined in ICHQ6B.Citation88
Studies in human antigen transgenic mice.
If human target antigen transgenic mice are available, the human drug product mAb could be tested in these mice.Citation89 The advantage, compared with use of a mAb surrogate, is in the use of the human drug product mAb that binds the human target, allowing the simultaneous assessment of pharmacology-related toxicity, non-specific toxicity and local tolerance effects. As described above for the surrogate mAb, studies should be performed to assess whether the human transgene is expressed in the same cells and tissues, and at the same level in the mouse as the human target is expressed in humans, and whether the mAb has the same pharmacological activity in these mice compared to humans.
A fully human, humanized or chimeric mAb is likely to be immunogenic in human antigen transgenic mice after repeated dosing, reducing mAb exposure and compromising toxicology assessment. The drug product might only be evaluable in studies of limited duration, e.g., 4 weeks, in these mice. While this may be sufficient to support FIH studies, chronic dosing studies might be required to support longer-term clinical studies and market authorization. In this case, a surrogate mAb (mouse anti-human target) would be required for chronic studies in these transgenic mice to avoid or reduce immunogenicity. When the drug product is a chimeric or humanized mAb, the parent mouse mAb (upon which the human drug product is based and which expresses the same CDR regions as the drug product) could be considered.
Consideration of differences in human and primate immune systems.
In humans and animals, the immune system is regulated by a tightly-controlled balance of signals transmitted by stimulatory and inhibitory receptors; however, the immune systems of humans and NHPs show some important differences. Compared with chimpanzee and macaque T cells, human T cells exhibit stronger proliferative responses upon activation via the T cell receptor, a response that is attributed to the loss of T cell Siglec expression from human T cells.Citation90 CD33-related Siglecs are inhibitory signaling molecules expressed on most immune cells that downregulate cellular activation pathways via cytosolic immunoreceptor tyrosine-based inhibitory motifs.Citation91 Concordant with this species-related difference in Siglec expression is the observation that several common human T cell-mediated diseases, such as bronchial asthma, RA and type 1 diabetes, have not been reported in chimpanzees or other Great Apes. In addition, cynomolgus monkeys have a higher prevalence of CD4+/CD8+ (double positive) blood T cells than in humans.Citation92 Double positive T-cells exhibit a resting memory phenotype that increases proportionally with age, and CD28 expression also changes in relation to age. CD28-mediated T cell activation and cytokine release has also been shown to be different in young and adult cynomolgus monkeys. Since young monkeys 2–3 years of age are commonly used in toxicology studies, the T cell phenotype in these animals is an important consideration for testing some immunomodulatory and T cell-targeting mAbs.Citation93 Fc receptor expression also differs between human and animals. In humans, FcγRIIIA (CD16A) is expressed on monocytes, macrophages and NK cells whereas the FcγRIIIB (CD16B) isoform is expressed on neutrophils, eosinophils and other cells. In NHPs, there is only one CD16 gene, homologous to the human CD16A, which is restricted to NK cells and monocytes.Citation94 Further differences in humans and animal immune systems have been reviewed.Citation95 These immunological differences between human and animals should be considered during safety assessment of immunomodulatory mAbs.
In Vivo Studies with Immunomodulatory mAbs—Immunotoxicity Assessment within GLP Toxicity Studies and Animal Disease Models
General toxicity studies.
Study design and dose selection for toxicology studies with mAbs have been described in detail previously.Citation12,Citation36 Within toxicology studies, usually in cynomolgus monkeys and sometimes also rodents, it is important to assess the nature and extent of the immunological effects of the mAb. This is not only to confirm that the desired immunopharmacological activity of the mAb is occurring in the toxicology animals, thereby validating the study, but also to determine if any other undesirable or unpredicted immunological activity (immunotoxicity) is observed. For NCEs, immunotoxicity testing normally involves assessment of unintentional effects on the immune system and ICHS8,Citation40 recommends that primary immunotoxicity endpoints are included within standard toxicology studies. Inclusion of secondary endpoints/follow-up studies, e.g., immune function tests or host defense assays, is recommended only if justified following a weight-of-evidence review indicating a cause for concern. Factors prompting specific immunotoxicology studies include findings from standard toxicity studies, the pharmacological properties of the drug, the intended patient population, structural similarities to known immunomodulators, disposition of the drug in lymphoid organs and clinical information including known immunomodulatory effects. This ‘weight of evidence’ approach is also relevant for mAbs. Immunopharmacology/immunotoxicity testing of mAbs should focus on the specific cells and immunological pathways targeted by the mAb. A tiered approach to immunotoxicity assessment of mAbs should be used in which immune status is first assessed (primary tests) followed by an assessment of immune function (secondary tests) if the mAb targets the immune system or has effects in the primary screens. Importantly, it should be demonstrated that the immune system returns to normal on cessation of dosing and there are no long-lasting or irreversible effects on immune function or toxicological or pathological effects resulting from the immune modification. The long half-life of mAbs, e.g., 10–14 days in cynomolgus monkeys, necessitates the need for a long recovery to allow mAb clearance and ‘true’ recovery.
Some assessment of immune effects should be made for all mAbs whether immunomodulatory or not. Primary tests (immune status/descriptive endpoints) can be included in all toxicity studies (). These include standard hematology assessment total and absolute differential leukocyte counts (including macrophages), clinical chemistry (globulin levels and albumin:globulin ratios), gross pathology (lymphoid organs and tissues), organ weights (thymus, spleen, lymph nodes) and extended histopathology of lymphoid organs (thymus, spleen, bone marrow, lymph nodes, including both draining and those distal to injection sites).Citation96,Citation97 A semi-quantitative assessment of lymphoid tissue compartments with respect to both the lymphocyte and non-lymphocyte components can be performed. The architecture and size of different compartments and cellularity of the organs is examined and described if different from control. Identification of lymphoid changes is largely dependent on the severity of the lesion, i.e., whether it is minimal, which is often observed in control animals, mild, moderate or marked.Citation98
For mAbs, researchers generally want to demonstrate desirable immunopharmacology and lack of effects on the rest of the immune system, so what to look for is usually known (not trying to detect and unintentional NCE ‘immunotoxicant’). Such evaluations are more likely to detect expected (primary pharmacology-driven) significant direct effects on specific cell type, e.g., B cell depletion or activation of major T cell population, but should be comprehensive and careful enough to detect subtle, minor or “off-target“ effects that may be unanticipated effects related to the primary pharmacology. Normal animals in toxicology studies may express only low levels of the target and there may be compensatory/redundant mechanisms that could mask effects.
Effects in the thymus appear to be easier to detect than those in the spleen or lymph node,Citation97 although this might not always be the case in sexually-mature animals. There appears to be good agreement by pathologists in reading cellularity of thymus cortex and spleen follicle, spleen and lymph node germinal centre development, but not in the reading of spleen red pulp changes. There is also no agreement on the level of histopathological change (number of endpoints altered or severity of lesion) that constitutes a biologically-significant immune effect.Citation98 The correlation between histopathologic effects and other immune assays such as immunophenotyping and immune function is not well established, although in some cases correlations between histopathologic findings and other immune tests have been observed, e.g., thymic cortex effects and cell-mediated host resistance.Citation99
If mAb-mediated histopathologic changes are observed, immunohistochemical immunophenotyping of the affected tissues to identify the affected cell types should be considered. Use of flow cytometry to immunophenotype lymphocytes, e.g., T, B and NK cells from both blood and lymphoid organs and to evaluate specific cell subsets and determine activation status can also be included in the toxicology assessment, depending on MoA and species to evaluated. Many reagents are now available in NHPs for immunophenotyping of naïve, effector, memory and regulatory T cells and a range of B cell subsets also. Flow cytometry is unlikely to detect minor/subtle immunological effects due to the variability in lymphocyte counts over time within the same animal, e.g., stress-related glucocorticoids or adrenaline affects lymphocyte re-circulation. A parallel untreated control group, as well as multiple sampling of mAb-treated and control groups prior to dosing, will enhance the chances of seeing a mAb-related change. It is unclear how small/large a change is necessary to predict a biologically-significant consequence/clinical concern and what relationship exists between immunophenotypic change and effects on immune function. Abatacept (CTLA-4-Ig) is immunosuppressive and inhibits a T cell-dependent antibody response (TDAR) in monkeys and rodents, and even ADA production in rodents; however, it had no effects on the numbers of T or B cells in either species.Citation100 Conversely, alefacept (LFA3-Ig) causes T cell depletion in blood and tissues of monkeys and yet has no effect on the TDAR responses to human serum albumin (HSA) and only a minimal effect on the keyhole limpet hemocyanin (KLH) response.Citation101 Efalizumab (anti-CD11a) depletes T cells and has also significant effects on the TDAR response in chimpanzees, as does the surrogate anti-mouse CD11a mAb in mice.Citation102
Evaluation of other product-relevant immune parameters should be considered on a case-by-case basis, depending on the MoA, e.g., total Ig measurements (for mAbs targeting B cells or if effects are observed in total globulin levels), serum cytokines (for mAbs such as IgG1 that bind to the surface of immune cells and with strong effector function), acute phase proteins, complement components, clotting factors, ex-vivo lymphocyte Stat-6 activation, ex-vivo T cell proliferation, receptor occupancy (RO). Electrocardiogram (ECG) assessment can be timed to coincide with cytokine release sampling to assess whether any observed increased cytokine levels correlate with cardiovascular effects. It is useful to include a range of immunological markers in preliminary dose range-finding (DRF) studies to assess the value of inclusion in subsequent regulatory-compliant GLP studies. All data from the above assessments should be considered as a whole and not as individual endpoints, e.g., any histopathology findings should be considered together with the organ weights and immunophenotyping data.
For mAbs that target the immune system, secondary tests (immune function tests) should be included within the 4- or 13-week GLP toxicology studies in the primary species (), even if no effects are observed in the primary screens described above. The functional assays should reflect the cells/pathways targeted by the mAb (T, B, NK, macrophage etc.) and the MoA (immunosuppression or activation). Assessment of the effects of a mAb on the TDAR to KLH or Tetanus Toxoid (TT) in cynomolgus monkeys, or sheep red blood cells (SRBC)/KLH in rodents, is a common functional endpointCitation103 unless not indicated by the MoA. Both the primary (IgM, IgG) and secondary (IgG) responses to antigen(s) administered during dosing and recovery can be determined to assess the effect of the mAb on immune priming and boosting (immune memory) and recovery from any effects. An effect on the TDAR means possible effects on APCs, B cells and T cells, hence a positive effects in the TDAR could be followed up with other functional tests to further define the target cells/mechanism, such as specific assessment of T/B cell or APC function, e.g., delayed-type hypersensitivity (DTH) responses, proliferation in response to B and T cell mitogens, e.g., conA, PHA, anti-CD3, LPS or antigen, cytokines/Ig responses to stimuli (antigens, infective agents), in vitro APC function etc. If the TDAR is not relevant, other functional assays should be considered depending on the target and MoA, e.g., CTL killing of P815 cells as measured by Cr51 release or flow cytometry, NK cell killing assay or macrophage/polymorphonuclear cell function assessments such as phagocytosis and chemotaxis assessment by flow cytometry, although, as with the other tests, there is no real understanding of the extent reduced immune function required to have significant biological effect, e.g., increased risk of infection and tumor development, in humans. A weight-of-evidence approach where all immunotoxicity data is considered as a whole (and in consideration of the MoA of the mAb, the predicted extent and duration of human exposure, the clinical population, disease status, concomitant medication etc.) is recommended when interpreting the findings of immunotoxicity assays and in considering the risk of clinically-significant immunotoxicity occurring in humans.
In chronic studies of up to 26 weeks duration, one could consider only performing TDAR or other immune function tests if effects are seen in the 4/13-week studies to increase the size of the dataset. If immunosuppressive effects are seen in the 4/13-week studies, detailed histopathology/IHC assessment to look for early signs of lymphoproliferative disease and possible increased risk of tumors could be included in the chronic toxicity studies. Monitoring for the effects of the mAb on titers of endogenous tumor-promoting viruses, e.g., Lymphocryptovirus (LCV) in monkeys should also be considered, as has been done for the immunosuppressive Fc fusion proteins alefaceptCitation101 and abatacept.Citation100 LCV and other tumor-promoting viruses induce polymorphic B cell hyperplasia or plasmacytoid hyperplasia that could result in lymphoproliferative changes and potentially to lymphoma. Indicators of viral load (viral DNA/gene products) could be monitored during chronic toxicity studies (in addition to any clinical manifestations of viral infection) to determine whether they are increased following treatment with an immunosuppressive mAb. Increased titers of LCV were observed after chronic treatment of monkeys with alefacept and lymphoma was observed in a single monkey although the relevance of this finding for humans is not clear (no mAb-induced lymphomas have been reported with alefacept to-date in humans).Citation101 With abatacept, no change in viral infection status was observed in a 52-week NHP study whereas virus-induced tumors were observed in a 2 year mouse carcinogenicity study. It is not known whether an effect on tumor-promoting viruses or occurrence of lymphoma in animals within a chronic toxicity study in any way predicts effects on human tumor-promoting viruses and the risk of human lymphoma and other neoplasms. Human lymphoma is caused by human viruses, e.g., EBV, HTLV-1, HHV-8, HPV, which are different from the animal viruses. The endogenous levels of these human viruses are also expected to be different from the animal viruses present in normal toxicology species. The immunological status of human patients and viral control mechanisms are also likely to differ from normal toxicology animals. In addition, it may be that lymphoma will only be observed in humans after longer exposure (years) to an immunosuppressive mAb, an effect that cannot not detected in a 26-week toxicity study. However, viral monitoring in animals might add to the overall weight-of-evidence for immunosuppression and decreased host resistance.
Reproductive/developmental toxicity studies.
Studies to assess embryo-fetal and peri-/post-natal development (EFD-PPND) are required for novel immunomodulatory mAbs indicated for the treatment of women of child-bearing potential with a non life-threatening disease. Immunomodulatory mAbs have the potential to affect different aspects of pregnancy and fetal development. During pregnancy there is a delicate balance of innate and adaptive immune responses at the maternal-fetal interface that promotes survival of the semi-allogeneic embryo and also protects the mother from environmental pathogens. Inadequate recognition of fetal antigens might result in failed pregnancy. Immune cells, e.g., T cells, NK cells, DCs, macrophages at the maternal-fetal interface may play a key role in maintenance of pregnancy, and cytokines such as TNFα, TGFβ, IL-2 and IFNγ are known to be involved in organ development and affect gene expression and apoptosis.Citation104–Citation106 There appears to be a reduced Th1 and NK cell function in the mother to prevent rejection of the paternal antigens of the fetus.Citation104 Hence effects on cellular immune function and direct neutralization of these cytokines by a mAb could affect these processes and impact pregnancy
In humans and animals, there is active transfer of IgG from mother to fetus via FcRn,Citation107 and the long half-life of many therapeutic mAbs could result in prolonged pharmacological activity and effects on the developing fetus, including the immune system (developmental immunotoxicity). As with general toxicity studies, the NHP is often the only relevant species for study of mAbs, and it is similar to humans in reproductive physiology, endocrine control and placental transport. Drawbacks in use of NHPs are the small number of animals available for studies, which may prevent adequate risk assessment; low background data; high spontaneous abortion; and issues with ethical use.Citation108 If rodents or rabbits are also relevant species, then these should be used for reproductive toxicity studies.Citation37,Citation85
The traditional dosing period in an embryo-fetal development study in cynomolgus monkeys begins on about gestation day (GD) 20, when pregnancy can be detected by ultrasound, and ends on GD50 (the end of the period of organogenesis).Citation108 Exposure to the developing fetus under these conditions is limited since the majority of mAbs are transferred via FcRn in the second and third trimestersCitation109 and fetal CD3+ T cell development in the thymus only begins on GD60, with immunoreactive B cells appearing around GD85.Citation108 Hence it is critical that pregnant animals are dosed throughout gestation to optimally expose the fetus and the developing immune system.Citation110 A new developmental toxicity study design in monkeys (combined EFD-PPND study rather than separate EFD and PPND studies) has recently been proposed and is starting to be used throughout the industry and gaining regulatory acceptance.Citation108,Citation111 This design uses a single cohort of mAb-treated dams (no separate sub-group for c-section group at the end of gestation) and gestationally-exposed offspring for all endpoints at each dose level and generates at least as much relevant information than the previous 2-study design while using fewer animals (and the mothers can be placed back in the colony). In addition to the developmental readouts of fetal growth throughout gestation by ultrasound and post-natal assessment of fetal survival, birthweight, external observations and skeletal morphology (by X-ray), immunotoxicity endpoints such as lymphocyte immunophenotyping (from one-month after birth), TDAR (from 3–6 months after birth) and other immune function tests can be included in the post-natal assessment depending on the MoA of the mAb.Citation108,Citation111 Infant lymphoid organ weights and histopathology can be performed at termination. The question arises as to whether it is necessary to include developmental immunotoxicity measurements within an EFD-PPND study (and potentially extend the duration of the study for up to 6 months) if a mAb has no effects on the immune system in adult animals. The need for developmental immunotoxicity assessment is based on the premise that the developing immune system may be more susceptible to immune perturbation than the adult immune system; however, currently there is little evidence to support this.Citation112,Citation113
Juvenile toxicity studies.
When immunomodulatory mAbs are developed for pediatric clinical indications, an important question is whether existing toxicology data developed in sexually-mature or peri-pubertal (2–3 years) animals can be extrapolated to children. The answer depends on both the MoA of the drug and the intended patient population. Clinical trials of immunomodulatory mAbs in children ≥2 years-old may generally be supported by toxicology studies conducted to support study of adult patients. Clinical studies in children and infants below the age of 2 may require specific juvenile toxicity studies to evaluate effects of the mAb on the immune system, which is not mature at birth and continues to develop post-natally, particularly in the first six months of life.Citation113 Immunotoxicity endpoints can be included as with adult animals, but the age/size-related limitations on blood sampling may limit the number of endpoints that can be assessed. If juvenile toxicity studies are necessary to support clinical trials in infants or children, it is desirable to conduct these studies in rats provided that the rat is a pharmacologically-responsive species. If the only pharmacologically-responsive species is the NHP, as frequently occurs with mAbs against human immune system targets, conducting the juvenile toxicology study in a breeding facility may be the only option since infant NHPs (<9–12 months old) cannot generally be shipped between sites. Another option is to evaluate effects in neonatal rodents using a surrogate molecule. The decision as to which approach is best for risk assessment should be made on a case-by-case basis reflecting the MoA and the age of children to be included in trials.
Host resistance assays.
Host resistance assays, i.e., effect of the mAbs on growth and pathogenesis/mortality following challenge with bacteria, viruses, fungi, parasites or tumors should be considered on a case-by-case basis for mAbs with broad-spectrum immunosuppressive activity or have shown suppressive effects on the TDAR or other immune function tests. Host resistance assays might potentially be useful in determining if the immune system is compromised to the point of ineffectiveness in providing protection from specific organisms, in determining the length of reduced resistance and relationship with the PK/PD and might confirm the affected cell population from immune function tests by decreasing resistance to organism controlled by certain cell types.Citation114 Host resistance assays might also help to rank a reduced host defense effect versus an immunosuppressive agent with clinical experience and might help to avoid the requirement for clinical infection studies. If such studies are deemed useful then the choice of host resistance model is dependent on the MoA, i.e., the particular immune cells/pathways targeted by the mAb. For example, for a mAb that affects NK cell function, models of CMV infection or tumors known to be controlled by NK cells would be considered. For a mAb that affects cell-mediated immunity or macrophage function then challenge with a facultative intracellular parasite such as Listeria might be relevant. However, host resistance assays are not routinely performed within the industry since their predictive value for humans is unproven, although rodent influenza and Candida albicans models and others have been used with some mAbs.Citation89,Citation100,Citation114,Citation115 Even if a mAb shows no effects in a range of host resistance assays, one cannot conclude that no such effects will occur in humans. In addition, a mAb with an immunosuppressive MoA or that neutralizes cytokines/cell types involved in host defense is likely to get a general label of potential increased risk of infection and cancer, even if host defense studies prove negative. Many investigators instead choose to address the potential impaired resistance to microbial pathogens in clinical trials and in the subsequent clinical risk management and pharmacovigilence plans. Host resistance assays are rarely performed in the NHP due to lack of qualified models, low animal numbers, high inter-animal variation and lack of Specific Pathogen-Free (SPF) animals, hence NHPs carry different pathogens. Rodent models are available, usually requiring the use of a surrogate mAb. These rodent models are time-consuming and expensive, require specialist external CRO expertise and, due to overlapping and compensatory immune pathways, effects on immune function may not result in decreased host resistance unless multiple host resistance models (a combination of bacterial, viral and tumor models) and immune function tests are utilized to increase the weight-of-evidence.Citation99 In these models, the primary endpoint is often mortality, which is insensitive and of debatable utility as a predictor of immunosuppression. However, continuous endpoints, e.g., colony/plaque-forming units, are now being used to increase sensitivity.Citation116 In addition, the susceptibility to infection in animals is dependent both on the degree of immunosuppression and number of challenge organisms. The predictivity of such models for humans, where the degree of immunosuppression may be variable in the out-bred population and the number/nature of challenge organisms cannot be controlled, is further questioned. Infection in humans occurs on a background of concomitant medication and underlying disease, e.g., RA, psoriasis, variables not tested in host resistance models. The available host resistance database is limited to a small number of usually high immunosuppressive drugs and hence the question remains as to whether these models can detect the effect of a mild/moderate immunosuppressant on host defence.
One should first consider whether the target is involved in mediating defense against particular organisms that might be a risk in humans and if existing ‘class effect’ data is known in animals or humans or whether infectious agent/tumor challenge data exists from animals treated with a mAb against the same target or from target knockout mice. In these cases host resistance studies may be of little value since a negative result in a challenge model would not negate the existing data. In many cases it is more relevant to address the risk of infection in the clinical risk management program.
Autoimmune disease, hypersensitivity and allergy models.
Diseases such as autoimmunity (arthritis, multiple sclerosis (MS), thyroiditis, diabetes, lupus) and allergy/hypersensitivity, e.g., anaphylaxis, glomerulonephritis, vasculitis, could be induced or exacerbated by mAbs.Citation32,Citation33 For most mAbs, the incidence is likely to be very low and dependent on factors in addition to the MoA such as patient disease state, genetics, ethnicity, age, environmental exposure, immune status etc., which are difficult to replicate in animals. Existing animal models for autoimmunity, e.g., genetically-susceptible rodent models of spontaneous autoimmune disease and autoantigen-induced autoimmunity in rodents, are not standardized and validated to predict risk of autoimmunity with mAbs in humans, and major discrepancies in the data obtained from these models and human data have been observed. Hence they are not recommended.Citation117 It is possible that autoimmune effects seen in humans might allow specific animal models to be re-investigated and modified to increase predictivity so they can be used to assess effects of other mAbs with a similar MoA. There are also no validated in vivo models for assessing hypersensitivity/allergy to mAbs, i.e., ADA leading to anaphylaxis or immune-complex disease, that are predictive of effects in man. mAbs that are non-immunogenic in humans induce severe anaphylaxis in existing guinea-pig anaphylaxis models.Citation117 Animals models that are more relevant and in silico and in vitro tests for predicting immunogenicity/hypersensitivity of mAbs were discussed previously. Existing models of allergen-induced allergy/asthma, e.g., with ovalbumin, house-dust mite, cat dander, are also not validated for predicting effects of mAbs on human allergic disease.
Immunopharmacology and Immunotoxicology Data Analysis and Impact on the Clinical Risk Management Plan
In performing these immunotoxicity tests and reviewing the obtained data, one must consider the nature, severity, frequency, dose dependency and reversibility of any immunotoxic effect in animals and their relevance to humans. Certain potential immunotoxic effects might be better assessed in the clinical risk management plan rather than in additional non-clinical studies. The immunopharmacology, immunotoxicology and host defense data should help clinicians understand what immunopharmacology is desirable, and what risks are may be involved in undesirable immunotoxicity and decreased host defense. The data can be used to help set inclusion/exclusion criteria for patients and guidelines for the use of concomitant medication, e.g., certain mAbs should not be administered with other immunomodulatory biologicals or NCEs. The data may assist in setting the clinical dosing regimen, e.g., on-and-off dosing to reduce chances of infection/tumors. The data might help identify patient subgroups for pharmacovigilence or infective organisms to be closely monitored for. The recovery period from any immunotoxicity, if PK/PD related, might inform the clinician about a suitable period of post-treatment monitoring for infections, autoimmunity or other effects. Consider also whether any immune tests/immune biomarkers have been identified that could be used to detect signs of immunotoxicity in the clinic.
Use of Immunopharmacology and Immunotoxicity Data in Selection of a Safe Starting Dose in Humans
With TGN1412, the life-threatening events were related to the pharmacology of the mAb and were not predicted from monkey toxicology studies since subsequent studies have shown TGN1412 to be minimally responsive at activating T cells in NHPs compared with humans. This illustrates the dangers of failing to understand the relative immunopharmacology (especially potency and downstream effects of signaling) between animals and humans. In response to the TGN1412 incident, a guideline was issued by the EMACitation11 which presents steps that can be taken as part of a risk mitigation strategy when conducting FIH studies. It emphasizes the importance of not just determining a pharmacologically-active dose (PAD), as recommended in the FDA guideline,Citation10 but also exploring the full pharmacological dose/concentration-response curve. The EMA guideline also introduces the concept of defining the minimal anticipated biological effect level (MABEL) and its consideration in the selection of a safe maximum recommended starting dose (MRSD) in humans. The MABEL represents the lowest animal dose or concentration required to produce pharmacological activity in vivo or in vitro in animal/human systems. The MRSD should be selected based on demonstration of an adequate safety margin compared with doses which cause toxicity, or the highest safe dose (NOAEL) tested in the case of mAbs with low toxicity, in non-clinical testing, as well as consideration of the MABEL. The calculation of the MABEL for mAbs has recently been reviewed,Citation12,Citation13 and should utilize all relevant biological and pharmacological information and consider the novelty of the agent and its MoA (agonist or antagonist), the degree of species-specificity of the mAb for both target binding and in some cases FcγR binding, target distribution and expression level and pharmacological activity. The full concentration:(immuno)pharmacology response curves in human and animal cells in vitro should be characterized to include a quantitative comparison of binding and resulting (immuno)pharmacological activity. Differences in immunopharmacological activity and relative potency between humans and the chosen toxicology species should be accounted for when extrapolating the immunotoxicological and immunopharmacological responses observed in animals to those predicted in humans and in calculating the MABEL. PK/PD modeling can be used to integrate mAb concentrations in blood and tissue with immunopharmacological or immunotoxicological properties of the mAb in animals and enables the prediction of immune target binding/immunopharmacology in humans based upon adjusted animal parameters.Citation118,Citation119
Immunotoxicity Assessment in Humans
As described here, a range of in vivo immunopharmacology studies with human blood and cells, as well as toxicology studies in pharmacologically-relevant species, will help to characterize the immunological effects of a mAb and some aspects of potential immunotoxicity prior to human dosing. Sensitive methods to predict and prevent acute life-threatening effects like cytokine storms, as well as hypersensitivity responses, should continue to be explored and developed. In addition, a number of the in vivo immune endpoints for use in non-clinical animal studies, such as standard hematology assessment (total and absolute differential leukocyte counts), clinical chemistry (globulin levels and albumin:globulin ratios, acute phase proteins), as well as serum cytokine, complement and immunoglobulin measurements and immunophenotyping of peripheral blood cells, including specific subsets of interest and markers of activation, can be performed with blood from clinical trial subjects treated with the mAb. Humans can also be immunized with antigens such Hepatitis B surface antigen, influenza and KLH to assess the effect of a mAb on the TDAR; however the prior infection status of the subjects needs to be considered. Depending on the MoA of the mAb, an ex vivo functional assessment of the effects of a mAb on a range of immune cell types such as T cells, B cells and NK cells and macrophages can be performed. For immunosuppressive mAbs, the incidence of infections within mAb-treated subjects should be compared with control-treated subjects following specifically-designed protocols and techniques for microbiological identification. To increase the chances of early detection of immunotoxicity in humans, it is recommended that, where possible, all immunopharmacological and immunotoxicological effects suspected based on mechanism of action or results of non-clinical studies be assessed in the clinic. Although the relevance of many of the aforementioned immunological parameters for the detection of immunotoxicity in humans is largely unknown at present, maximizing the number of immunological parameters measured might increase the chance of early detection of immunotoxicity. However, there is a pressing need to identify and validate novel clinical biomarkers of immunotoxicity so that relatively rare immunotoxic events observed with mAbs, such as the development of autoimmune disease and hypersensitivity reactions, can be detected early.Citation23
Conclusion
Based on their structure, MoA and treatment populations, many therapeutic mAbs will directly interact with the immune system. Immunotoxicity of immunomodulatory mAbs is largely due to exaggerated immunopharmacology. A range of tests can be performed with human blood/cells/tissue and in pharmacologically-relevant animal models to both reduce the risk of some types of immunotoxicity occurring in humans and to characterize the immunopharmacological and immunotoxicological effects of mAbs. This will allow a better understanding of the expected immunological effects in humans and the likely immunotoxicological consequences of any exaggerated pharmacology in FIH studies and beyond. It is recognized that not all immunotoxic effects can be predicted from non-clinical tests and that there is a need for the development of further tests for the prevention or early detection of immunotoxicity in humans.
Abbreviations
| ADA | = | anti-drug antibody |
| ADCC | = | antibody-dependent cellular cytotoxicity |
| ADME | = | absorption, distribution, metabolism and excretion |
| APC | = | antigen-presenting cell |
| AS | = | ankylosing spondylitis |
| CAPS | = | cropyrin-associated periodic syndromes |
| CD | = | cluster of differentiation |
| CDC | = | complement-dependent cytotoxicity |
| CDR | = | complementarity-determining region |
| CMV | = | cytomegalovirus |
| COPD | = | chronic obstructive pulmonary disease |
| CRA | = | cytokine release assay |
| CrD | = | Crohn disease |
| CRS | = | cytokine release syndrome |
| CTLA-4 | = | cytotoxic T lymphocyte antigen-4 |
| DAMPs | = | damage-associated molecular patterns |
| DC | = | dendritic cell |
| DTH | = | delayed-type hypersensitivity |
| EBV | = | Epstein Barr virus |
| EFD-PPND | = | embryo-fetal development and peri-/post-natal development |
| EMA | = | European Medicines Agency |
| EPAR | = | European Public Assessment Report |
| EPO | = | erythropoietin |
| ESG | = | Expert Scientific Group |
| FDA | = | Food and Drug Administration |
| FIH | = | first-in-human |
| GD | = | gestation day |
| GLP | = | good laboratory practice |
| HED | = | human equivalent dose |
| HHV-8 | = | human herpes virus-8 |
| HLA | = | human leukocyte antigen |
| HSA | = | human serum albumin |
| HSP | = | heat shock protein |
| HTLV-1 | = | human T cell leukemia virus-1 |
| ICH | = | International Conference on Harmonization |
| IHC | = | immunohistochemistry |
| KLH | = | keyhole limpet hemocyanin |
| LCV | = | lymphocryptovirus |
| LFA-1 | = | leukocyte function antigen-1 |
| LPS | = | lipopolysaccharide |
| mAb | = | monoclonal antibody |
| MABEL | = | minimum anticipated biological effect level |
| MHC | = | major histocompatibility comlex |
| MoA | = | mechanism of action |
| MRSD | = | maximum recommended starting dose |
| MS | = | multiple sclerosis |
| NCE | = | new chemical entity |
| NHP | = | non-human primate |
| NK | = | natural killer |
| NLR | = | nod-like receptor |
| NOAEL | = | no observed adverse effect level |
| PAD | = | pharmacologically-active dose |
| PAMPs | = | pathogen-associated molecular patterns |
| PEG-MGDF | = | pegylated megakaryocyte growth and development factor |
| PD | = | pharmacodynamic |
| PHA | = | phytohemaglutinin |
| PK | = | pharmacokinetic |
| PML | = | progressive multifocal leukoencephalopathy |
| PsA | = | psoriatic arthritis |
| RA | = | rheumatoid arthritis |
| RMP | = | risk management plan |
| RO | = | receptor occupancy |
| RSV | = | respiratory syncytial virus |
| SBA | = | summary basis of approval |
| SLE | = | systemic lupus erythromatosus |
| SPC | = | summary of product characteristics |
| SRBC | = | sheep red blood cell |
| TCR | = | tissue cross reactivity |
| TDAR | = | T cell-dependent antibody response |
| TLR | = | toll-like receptor |
| TT | = | tetanus toxoid |
| UC | = | ulcerative colitis |
| VLA-4 | = | very late antigen-4 |
Figures and Tables
Figure 1 Key immune system interactions are targeted by approved therapeutic mAbs. This figure illustrates the immunological pathways targeted by the approved mAbs and Fc-fusion proteins summarized in Table 1. CD, cluster of differentiation; CTLA-4, cytotoxic T-lymphocyte antigen-4; EpCAM, epithelial cell adhesion molecule; GM-CSF, granulocyte macrophage-colony stimulating factor; HLA, human leukocyte antigen; ICAM, intercellular adhesion molecule; IFN, interferon; Ig, immunoglobulin; IL, interleukin; LFA, lymphocyte function-associated antigen; TNF, tumor necrosis factor; LT, lymphotoxin; RANKL, receptor activator of nuclear factor kappaB ligand; TH cell, T helper cell; TRAIL, TNF-related apoptosis-inducing ligand; VCAM, vascular cell adhesion molecule; VLA, very late antigen.
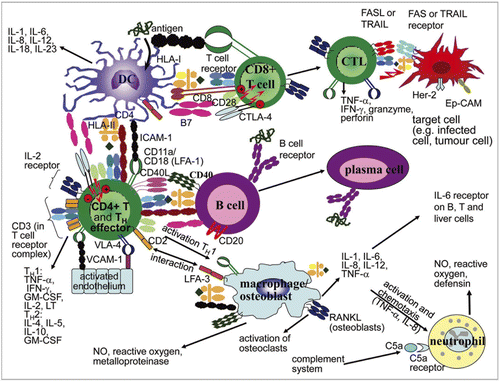
Figure 2 Strategy and timing for assessment of immunotoxicity with immunomodulatory mAbs. A range of immunological tests can be applied at different stages of the product development program of a mAb. *May be required depending on the MoA of the mAb and/or to investigate the mechanism of any immune function defects observed in non-clinical or clinical studies.
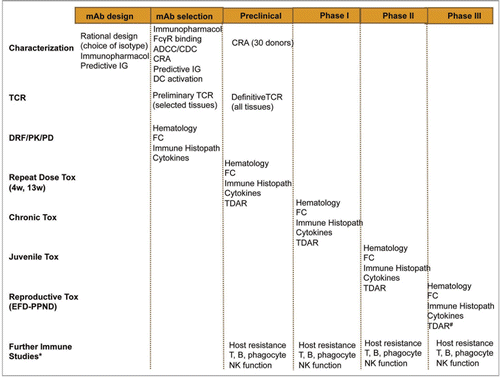
Figure 3 T cell-dependent and -independent induction of anti-drug antibody formation. In most cases, formation of anti-drug antibodies is T cell-dependent (A). T cell activation requires preceding activation of professional APCs such as DCs. Immature DCs (im DC) scan their direct environment constantly for danger signals, while they ingest the surrounding matrix by fluid phase or receptor mediated endocytosis. Ligation of pattern recognition receptors (PRRs) by danger-associated molecular patterns (DAMPs) such as exposed hydrophobic structures of aggregated proteins or pathogen-associated molecular patterns (PAMPs) such as LPS and ligation of Fcγ receptors (FcγR) induce immature DCs to mature and migrate to draining lymph nodes. Drugs that were taken up by the immature DCs will then get processed to small peptides. If these peptides have the appropriate primary structure, they form a complex with HLA class II molecules (HLA II:peptide). In the lymph node, the mature DCs (mat DC) will present the HLA II:peptide complexes to T cells, while also providing the additional signals required to prime naïve CD4+ T cells (co-stimulatory molecules such as C80 and CD86, as well as cytokines such as IL-10 and IL-4). Naïve B cells expressing a B cell receptor that recognize the drug will specifically endocytose the drug and process it to the same peptides, which are generated by the DCs. This allows the activated drug specific CD4+ T cells to induce drug specific B cells to proliferate and differentiate into memory B cells and plasma cells. The same cellular activation steps are required to react to neo antigens derived from non-human or modified protein and to break down tolerance to drugs with fully human protein sequences. In contrast, aggregated drug may induce B cell activation directly by cross-linking of the drug or drug aggregate specific B cell receptor on the surface of B cells (B). However, in this case typically no isotype switching or memory formation is observed. As aggregated drug can also induce the T cell-dependent B cell activation by affecting the DC activation, the two pathways may also be synergistic in some cases. The presence of DAMPs and PAMPs in drug substance or drug product can be investigated in vitro by using monocyte-derived DCs. In silico immunogenicity prediction tools focus on the capability of a defined protein sequence to bind to HLA class II molecules. A more reliable approach to identify drug-derived peptides which are presented on APCs is to sequence the peptides which were eluted from HLA class II molecules expressed by monocyte-derived DCs after challenge with the protein drug. Furthermore, in vitro T cell activation assays allow investigation of the whole process from antigen uptake, through antigen processing, to the capability of the generated peptides to activate naïve T cells. As there is currently no in vitro tool available to investigate T cell-dependent or -independent B cell activation, the whole process can only be investigated by using transgenic or double transgenic mouse models, with the drawback that the immune system of mice and humans appears to be quite different.
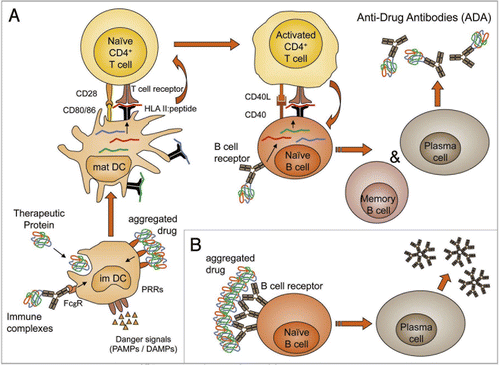
Table 1 FDA and/or EMA-Approved mABs and Fc-fusion proteins
Table 2 Key guidelines relevant to non-clinical safety testing of mAbs
Table 3 Key functional characteristics of human IgG subclasses
References
- St. Clair EW. Novel targeted therapies for autoimmunity. Curr Opin Immunol 2009; 21:648 - 657
- Genovese MC. Biologic therapies in clinical development for the treatment of rheumatoid arthritis. J Clin Rheumatol 2005; 11:45 - 54
- Menter A. The status of biologic therapies in the treatment of moderate to severe psoriasis. Cutis 2009; 84:14 - 24
- Rommer PS, Stüve O, Goertsches R, Mix E, Zettl UK. Monoclonal antibodies in the therapy of multiple sclerosis:an overview. J Neurol 2008; 255:28 - 35
- Isaacs JD. Therapeutic agents for patients with rheumatoid arthritis and an inadequate response to tumor necrosis factor-alpha antagonists. Expert Opin Biol Ther 2009; 9:1463 - 1475
- Melero I, Hervas-Stubbs S, Glennie M, Pardoll DM, Chen L. Immunostimulatory monoclonal antibodies for cancer therapy. Nat Rev Cancer 2007; 7:95 - 106
- Deonarain MP. Recombinant antibodies for cancer therapy. Expert Opin Biol Ther 2008; 8:1123 - 1141
- Griggs J, Zinkewich-Peotti K. The state of the art: immune-mediated mechanisms of monoclonal antibodies in cancer therapy. Br J Cancer 2009; 101:1807 - 1812
- Suntharalingam G, Perry MR, Ward S, Brett SJ, Castello-Cortes A, Brunner MD, et al. Cytokine storm in a phase 1 trial of the anti-CD28 monoclonal antibody TGN1412. N Engl J Med 2006; 355:1018 - 1028
- CDER/FDA. Estimating the Maximum Safe Starting Dose in Initial Clinical Trials for Therapeutics in Adult Healthy Volunteers 2005;
- EMEA/CHMP. Guideline on Strategies to Identify and Mitigate Risks for First-In-Human Clinical Trials with Investigational Medicinal Products 2007;
- Muller PY, Brennan FR. Safety assessment and dose selection for first-in-human clinical trials with immunomodulatory monoclonal antibodies. Clin Pharmacol Ther 2009; 85:247 - 258
- Muller PY, Milton M, Lloyd P, Sims J, Brennan FR. The Minimum Anticipated Biological Effect Level (MABEL) for selection of first human dose in clinical trials with monoclonal antibodies. Curr Opin Biotechnol 2009; 20:1 - 8
- Su X, Lacouture ME, Jia Y, Wu S. Risk of high-grade skin rash in cancer patients treated with cetuximab-an antibody against epidermal growth factor receptor:systemic review and meta-analysis. Oncology 2009; 77:124 - 133
- Bria E, Cuppone F, Milella M, Verma S, Carlini P, Nistico C, et al. Trastuzumab cardiotoxicity: biological hypotheses and clinical open issues. Expert Opin Biol Ther 2008; 8:1963 - 1971
- Zareba KM. Eculizumab: A novel therapy for paroxysmal nocturnal hemoglobinuria. Drugs Today 2007; 43:539 - 546
- Sabashi R, Anolik JH. B-cell-targeted therapy for systemic lupus erythematosus. Drugs 2006; 66:1933 - 1948
- Quintás-Cardama A, Wierda W, O’Brien S. Investigational immunotherapeutics for B cell malignancies. J Clin Oncol 2010; 28:884 - 892
- Cranmer LD, Hersh E. The role of CTLA4 blockade in the treatment of malignant melanoma. Cancer Invest 2007; 25:613 - 631
- Valzasina B, Guiducci C, Dislich H, Killeen N, Weinberg AD, Colombo MP. Triggering of OX40 (CD134) on CD4(+)CD25+ T cells blocks their inhibitory activity: a novel regulatory role for OX40 and its comparison with GITR. Blood 2005; 105:2845 - 2851
- Khalil M, Vondeheide RH. Anti-CD40 agonist antibodies: preclinical and clinical experience. Update Cancer Ther 2007; 2:61 - 65
- Ponce R. Adverse consequences of immunostimulation. J Immunotoxicol 2008; 33 - 41
- Descotes J. Immunotoxicity of monoclonal antibodies. mAbs 2009; 1:104 - 111
- Rychly DJ, Dipiro JT. Infections associated with tumor necrosis factor-alpha antagonists. Pharmacotherapy 2005; 25:1191 - 1192
- Fraser G, Smith CA, Imrie K, Meyer R. the Hematology Disease Site Group of Cancer Care Ontario’s Program in Evidence-Based Care. Alemtuzumab in chronic lymphocytic leukemia. Curr Oncol 2007; 14:96 - 109
- Kimby E. Tolerability and safety of rituximab (Mabthera). Cancer Treat Rev 2005; 31:456 - 473
- Berger JR. Natalizumab and progressive multifocal leucoencephalopathy. Ann Rheum Dis 2006; 65:48 - 53
- Lindå H, von Heijne A, Major EO, Ryschkewitsch C, Berg J, Olsson T, et al. Progressive multifocal leukoencephalopathy after natalizumab monotherapy. N Engl J Med 2009; 361:1081 - 1087
- Stüve O, Marra CM, Jerome KR, Cook L, Cravens PD, Cepok S, et al. Immune surveillance in multiple sclerosis patients treated with natalizumab. Ann Neurol 2006; 59:743 - 747
- Major EO. Progressive multifocal leukoencephalopathy in patients on immunomodulatory therapies. Ann Rev Med 2010; 61:35 - 47
- Coles AJ, Wing M, Smith S, Coraddu F, Greer S, Taylor C, et al. Pulsed monoclonal antibody treatment and autoimmune thyroid disease in multiple sclerosis. Lancet 1999; 354:1691 - 1695
- Yang JC, Hughes M, Kammula U, Royal R, Sherry RM, Topalian SL, et al. Ipilimumab (anti-CTLA4 antibody) causes regression of metastatic renal cell cancer associated with enteritis and hypophysitis. J Immunother 2007; 30:825 - 830
- Wetter DA, Davis MD. Lupus-like syndrome attributable to anti-tumor necrosis factor alpha therapy in 14 patients during an 8-year period at the Mayo Clinic. Mayo Clin Proc 2009; 84:979 - 984
- Kang SP, Saif MW. Infusion-related and hypersensitivity reactions of monoclonal antibodies used to treat colorectal cancer-identification prevention and management. J Support Oncol 2007; 5:451 - 457
- Chung CH, Mirakhur B, Chan E, Le QT, Berlin J, Morse M, et al. Cetuximab-induced anaphylaxis and IgE specific for galactose α-1,3-galactose. N Engl J Med 2008; 358:1109 - 1117
- Lynch C, Hart BW, Grewal IS. Practical considerations for nonclinical safety evaluation of therapeutic monoclonal antibodies. mAbs 2009; 1:2 - 11
- ICH M3 (R2): Nonclinical safety studies for the conduct of human clinical trials with pharmaceuticals (2009) available at http://www.ema.europa.eu/pdfs/human/ich/028695en.pdf.
- ICH guideline S6: Preclinical Safety Evaluation of Biotechnology-Derived Pharmaceuticals (1998) available at http://www.ema.europa.eu/pdfs/human/ich/030295en.pdf.
- ICH guideline S6 (R1): Preclinical Safety Evaluation of Biotechnology-Derived Pharmaceuticals. Step 2 Addendum (2009) available at http://www.ich.org/LOB/media/MEDIA5784.pdf.
- Points to Consider in the Manufacture and Testing of Monoclonal Antibody Products for Human Use FDA, 2000 available at http://www.fda.gov/downloads/BiologicsBloodVaccines/GuidanceComplianceRegulatoryInformation/OtherRecommendationsforManufacturers/UCM153182.pdf.
- ICH guideline S8: Notes for Guidance on Immunotoxicity Testing of Human Pharmaceuticals (2004) available at http://www.ema.europa.eu/pdfs/human/ich/16723504en.pdf.
- FDA (CDER): Immunotoxicology Evaluation of Investigational New Drugs (2002) available at http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM079239.pdf.
- Japanese MHLW/JPMA Draft Guidance for Immunotoxicity Testing (draft 2001)
- Guideline on Immunogenicity Assessment of Biotechnology-Derived Therapeutic proteins (2007) available at http://www.ema.europa.eu/pdfs/human/biosimilar/1432706enfin.pdf.
- Jefferis R. Antibody therapeutics: isotype and glycoform selection. Expert Opin Biol Ther 2007; 7:1401 - 1413
- Bruhns P, Iannascoli B, England P, Mancardi DA, Fernanedez N, Jorieux S, et al. Specificity and affinity of human Fcgamma receptors and their polymorphic variants for human IgG subclasses. Blood 2009; 113:3716 - 3725
- Nimmerjahn F, Ravetsch JV. Fcγ receptors as regulators of immune responses. Nat Rev Immunol 2 2008; 8:34 - 47
- Labrijn AF, Buijsse AO, van den Brener ETJ, Verwilligen AYW, Bleeker WK, Thorpe SJ, et al. Therapeutic IgG4 antibodies engage in Fab-arm exchange with endogenous human IgG4 in vivo. Nat Biotechnol 2009; 27:767 - 771
- Oganesyan V, Gao C, Shirinian L, Wu H, Dall’Acqua WF. Structural characterization of a human Fc fragment engineered for lack of effector function. Acta Crystallogr D Biol Crystallogr 2008; 64:700 - 704
- Panelli MC, White R, Foster M, Martin B, Wang E, Smith K, et al. Forecasting the cytokine storm following systemic interleukin (IL)-2 administration. J Transl Med 2004; 2:17
- Wing M. Monoclonal Antibody First Dose Cytokine Release Syndromes-Mechanisms and Prediction. J Immunotoxicology 2008; 5:11 - 15
- Stebbings R, Findlay L, Edwards C, Eastwood D, Bird C, North D, et al. “Cytokine storm“ in the phase I trial of monoclonal antibody TGN1412: better understanding the causes to improve preclinical testing of immunotherapeutics. J Immunol 2006; 179:3325 - 3331
- Hsu DH, Shi JD, Homola M, Rowell TJ, Moran J, Levitt, et al. A humanized anti-CD3 antibody, HUM291, with low mitogenic activity, mediates complete and reversible T-cell depletion in chimpanzees. Transplantation 1999; 68:545 - 554
- Langezaal I, Coecke S, Hartunga T. Whole blood cytokine response as a measure of immunotoxicity. Toxicology in Vitro 2001; 15:313 - 318
- Bugelski PJ. Cytokine Release Syndrome: Strategy for a Primary Screen. Slide Presentation, FDA Immunotoxicology Symposium, Cytokine Release: What Does It Mean? 2008;
- Final Report of the Expert Scientific Group on Phase I Clinical Trials and the subsequent EMEA Guideline on Strategies to Identify and Mitigate Risk for FIH Clinical Trials with Investigational Medicinal Products (EMEA/CHMP/SWP/28367/07)
- Rodriguez-Palmero M, Hara T, Thumbs A, Hünig T. Triggering of T cell proliferation through CD28 induces GATA-3 and promotes T helper type 2 differentiation in vitro and in vivo. Eur J Immunol 1999; 29:3914 - 24
- Jäger M, Hara T, Thumbs A, Hünig T. The trifunctional antibody ertumaxomab destroys tumor cells that express low levels of human epidermal growth factor receptor. Cancer Res 2009; 69:4270 - 4276
- Ju X, Clark G, Hart DN. Review of human DC subtypes. Methods Mol Biol 2010; 595:3 - 20
- Fukata M, Vamadevan AS, Abreu MR. Toll-like receptors (TLRs) and Nod-like receptors (NLRs) in inflammatory disorders. Semin Immunol 2009; 21:242 - 253
- Fujii S, Liu K, Smith C, Bonito AJ, Steinman RM. The linkage of innate to adaptive immunity via maturing dendritic cells in vivo requires CD40 ligation in addition to antigen presentation and Cd80/86 costimulation. J Exp Med 2004; 199:1607 - 1618
- Geissmann F, Revy P, Regnault A, Lepelletier Y, Dy M, Brousse N, et al. TGFbeta prevents the noncognate maturation of human dendritic Langerhans cells. J Immunol 1999; 162:4567 - 4575
- Schuurhuis DH, Ioan-Facsinay A, Nagelkerken B, van Schip JJ, Sedlik C, Melief CJ, et al. Antigen-antibody immune complexes empower dendritic cells to efficiently prime CD8+ CTL responses in vivo. J Immunol 2002; 168:2240 - 2246
- Mueller R, Karle A, Vogt A, Kropshofer H, Ross A, Maeder K, et al. Evaluation of the immuno-stimulatory potential of stopper extractable and leachables by using dendritic cells as readout. J Pharm Sci 2009; 98:3548 - 3561
- Descotes J, Gouraud A. Clinical immunotoxicity of therapeutic proteins. Expert Opin Drug Metab Toxicol 2008; 4:1537 - 1549
- Li J, Yang C, Xia Y, Bertino A, Glaspy J, Roberts M, et al. Thrombocytopenia caused by the development of antibodies to thrombopoietin. Blood 2007; 98:3241 - 3248
- Casadevall N, Nataf J, Viron B, Kolta A, Kiladjian JJ, Martin-Dupont P, et al. Pure red-cell aplasia and anti-erythropoietin antibodies in patients treated with recombinant erythropoietin. N Engl J Med 2002; 346:469 - 475
- Hermeling S, Aranha L, Damen JMA, Slijper M, Schellekens H, Crommelin DJA, et al. Structural characterization and immunogenicity in wild-type and immune tolerant mice of degraded recombinant human interferon alpha2b. Pharm Res 2005; 22:1997 - 2006
- Weinblatt ME, Keystone EC, Furst DE, Moreland LW, Weisman MH, Birbara CA, et al. Adalimumab, a fully human anti-tumor necrosis factor alpha monoclonal antibody, for the treatment of rheumatoid arthritis in patients taking concomitant methotrexate: the ARMADA trial. Arthritis Rheum 2003; 48:35 - 45
- Van de Putte LBA, Atkins C, Malaise M, Sany J, Russell AS, van Riel PLCM, et al. Efficacy and safety of adalimumab as monotherapy in patients with rheumatoid arthritis for whom previous disease modifying antirheumatic drug treatment has failed. Ann Rheum Dis 2004; 63:508 - 516
- Rosloniec EF, Brand DD, Myers LK, Whittington KB, Gumanovskaya M, Zaller DM, et al. An HLA-DR1 transgene confers susceptibility to collagen-induced arthritis elicited with human type II collagen. J Exp Med 1997; 185:1113 - 1122
- Gonzalez-Gay MA, Zanelli E, Khare SD, Krco CJ, Zhou P, Inoko H, et al. Human leukocyte antigen-DRB1*1502 (DR2Dw12) transgene reduces incidence and severity of arthritis in mice. Hum Immunol 1996; 50:54 - 60
- Strauss G, Vignali DA, Schonrich G, Hammerling GJ. Negative and positive selection by HLA-DR3(DRw17) molecules in transgenic mice. Immunogenetics 1994; 40:104 - 108
- Kouskoff V, Fehling HJ, Lemeur M, Benoist C, Mathis DA. A vector driving the expression of foreign cDNAs in the MHC class II-positive cells of transgenic mice. J Immunol Methods 1993; 166:287 - 291
- Braud VM, McMichael AJ, Cerundolo V. Differential processing of influenza nucleoprotein in human and mouse cells. Eur J Immunol 1998; 28:625 - 635
- Ito M, Hiramatsu H, Kobayashi K, Suzue K, Kawahata M, Hioki K, et al. NOD/SCID/gamma(c)(null) mouse: an excellent recipient mouse model for engraftment of human cells. Blood 2002; 100:3175 - 3182
- Traggiai E, Chicca L, Mazzucchelli L, Bronz L, Piffaretti JC, Lanzavecchia A, et al. Development of a human adaptive immune system in cord blood cell-transplanted mice. Science 2004; 304:104 - 107
- Sturniolo T, Bono E, Ding J, Raddrizzani L, Tuereci O, Sahin U, et al. Generation of tissue-specific and promiscuous HLA ligand databases using DNA microarrays and virtual HLA class II matrices. Nat Biotechnol 1999; 17:555 - 561
- Peters B, Sette A. Generating quantitative models describing the sequence specificity of biological processes with the stabilized matrix method. BMC Bioinformatics 2005; 6:132
- Nielsen M, Lundegaard C, Worning P, Hvid CS, Lamberth K, Buus S, et al. Improved prediction of MHC class I and class II epitopes using a novel Gibbs sampling approach. Bioinformatics 2004; 20:1388 - 1397
- Desmet J, Meersseman G, Boutonnet N, Pletinckx J, De Clercq K, Debulpaep M, et al. Anchor profiles of HLA-specific peptides: analysis by a novel affinity scoring method and experimental validation. Proteins 2005; 58:53 - 69
- Kropshofer H, Spindeldreher S. Kropshofer Harald, Vogt Anne B. Naturally processed self-peptides of MHC molecules. Antigen Presenting Cells 2005; Weinheim Wiley-VCH 159 - 198
- Perry LC, Jones TD, Baker MP. New Approaches to prediction of immune responses to therapeutic proteins during preclinical development. Drugs R D 2008; 9:385 - 396
- Koren E, Smith HW, Shores E, Shankar G, Finco-Kent D, Rup B, et al. Recommendations on risk-based strategies for detection and characterization of antibodies against biotechnology products. J Immunol Methods 2008; 333:1 - 9
- Chapman K, Pullen N, Coney L, Dempster M, Andrew L, Bajramovic J, et al. Preclinical development of monoclonal antibodies. Considerations for the use of non-human primates. mAbs 2009; 1:505 - 516
- Treacy G. Using an analogous monoclonal antibody to evaluate the reproductive and chronic toxicity potential for a humanized anti-TNF-a monoclonal antibody. Hum Exp Toxicol 2000; 19:226 - 229
- Clarke J, Leach W, Pippig S, Joshi A, Wu B, House R, et al. Evaluation of a surrogate antibody for preclinical safety testing of an anti-CD11a monoclonal antibody. Regul Toxicol Pharmacol 2004; 40:219 - 226
- ICHQ6B: Specifications: Test Procedures and Acceptance Criteria for Biotechnological/Biological Products Available at http://www.ema.europa.eu/pdfs/human/ich/036596en.pdf.
- Bugelski PJ, Herzyk DJ, Rehm S, Harmsen AG, Gore EV, Williams DM, et al. Preclinical development of keliximab, a primatized anti-CD4 monoclonal antibody, in human CD4 trangenic mice: characterization of the model and safety studies. Hum Exp Toxicol 2000; 19:230 - 243
- Nguyen DH, Hurtado-Ziola N, Gagneux P, Varki A. Loss of Siglec expression on T lymphocytes during human evolution. Proc Natl Acad Sci USA 2006; 103:7765 - 7770
- Crocker PR. Siglecs in innate immunity. Curr Opin Pharmacol 2005; 5:431 - 437
- Akari H, Terao K, Murayama Y, Nam K-H, Yoshikawa Y. Peripheral blood CD4+CD8+ lymphocytes in cynomolgus monkeys are of resting memory T lineage. Internat Immunol 1997; 9:591 - 597
- Teroa K. Essentials for starting a pediatric clinical study (3): dynamic changes in early development of immune system in macaque monkeys—the significance from standpoint of preclinical toxicity test using nonhuman primates. J Toxicol Sci 2009; 34:321 - 325
- Rogers KA, Scinicariello F, Attanasio R. IgG Fc receptor III homologues in nonhuman primate species: genetic characterization and ligand interactions. J Immunol 2006; 177:3848 - 3856
- Haley PJ. Species Differences in the Structure and the Function of the Immune System. Toxicology 2003; 188:49 - 71
- Haley P, Perry R, Ennulat D, Frame S, Johnson C, Lapointe JM, et al. STP Immunotoxicology Working Group. STP position paper: best practice guideline for the routine pathology evaluation of the immune system. Tox Path 2005; 33:404 - 407
- Kuper CF, Harleman JH, Richter-Reichelm HB, Vos JG. Histopathologic approaches to detect changes indicative of immunotoxicity. Toxicol Pathol 2000; 28:454 - 466
- Germolec DR, Kashon M, Nyska A, Kuper CF, Portier C, Kommineni C, et al. The accuracy of extended histopathology to detect immunotoxic chemicals. Toxicol Sci 2004; 82:504 - 514
- Luster MI, Portier C, Pait DG, Rosenthal GJ, Germolec DR, Corsini E, et al. Risk assessment in immunotoxicology II. Relationships between immune and host resistance tests. Fundam Appl Toxicol 1993; 71 - 82
- Orencia European Public Assessment Report; Scientific Discussion, EMEA, 2007 available at http://www.ema.europa.eu/humandocs/PDFs/EPAR/orencia/H-701-en6.pdf.
- Amevive BLA review: Pharmacology and Toxicology Assessment, FDA 2002 available at http://www.fda.gov/downloads/Drugs/DevelopmentApprovalProcess/HowDrugsareDevelopedandApproved/ApprovalApplications/TherapeuticBiologicApplications/ucm086014.pdf.
- Herzyk D, Holsapple M. Immunotoxicity evaluation by immune function tests: focus on the T-dependent antibody response. J Immunotoxicol 2007; 4:143 - 147
- Raptiva European Public Assessment Report; Scientific Discussion, EMEA, 2004 available at http://www.ema.europa.eu/humandocs/PDFs/EPAR/raptiva/6565604en6.pdf.
- Szekeres-Bartho J. Immunological relationship between the mother and fetus. Int Rev Immunol 2002; 21:471 - 495
- Laskarin G, Kammerer U, Rukavina D, Thomson AW, Fernandez N, Blois SM. Antigen-presenting cells and materno-fetal tolerance:an emerging role for dendritic cells. Am J Reprod Immunol 2007; 58:255 - 267
- Riley JK, Yokoyama WM. NK cell tolerance and the materno-fetal interface. Am J Reprod Immunol 2008; 59:371 - 387
- Roopenian DC, Akilesh S. FcRn: the neonatal Fc receptor comes of age. Nat Rev Immunol 2007; 7:715 - 725
- Weinbauer GF, Frings W, Fuchs A, Niehaus M, Osterburg I. Cavagnaro JA. Reproductive/developmental toxicity assessment of biopharmaceuticals in nonhuman primates. Preclinical safety evaluation of biopharmaceuticals 2008; New Jersey John Wiley & Son 379 - 398
- Pentsuk N, Van der Laan JW. An interspecies comparison of placental antibody transfer-implications for developmental toxicity testing of monoclonal antibodies and for human pregnancy outcome. Birth Defects Res B 2009; 86:328 - 344
- Martin PL, Oneda S, Treacy G. Effects of an anti-TNFα monoclonal antibody, administered throughout pregnancy and lactation, on the development of the macaque immune system. Am J Reprod Immunol 2007; 58:138 - 149
- Stewart J. Developmental toxicity testing of monoclonal antibodies: An enhanced pre- and post-natal study design option. Reproductive Toxicol 2009; 28:220 - 225
- Dietert RR. Developmental Immunotoxicology (DIT): windows of vulnerability, immune dysfunction and safety assessment. J Immunotoxicol 2008; 5:401 - 412
- Burns-Naas LA, Hastings KL, Ladics GS, Makris SL, Parker GA, Holsapple MP. What’s so special about the developing immune system?. Int J Toxicol 2008; 27:223 - 254
- Burleson GR, Burleson FG. Testing human biologicals in animal host resistance models. J Immunotoxicol 5:23 - 31
- Burleson GR, Burleson FG. Influenza virus host resistance model. Methods 2007; 41:31 - 37
- Cohen MD. Bacterial host resistance models in the evaluation of immunotoxicity. Methods 2007; 41:20 - 30
- Descotes J. Cavagnaro JA. Assessment of autoimmunity and hypersensitivity. Preclinical safety evaluation of biopharmaceuticals 2008; New Jersey John Wiley & Son 487 - 497
- Agoram BM. Use of pharmacokinetic/pharmacodynamic modeling for starting dose selection in first-in-human trials of high-risk biologics. Br J Clin Pharmacol 2009; 67:153 - 160
- Lowe PJ, Hijazi Y, Luttringer O, Yin H, Sarangapani R, Howard D. On the anticipation of the human dose in first-in-man trials from preclinical and prior clinical information in early drug development. Xenobiotica 2007; 37:1331 - 1354