Abstract
The Triomab® family of trifunctional, bispecific antibodies that maintain an IgG-like shape are novel tumor targeting agents. These chimeras consist of two half antibodies, each with one light and one heavy chain, that originate from parental mouse IgG2a and rat IgG2b isotypes. This combination allows cost-effective biopharmaceutical manufacturing at an industrial scale since this specific mouse/rat isotype combination favors matching of corresponding antibody halves during production by means of quadroma technology. Whereas every Triomab® family member is composed of an anti-CD3 rat IgG2b half antibody for T cell recognition, the antigen binding site presented by the mouse IgG2a isotype is exchangeable. Several Triomab® antibodies have been generated that bind to tumor-associated antigens, e.g., EpCAM (catumaxomab), HER2/neu (ertumaxomab), CD20 (FBTA05), gangliosides GD2/GD3 (Ektomun®), on appropriate tumor target cells associated with carcinomas, lymphomas or melanomas. Catumaxomab (Removab®) was launched in Europe for treatment of malignant ascites in April 2009. Here, we report the structural and functional characterization this product. Mass spectrometry revealed an intact mass of 150511 Dalton (Da) and 23717 Da, 24716 Da, 51957 Da and 52019 Da of the reduced and alkylated rat light chain, mouse light chain, rat heavy chain, mouse heavy chain chains, respectively. The observed masses were in agreement with the expected masses based on the amino acid sequence obtained from cDNA sequencing. The glycosylation profile was similar to other human IgG consisting of biantennary oligosaccharides with different numbers of terminal galactose. CD spectroscopy showed mainly β-sheets secondary structure that is typical for IgG antibodies. Binding measurement revealed the unique trifunctional features of catumaxomab. Other analytical tools were used to evaluate characteristics of catumaxomab preparations, including the presence of isoforms and aggregates.
Introduction
The success of immunotherapeutics as cancer drugs critically depends on the number of recruitable effector cells. The Triomab® antibody format was therefore designed to simultaneously induce several anti-tumor defense mechanisms by directing natural killer cells, cytotoxic T cells, macrophages/monocytes and dendritic cells to the site of tumor growth.Citation1–Citation4 Triomab® antibodies consist of two different full-size IgG-like half antibodies, i.e., mouse IgG2a and rat IgG2b isotypes; the unique structure of which allows binding to two distinct antigenic structures such as tumor-associated antigens and the CD3 molecule on T-cells, as well as binding to Fcγ-receptors via the Fc-region on accessory cells ().
Triomab® molecules mediate formation of tri-cell complexes, and in this manner classical antibody therapy, i.e., passive immunization, can be transformed into active in situ immunization.Citation5 Directed cytotoxic activity of T cells, participation of accessory cells and their costimulatory signaling, and Th1-type cytokine induced pro-inflammatory responses represent the major hallmarks of a Triomab®-based cancer treatment concept.Citation3,Citation4 Thus, use of Triomab® antibodies may immunize patients specifically against their own primary tumors, thereby protecting them from future relapses.
It has been shown that monoclonal antibodies (mAbs) pre-bound to tumor cells are superior to tumor cells alone in the priming of anti-tumor responses, with an emphasis on CD8 T cell immunity.Citation6 In this context, it is noteworthy that the chimeric mouse/rat Fc portion binds not or only marginally to the inhibitory low-affinity receptor FcγRIIb (CD32b), e.g., on B lymphocytes and platelets.Citation4 As the issue of abrogating the inhibitory FcγRIIb-engagement is central for improving effector mechanisms induced by mAbs such trastuzumab (anti-HER2/neu) or rituximab (anti-CD20), this feature of Triomab® Fc-region and Fcγ-receptor engagement is even more important.Citation7,Citation8 In general, most of the approved therapeutic antibodies exert their anti-tumor efficacy via antibody-dependent cellular cytotoxicity mainly evoked by Fcγ-receptor binding on natural killer cells. Taken together, the combined and highly concerted anti-tumor responses mediated by Triomab® antibodies lead to the elimination of tumor cells by necrosis,Citation9 phagocytosisCitation4 and enduring humoral, as well as cell-mediated, immunity in relapse situations.
Triomab® therapeutics are distinguished by their excellent dose-response profile. Four intraperitoneal catumaxomab infusions totaling 230 µg administered within 11 days sufficed for clinical benefit against malignant ascites (MA) in a pivotal Phase 2/3 study.Citation10,Citation11 Conventional antibody therapies against cancer diseases require cumulative antibody amounts ranging from 5–20 g per patient and year. MA is a manifestation of advanced disease in a variety of indications such as ovarian, breast or gastric cancer, and is characterized by strong fluid influx and tumor growth into the peritoneal cavity. The onset and progression of MA is associated with significant morbidity and deterioration in quality of life. Even in patients suffering from end-stage cancer disease, catumaxomab showed strong anti-tumor efficacy in a pivotal Phase 2/3 trial.Citation12,Citation13
Here we describe for the first time the physicochemical and functional characterization of a trifunctional antibody based on the Triomab® technology. Although marketed therapeutic antibodies do not have structures as complex as that of Triomab® products, which possess two different light and heavy chains, the characterization approach corresponds closely to those used for conventional therapeutic antibodies, with a few exceptions involving assessment of functional features.
Results
Regulatory guidelines define the characterization requirement of biopharmaceuticals.Citation14,Citation15 Comprising of a rat and a mouse half antibody, the unique structure of Triomab® antibodies results in some unique analytical challenges that need to be addressed, although most analytical methods used for the characterization of conventional mAbs can be applied to Triomab® antibodies as well. Analytical methods to assess the primary, secondary and tertiary structure, biological activity, and the presence of impurities such as aggregates were developed and used to characterize catumaxomab, an anti-EpCAM x anti-CD3 Triomab® antibody.
Primary structure.
Intact and reduced mass.
Liquid chromatography (LC) coupled with mass spectrometry (MS) has been used for the analysis of small organic and protein molecules for many years. The development of electrospray ionization in combination with new mass spectrometers allowed mass spectrometry of large molecules, including intact antibody molecules that are approximately 150 kiloDalton (kDa).Citation16,Citation17 In addition, new LC techniques have been developed for the analysis of intact antibody molecules and their individual chains, thereby improving the analytical capabilities significantly.Citation18 Using state of the art LC/MS technologies, mass accuracies of a few Daltons have been reported for intact antibody molecules.Citation19
Reversed phase HPLC coupled with an ESI-TOF mass spectrometer was used for the analysis of intact and reduced catumaxomab samples. The mass spectrum of the intact antibody and the deconvoluted spectrum are shown in . The observed molecular weight correlated well with the theoretical molecular weight of the molecule based on the amino acid sequence information deduced from the cDNA sequence. A mass accuracy of ±20 Da (∼100 ppm) was routinely achieved using external mass calibration. The intact mass analysis was used for the assessment of the integrity of the antibody. Minor modifications, e.g., oxidation or the status of the N-termini, cannot be assessed with this technology.
The different glycoforms of the molecule are clearly separated in the deconvoluted mass spectrum and therefore can be determined with this technology. A more detailed glycosylation analysis is shown later in the text. The raw data (insert in ) compares well to the deconvoluted mass spectra and can be used to assess the proper use of the deconvolution algorithm.
In order to improve the accuracy of mass determination, catumaxomab was chemically reduced, resulting in four individual peptide chains. The UV-chromatogram of the reduced and alkylated catumaxomab antibody is shown in . In contrast to conventional mAbs, four different chains are detectable (2 light and 2 heavy chains). The results of the ESI-TOF mass spectrometry analysis of the four chains are summarized in . The mass accuracy was approximately 10 ppm for the light chains and 20 ppm for the heavy chains. This mass accuracy was obtained using external calibration and could be improved to <2 ppm for the light and heavy chains using internal calibration. Covalent modifications like the status of the N-termini (pyroglutamic acid formation: −17 Da), oxidation (+16 Da), or the status of the C-terminal lysine of the heavy chains (+128 Da) can be assessed with this technology.
Amino acid sequence.
Peptide mapping has been the method of choice for the analysis of the amino acid sequence and disulfide bonds in biopharmaceuticals. Although Edman degradation is still used to determine the N-terminal sequence of proteins, sequencing using MS/MS data is rapidly replacing this time-consuming technology. MS/MS can reveal the N-terminal sequence even when the N-terminus is blocked for Edman sequencing, e.g., via pyroglutamate formation or acetylation. Sequencing via MS/MS is also convenient because all four N-termini can be analyzed simultaneously without previous chain separation. A large number of chemical and enzymatic cleavage reagents are available.Citation20,Citation21 The most common technique is the enzymatic digestion of the reduced and alkylated antibodies with trypsin for the generation of peptides for LC/MS analysis.Citation22,Citation23
Peptide mapping using Lys-C was used for the analysis of catumaxomab because Lys-C, in contrast to most other enzymes like trypsin, is also able to digest antibodies without prior unfolding, reduction and alkylation, thereby making it possible to generate non-reduced peptide maps for the analysis of the disulfide linkage as well.
Catumaxomab samples were digested by Lys-C and the resulting peptides were separated by HPLC. The HPLC system was directly coupled to an ESI-ion trap mass spectrometer or an ESI-TOF mass spectrometer. Peptides were identified manually by comparing the observed tandem mass spectra to the theoretical mass spectra generated from the known amino acid sequence, or automatically by Mascot software. Large peptides were identified manually by comparing the observed mass with the theoretical mass of the peptides using accurate mass information from ESI-TOF experiments. Ninety-nine percent sequence coverage could be achieved using this single digest. Glu-C and Asp-N digests were used to map the remaining amino acids. Importantly, all methionines susceptible to oxidation and asparagines susceptible to deamidation were covered by the Lys-C peptide method, allowing this method to monitor oxidation and deamidation.Citation24
Glycosylation.
The analysis of the glycosylation state of therapeutic antibodies has become increasingly important, since profound effects of glycosylation differences on Fc receptor binding, circulation half life, and immunogenicity have been described.Citation25–Citation27 Several analytical methods can be used for the analysis of glycosylation, including methods for the analysis of the monosaccharide content, the oligosaccharide content and detailed linkage analysis of the oligosaccharides.
For glycosylation analysis of mAbs, high performance anion exchange chromatography with pulsed amperometric detection (HPAEC-PAD), fluorescence labeled HPLC, and capillary electrophoresis with laser induced fluorescence (CGE-LIF) of the released glycans are the most common techniques.Citation28–Citation30 A detailed linkage analysis of the glycan structure is often not required, since the glycan structure of mammalian expression systems is well-documented in the literature.Citation31 Monosaccharide analysis is frequently required by the regulatory agencies, although reproducible and accurate methods for the quantification of the monosaccharides, which require hydrolysis of the oligosaccharides, are notoriously difficult to develop.Citation32
Mass spectrometry based analytical methods have also been used for the analysis of glycosylation.Citation33–Citation37 Technologies allowing the analysis of oligosaccharides attached to the proteins provide especially useful information.Citation38,Citation39 In the case of catumaxomab, two different glycosylation sites exist—one on the mouse and one on the rat half molecule. These techniques allow the site-specific quantification of glycosylation and also address the levels of non-glycosylated variants. A comparison of the glycosylation analysis from HPAEC-PAD analysis used as a routine method and a LC/MS peptide mapping method is shown in . No non-glycosylated peptides were detected with the LC/MS method, indicating that catumaxomab is fully glycosylated.
Secondary and tertiary structure
Spectroscopic methods like circular dichroism (CD) and fluorescence spectroscopy are applied for the analysis of protein conformations in solution. CD is an excellent method for rapidly evaluating the secondary structure of proteins.Citation40,Citation41 Left and right circularly polarized light in the far-UV region is absorbed by the periodic repeated binding angles of the secondary structure elements to a different extent. Alpha-helical secondary structure elements lead to a stronger absorption of the light compared to β-strands.Citation42–Citation44 Normally, the CD spectrum of a native protein represents the sum of the signals for the different secondary structure elements, e.g., turns and loops. There are many different methods to analyze CD spectra to estimate secondary structure.Citation41
In the near-UV region the asymmetric environment of immobilized aromatic amino acids defines the CD-signal and spectra depend strongly on the environment of the aromatic amino acids. Therefore, they have fewer common characteristics than far UV-spectra and are a fingerprint of the protein. Near UV-spectra are a sensitive characteristic for the native state of a protein.
Fluorescence spectroscopy is well-suited to analyze the three dimensional structure of proteins. Within proteins, the side chains of the amino acids Phe, Tyr and Trp exhibit fluorescence upon excitation. In proteins, which have all three aromatic amino acids, the fluorescence is usually dominated by the Trp side chains because both the absorption and the intensity of the emission are clearly stronger than those of Phe or Tyr. Changes of the protein structure often lead to pronounced changes of the fluorescence emission, because the fluorescence of the aromatic amino acids depends on the relative environment. Both changes in the emission wavelength and in the intensity of fluorescence can be observed.Citation45–Citation47
Triomab® antibody preparations were analyzed by near- and far-CD spectroscopy, the melting points were determined and the fluorescence change upon unfolding was analyzed using fluorescence measurements. The fluorescence spectrum of a catumaxomab sample is shown in . The fluorescence spectra confirmed that the antibody is structured. Tryptophanes are in a hydrophobic surrounding in the native protein. After denaturation with guanidine-HCl, they become solvent-exposed. This becomes evident by a shift of the maximum emission wavelength and an increase in signal intensity. The signal changes in the presence of DTT show that upon reduction of the disulfide bonds within the antibody domains, the quenching of adjacent tryptophane is abolished. Accordingly, the signal intensity increases. The near and far-UV-CD spectra of catumaxomab are shown in . The far-UV-CD data () was used to calculate the predicted secondary structure elements for the antibody sample, analyzed by the CDNN software ().Citation48 The near-UV-CD spectrum clearly showed that the protein possesses a tertiary structure (). The local minimum at 283 nm is caused by tryptophane absorption. The melting point of the molecule was also evaluated using CD spectroscopy. The results are shown in , indicating a conversion of a structured molecule to an unfolded, unstructured molecule at approximately 65°C. All results obtained using CD and fluorescence spectroscopy are in good agreement with the expected secondary and tertiary structural properties of a mAb.Citation49,Citation50
Functional characterization.
Therapeutic antibody variants that are likely to affect biological activity can occur during the manufacturing process. In addition to assessment of structural changes, it is important to address the functional activity of these biopharmaceuticals by means such as proliferation, inhibition of proliferation, or apoptosis assays. Since Triomab® antibodies direct several cytotoxic effectors to tumor cells, the binding capacity of the relevant catumaxomab to each respective target molecule, i.e., EpCAM, CD3 and the high affinity receptor FcγRI (CD64) was determined by flow cytometry.Citation3,Citation4 Secondly, the cumulative cytotoxicity exerted on an allogeneic tumor cell line by Triomab®-stimulated natural killer cells, T cells or macrophages/monocytes was assessed by a proliferation assay for remaining tumor cells.
Trifunctional binding features.
As a trifunctional antibody, catumaxomab has the capacity to bind to the surface antigen EpCAM on tumor cells, the CD3 molecule exposed on the surface of T cells, and Fcγ receptors such as FcγRI (CD64) on accessory cells. To characterize the binding capacities of each antigen binding site of catumaxomab, the attachment behavior to the respective single antigen, e.g., EpCAM, CD3 or CD64 exposed on different cell surfaces was determined by immunofluorescent flow cytometry (). In summary, flow cytometric staining represents a robust method for evaluating Triomab® binding on viable cells with native surface antigen expression that is similar to the in vivo situation. In our hands, flow cytometry was superior to other methods such as ELISA or Biacore that largely depend on the quality of the recombinant antigen purified.
Allogeneic cytotoxicity assay.
The cellular cytotoxic impact on tumor cell lines induced by catumaxomab in comparison to conventional monoclonal antibodies has been described.Citation51 Here, we show the destructive effect of catumaxomab on EpCAM positive HCT-8 tumor cells. The robustness of the method is demonstrated by obtaining similar killing curves with different PBMC donors (). Highly effective tumor cell elimination is shown by EC50 values ranging between 0.07 and 0.21 ng/ml. This finding holds true for other Triomab® product candidates such as ertumaxomabCitation52 or FBTA05.Citation53 The in vitro assessment of allogeneic cytotoxicity thus suggests that structural preciseness of trifunctional antibodies provides the expected functional properties required for Triomab® antibodies.
Aggregates.
Aggregates are generally considered impurities. Size exclusion chromatography (SEC)-HPLC for the analysis of aggregates is a fast, robust method that can be easily transferred into a quality control (QC) environment. Analytical ultracentrifugation and field flow fractionation have been used as orthogonal methods to SEC for the evaluation of aggregates in biopharmaceuticals.Citation54–Citation56
Analytical ultracentrifugation (AUC) is a powerful tool to characterize protein aggregation.Citation54 Aggregation analysis of proteins almost exclusively uses sedimentation velocity in contrast to sedimentation equilibrium. Sedimentation velocity causes physical separation of molecular species of different mass or shape, generating a wealth of information, including heterogeneity or homogeneity of hydrodynamically distinct molecular species and the sedimentation coefficients and diffusion coefficients of the solutes. It is possible to calculate their molecular weights based on the Svedberg equation.Citation58,Citation59 Additional algorithms have been developed for better quantification of the fraction of each species.Citation60
Field flow fractionation (FFF) is a family of separation techniques that can be compared with liquid chromatography LC. Whereas LC is primarily suitable for the separation of small molecules, FFF is typically used for the separation of large molecules and particles. In asymmetrical flow FFF (AF4), the separation force is made by a cross flow perpendicular to the channel flow along the channel. AF4 has been successfully used for the separation of large biomolecules and aggregates.Citation61,Citation62
Gabrielson et al. compared SEC, FFF and AUC for the analysis of aggregates in humanized monoclonal antibody formulations.Citation55 They concluded that an orthogonal technique such as SV-AUC or potentially AF4 should be considered as primary calibration technique in an analysis of protein systems with soluble aggregates.Citation55
The levels of aggregates in catumaxomab preparations are routinely analyzed using SEC. To confirm the SEC data, AUC and FFF have been performed. FFF was performed using an AF2000 FOCUS separation system equipped with a UV and light scattering detection system (Postnova Analytics) and a Eclipse 2 system (Wyatt). Sedimentation velocity AUC was performed using a Optima XL-A (Beckman) centrifuge. Data analysis of the AUC experiments was performed using the SEDFIT algorithm.Citation60 The results are summarized in . The data generated using FFF and AUC provided valuable information and validated our SEC data; however, in our hands, neither AUC nor FFF could provide the sensitivity or the reproducibility of SEC for analysis of soluble aggregates.
Isoforms.
MAbs are heterogeneous molecules due to glycosylation, C- and N-terminal variants, and other posttranslational modifications. The heterogeneity often results in different pI values of the different species. Characterization and quantification of these charged variants is often one of the most challenging tasks for the analytical method development. Several analytical methods are available for the characterization of charged variants. The most common separation technique is ion exchange chromatography (IEX), which has been used successfully for the characterization of antibody isoforms.Citation63–Citation66 Other techniques for the separation of charged protein variants are isoelectric focusing (IEF), hydrophobic interaction chromatography (HIC), reversed-phase HPLC, and free flow electrophoresis (FFE).Citation65–Citation72
Catumaxomab charged variants were separated and isolated using FFE fractionation and analyzed using LC/MS methods. We identified a deamidation site in catumaxomab that is almost exclusively responsible for the appearance of different charged variants.Citation24 The deamidation site is located in the constant region of the antibody and deamidation had no effect on the biological activity of the molecule.
Discussion
Like other mAb therapeutics, Triomab® antibodies are complex molecules, requiring an extensive panel of analytical methods for detailed characterization. A battery of analytical technologies developed for the characterization of mAbs can be readily applied to Triomab® antibodies as well. In our opinion, mass spectrometry has become the most powerful tool for the analysis of these complex molecules, providing new insights in the protein chemistry of biopharmaceuticals. Information about modifications like oxidation,Citation73–Citation75 deamidation,Citation76–Citation78 isomerization,Citation79,Citation80 C- and N-terminal variants,Citation81–Citation83 as well as some unexpected modificationsCitation84–Citation86 would not have been possible without the development of new mass spectrometers with increased sensitivity, resolution, mass accuracy, and mass range. However, “older” techniques such as SDS-PAGE or SEC still provide useful information and are important for routine analysis in a Good Manufacturing Practice (GMP) environment.
Materials and Methods
Intact and reduced mass analysis.
Sample preparation for intact mass analysis.
For intact mass analysis of the antibody, and identification and quantification of the impurities, catumaxomab samples (1 mL, 0.1 mg/mL) were concentrated by a factor of 10 (1 mg/mL) by filtration through 10 kDa MWCO membranes. 40 µL (40 µg) of the catumaxomab samples were injected and analyzed by means of RP-HPLC ESI-TOF-MS.
Sample preparation for reduced mass analysis.
For reduced mass analysis of the antibody chains, catumaxomab samples (500 µL, 0.1 mg/mL) were concentrated by a factor of 5 (0.5 mg/mL) by filtration through 10 kDa MWCO membranes. Samples were buffer exchanged four times to 6 M guanidine hydrochloride, 250 mM ammonium hydrogencarbonate, pH 7.8. The samples were first reduced by adding 2 µL of a 0.2 M DTT-solution and incubated for 30 min at 37°C. In the second step the samples were alkylated by adding 2 µL of a 0.5 M IAA-solution and incubated for 40 min at room temperature in the dark. 40 µL (20 µg) of the reduced ertumaxomab samples were injected for RP-HPLC ESI-TOF-MS analysis.
RP-HPLC of intact and reduced samples.
The samples were separated by RP-HPLC on a 150 mm × 2.1 mm PLRP-S column (Varian, Darmstadt, Germany) packed with 5 µm particles (pore size 300 Å); eluents were 0.1% TFA in water (eluent A) and 0.1% TFA in acetonitrile (eluent B) and the flow rate was 200 µL/min. The column was heated at 65°C to enhance separation was initially equilibrated with 27% eluent B. Ten minutes after sample injection, the concentration of eluent B was increased to 42% within 40 minutes. The column was re-equilibrated by ramping up eluent B to 100% within one minute, holding for 15 minutes at 100% eluent B, and dropping to 27% eluent B over 1 minute, which was followed by 27% eluent B for 15 minutes. UV absorption was monitored at 214 and 280 nm.
Mass spectrometry of intact and reduced samples.
The HPLC was directly coupled with an Agilent 6220 ESI-TOF mass spectrometer. The ESI-TOF mass spectrometer was set to run in positive ion mode. The capillary voltage was set to 5,000 V and the fragmentor voltage was 350 V.
Data analysis for intact and reduced analysis.
The deconvolution of ESI mass spectra was carried out using the MaxEnt algorithm, a part of the MassHunter analysis software (Agilent, Palo Alto, CA).
Peptide mapping.
Reduction, alkylation and enzymatic digestion.
Samples (1 mL, 0.1 mg/mL) were concentrated by a factor of 5 (0.2 mL, 0.5 mg/mL) by filtration through 10 kDa MWCO membranes (Millipore, Billerica, MA). During this procedure, the buffer was changed to 4 M urea in 100 mM tris(hydroxymethyl)aminomethane/hydrochloric acid pH 8.4. After addition of hydroxylamine hydrochloride to a final concentration of 10 mM (to prevent carbamylation) and endoproteinase Lys-C (Wako, Osaka, Japan) at a ratio of 1:20 enzyme to substrate, samples were incubated at 37°C overnight. DTT (2 µL, 0.2 M) was then added and the mixture was incubated at 37°C for 45 minutes. Afterwards, the samples were alkylated by adding 2 µL IAA (0.5 M) and incubation at room temperature for 45 minutes in the dark. The samples were analyzed immediately after the alkylation procedure or stored at −30°C while waiting for analysis.
Prior to the reduction, alkylation and enzymatic digestion procedure, the FFE fractions were incubated at 65°C for 30 minutes and the cellulose was removed by centrifugation for a few seconds.
HPLC separation of peptides generated by the Lys-C digest.
The peptides were separated on a reversed-phase HPLC using an Agilent 1200 HPLC system equipped with a diode-array detector, an auto-sampler, and a temperature controlled column compartment (Agilent, Waldbronn, Germany). The eluents were A: 0.1% TFA in water (Merck, Darmstadt, Germany) and B: 0.1% TFA in acetonitrile (Merck, Darmstadt, Germany). Separation was performed on a Jupiter C5 column packed with 5 µm particles, 300 Å pore size (Phenomenex, Torrance, CA). The column was heated at 50°C to enhance separation and the flow rate was 200 µL/min. The column was initially equilibrated at 2% solvent B. Three minutes after sample injection, the concentration of mobile phase B was increased to 50% over 127 minutes. The column was re-equilibrated by ramping up mobile phase B to 100% over 5 minutes, holding for 5 minutes at 100% B, dropping down to 2% B over 5 minutes followed by 15 minutes at 2% B. UV absorption was monitored at 214 and 280 nm.
Mass spectrometry analysis of the separated peptides.
The HPLC was directly coupled to an Agilent 6330 ESI-ion trap (ESI-IT) mass spectrometer or an Agilent 6220 ESI-TOF mass spectrometer (Agilent, Santa Clara, CA). The ESI voltage for the ion trap and TOF mass spectrometer were set at 3,500 V and 4,000 V respectively in positive ion mode. The capillary temperature was 250°C for both instruments. The fragmentation in positive mode was obtained using ion trap collision energies of 1 V. Each full-scan mass spectrum in the ESI-IT analysis was followed by two data-dependent MS/MS scans of the most intense ions. Both instruments were calibrated and tuned by infusion of calibration solution.
Data analysis.
Peptides were identified manually by comparing the observed tandem mass spectra with the theoretical mass spectra generated from the known amino acid sequence, or automatically by Mascot software. Large peptides were identified manually by comparing the observed mass with the theoretical mass of the peptides using accurate mass information from ESI-TOF experiments.
CD spectroscopy.
All spectra were recorded in a 715 CD-spectropolarimeter (Jasco, Easton, MD) at 20°C. All spectra are average values of 15 accumulated measurements. The Y-axis represents the mean residual weight ellipticity in degree × cm-2 × dmol-1. The path length of the cuvette was 0.1 cm for the far-UV spectra and 0.5 cm for near-UV spectra. Near-UV spectra were measured with a protein concentration of 0.5 mg/mL from 250–340 nm, far-UV spectra with a concentration of 0.25 mg/mL between 190–260 nm. Near- and far-UV spectra were measured exclusively in PBS buffer. The secondary structure content was calculated for the far UV-spectra using CDNN (http://bioinformatik.biochemtech.uni-halle/cdnn/).
Fluorescence spectroscopy.
Fluorescence spectroscopy was carried out in a Fluoromax-2 spectrometer with a path length of 1 cm. Emission spectra of the samples were recorded at a temperature of 20°C in the range of 300–450 nm with an excitation wavelength of 295 nm. The slits for excitation were 5 nm and for emission 10 nm. All samples were diluted to 0.25 mg/mL with sample buffer. Samples were denatured in 6 M guanidinium hydrochloride. Samples, which had to be reduced in addition, were incubated with DTT at a final concentration of 20 mM. For complete denaturation, samples were incubated for 2 hours at room temperature.
Flow cytometry.
The trifunctional binding feature of catumaxomab was analyzed by flow cytometry with established cell lines that express the respective single antigen on the cell surface. EpCAM positive HCT-8 (human ileocecal colorectal adenocarcinoma cell line, ATCC: CCL-224), CD3 positive Jurkat (human acute T cell leukemia cell line, ATCC: TIB-152) and CD64 positive THP-1 cells (human acute monocytic leukemia cell line, ATCC: TIB-202) were used. In each case 5 x 105 target cells were stained with 1.2–1.6 µg/mL catumaxomab for 60 minutes at ambient temperature. After a washing step cell bound catumaxomab was detected by 30 minutes incubation time with phycoerythrin (PE) labelled secondary antibodies. Goat anti-mouse IgG and goat-anti-rat IgG specific antibodies (Jackson Immuno Research) were used to stain cell bound catumaxomab on Jurkat, respectively HCT-8 cells. In the case of THP-1 cells F(ab′)2 fragments of PE-labelled donkey-anti-rat IgG (Jackson Immuno Research) were preferred to avoid binding interference of secondary detection antibody with FcγR. Data acquisition was carried out with FACS Calibur flow cytometer and CellQuest Pro Software version 4.0.2 (BD Biosciences).
Allogeneic cytotoxicity assay.
Catumaxomab mediated tumor cell destruction was evaluated by an allogeneic cytotoxicity assay as described previously.Citation48 Briefly, peripheral blood mononuclear cells (PBMC) from healthy donors were isolated by Ficoll density centrifugation and subsequently mixed with 1 × 104 HCT-8 tumor cells in the presence of the indicated amounts of antibodies. After 3 days of co-cultivation, PBMC were removed and adherent tumor cells were then stained with tetrazolium hydroxide (XTT cell proliferation kit II, Roche). Tumor cell killing (%) was calculated according to the formula:
.
Size exclusion chromatography.
Size exclusion chromatography was performed using an Agilent 1100 HPLC system equipped with a diode-array detector, an auto-sampler, a temperature controlled column compartment, and a fluorescence detector. (Agilent, Waldbronn, Germany). A 50 µL (0.1 mg/mL) aliquot of the sample was injected for analysis. Separation was performed on a 7.8 × 300 mm TSK-GEL G3000SWXL column (TOSOH Biosciences, Tokio, Japan). The mobile phase was 100 mM citrate buffer pH 6.7, 0.2 M ammonia sulfate, 0.01% sodium azide, 0.02% Tween-80, and the flow rate was 0.5 mL/min. The amounts of high molecular weight species were calculated based on the peak areas of the fluorescence chromatogram.
Analytical ultracentrifugation.
Sedimentation experiments were performed with an analytical ultracentrifuge Optima-XLA (Beckman), which was equipped with an absorption optical system, an 8-hole analytical rotor An-50-Ti, and centrifuge cells with quartz windows and 2-channel charcoal filled centerpieces and an optical path length of d = 1.2 cm. Experiments were performed at 42,000 rpm at 22°C and scan intervals of 3 minutes. The absorbance difference at 280 nm between sample and reference channels was measured. The experimental data were analyzed with the program SEDFIT.Citation57 The distribution of apparent sedimentation coefficients and of molecular weights were determined with the models c(s) and c(M), “continuous distribution of sedimentation coefficients” and “continuous distribution of molecular weights”, respectively. These are based on direct least-squares modeling of the sedimentation boundary using linear combinations of Lamm equations and take diffusion into account.Citation57
Field flow fractionation.
The AF2000 FOCUS system was equipped with a ternary detection system, combining multi-angle light scattering (MALS), UV spectroscopy, and RI detection (Postnova Analytics, Landsberg, Germany). Separation was performed on a PES 4 kDa membrane using a mobile phase of 0.14 M NaCl + 0.02 M Na2HPO4 pH 6. A 250 µm spacer was used and the flow rate at the outlet was set to 0.8 mL/min. Separation was achieved by applying a 1.5 mL cross flow for 9 minutes. During that time the sample (200 µL; 0.1 mg/mL) was loaded and focused. The cross flow was then reduced to 0.2 mL/min over 30 minutes and held at 0.2 mL/min for 15 minutes.
The Eclipse 2 (Wyatt Technology, Santa Barbara, CA) system was equipped with an 1100 HPLC pump (Agilent, Waldbronn, Germany), an autosampler (Shimadzu, Tokio, Japan) and an UV detector (SOMA, Tokio Japan). Fractionation of the sample (100 µL; 0.1 mg/mL) was achieved using a 10 kDa cellulose membrane with a 0.1 M sodium citrate pH 5.6 buffer. The injection flow was 0.3 mL/min, the cross flow was 2.0 mL/min and the flow rate was 0.5 mL/min. A 490 µm spacer was used and separation was achieved by a linear reduction of the cross flow from 2.0 mL/min to 0.0 mL/min over 25 minutes.
The amounts of high molecular weight species were calculated based on the peak areas of the UV chromatogram for both instruments.
Abbreviations
| HPLC | = | high performance liquid chromatography |
| LC/MS | = | liquid chromatography mass spectrometry |
| ESI | = | electrospray ionization |
| FFE | = | free flow electrophoresis |
| IEF | = | isoelectric focusing |
| TOF | = | time of flight |
| SV | = | sedimentation velocity |
| AUC | = | analytical ultracentrifugation |
| MA | = | malignant ascites |
| DTT | = | dithiothreitol |
| IAA | = | iodacetamid |
Figures and Tables
Figure 1 Triomab’s proposed mechanism of action. Abbreviation: TAA, tumor-associated antigens.
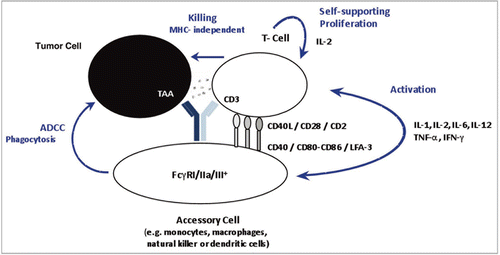
Figure 2 The catumaxomab sample was analyzed using RP-HPLC mass spectrometry using a PLRP-S column (Varian) with an acetonitrile gradient from 27% to 42% over 40 minutes on a 1200 HPLC system (Agilent) for separation, coupled with an Agilent 6220 ESI-TOF mass spectrometer. The column temperature was 65°C and the flow rate was 200 µL/min. (A) Combined raw spectrum of approximately 60 scans (1 minute) at a resolution of approximately 14,000. (B) The raw data was converted using MassHunter deconvolution software.
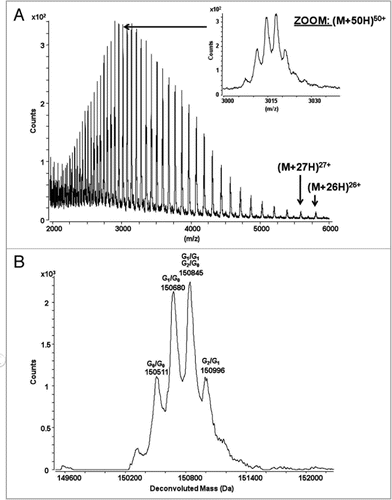
Figure 3 Reversed-phase chromatogram with UV absorbance at 214 nm of the reduced antibody. The catumaxomab sample was denatured using 6 M guanidine, reduced with DTTand alkylated using iodoaceteamide. The reduced and alkylated antibody chains were separated on a PLRP-S column (Varian) using an acetonitrile gradient from 27% to 42% over 40 minutes on a 1200 HPLC system (Agilent) coupled with an Agilent 6220 ESI-TOF mass spectrometer. The column temperature was 65°C and the flow rate was 200 µL/min. The raw mass spectra of the 4 antibody chains were converted using MassHunter deconvolution software for the calculation of the observed masses (see ).
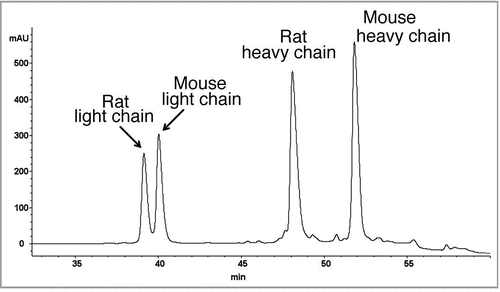
Figure 4 Fluorescence spectroscopy was carried out in a Fluoromax-2 spectrometer with a path length of 1 cm. Emission spectra of the samples were recorded at a temperature of 20°C in the range of 300–450 nm with an excitation wavelength of 295 nm. The slits for excitation were 5 nm and for emission 10 nm. The catumaxomab sample was diluted to 0.24 mg/ml with sample buffer (native sample shown as solid line). Samples were denatured in 6 M guanidine hydrochloride (denatured sample shown as dashed line). Samples, which had to be reduced in addition, were incubated with DTT at a final concentration of 20 mM (denatured and reduced samples is shown as dotted line). For complete denaturation, samples were incubated for 2 hours at room temperature.
Figure 5 Far-UV CD spectrum (A), near-UV CD spectrum (B) and the thermal transition spectrum (C) of a catumaxomab sample. All spectra were recorded in a Jasco 715 CD-spectropolarimeter at 20°C. The spectra are average values of 15 accumulated measurements. The Y-axis represents the mean residual weight ellipticity in degree x cm-2 x dmol-1. (A) The path length of the cuvette was 0.1 cm for the far-UV spectra and 0.5 cm for near-UV spectra. (B) Near-UV spectra were measured with a protein concentration of 0.6 mg/ml from 250–340 nm, far-UV spectra with a concentration of 0.3 mg/ml between 190–260 nm. (A and B) Near- and far-UV spectra were measured exclusively in PBS buffer. The secondary structure content was calculated for the far-UV-spectra using CDNN.Citation45 (C) Thermal transitions were recorded in a Jasco 715 CD-spectropolarimeter at 220 nm. The path length of the cuvette was 0.1 cm. The experiments were performed with a constant heating rate of 15°C/h from 20°C to 80°C with a protein concentration of 0.2 mg/ml.
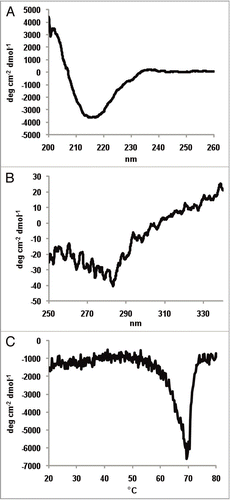
Figure 6 Exemplary samples of trifunctional binding of catumaxomab to CD3, EpCAM and FcγR on the target cells Jurkat, HCT-8 and THP-1. Antibody binding was assessed by flow cytometry at catumaxomab concentrations of 1.2–1.6 µg/ml. Negative controls were performed in the absence of catumaxomab (filled histograms). Cell bound catumaxomab was detected via phycoerythrin (PE) labelled goat anti-mouse IgG (Jurkat cells) or goat-anti-rat IgG (HCT-8 cells) secondary detection antibodies. For THP-1 cells F(ab′)2 fragments of PE-labelled donkey-anti-rat IgG were used to avoid binding interference of secondary detection antibody with FcγR.
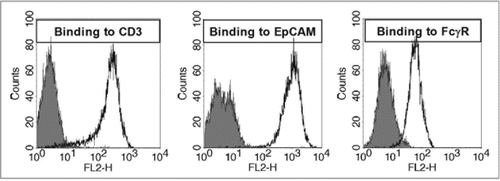
Figure 7 Dose response curve of catumaxomab mediated lysis of HCT-8 tumor cells using three different PBMC donors. The allogeneic cytotoxicity bioassay was performed as described previously.Citation48 Approaches were carried out in duplicates and five parameter curve fitting of dose response curves was executed by Prism Software version 5.02 (Graph Pad Software, San Diego, CA).
Table 1 The intact antibody and the reduced antibody samples were analyzed by LC/MS observed masses were calculated from the raw data using the MassHunter deconvolution algorithm.
Table 2 The antibody sample was enzymatically cleaved using Lys-C followed by reduction (DTT) and alkylation (iodoacetamide). The resulting peptides were separated on a C5 reversed-phase column (Phenomenex) using an acetonitrile gradient from 0% to 50% over 127 minutes on a 1200 HPLC system (Agilent) and analyzed by mass spectrometry using an ESI TOF (Agilent 6220) mass spectrometer. The amounts of the different glycoforms were calculated from the ion intensities of the most abundant charge state. PNGase released glycans were analyzed by HPAEC-PAD on a CarboPac PA1 column using a DX 500 system (Dionex) equipped with an ED40 detector.
Table 3 Far UV-CD spectra were recorded using a jasco 715 CD-spectropolarimeter from 190 to 260 nm at 20°C and accumulated 15 times in PBS-buffer (). The protein concentration was 0.321 mg/ml. The analysis of the secondary structure was performed with the CD deconvolution software CDNN.Citation45
Table 4 Percentage of aggregates measured using SEC, AUC and FFF technologies. SEC was performed on a TSK gel G3000 SWXL column (Tosoh Bioscience LLC) using an Agilent 1100 HPLC system. the AUC experiments were performed on a Optima XL-A Beckman analytical ultracentrifuge (Beckman) equipped with an absorption optical system, an 8-hole analytical rotor An-50-Ti, and centrifuge cells with quartz windows and 2-channel charcoal filled centerpieces with an optical path length of d = 1.2 cm. using a centrifugation speed of 42,000 rpm. The data was analyzed using SEDFIT algorithm.Citation57 FFF was performed on a Eclipse 2 (Wyatt) system using a 10 kDa cellulose membrane (control sample) or on a AF2000 FOCUS system (Postnova) using a PES 4 kDa membrane (stressed and in-process sample). the stressed sample was generated by incubation at 37°C and the in-process sample was from an protein-A purification step.
Acknowledgements
We like to thank Erika Schmidt (Solvias) for the generation of the AUC data and Prof. Buchner (Technical University Munich) for the generation of the CD and fluorescence data. Eveline Moldenhauer (Postnova) and Franz-Rudolf Kunz (AQura) we thank for the generation of the FFF data. We also like to thank Afsaneh Abdolzade (BD Diagnostics) for the FFE separation. Jens Herold we thank for careful proofreading of the manuscript and valuable suggestions.
References
- Lindhofer H. Trifunctional antibody format: Concept and first drug candidate. Pharma Focus Asia 2008; 9:51 - 54
- Chames P, Baty D. Bispecific antibodies for cancer therapy: the light at the end of the tunnel?. MAbs 2009; 1:1 - 9
- Zeidler R, Reisbach G, Wollenberg B, Lang S, Chaubal S, Schmitt B, et al. Simultaneous activation of T cells and accessory cells by a new class of intact bispecific antibody results in efficient tumor cell killing. J Immunol 1999; 163:1246 - 1252
- Zeidler R, Mysliwietz J, Csánady M, Walz A, Ziegler I, Schmitt B, et al. The Fc-region of a new class of intact bispecific antibody mediates activation of accessory cells and NK cells and induces direct phagocytosis of tumour cells. Br J Cancer 2000; 83:261 - 266
- Ruf P, Lindhofer H. Induction of a long-lasting antitumor immunity by a trifunctional bispecific antibody. Blood 2001; 98:2526 - 2534
- Franki SN, Steward KK, Betting DJ, Kafi K, Yamada RE, Timmerman JM. Dendritic cells loaded with apoptotic antibody-coated tumor cells provide protective immunity against B-cell lymphoma in vivo. Blood 2008; 111:1504 - 1511
- Clynes RA, Towers TL, Presta LG, Ravetch JV. Inhibitory Fc receptors modulate in vivo cytoxicity against tumor targets. Nat Med 2000; 6:443 - 446
- Shields RL, Namenuk AK, Hong K, Meng YG, Rae J, Briggs J, et al. High resolution mapping of the binding site on human IgG1 for FcgammaRI, FcgammaRII, FcgammaRIII and FcRn and design of IgG1 variants with improved binding to the FcgammaR. J Biol Chem 2001; 276:6591 - 6604
- Riesenberg R, Buchner A, Pohla H, Lindhofer H. Lysis of prostate carcinoma cells by trifunctional bispecific antibodies (alpha EpCAM x alpha CD3). J Histochem Cytochem 2001; 49:911 - 917
- Shen J, Zhu Z. Catumaxomab, a rat/murine hybrid trifunctional bispecific monoclonal antibody for the treatment of cancer. Curr Opin Mol Ther 2008; 10:273 - 284
- Sebastian M, Kuemmel A, Schmidt M, Schmittel A. Catumaxomab: a bispecific trifunctional antibody. Drugs Today 2009; 45:589 - 597
- Parsons S, Murawa PX, Koralewski P, Kutarska E, Kolesnik OO, Stroehlein MA, et al. Intraperitoneal treatment of malignant ascites due to epithelial tumors with catumaxomab: A phase II/III study. J Clin Oncol 2008; 26:3000
- Parsons S, Hennig M, Linke R, Klein A, Lahr AS, Lindhofer H, et al. Clinical benefit of catumaxomab in malignant ascites in patient subpopulations in a pivotal phase II/III trial. J Clin Oncol 2009; 27:14000
- ICH Q6B “Test procedures and acceptance criteria for biotechnological/biological products” (CPMP/ICH/365/96) http://www.ema.europa.eu/pdfs/human/ich/036596en.pdf.
- EMEA “Guideline on development, production, characterization and specifications for monoclonal antibodies and related products” (CHMP/BWP/157653/2007) http://www.ema.europa.eu/pdfs/human/bwp/15765307enfin.pdf.
- Fenn JB, Mann M, Meng CK, Wong SF, Whitehouse CM. Electrospray ionization for mass spectrometry of large biomolecules. Science 1989; 246:64 - 71
- Verentchikov AN, Ens W, Standing KG. Reflecting time-of-flight mass spectrometer with an electrospray ion source and orthogonal extraction. Anal Chem 1994; 66:126 - 133
- Dillon TM, Bondarenko PV, Speed Ricci M. Development of an analytical reversed-phase high-performance liquid chromatography-electrospray ionization mass spectrometry method for characterization of recombinant antibodies. J Chromatogr A 2004; 1053:299 - 305
- Bondarenko PV, Second TP, Zabrouskov V, Makarov AA, Zhang Z. Mass measurement and top-down HPLC/MS analysis of intact monoclonal antibodies on a hybrid linear quadrupole ion trap-Orbitrap mass spectrometer. J Am Soc Mass Spectrom 2009; 20:1415 - 424
- Henzel WJ, Stults JT, Hsu CA, Aswad DW. The primary structure of a protein carboxyl methyltransferase from bovine brain that selectively methylates L-isoaspartyl sites. J Biol Chem 1989; 264:15905 - 15911
- Stults JT. Peptide sequencing by mass spectrometry. Methods Biochem Anal 1990; 34:145 - 201
- Chloupek RC, Harris RJ, Leonard CK, Keck RG, Keyt BA, Spellman MW, et al. Study of the primary structure of recombinant tissue plasminogen activator by reversed-phase high-performance liquid chromatographic tryptic mapping. J Chromatogr 1989; 463:375 - 396
- Ling V, Guzzetta AW, Canova-Davis E, Stults JT, Hancock WS, Covey TR, et al. Characterization of the tryptic map of recombinant DNA derived tissue plasminogen activator by high-performance liquid chromatography-electrospray ionization mass spectrometry. Anal Chem 1991; 63:2909 - 2915
- Timm V, Gruber P, Wasiliu M, Lindhofer H, Chelius D. Identification and characterization of oxidation and deamidation sites in monoclonal rat/mouse hybrid antibodies. J Chromatogr B 2010; In press
- Kaneko Y, Nimmerjahn F, Ravetch JV. Anti-Inflammatory Activity of Immunoglobulin G Resulting from Fc Sialylation. Science 2006; 313:670 - 673
- Scanlan CN, Burton DR, Dwek RA. Making autoantibodies safe. Proc Natl Acad Sci USA 2008; 105:4081 - 4082
- Jefferis R. Antibody therapeutics: isotype and glycoform selection. Expert Opin Biol Ther 2007; 7:1401 - 1413
- Hardy MR, Townsend RR. Separation of positional isomers of oligosaccharides and glycopeptides by high-performance anion-exchange chromatography with pulsed amperometric detection. Proc Natl Acad Sci USA 1988; 85:3289 - 3293
- Guile GR, Rudd PM, Wing DR, Prime SB, Dwek RA. A rapid high-resolution high-performance liquid chromatographic method for separating glycan mixtures and analyzing oligosaccharide profiles. Anal Biochem 1996; 240:210 - 226
- Guttman A, Chen FT, Evangelista RA, Cooke N. High-resolution capillary gel electrophoresis of reducing oligosaccharides labeled with 1-aminopyrene-3,6,8-trisulfonate. Anal Biochem 1996; 233:234 - 242
- Tomiya N, Kurono M, Ishihara H, Tejima S, Endo S, Arata Y, et al. Structural analysis of N-linked oligosaccharides by a combination of glycopeptidase, exoglycosidases, and high-performance liquid chromatography. Anal Biochem 1987; 163:489 - 499
- Townsend RR, Manzi A, Merkle RK, Rohde MF, Spellman M, Smith A, et al. Quantitative Monosaccharide Analysis: A Multi-Center Study ABRF. News 1997; 8
- Siemiatkoski J, Lyubarskaya Y, Houde D, Tep S, Mhatre R. A comparison of three techniques for quantitative carbohydrate analysis used in characterization of therapeutic antibodies. Carbohydr Res 2006; 341:410 - 419
- Viseux N, Hronowski X, Delaney J, Domon B. Qualitative and quantitative analysis of the glycosylation pattern of recombinant proteins. Anal Chem 2001; 73:4755 - 4762
- Field M, Papac D, Jones A. The use of high-performance anion-exchange chromatography and matrix-assisted laser desorption/ionization time-of-flight mass spectrometry to monitor and identify oligosaccharide degradation. Anal Biochem 1996; 239:92 - 98
- Papac DI, Briggs JB, Chin ET, Jones AJ. A high-throughput microscale method to release N-linked oligosaccharides from glycoproteins for matrix-assisted laser desorption/ionization time-of-flight mass spectrometric analysis. Glycobiology 1998; 8:445 - 454
- Harvey DJ. Analysis of carbohydrates and glycoconjugates by matrix-assisted laser desorption/ionization mass spectrometry: An update for 2003–2004. Mass Spectrom Rev 2009; 28:273 - 361
- Thaysen-Andersen M, Mysling S, Højrup P. Site-specific glycoprofiling of N-linked glycopeptides using MALDI-TOF MS: strong correlation between signal strength and glycoform quantities. Anal Chem 2009; 81:3933 - 3943
- Sinha S, Pipes G, Topp EM, Bondarenko PV, Treuheit MJ, Gadgil HS. Comparison of LC and LC/MS methods for quantifying N-glycosylation in recombinant IgGs. J Am Soc Mass Spectrom 2008; 19:1643 - 1654
- Sreerama N, Woody RW. Computation and analysis of protein circular dichroism spectra. Methods Enzymol 2004; 383:318 - 351
- Greenfield NJ. Using circular dichroism spectra to estimate protein secondary structure. Nat Protoc 2006; 1:2876 - 2890
- Holzwarth G, Doty P. The ultraviolet circular dichroism of polypeptides. J Am Chem Soc 1965; 87:218 - 228
- Greenfield N, Fasman GD. Computed circular dichroism spectra for the evaluation of protein conformation. Biochemistry 1969; 8:4108 - 4116
- Venyaminov S, Baikalov IA, Shen ZM, Wu CS, Yang JT. Circular dichroic analysis of denatured proteins: inclusion of denatured proteins in the reference set. Anal Biochem 1993; 214:17 - 24
- Eftink MR. Fluorescence techniques for studying protein structure. Methods Biochem Anal 1991; 35:127 - 205
- Vivian JT, Callis PR. Mechanisms of tryptophan fluorescence shifts in proteins. Biophys J 2001; 80:2093 - 2109
- Frey S, Haslbeck M, Hainzl O, Buchner J. Synthesis and characterization of a functional intact IgG in a prokaryotic cell-free expression system. Biol Chem 2008; 389:37 - 45
- Böhm G, Muhr R, Jaenicke R. Quantitative analysis of protein far UV circular dichroism spectra by neural networks. Protein Eng 1992; 5:191 - 195
- Tetin SY, Prendergast FG, Venyaminov SY. Accuracy of protein secondary structure determination from circular dichroism spectra based on immunoglobulin examples. Anal Biochem 2003; 321:183 - 187
- Saphire EO, Parren PW, Pantophlet R, Zwick MB, Morris GM, Rudd PM, et al. Crystal structure of a neutralizing human IGG against HIV-1: a template for vaccine design. Science 2001; 293:1155 - 1159
- Ruf P, Gires O, Jäger M, Fellinger K, Atz J, Lindhofer H. Characterisation of the new EpCAM-specific antibody HO-3: implications for trifunctional antibody immunotherapy of cancer. Br J Cancer 2007; 97:315 - 321
- Jäager M, Schoberth A, Ruf P, Hess J, Lindhofer H. The trifunctional antibody ertumaxomab destroys tumor cells that express low levels of human epidermal growth factor receptor 2. Cancer Res 2009; 69:4270 - 4276
- Stanglmaier M, Faltin M, Ruf P, Bodenhausen A, Schröder P, Lindhofer H. Bi20 (FBTA05), a novel trifunctional bispecific antibody (anti-CD20 × anti-CD3), mediates efficient killing of B-cell lymphoma cells even with very low CD20 expression levels. Int J Cancer 2008; 123:1181 - 1189
- Pekar A, Sukumar M. Quantitation of aggregates in therapeutic proteins using sedimentation velocity analytical ultracentrifugation: practical considerations that affect precision and accuracy. Anal Biochem 2007; 367:225 - 237
- Gabrielson JP, Brader ML, Pekar AH, Mathis KB, Winter G, Carpenter JF, et al. Quantitation of aggregate levels in a recombinant humanized monoclonal antibody formulation by size-exclusion chromatography, asymmetrical flow field flow fractionation, and sedimentation velocity. J Pharm Sci 2007; 96:268 - 279
- Arakawa T, Philo JS, Ejima D, Tsumoto K, Arisaka F. Aggregation analysis of therapeutic proteins, part 1: General aspects and techniques for assessment. Bioprocess International 2006; 4:42 - 49
- Arakawa T, Philo JS, Ejima D, Tsumoto K, Arisaka F. Aggregation analysis of therapeutic proteins, part 2: Analytical ultracentrifugation and dynamic light scattering. Bioprocess International 2007; 5:36 - 47
- Svedberg T, Pedersen KO. The Ultracentrifuge 1940; Oxford Clarendon Press
- Philo JS. Schuster TM, Laue TM. Measuring sedimentation, diffusion and molecular weights of small molecules by direct fitting of sedimentation velocity concentration profiles. Modern Analytical Ultracentrifugation 1994; Boston Birkhauser 156 - 170
- Schuck P. Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and Lamm equation modeling. Biophys J 2000; 78:1606 - 1619
- Wahlund KG, Litzén A. Application of an asymmetrical flow field-flow fractionation channel to the separation and characterization of proteins, plasmids, plasmid fragments, polysaccharides and unicellular algae. J Chromatogr 1989; 6:73 - 87
- Liu MK, Li P, Giddings JC. Rapid protein separation and diffusion coefficient measurement by frit inlet flow field-flow fractionation. Protein Sci 1993; 2:1520 - 1531
- Harris RJ, Kabakoff B, Macchi FD, Shen FJ, Kwong M, Andya JD, et al. Identification of multiple sources of charge heterogeneity in a recombinant antibody. J Chromatogr B Biomed Sci Appl 2001; 752:233 - 245
- Chu GC, Chelius D, Xiao G, Khor HK, Coulibaly S, Bondarenko PV. Accumulation of succinimide in a recombinant monoclonal antibody in mildly acidic buffers under elevated temperatures. Pharm Res 2007; 24:1145 - 1156
- Perkins M, Theiler R, Lunte S, Jeschke M. Determination of the origin of charge heterogeneity in a murine monoclonal antibody. Pharm Res 2000; 17:1110 - 1117
- Zhang W, Czupryn MJ. Analysis of isoaspartate in a recombinant monoclonal antibody and its charge isoforms. J Pharm Biomed Anal 2003; 30:1479 - 1490
- Dick LW Jr, Qiu D, Cheng KC. Identification and measurement of isoaspartic acid formation in the complementarity determining region of a fully human monoclonal antibody. J Chromatogr B Analyt Technol Biomed Life Sci 2009; 877:3841 - 3849
- Dillon TM, Bondarenko PV, Rehder DS, Pipes GD, Kleemann GR, Ricci MS. Optimization of a reversed-phase high-performance liquid chromatography/mass spectrometry method for characterizing recombinant antibody heterogeneity and stability. J Chromatogr A 2006; 1120:112 - 120
- Ren D, Pipes GD, Hambly DM, Bondarenko PV, Treuheit MJ, Brems DN, et al. Reversed-phase liquid chromatography of immunoglobulin G molecules and their fragments with the diphenyl column. J Chromatogr A 2007; 1175:63 - 68
- Ren D, Pipes G, Xiao G, Kleemann GR, Bondarenko PV, Treuheit MJ, Gadgil HS. Reversed-phase liquid chromatography-mass spectrometry of site-specific chemical modifications in intact immunoglobulin molecules and their fragments. J Chromatogr A 2008; 1179:198 - 204
- Gangopadhyay A, Saravis CA, van den Abbeele AD, Kassis AI. pI-dependent isolation of antibody isoforms by semipreparative isoelectric focusing. Q J Nucl Med 1995; 39:129 - 133
- Vlasak J, Ionescu R. Heterogeneity of monoclonal antibodies revealed by charge-sensitive methods. Curr Pharm Biotechnol 2008; 9:468 - 481
- Lam XM, Yang JY, Cleland JL. Antioxidants for Prevention of Methionine Oxidation in Recombinant Monoclonal Antibody HER2. J Pharm Sci 1997; 86:1250 - 1255
- Pan H, Chen K, Chu L, Kinderman F, Apostol I, Huang G. Methionine oxidation in human IgG2 Fc decreases binding affinities to protein A and FcRn. Protein Sci 2009; 18:424 - 433
- Liu D, Ren D, Huang H, Dankberg J, Rosenfeld R, Cocco MJ, et al. Structure and stability changes of human IgG1 Fc as a consequence of methionine oxidation. Biochemistry 2008; 47:5088 - 5100
- Chelius D, Rehder DS, Bondarenko PV. Identification and characterization of deamidation sites in the conserved regions of human immunoglobulin gamma antibodies. Anal Chem 2005; 77:6004 - 6011
- Liu H, Gaza-Bulseco G, Chumsae C. Glutamine deamidation of a recombinant monoclonal antibody. Rapid Commun Mass Spectrom 2008; 22:4081 - 4088
- Vlasak J, Bussat MC, Wang S, Wagner-Rousset E, Schaefer M, Klinguer-Hamour C, et al. Identification and characterization of asparagine deamidation in the light chain CDR1 of a humanized IgG1 antibody. Anal Biochem 2009; 392:145 - 154
- Rehder DS, Chelius D, McAuley A, Dillon TM, Xiao G, Crouse-Zeineddini J, et al. Isomerization of a single aspartyl residue of anti-epidermal growth factor receptor immunoglobulin gamma2 antibody highlights the role avidity plays in antibody activity. Biochemistry 2008; 47:2518 - 2530
- Wakankar AA, Borchardt RT, Eigenbrot C, Shia S, Wang YJ, Shire SJ, et al. Aspartate isomerization in the complementarity-determining regions of two closely related monoclonal antibodies. Biochemistry 2007; 46:1534 - 1544
- Chelius D, Jing K, Lueras A, Rehder DS, Dillon TM, Vizel A, et al. Formation of pyroglutamic acid from N-terminal glutamic acid in immunoglobulin gamma antibodies. Anal Chem 2006; 78:2370 - 2376
- Rehder DS, Dillon TM, Pipes GD, Bondarenko PV. Reversed-phase liquid chromatography/mass spectrometry analysis of reduced monoclonal antibodies in pharmaceutics. J Chromatogr A 2006; 1102:164 - 175
- Dick LW Jr, Qiu D, Mahon D, Adamo M, Cheng KC. C-terminal lysine variants in fully human monoclonal antibodies: investigation of test methods and possible causes. Biotechnol Bioeng 2008; 100:1132 - 1143
- Pristatsky P, Cohen SL, Krantz D, Acevedo J, Ionescu R, Vlasak J. Evidence for trisulfide bonds in a recombinant variant of a human IgG2 monoclonal antibody. Anal Chem 2009; 81:6148 - 6155
- Dillon TM, Ricci MS, Vezina C, Flynn GC, Liu YD, Rehder DS, et al. Structural and functional characterization of disulfide isoforms of the human IgG2 subclass. J Biol Chem 2008; 283:16206 - 16215
- Wypych J, Li M, Guo A, Zhang Z, Martinez T, Allen MJ, et al. Human IgG2 antibodies display disulfide-mediated structural isoforms. J Biol Chem 2008; 283:16194 - 16205