Abstract
The American Association of Pharmaceutical Scientists (AAPS) National Biotechnology Conference Short Course “Translational Challenges in Developing Antibody-Drug Conjugates (ADCs),” held May 24, 2012 in San Diego, CA, was organized by members of the Pharmacokinetics, Pharmacodynamics and Drug Metabolism section of AAPS. Representatives from the pharmaceutical industry, regulatory authorities, and academia in the US and Europe attended this short course to discuss the translational challenges in ADC development and the importance of characterizing these molecules early in development to achieve therapeutic utility in patients. Other areas of discussion included selection of target antigens; characterization of absorption, distribution, metabolism, and excretion; assay development and hot topics like regulatory perspectives and the role of pharmacometrics in ADC development. MUC16-targeted ADCs were discussed to illustrate challenges in preclinical development; experiences with trastuzumab emtansine (T-DM1; Genentech) and the recently approved brentuximab vedotin (Adcetris®; Seattle Genetics) were presented in depth to demonstrate considerations in clinical development. The views expressed in this report are those of the participants and do not necessarily represent those of their affiliations.
Introduction
Antibody drug conjugates (ADCs) are an emerging class of novel biotherapeutic agents comprised of 0 to 8 cytotoxic payloads that are covalently bound via a linker to a targeted monoclonal antibody (mAb). ADCs have an average of 2–3 payloads per mAb () and are thus heterogeneous mixtures of conjugates. ADCs represent promising therapeutic options in the treatment of various malignancies. Their development has generated substantial enthusiasm across industry, academia, and regulatory authorities in recent years, and more than 20 ADCs are in clinical development (). The rising level of interest in ADCs was evidenced by the increased presence of ADC-focused sessions at the 2012 American Association of Pharmaceutical Scientists National Biotechnology Conference (NBC) held in San Diego, which included three dedicated sessions and numerous contributed papers on ADCs. Following the NBC Conference, the short course “Translational Challenges in Developing Antibody Drug Conjugates” was held on May 24, 2012. This one-day course provided a forum for participants to interact with speakers with expertise in preclinical and clinical ADC development, as well as a representative from the US. Food and Drug Administration (FDA). The speakers provided their perspectives and shared their current thinking on ADC development. The day concluded with a panel session in which panel experts addressed questions posed by the audience. Participants included industry professionals involved in preclinical and clinical pharmacology, manufacturing, and patent law, as well as clinicians and individuals representing regulatory authorities. We present here a summary of the topics discussed during this short-course, with sections for each of the topics followed by key questions that were addressed during the panel session, and conclude with an evaluation of the short-course from the viewpoint of a participant.
Figure 1. Two major mechanisms of action have been described for ADC, cytotoxic targets microtubules disrupting the microtubule network (5a) or DNA targeted cytotoxic enter target cell’s nucleus and binds to the minor groove of the DNA blocking replication (5b); some cytotoxic payloads are released from the cell and may cross the membrane of neighboring cells causing bystander effect killing while others do not (e.g. DM1). The ADC first enters the cell upon binding to the tumor target cell’s antigen (1); whereby the ADC-antigen complex undergoes internalization into the endosome (2); lysosomes then merge with the endosome inducing acidification and enzymatic reactions (3); the acidic environment and enzymes mediate cleavage of linkers, releasing payloads into target cell cytosol (4); Whereby the cytotoxic works on the microtubule (5a) or DNA minor groove (5b). Ultimately the damaged caused to the target cells results in apoptosis (6).
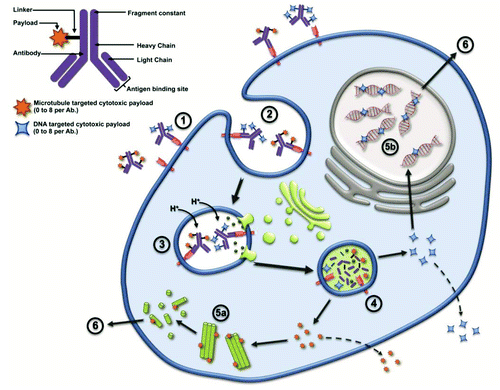
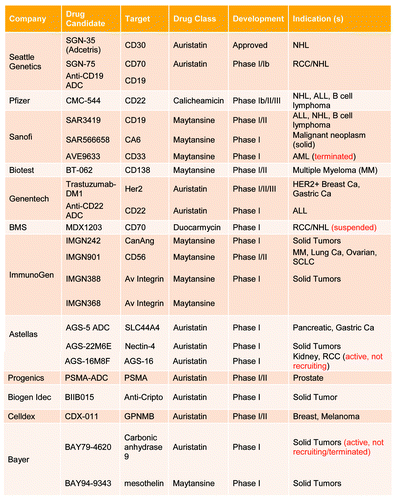
Figure 2. Summary of ADC targets under clinical development. Source: Clinicaltrials.gov as of October 3, 2012.
Morning Sessions
The morning sessions included an overview of ADCs with a specific focus on the impact of target biology on the selection of appropriate linkers and payloads, as well as the importance of developing adequate assays to allow appropriate translation. The mid-morning sessions focused on the absorption, distribution, metabolism, and excretion (ADME) characterization of ADCs and the translational challenges in pharmacokinetics (PK), safety, and efficacy.
Overview of ADCs
Sanela Bilic (Novartis Pharmaceuticals), a co-moderator for this workshop, presented the ADC landscape. This session provided a summary of the key elements for the short-course, including, aspects of translation that are unique to ADCs, elements for successful translation of animal data to human, description of ADC analytes, and the importance of bioanalytical assays for PK and immunogenicity, as well as the ADME and biodistribution of ADCs. The composition and two main mechanism of action for ADCs whereby the antibody specifically recognizes and attaches to the receptor target on tumor cells were introduced. The formation of an ADC-receptor complex induces its internalization into the target cell via a clathrin-coated pit, calveolae, or pinocytosis mechanisms (). Proteases in the acidic environment of the late endosome digest the antibody and potentially the linker, thus releasing the cytotoxic payload. The free cytotoxic agent then crosses the late endosomal membrane, entering the cytoplasm where it binds to its molecular target, which leads to cell cycle arrest or apoptosis. In some instances, a portion of the cytotoxic agent may be effluxed from the cell by passive diffusion, active transport, or leakage from dying cells. If the effluxed cytotoxic payload is cell permeable, it may enter neighboring cells and cause bystander cell killing. The payload may then be metabolized; resulting metabolites may exhibit different tumor cell killing capability and efflux potentials compared with the parent payload. ADCs currently in clinical development () were discussed to illustrate the number of companies pursuing ADC molecules across all stages of development.
Selection of Target Antigens for ADCs
William Mallet (Novartis Institutes for BioMedical Research) discussed the selection of target antigens for ADCs. ADC target identification strategies have evolved in parallel with ADC technology. The MUC16 example was presented to illustrate the point that mRNA expression profiling demonstrated the overexpression of MUC16 in ovarian cancers, and then immunohistochemistry (IHC) was used to confirm expression of MUC16 on the surface of serous ovarian adenocarcinoma cells. Additionally, the in vitro and in vivo potency of MUC16 targeted ADCs (MUC16 vc-MMAE and mc-MMAF) was emphasized. A clear disconnect between in vitro and in vivo activity of MMAE and MMAF ADCs was observed whereby similar in vitro potency between these ADCs was observed. In vivo, however, the MMAE ADC demonstrated 8 times greater efficacy than the MMAF ADC, which underscores the importance of understanding how in vitro properties will translate to in vivo activity.
The importance of antibody and linker selection and their dependence on factors like antigen expression level and internalization were emphasized in this session; beyond expression profiling, the biology of the target antigen must be taken into account. Several examples involving ADC targets that have led to clinical promise or disappointing results were described, and the fact that target identification strategies (such as mRNA profiling) are not sufficient to predict a successful ADC program was emphasized. Preclinical ADC development must involve a continual re-validation of the target through different experimental strategies. An additional important consideration in the development of ADCs is the understanding of the shedding of cell-surface epitopes and the affect that this may have on efficacy and tolerability of the ADC. Preclinical models and studies were described that may help assess and mitigate the risk posed by shed epitopes. Finally, this session alluded to Novartis’ strategy to explore emerging cancer genetics data sets to identify novel ADC targets through collaborations with The Broad Institute and Dana Farber Cancer Institute.
ADME characterization of ADCs
A discussion on the ADME of ADCs was led by Hans Erickson (ImmunoGen, Inc.) Several examples, including T-DM1, SAR3419, and lorvotuzumab mertansine (IMGN901), were presented to underscore the importance of in vitro and in vivo characterization of ADME. This session included a brief discussion of ImmunoGen’s relationship with Genentech and Sanofi Aventis while characterizing T-DM1 and SAR3419, respectively. Several highlights of the ongoing clinical programs for T-DM1 (Genentech), SAR3419 (Sanofi) and IMGN901 (ImmunoGen) were cited to illustrate the therapeutic utility of these agents. Although T-DM1, SAR3419, and IMGN901 each employ different linker formats, the ImmunoGen technology has demonstrated that these three molecules exhibit targeted delivery of maytansinoids to tumors and that conjugations have minimal effects on the properties of the antibody (as determined by PK and biodistribution studies). The chemical nature of the linker influences the plasma PK properties of the intact ADC primarily by modulating the rate of linker cleavage. For example, the disulfide-linked IMGN901 is cleared from plasma faster than the thioether-linked T-DM1 in preclinical tumor-free mouse models.
The complex relationship between plasma pharmacokinetics and tumor delivery is exemplified by the similar payload delivery (and efficacy) in mouse xenograft models achieved for T-DM1 and a trastuzumab-DM1 conjugate prepared using the cleavable disulfide linker used in IMGN901, despite the faster plasma clearance of the disulfide-linked version. The accumulation of total maytansinoid at the tumor following administration of the IMGN901, SAR3419, and T-DM1to tumor-bearing mice was found to be about 2-fold higher than non-targeting controls. The relatively small differential between the accumulation of the targeting and non-targeting ADCs is consistent with several reports in the literature that show significant accumulation of nonbinding IgG in tumor tissues. Analysis of the active maytansinoid catabolites of the ADCs in the tumor tissues reveals a larger differential between the targeted and non-targeted levels, which suggests an additional role of target binding and processing on ADC specificity. It was noted that ADCs utilizing the three linkers selected for SAR3419, T-DM1, and IMGN901 all undergo hepatic metabolism in mice to yield maytansinoids metabolites with low cytotoxic potencies. The promising data that has begun to emerge from clinical evaluations of ADCs utilizing ImmunoGen’s technology are consistent with the favorable ADME properties of ADCs observed in preclinical evaluations.
ADC Assay Translation Interpretation and Challenges
Bioanalytical strategies and challenges in ADC development were discussed by Surinder Kaur (Genentech). The molecular characteristics of ADCs were reviewed and noted to encompass both large molecule (e.g., mAb) and small molecule characteristics. In addition, the heterogeneity of ADCs and the potential for biotransformations in vivo was discussed. The structural characteristics and heterogeneity of ADCs drives bioanalytical strategies and requires integration of existing methods and novel technologies for PK, immunogenicity and biotransformation assessment. Specific emphasis was placed on the need to include a multi-disciplinary bioanalytical group with expertise in quantitative immunoassays, LC-MS/MS, and novel protein structural characterization methods such as affinity capture hydrophobic interaction chromatograph (HIC) and affinity capture capillary LC-MS. The structural characterization of ADCs identifies the distribution of drug/antibody (DAR) species in serum/plasma to understand the key analytes in circulation. This information is important to develop appropriate quantitative assays and provides an overview of the fate of ADCs in vivo. Examples of DAR characterization in vitro and in vivo showing the impact of site of drug conjugation on the ADC stability were shown.
An example of the T-DM1 cynomolgus monkey PK assay strategy was provided. The characterization of T-DM1 DAR distribution in vivo by HER2 extracellular domain (ECD) affinity capture capillary LC-MS was shown. These data suggest that T-DM1 DAR distribution was relatively unchanged for the first 7 d, after which the relative abundance of higher DARs gradually decreased over time and showed a DAR of 1 for the last study sample at day 28. Data from multiple PK assays that assessed the PK of T-DM1 were shown, including ELISA to measure conjugated-trastuzumab and total-trastuzumab and LC-MS/MS assays to measure small molecule catabolites. The ELISA ligand binding reagents (anti-DM1 mAb and HER2 ECD) were characterized by affinity capture LC-MS and shown to bind all DARs properly. Assays were further characterized using enriched DAR fractions to show accurate quantification. Data from ELISA and LC-MS/MS clinical studies were also shown. The similar clinical PK across three Phase 2 studies was discussed for T-DM1 at 3.6 mg/kg administered every 3 weeks, and the consistent low exposure to DM1 was noted across these studies.
An alternative bioanalytical strategy was shown for ADCs with cleavable linkers. In this case, the DAR characterization in plasma showed the formation of new DARs during stability studies (e.g., DAR1) that were not present in the reference standard. It was noted that the anti-drug mAb reagents used in ELISA, which are designed to measure conjugated-antibody, were not capable of measuring DAR1 accurately. The alternate PK assay measured antibody-conjugated drug and involved affinity capture of the ADC from plasma using protein A, cleavage of the linker, followed by LC-MS/MS quantification of the conjugated-drug. In vitro plasma stability across multiple species and clinical PK antibody-conjugated drug data from four dose cohorts was presented to show utility of the assay.
The importance of developing sensitive and robust immunogenicity assays capable of detecting an immune response to all ADC components, including: antibody, linker, cytotoxic drug, and epitopes involving multiple components, was highlighted. As for all biologics, these molecules have the potential to elicit immune responses in vivo that may affect PK, PD, safety, or efficacy. Nonclinical anti-therapeutic antibody (ATA) data for T-DM1 and another trastuzumab ADC with a different linker-drug was shared across four nonclinical studies. Overall, T-DM1 showed a positive ATA response in 2 out of 84 cynomolgus monkeys, whereas trastuzumab conjugated with a different linker-drug showed an immune response in 28 out of 84 monkeys. The example highlighted how the choice of linker-drug can result in very different immune responses for ADCs. The T-DM1 anti-therapeutic antibody (ATA) rate data was shared from several toxicology studies,
Afternoon Sessions
The afternoon sessions started with examination of the translational challenges in PK, safety, and efficacy of ADCs. The focus then shifted to the role of clinical pharmacology and pharmacometric strategies in the development of ADCs, followed by a regulatory perspective on the clinical development of ADCs.
Translational Challenges in PK, Safety, and Efficacy of ADCs
The increase in the number of first–in-man clinical trials for ADCs has drawn attention to the need for better nonclinical-to-clinical translation. Jay Tibbitts (Genentech, Inc.) discussed opportunities to use PK, safety, and efficacy data to drive ADC success. He discussed lack of efficacy or unacceptable toxicity as reasons for oncology trial failure, and target relevance, drug suitability and sound strategy as critical factors for success. The importance of nonclinical data in providing valuable insight into pharmacology, efficacy, preclinical validation biomarkers, and exposure-response relationships was noted. The Phase 1 data should confirm and refine data obtained in the nonclinical studies. Dr. Tibbitts suggested that both nonclinical and Phase 1 clinical data should be merged to develop the optimal Phase 2 strategy. The challenge with this approach is understanding the effects of differences between humans and nonclinical species with respect to physiology, tumor biology, target characteristics, efficacy endpoints, PK, and linker stability.
Dr. Tibbitts discussed a recent publicationCitation1 describing how species-invariant time scaling applied to monkey PK data was used to generate estimates of human PK that were in good agreement with those observed in clinical studies. Because ADCs are composed of multiple elements, it is important to understand the properties of the drug component causing the pharmacologic effects so that the effects can be translated from a nonclinical to clinical setting. He noted the importance of understanding the linker stability between species, as this may lead to differences in drug effect. The more challenging issue associated with translation, i.e., efficacy, was also discussed PK/PD modeling was suggested as a potential tool to account for inter-species differences (e.g., receptor expression, internalization rates, proteolysis). Although these strategies do not provide a solution to the translational challenges, this session focused on methods to improve our basic understanding of how to appropriately use preclinical and Phase 1 clinical data to inform Phase 2 and beyond.
Clinical pharmacology and pharmacometric strategies in development of ADCs
Manish Gupta (Bristol Myers-Squibb) discussed clinical pharmacology and pharmacometric strategies employed in the development of ADCs. The differences in physio-chemical properties between small molecules, mAbs, and ADCs were discussed. Case examples of trastuzumab-emtansine molecule (T-DM1) and brentuximab vedotin, currently the only commercially available ADC, were presented to illustrate their mechanisms of action, metabolic fate, and components of the preclinical/clinical pharmacology development program. A semi-mechanistic model of trastuzumab emtansine, which illustrates the disposition of trastuzumab and DM1, was also discussed.Citation2 Dr. Gupta introduced the use of PK/PD modeling to assist in the selection of efficacious human doses, along with the use of physiologically-based pharmacokinetic (PBPK) models to quantitatively characterize the biodistribution of ADC. He noted that model-based approaches can be used to aid in the selection of linker types, cytotoxic molecules, and to understand the inter-individual variability for ADCs and their byproducts. The use of exposure-response (E-R) modeling was cited as an important determinant for the drivers of efficacy and safety and a means to optimize the dose and schedule.
Regulatory Perspectives on the Clinical Development of ADCs
The afternoon sessions concluded with a regulatory perspective on the preclinical and clinical challenges in ADC development provided by Hong Zhao (US. FDA). Preclinical studies recommended prior to first-in-human studies included: 1) target delivery assessment of the mAb component in two relevant species (or one species, if justified); 2) general toxicity study to understand the cytotoxic potential of the payload; 3) evaluation of plasma stability of the linker conjugated to the mAb and payload. Safety pharmacology, repeated dose toxicology, developmental and reproductive toxicology, genetic toxicology, and carcinogenicity studies as outlined in the ICH9 and ICH M3 (R2) guidances were recommended for ADC preclinical studies. Participants were advised, however, to engage specific FDA Review Divisions in the Center for Drug Evaluation and Research to determine what preclinical studies should be conducted. It is recommended to measure concentrations of all ADC components in circulation to characterize the PK of these molecules in totality (conjugated antibody, total antibody, total drug, free drug). Brentuximab vedotin was cited throughout the session to illustrate the information included in its label, e.g., preclinical studies and clinical PK, ADME, organ dysfunction, drug-drug interactions (DDI), immunogenicity, and QT/QTc prolongation potential assessment. Dr. Zhao recommended merging development approaches of small molecule drugs and mAbs to build a successful clinical pharmacology program for ADCs. Based on the experience with brentuximab vedotin, the current regulatory expectation is that data showing that PK was characterized, metabolism and DDI potential evaluated, QT/QTc interval prolongation potential assessed, immunogenicity assessment was performed, and use in specific populations was evaluated (e.g., renal/hepatic impairment) will be included in marketing applications of ADCs.
Panel Discussion
The ADC short course concluded with a panel discussion that provided attendees with an opportunity to discuss developmental challenges, but it also opened dialog on unanswered questions in ADC development from the perspective of thought leaders in ADC development across the industry. The panel of experts entertained a broad range of questions ranging from preclinical and translational to clinical development and beyond.
Bioanalytical assay submission package requirements
The diversity of the panel allowed participants to better understand the perspective from scientists experienced with ADC development, but also receive general outlooks from the FDA representative. Attendees benefited from comments from Hong Zhao, who fielded several questions on submission strategies. One specific point of interest and discussion was about what specific bioanalytical assays should be developed for inclusion in submission packages. ADC submission packages should closely mirror those of mAb submissions with the additional PK assessment for total antibody, ADC, and small molecule payload. Additionally, immunogenicity assays should be developed in accordance with FDA recommendations for mAbs.
Biodistribution, biotransformation, stability studies and clinical implication
Biodistribution studies are routine in small molecule development, but the body of literature on such studies with ADCs is limited. Understanding the role of biodistribution in the context of ADC development led to a more fundamental question posed by the panel – what is the value of biodistribution studies if they may introduce inter-species differences in plasma stability between species? If the goal of a biodistribution study is merely to understand the time course of exposure for the total antibody, ADC, and payload, one could run biodistribution studies; however, the concern is that biodistribution studies that are actually unnecessary may be required for submission of a marketing application. Understanding the utility of biodistribution and biotransformation pathways of ADCs were key topics of discussion during the panel session.
It was clearly stated that plasma instability across species may lead to erroneous translation to man; companies developing ADCs should therefore emphasize the importance of evaluating ADC plasma stability. This specific dialog on the importance of ADC plasma stability was led by Jay Tibbitts, who pointed out that, based on the literature reports, ADC plasma stability has been observed across different species. Mechanisms for instability, however, may differ between species, and thus biodistribution studies should only be warranted after plasma stability has been thoroughly defined across relevant species. In response, it was suggested that when there is a loss of the cytotoxic payload, we need to understand where it is distributing and the mechanism for release because these may help identify the ADCs instability.
The clinical implications of this question were later addressed using brentuximab vedotin and trastuzumab emtansine as examples. Literature to date has not suggested a mechanism-based rationale for brentuximab vedotin-associated neutropenia. It remains unclear whether it is the result of the payload falling off systemically; whether it is related to the metabolism of the payload in the tumor, or some other tissue; or whether the stability of the ADC plays a role in the observed toxicity. The neutropenia observed with brentuximab vedotin does not appear to be specific to all ADCs; therefore, it is hard to mitigate such toxicities in the development of ADCs when they appear to be an ADC-specific, and not class, effect. As noted by members of the audience, thrombocytopenia seems to be a common toxicity because it has been observed with different payloads and different antibodies. Attendees questioned whether the mechanism of this toxicity is known and whether blood partitioning would be a useful tool to assess specificity. Jay Tibbitts cited trastuzumab emtansine and several recent publicationsCitation3 to address this question.Citation4-Citation6 Trastuzumab emtansine-induced thrombocytopenia appears to be pure thrombocytopenia, not pancytopenia. Other ADCs have shown pancytopenia toxicity profiles with more prominent neutropenia and effects on platelets suggesting overall marrow toxicity; however, this was not the case with trastuzumab emtansine. As a proof of concept, an ex vivo mechanistic study was conducted to explore the mechanism for trastuzumab emtansine transient thrombocytopenia observed in the clinic in HER2-positive breast cancer patients.Citation6 This study suggests that the toxicity is primarily driven by non-specific uptake of trastuzumab emtansine, and not by free drug, because T-DM1 entered mouse megakaryocytes, where it undergoes catabolism to release lysine-MCC-DM1, which disrupts the microtubule cytoskeleton and causes inhibition of pro- platelet production. It has also been shown that trastuzumab emtansine goes through typical biotransformation pathways such that it is catabolized to form lysine-MCC-DM1. The results of this work suggested that defining pathways whereby molecules such as trastuzumab emtansine affect megakaryocyte differentiation and pro-platelet production may yield strategies to manage ADC-induced thrombocytopenia.
Affects of drug-to-antibody ratios on efficacy
The affect of the site of conjugation on the efficacy and PK of ADCs was discussed to clarify the importance of high drug to antibody ratio (DAR) in ADC program programs. It was suggested that high DAR conjugates may not be ideal for ADC programs where high antibody doses are desired, such as in cases where the activities associated with the naked antibody may be realized. Surinder Kaur recommended that the molecular and structural properties of the molecules should be considered, and cited the notion that an eight DAR molecule will have different properties than a molecule with a more concentrated DAR. While drug loading has been shown to negatively impact the PK and other properties associated with some ADC technologies, it was noted that other technologies may be more amenable to achieving higher DAR. Available literature does not provide a clear understanding on DAR impact on molecules using the various technologies. The panel recommended that DAR impact should be explored further.
Leveraging Prior Submissions for new applications
Several questions were posed to the panel to better understand the importance of how much information can be borrowed from prior submissions with respect to use of the same drug-linker. The panel recommended, as with any molecule, ADC or otherwise, each is considered for approval based on the individual molecule efficacy, safety and overall patient benefit. The consideration for inclusion of previously submitted data, for example, maytansinoid or auristatin free drug ADME or QTc data, would be based on a number of factors including, but not limited to, potential exposure differences in ADC or free toxin levels, extent of similarities in the characteristics of the ADC products, as well as sensitivity of the patient population to the free toxin. As with any submission, there always should be a balance of good, solid science and regulatory constraints that justify the leveraging of previously submitted ADC data.
Therapeutic Index (TI)
The discussion of the ADC TI was of particular interest among attendees, given the theoretical expectation of a wider TI with the ADCs due to targeted delivery of payloads. The current evidence generally supports the idea that more potent ADCs tend to be toxic, but also tend to be more efficacious. Several attendees questioned whether the use of less potent ADCs may confer some advantage in widening the TI, since the de-conjugation, or non-specific toxicity, of a less potent ADC would result in less toxicity. The panelists and attendees conceded that, although a less potent ADC would theoretically have lowered toxicity, this less potent ADC would also certainly be less efficacious. Thus, rather than narrowing the TI, the less potent ADC would only effectively move the TI to a higher dose range.
Payload Selection
The decision to use a more potent toxin vs. a less potent toxin may depend on a number of factors, including, but not limited to, target expression, target internalization/processing, and intrinsic sensitivity to the toxin. Choosing the best expression pattern or processing of the antigen may be the best way to maximize the TI. Similarly, understanding the intrinsic sensitivity of the toxin in nonclinical models (in vitro and in vivo), as well as in patients, may be a critical component in improving the TI of an ADC.
Maximum biologically effective dose vs. maximum tolerated dose for ADCs
The definition of the maximum tolerated dose (MTD) in a Phase 1 study was of great interest to companies that are currently developing Phase 1 ADC programs. The panel indicated that dose-response, maximum biologically effective dose (MBED) and MTD in Phase 1 of an ADC is typically done using an approach similar to that conventionally used for cytotoxic drugs. There is no clear differentiation, as of yet, to determine MTD or MBED beyond what is informed by Phase 1 data for an ADC. As more clinical data becomes available for ADCs with similar drugs/ linkers, it will be increasingly important that preclinical information, in tandem with information available from other ADCs (with same linker and cytotoxic molecule), be integrated to facilitate decision making and convince oncologists that they may not need to dose to the MTD for targeted therapies, including ADC, but to a MBED.
Characterizing PK of ADCs
Understanding the PK of ADCs was a hot topic of discussion; the panel discussed whether Cmax or AUC is the primary driver for ADC efficacy. The presence of multiple active species of ADC (ADC, small molecule drug, total antibody) in systemic circulation makes the identification of key analyte or PK metric (e.g., Cmax, AUC.) driving the drug effect very difficult. Typically, efficacy/ safety of ADC can be attributed to the overall systemic exposure of multiple moieties of ADC. In essence, the perception is that the safety is driven by three mechanisms: 1) ADC binding to it target on a normal cell causing on-target toxicity; 2) ADC getting engulfed by a normal cell and causing off-target toxicity; and 3) systemic availability of the cytotoxic moiety from the ADC, where the efficacy can be attributed to the on-target delivery of the ADC, antibody (if mAb has any activity) and payload component of an ADC.Citation7,Citation8
ADC drug-drug interaction (DDI) potential
The final topic discussed during the panel session was the relevance of understanding ADC DDI and the appropriate studies recommended during clinical ADC development. Manish Gupta led the discussion by indicating that there is a theoretical potential for ADC catabolites to engage in DDI with other small molecule therapeutics, which affects the serum or plasma concentrations of either the ADC catabolite or other co-administered medications. Collective in vitro metabolism and preclinical data can be used to determine whether ADC (or its catabolites) is likely to be a perpetrator or a victim of a drug interaction when co-administered with another drug;Citation9 the outcome will typically be mostly influenced by the pathways of elimination of the small molecule drug payload in an ADC. Risk for a PK-based drug interaction DDI between ADC and concomitant drug is considered low for mAb because of non-overlapping pathways of elimination with small molecule drug; the likelihood of an ADC acting as a “perpetrator” and having an effect on the concomitant drug in-spite of sharing the same drug metabolism pathway is relatively low, especially when the concentrations of the small molecule drug (of an ADC) in systemic circulation is relatively low.Citation10,Citation11 However, an ADC (or its payload) could still be a victim of DDI in combination with other drugs. This can be determined by additional DDI studies in clinic studies (e.g., with CYP3A4 substrates, inhibitors or inducers). These studies may not be necessary if metabolism has been characterized and DDI studies conducted for a payload that is an approved small molecule drug; this needs to be evaluated in the context of efficacy/ safety of a drug on a case-by-case basis.
The panel session provided a focused forum for attendees to discuss both preclinical and clinical development questions with the panel of experts. As with any molecule under development, a substantial amount of research effort goes into development, and ADCs are no exception.
Course Review
The short course’s organizing committee selected a well-balanced panel whose individual scientific expertise allowed for delivery of cutting-edge material that is applicable across line functions (e.g., PK, toxicology, formulation, regulatory, preclinical and clinical). ADCs represent an innovative and powerful biotherapeutic approach to treat different malignancies and are relatively new therapeutic entities; this short course successfully brought together many thought leaders driving ADC development in their respective institutions, allowing for a refreshing scientific exchange. This short course addressed the unique translational and clinical development challenges, including assay interpretation, translation of safety, PK and ADME in different species and in the clinic, chemistry, manufacturing, and control, and analytical chemistry, clinical pharmacology and, finally, regulatory landscape (current, future), to ADC development. Brentuximab vedotin and trastuzumab emtansine were frequently cited throughout the course, which provided specific examples of the translational challenges associated with their development; but also highlighted the impact these molecules have had on clinical outcomes. The high enrollment and attendance of this course suggests a strong interest within the industry to better understand the development path for these molecules. Additionally, this course spurred much discussion on the need to establish an ADC Focus Group within AAPS to continue healthy dialog/discussions between pharmaceutical companies, regulatory agencies and academia.
| Abbreviations: | ||
| ADC | = | antibody drug conjugate |
| mAb | = | monoclonal antibody |
| PK | = | pharmacokinetics |
| PD | = | pharmacodynamics |
| ADME | = | absorption distribution metabolism elimination |
Acknowledgments
The authors acknowledge and thank AAPS- NBC for supporting the short-course and recognize the scientists dedicated to the defining the ADC development path. The authors would like to acknowledge Benjamin Guiastrennec who assisted in the preparation of Figure 1.
Note
K.T. wrote and prepared the manuscript for publication. K.T, S.B., D.L., W.M., S.K., B.M., H.E., J.T., H.Z., and M.G. wrote and reviewed the manuscript. S.B., D.L, and M.G. organized and moderated this short course. S.B., D.L., W.M., S.K., H.E., J.T., H.Z., and M.G. presented during the short-course and served as panelists for the panel session.
Potential Conflicts of Interest
K.T., S.B., and W.M are employed by Novartis Pharmaceuticals and declare no competing financial interests. D.L., and S.K., are employed by Genentech. J.T. is currently employed at UCB Pharma Ltd and was formerly employed by Genentech during the development of Trastuzumab emtansine. H.E. is employed by ImmunoGen, Inc. H.Z. is employed by the US. Food and Drug Administration. M.G. is employed by Bristol-Myers Squibb and was formerly employed by Genentech during the development of trastuzumab emtansine. B.M. is employed by the University of Tennessee.
Related Research Data
References
- Deng R, Iyer S, Theil FP, Mortensen DL, Fielder PJ, Prabhu S. Projecting human pharmacokinetics of therapeutic antibodies from nonclinical data: what have we learned?. MAbs 2011; 3:61 - 6; http://dx.doi.org/10.4161/mabs.3.1.13799; PMID: 20962582
- Chudasama VL, Schaedeli Stark F, Harrold JM, Tibbitts J, Girish SR, Gupta M, et al. Semi-mechanistic population pharmacokinetic model of multivalent trastuzumab emtansine in patients with metastatic breast cancer. Clin Pharmacol Ther 2012; 92:520 - 7; http://dx.doi.org/10.1038/clpt.2012.153; PMID: 22968044
- Thon JN, Devine MT, Jurak Begonja A, Tibbitts J, Italiano JE Jr.. High-content live-cell imaging assay used to establish mechanism of trastuzumab emtansine (T-DM1)--mediated inhibition of platelet production. Blood 2012; 120:1975 - 84; http://dx.doi.org/10.1182/blood-2012-04-420968; PMID: 22665936
- Burris HA 3rd, Rugo HS, Vukelja SJ, Vogel CL, Borson RA, Limentani S, et al. Phase II study of the antibody drug conjugate trastuzumab-DM1 for the treatment of human epidermal growth factor receptor 2 (HER2)-positive breast cancer after prior HER2-directed therapy. J Clin Oncol 2011; 29:398 - 405; http://dx.doi.org/10.1200/JCO.2010.29.5865; PMID: 21172893
- Krop IE, Beeram M, Modi S, Jones SF, Holden SN, Yu W, et al. Phase I study of trastuzumab-DM1, an HER2 antibody-drug conjugate, given every 3 weeks to patients with HER2-positive metastatic breast cancer. J Clin Oncol 2010; 28:2698 - 704; http://dx.doi.org/10.1200/JCO.2009.26.2071; PMID: 20421541
- Mahapatra KDW, Bumbaca D, Shen B. 2011. T-DM1-induced thrombocytopenia results from impaired platelet production in a HER2-independent manner. AACR-NCI-EORTC, ed., San Francisco, California: AACR.
- Lin K, Tibbitts J. Pharmacokinetic considerations for antibody drug conjugates. Pharm Res 2012; 29:2354 - 66; http://dx.doi.org/10.1007/s11095-012-0800-y; PMID: 22740180
- Bender BC, Schaedeli-Stark F, Koch R, Joshi A, Chu YW, Rugo H, et al. A population pharmacokinetic/pharmacodynamic model of thrombocytopenia characterizing the effect of trastuzumab emtansine (T-DM1) on platelet counts in patients with HER2-positive metastatic breast cancer. Cancer Chemother Pharmacol 2012; 70:591 - 601; http://dx.doi.org/10.1007/s00280-012-1934-7; PMID: 22886072
- Shen BQ, Bumbaca D, Saad O, Yue Q, Pastuskovas CV, Khojasteh SC, et al. Catabolic fate and pharmacokinetic characterization of trastuzumab emtansine (T-DM1): an emphasis on preclinical and clinical catabolism. Curr Drug Metab 2012; 13:901 - 10; http://dx.doi.org/10.2174/138920012802138598; PMID: 22475269
- Girish S, Gupta M, Wang B, Lu D, Krop IE, Vogel CL, et al. Clinical pharmacology of trastuzumab emtansine (T-DM1): an antibody-drug conjugate in development for the treatment of HER2-positive cancer. Cancer Chemother Pharmacol 2012; 69:1229 - 40; http://dx.doi.org/10.1007/s00280-011-1817-3; PMID: 22271209
- Lu D, Burris HA 3rd, Wang B, Dees EC, Cortes J, Joshi A, et al. Drug interaction potential of trastuzumab emtansine (T-DM1) combined with pertuzumab in patients with HER2-positive metastatic breast cancer. Curr Drug Metab 2012; 13:911 - 22; http://dx.doi.org/10.2174/138920012802138688; PMID: 22475266