Abstract
Hydroxyl radical footprinting is a covalent labeling strategy used to probe the conformational properties of proteins in solution. We describe the first application of this high resolution technique for characterizing the structure of a therapeutic monoclonal antibody (mAb) dimer. As monitored by size-exclusion chromatography (SEC), therapeutic mAbs typically contain small amounts of a dimer species relative to the primary monomeric form in its drug substance or drug product. To determine its structural orientation, a sample enriched in an IgG1 mAb dimer was oxidized by hydroxyl radicals generated by exposure of the aqueous solution to synchrotron X-rays in millisecond timescales. The antibody monomer that served as a control was oxidized in a similar fashion. The oxidized samples were digested with trypsin and analyzed by RP-UHPLC-MS. The footprinting data show that peptides displaying decreased rates of oxidation (i.e., regions of increased protection) in the dimer are localized in the light and heavy chains of the Fab domain. The interface region for the monomers comprising the dimer was thus inferred to be between their Fab arms, allowing us to model two possible theoretical dimer orientations: a head-to-head, single arm-bound Fab-to-Fab dimer, and a head-to-head, double arm-bound Fabʹ2-to-Fabʹ2 dimer. Lower resolution fragment-SEC analysis of the dimer and monomer samples treated with papain or FabRICATOR® enzyme provided complimentary evidence to support the Fab/Fab orientation of the IgG1 dimer.
Introduction
Monoclonal antibodies (mAbs) have become widely accepted therapies for various forms of cancer and immunologic diseases.Citation1-Citation3 For product quality testing, methods for characterizing mAbs at the primary structure level have been described and are well established; common post-translational modifications such as deamidation, oxidation, glycosylation, or fragmentation can be detected and quantified by various chromatographic or mass spectrometry-based methods with high sensitivity.Citation4 The higher order structure of the therapeutic mAbs must also be considered because the activity and stability of the molecules will be influenced by their three dimensional structure. Changes in the manufacturing process, formulation, or storage conditions may affect higher order structure, thus driving the need to have methods for its analysis. Such methods may also play a critical role in the emerging biosimilars industry, where structural comparability to originators needs to be assessed.Citation5 Traditional analytical methods used for structural studies of proteins include analytical ultracentrifugation (AUC), FTIR, circular dichroism (CD) spectroscopy, and fluorescence spectroscopy.Citation6-Citation9 These methods may have utility for monitoring global changes in higher order structure; they are, however, not ideal for localizing specific areas or regions of structural change in the molecule, and can thus be considered relatively low resolution techniques.
Mass spectrometry (MS)-based methods for analyzing the higher order structure of proteins have emerged that can offer higher resolution of structural detail, providing regional (peptide level) or even site-specific (single residue level) information. Hydrogen-deuterium (H/D) exchange mass spectrometry is a reversible covalent labeling (CL) technique that has been increasingly employed to study biopharmaceutical products, including mAbs.Citation10-Citation13 The method is desirable due its non-invasive nature, the potential of labeling at almost all amino acids, and the acquisition of structural information at both peptide and residue-specific levels via “bottom up” analysis. A drawback of the H/D exchange method is the transient and labile nature of the labeling probe, and the possibility that some structural information can be lost. The required experimental conditions also limit the choices for enzymatic digestion/peptide mapping to proteases that are active in acidic conditions (e.g., pepsin). In contrast to H/D exchange, other covalent labeling methods impart permanent or stable modifications to amino acid side chains for probing or mapping protein structure. Structural information at both peptide and residue-specific levels can also be obtained, and a larger variety of proteases for peptide mapping can be utilized (e.g., trypsin, Asp-N, Glu-C). However, the modifications and reaction conditions themselves may perturb the very structure or conformation that one is attempting to characterize, and care must be given to ensure that the native structure is preserved during the labeling reaction. Various labels can be utilized, including amino acid side chain-specific labeling reagents such as iodoacetamide (cysteines), N-bromosuccinimide (tryptophan), diethylpyrocarbonate (carboxyl acids), and S,SCitation1-dimethylthiobutanoylhdroxysuccinimide (lysines).Citation14-Citation17 Structural information from these reagents may not be as comprehensive due to their inherent specificities.
Use of hydroxyl radical footprinting, a stable covalent labeling method that can modify a wider variety of side chains, is becoming more prominent for higher order protein studies. In this method, hydroxyl radicals produced in situ with the protein of interest are used to introduce oxidative modifications to its amino acid chains. The extent of a region’s oxidation reflects its extent of protection, and this protection level can be defined at either the peptide or individual amino acid residue level. The hydroxyl radicals are non-selective and highly reactive, producing a stable oxidative modification in milliseconds (MS); this reaction is kinetically favored over radical-mediated backbone cleavage or cross-linking.Citation18 The amount or rate of oxidation of a particular amino acid is ultimately a function of its solvent accessibility and its inherent reactivity. The rank order of reactivity of the common amino acids used for protein footprinting studies has been described previously, with sulfur-containing and aromatic amino acids being the most reactive, followed by aliphatic amino acids, and then the other charged and neutral amino acids.Citation19,Citation20 Hydroxyl radical oxidation typically produces peptides with mass additions of +16 Da or +14 Da due to formation of hydroxyl or carbonyl groups in the side chains, although other mass shifts specific to aromatic side chains are also possible. Guan and Chance have described the side-chain products possible from radiolytic footprinting experiments for twenty amino acids plus cystine,Citation21 showing this technique’s high potential for structural resolution. Once the oxidative labeling is completed, proteolytic cleavage and tandem MS (MS/MS) is used to identify and quantitate peptides that have been oxidized. Relative levels of protection (or accessibility) for each peptide are determined from their rates or amounts of oxidation. Structural interpretations can then be made based on differences or changes in these protection levels. Oxidative, and thus solvent accessibility, information at specific residues can also be extracted from the tandem MS data.
Though there are several methods to generate hydroxyl radicals in solution for footprinting assays, methods that can generate radicals in the millisecond or faster timescales are preferred for protein studies so that native structures under study are not unduly altered by the oxidative modifications. Recently, Hambly and Gross developed the Fast Photochemical Oxidation of Proteins (FPOP) method using an excimer laser to produce hydroxyl radicals in microsecond timescales from hydrogen peroxide added to the protein solution.Citation22,Citation23 At this timescale, it has been shown that the oxidative labeling occurs faster than protein unfolding.Citation24 The FPOP method was recently applied to therapeutic proteins, although the panel of samples consisted of small proteins rather than mAbs.Citation25 Another fast method to generate hydroxyl radicals that preceded FPOP, and the method that was employed in this current study, is the use of synchrotron radiation to generate radicals directly from water solvent molecules. Mark Chance and colleagues pioneered the early use of this method for protein footprinting studies,Citation24-Citation26 utilizing synchrotron X-rays to irradiate aqueous solutions containing the protein of interest, producing a high flux of radicals in millisecond timescales. In contrast to FPOP, hydrogen peroxide does not need to be added to the protein solution. The details of the synchrotron procedure and its time-resolved methodology have been well documented in reviews, and in studies focusing on protein folding and dynamics, and protein-ligand complexes.Citation27-Citation29 Although longer synchrotron irradiation times (>50ms) may begin to introduce oxidative-induced structural changes, the exposure times for this current study were kept below 20 ms.Citation30
To our knowledge, no studies using hydroxyl radical footprinting to characterize the higher order structure of therapeutic mAbs have been previously reported. We report here the first application of this footprinting method to study the structure of a therapeutic mAb, specifically a dimeric (or aggregated) form of the mAb. Therapeutic mAb aggregates, assemblies of mAb molecules other than the desired monomeric form, can be formed at different phases of the manufacturing processes, including cell culture, purification, and formulation.Citation31 These aggregates can be subvisible or visible, ranging in size from dimers to very large multimers, and can be influenced by factors such as pH, temperature, and light exposure.Citation32,Citation33 Due to their potential to trigger an immunogenic response and other adverse effects,Citation34 aggregates are closely monitored and characterized within the biopharmaceutical industry by several established analytical methods, including size-exclusion chromatography (SEC), analytical ultracentrifugation (AUC), capillary electrophoresis (CE-SDS), multi-angle laser light scattering (MALLS), and dynamic light scattering (DLS).Citation35 These methods, however, do not provide detailed information on the conformational properties of mAb aggregates, thus providing an opportunity to assess the utility of hydroxyl radical footprinting for this purpose.
For this study, a soluble dimer of a representative therapeutic IgG1 mAb present in low amounts in the final drug substance relative to the desired monomer was analyzed by hydroxyl radical footprinting to determine the probable structural orientation of its components. The IgG1 dimer and monomer fractions were enriched by SEC, and then labeled by hydroxyl radicals generated by synchrotron irradiation of their aqueous solutions. The samples were then subjected to tryptic digestion, reverse-phase ultra-high pressure liquid chromatography (RP-UHPLC) peptide map analysis, and tandem MS detection. Analysis of the MS data allowed for detection of oxidized residues, quantification of peptide oxidation levels, and determination of rate constants that defined regions of more or less protection in the dimer relative to the monomer control. Regions of increased protection in the dimer were observed in the light and heavy chains in the Fab domain, pointing to the likely interface areas of the two component monomeric units, and leading us to model possible structural orientations. The high resolution footprinting data suggests that this IgG1 dimer is interfaced through the Fab domains in either a head-to-head, single-arm Fab-to-Fab orientation or a head-to-head, double-arm bound Fab'2-to-Fab'2 orientation, with the main interface regions more localized in the light chains. Supplementary data from lower resolution fragment-SEC experiments using papain and FabRICATOR® enzymes supported the Fab/Fab interface model, showing that the hydroxyl radical footprinting technique holds promise for characterizing the higher order structures of therapeutic mAbs.
Results
Enrichment of IgG1 dimer and monomer forms
A IgG1 mAb drug substance in its final formulation was analyzed by SEC to resolve the minor dimer form, comprising ~4% of the total peak area, and the primary monomer form, comprising ~96% of the total peak area (). The drug substance was not subjected to any forced stress studies (e.g., thermal or pH stress), so this dimer was considered “native” in that its formation results from the normal manufacturing process conditions. Size-exclusion chromatography-multi-angle laser light scattering (SEC-MALLS) confirmed that the dimer species had a molar mass of approximately 300 × 103 g/mol, and the monomer species had the expected approximate molar mass of 150 × 103 g/mol.
Figure 1. SEC chromatograms of enriched fractions. IgG1 bulk drug substance starting material contains approximately 4% dimer. Enriched dimer is 92% pure, and enriched monomer is 96% pure.
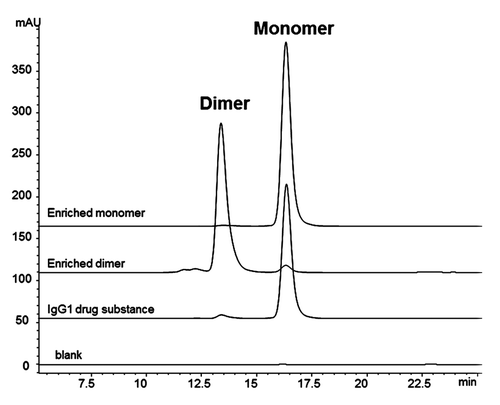
The peaks corresponding to the dimer and monomer components were enriched via fractionation from SEC. The monomer served as an experimental control for the footprinting analysis. Final concentrations for the dimer and monomer were 10 mg/mL and 30 mg/mL, respectively. Both the enriched dimer and monomer samples were re-analyzed by SEC to determine final purity; approximately 92% and 98% purity were achieved for the dimer and monomer samples, respectively ( and ). The dimer and monomer samples also had similar non-reduced CE-SDS profiles (Fig. S1 and Table S1), with comparable percent main peaks (). Since CE-SDS is a denaturing method, the comparable main peaks showed that the dimer was primarily a non-covalent association between two monomeric units. The majority of the dimer species survived the enrichment process without dissociating into monomers, indicating the stability of the non-covalent association. The enriched dimer and monomer samples were also assessed for potency in a cell-based anti-proliferation bioassay. The results of the potency assay showed a reduced potency of 42% for the dimer sample, compared to 97% for the monomer control ().
Table 1. Characterization data for SEC-enriched IgG1 dimer and monomer samples
Enriched fractions were stored frozen until needed for analysis. The enriched dimer was determined to be fairly stable for at least three freeze/thaw cycles. As determined by SEC re-analysis, the purity of the enriched dimer was still approximately 88% after three freeze/thaw cycles (data not shown). The monomer fraction showed no change in purity. After thawing, the enriched fractions showed no change in purity after storage at 2–8°C for one week.
Fragment-SEC Analysis of papain digested dimer and monomer samples
As a starting point for our structural studies, we utilized lower resolution methodologies as a supplementary tool for studying our therapeutic mAb dimer at the Fab/Fc domain level. A common strategy employed in previous studies involved cleaving the dimer with papain or Lys-C, and analyzing its associated Fab and Fc fragments to determine the domains involved in the dimer interface.Citation36,Citation37 Using a similar workflow strategy implemented by Remmele and co-workers,Citation36 we treated the enriched dimer and monomer samples with papain and subsequently analyzed the resulting fragments by SEC, MALLS, and LC-MS.
The SEC chromatograms of the dimer and monomer samples incubated for 2 hours at 37°C with papain are shown in . The three prominent peaks (Peaks 1, 2 and 3) observed in each chromatogram were characterized by SEC-MALLS, and by a previously described on-line LC-MS method operating in a two-dimensional format.Citation38 The characterization data, summarized in , show that Peak 1 is a non-covalently associated Fab/Fab dimerized species, Peak 2 is the disulfide-linked intact Fc species, and Peak 3 is the monomeric Fab species. Peaks 1 and 3 have the same observed mass by LC-MS analysis because the non-covalently bound Fab/Fab dimerized species of Peak 1 dissociates during the ionization process. No non-covalently Fab/Fc or Fc/Fc dimerized species were detected in the papain digests of either the dimer or monomer.
Figure 2. Fragment-SEC chromatograms of papain-digested dimer and monomer. (A) Papain digest of dimer and monomer. Peak 1 is a Fab/Fab associated species, Peak 2 is intact disulfide-linked Fc, and Peak 3 is the monomeric Fab (). (B) Papain digest incubation time course study of the dimer. Peak 1 (Fab/Fab species) increases with incubation time, while Peak 3 (Fab) decreases. Peak 2 (intact Fc) remains constant. (C) Papain digest incubation time course study of the monomer. Peak 1 (Fab/Fab species) increases with incubation time, while Peak 3 (Fab) decreases. Peak 2 is unchanged.
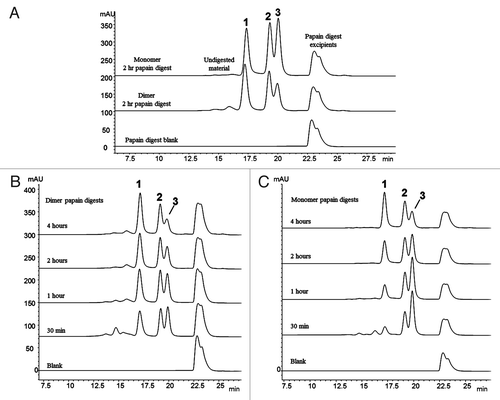
Table 2. Characterization data for papain-generated fragments
The presence of only Fab/Fab dimerized species (Peak 1) in the SEC profile of the enriched dimer after papain treatment suggests that the dimer interface is localized in the Fab region. Due to the stability of the dimer, we expected that most of the non-covalently interfaced regions of the intact dimer would survive the papain treatment. It is not surprising that some of these interface regions dissociated during the processing, thus leading to the presence of monomeric Fab (Peak 2) in the dimer’s profile (the small amount of monomer still present in this enriched fraction also contributes to the amount of Peak 2). Interestingly, though, the chromatograms showed that Peak 1 is notably present in both the dimer and monomer samples after a 2 hour incubation, leading to the question of why the monomer sample would also contain a Fab/Fab dimerized species. A subsequent study varying the incubation times with papain at 37°C showed that dimerization of papain-generated Fab molecules occurs in-situ as the enzymatic reaction proceeds over time.
The products of the papain cleavage at increasing incubation times of 30 minutes, 1 hour, 2 hour, and 4 hours were monitored by SEC. show the chromatograms of the papain time course experiments for the dimer and monomer samples, respectively. As the incubation time increases, the Fab/Fab dimerized species (Peak 1) increases, while the corresponding free Fab species (Peak 3) decreases in both samples. The amount of intact Fc species (Peak 2) remains constant. This demonstrates that the Fab species resulting from papain digestion have a strong affinity to form homodimers. No heterodimers involving Fab/Fc fragments were observed. The tendency for the monomeric Fab arms to dimerize suggests that this is an aggregation-prone region in this IgG1, and lends support to the Fab’s involvement in the intact dimer’s interface region. It should be noted that the observed Fab/Fab aggregate never grew larger than a dimer (i.e., two associated Fab units). Fab trimers and larger multimers did not form over time; thus, the Fab/Fab interaction must involve a specific orientation and region that does not allow for additional Fab molecules to add to the two-unit structure.
Although the Fab/Fab species (Peak 1) was present in both the dimer and monomer samples, Peak 1 in the dimer after 30 minute incubation with papain at 37°C was approximately 3 times larger than in the corresponding monomer sample (). This difference reflects the a priori amount of “native” Fab/Fab species involved in the dimer interface region that survived the papain treatment, before in-situ Fab dimerization begins to occur in solution. The monomeric Fab species (Peak 3) that was present in the dimer sample after the 30 minute incubation likely represents the “native” Fab/Fab species that dissociated as a result of the digestion process. Thus, there appear to be two competing reactions occurring in solution for the dimer: (1) the dissociation of “native” Fab/Fab into monomeric Fabs; and (2) the in-situ (re)dimerization of the monomeric Fabs into Fab/Fab species. For the monomer, only the in-situ dimerization of the monomeric Fabs into Fab/Fab species occurred. After 4 hours at 37°C, the amount of Fab/Fab species (Peak 1) and monomeric Fab species (Peak 3) in both the monomer and dimer samples were fairly similar, suggesting that the in-situ dimerization reaction may have approached an equilibrium state.
Fragment-SEC Analysis of dimer and monomer samples treated with FabRICATOR®
In an effort to further investigate the Fab domain interactions suggested by the papain cleavage data, we performed a second fragment-SEC analysis using an alternative enzyme to papain: FabRICATOR® enzyme (Bulldog Bio).Citation39 The products of FabRICATOR® digestion are similar to those produced by pepsin (i.e., cleavage below the hinge region disulfides), producing divalent Fabʹ2 and Fc fragments, instead of the monovalent Fab fragments and intact, disulfide-linked Fc produced by papain. Using the same workflow as the papain digest analysis, dimer and monomer samples were treated with FabRICATOR® and the resulting fragments analyzed by SEC, MALLS, and LC-MS.
The SEC chromatograms of the dimer and monomer samples incubated for 3 hours at 37°C with FabRICATOR® are shown in . The three prominent peaks (Peaks 1, 2 and 3) are observed in the dimer, whereas only two prominent peaks (Peaks 2 and 3) are observed in the monomer chromatogram. The characterization data are summarized in , showing that Peak 1 is a non-covalently associated Fabʹ/Fabʹ2 dimerized species, Peak 2 is monomeric Fabʹ2 species, and Peak 3 is a dimerized Fc/Fc fragment species. Similar to the papain digest data, Peaks 1 and 2 have the same observed mass by LC-MS since the Fabʹ2/Fabʹ2 dimerized species of Peak 1 dissociates upon electrospray ionization.
Figure 3. Fragment-SEC chromatograms of FabRICATOR®-digested dimer and monomer. FabRICATOR® digest of dimer and monomer samples. Peak 1 is a Fabʹ2/Fabʹ2 associated species, Peak 2 is monomeric Fabʹ2, and Peak 3 is an Fc/Fc fragment species. Peak 1 is only prominent in the dimer sample. Peak 3 is equally present in both the dimer and monomer sample. Refer to for characterization data for FabRICATOR®-generated fragments.
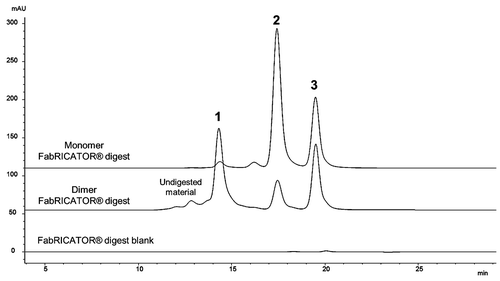
Table 3. Characterization data for FabRICATOR®-generated fragments
The Fabʹ2/Fabʹ2 dimerized species (Peak 1) is prominent only in the chromatogram of the dimer, clearly pointing to the Fab'2 as indicative of the intact dimer’s interface region; this result was consistent with the observations from the papain cleavage analysis. Some monomeric Fabʹ2 species (Peak 2) was present in the dimer’s chromatogram, likely due to some dissociation of its interface region during the enzymatic processing. Interestingly, the cleaved Fc fragments formed non-covalently associated homodimers (Peak 3) in solution in both the dimer and monomer sample; no monomeric Fc fragments were observed. Since Fc/Fc fragment dimerization was observed equally in both the dimer and monomer, indicating a property inherent to this IgG1, the Fc is not implicated as an interface region for the intact dimer. We also observed in an incubation time course study, similar to the one performed for the papain digestion, that the Fabʹ2 does not have a tendency to dimerize in-situ over time (data not shown). Since partial unfolding is believed to play a role in protein aggregation,Citation32,Citation40 it is possible that the FabRICATOR®-generated, native Fab'2 are not afforded a level of unfolding sufficient to promote self-association, as observed for papain-generated Fabs. It should be noted that Fc/Fc fragment dimerization also does not increase or proceed over time, with Peak 3 maintaining a constant amount regardless of incubation time.
Both the papain cleavage and FabRICATOR® cleavage data implicate the Fab domain as the region involved in the intact dimer’s interface. These lower resolution methodologies helped to localize dimeric interface regions to the domain level (e.g., Fab and Fc interactions). They also provide good supportive evidence to higher resolution techniques, such as hydroxyl radical footprinting, which can more precisely define structural details down to peptide or residue levels.
Hydroxyl radical footprinting via synchrotron irradiation
The enriched IgG1 dimer and monomer samples were kept frozen and sent to the National Synchrotron Light Source (NSLS) for the hydroxyl radical labeling procedure. Synchrotron irradiation was performed by NeoProteomics, Inc. using the X28C Beamline at the NSLS. Exposure conditions were initially determined by dose-response analysis of a fluorescent compound (Alexa 488), as described previously.Citation41 Results of the fluorophore dose analysis are shown in Figure S2. For radiolysis of the dimer and the monomer, samples were initially thawed and diluted 10× with a phosphate dilution buffer. The samples were then placed in a beamline flow set-up,Citation41,Citation42 and irradiated for 0ms, 3.75ms, 8.75ms and 17.5ms at ambient temperature with synchrotron X-rays to produce a high flux of hydroxyl radicals in situ. The irradiated samples were then collected in Eppendorf tubes containing methionine amide; this served to quench unreacted radicals and prevent secondary oxidation events resulting from hydrogen peroxide and other oxidative species generated during radiolysis.Citation43 The irradiated samples were frozen and shipped back to Genentech for downstream processing.
After receipt, the samples were thawed and subjected to a dithiothreitol (DTT) reduction and iodoacetamide carboxymethylation procedure, followed by digestion with trypsin. Tryptic peptides were then separated by reverse-phase ultra-high pressure liquid chromatography (RP-UHPLC), and detected online by a Thermo Orbitrap XL mass spectrometer. No gross peak differences were observed in the UV chromatograms between the graduated exposure times, suggesting overall low levels of oxidation on a global level (Fig. S3). Previous studies utilizing synchrotron irradiation have shown that the extent of total oxidation is expected to be limited due to the fast timescales involved and the minimized risk of secondary oxidation reactions.Citation30,Citation43 MS and tandem MS data from the tryptic peptide map were used to identify oxidized peptides and their unmodified, non-oxidized forms.
Mass spectrometry data analysis was primarily carried out using ProtMap MS, a commercial software package developed by NeoProteomics specifically for the automated analysis of oxidative footprinting data.Citation44 ProtMap MS uses the tandem MS data for identification of oxidized residues, and extracts the ion chromatograms from the MS data for quantitation of peptide oxidation. The +16 Da mass shift represented the most commonly observed, and most abundant oxidative modification. Other observed modifications include: the +14 Da carbonyl forms, the +32 Da forms corresponding to two oxidized residues in the same peptide, and some of the diverse mass shifts associated with aromatic side chains (e.g., -22 Da, -43Da).Citation21 For each sample and exposure time, ProtMap MS identified the oxidized peptides and their unmodified, non-oxidized forms using the tandem MS data. and summarize the tryptic peptides found to have oxidized residues in the IgG1 dimer and monomer samples. Overall sequence coverage by tryptic peptide mapping was 95%. Of the expected tryptic peptides ≥ 3 residues, 70% were identified as having oxidized forms resulting from hydroxyl radical exposure. Approximately 15% of the total number of residues were confirmed by the tandem MS data to be oxidized, with 14 different amino acids (Tyr, Pro, Val, His, Glu, Gln, Thr, Ile, Met, Leu, Ser, Ala, Phe, Asn) comprising the oxidized residue population.
Table 4A. Summary of oxidized heavy chain tryptic peptide data
Table 4B. Summary of oxidized light chain tryptic peptide data
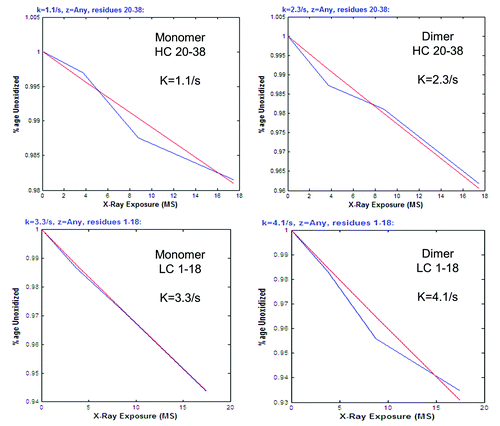
Extracted ion chromatograms (EIC) of the oxidized and non-oxidized peptide forms were derived from MS data, and peak areas were integrated from the EIC by ProtMap MS. The fraction of the non-oxidized peptide, relative to the sum total area of all its detected forms, was calculated at each exposure time. An example EIC data output from ProtMap MS is shown in Figure S4A and B. A dose response curve was then generated by plotting the fraction of the non-oxidized peptide versus exposure time; example dose response curves generated by trhe software are shown in . The curve or plot is used to assess and confirm the pseudo-first order reaction kinetics that the hydroxyl radical labeling reaction is expected to follow; this reaction is described by the following equation: y(t) = e-kt, where y(t) is the fraction of the non-oxidized peptide, k is the rate constant in s−1, and t is the exposure time in seconds.Citation38,Citation39 The rate constants (k) were also automatically calculated by the software ().
Figure 4. Examples of dose response curves and rate constants for oxidized tryptic peptides. Data generated by ProtMap MS software. Linear curves reflect the pseudo first order reaction kinetics of the hydroxyl radical oxidation. Rate constants (k) are also calculated by the software. Blue lines are point-to-point plots of the rate constants (k) at each exposure time. Red lines represent the lines of best fit.
ProtMap MS assignments of oxidation were verified by manual investigation of the tandem MS data, as were correlations with retention times (oxidized peptides typically eluted earlier, and within 2 minutes of their non-oxidized forms), and patterns of increasing levels of oxidation with increasing irradiation times. In addition, some oxidized peptides had to be interrogated manually due to inconclusive tandem MS data. For example, very large peptides or glycopeptides could not be assigned accurately by ProtMap MS, and were therefore data-processed manually to derive dose-response curves and rate constants. In these cases, the EIC and peak integration of the oxidized forms (+16 Da, +14 Da, +32 Da, etc.) and non-oxidized forms were performed manually using Thermo Excalibur software; the rate constants and dose response curves were then generated via Excel spreadsheets. It should also be noted that missed cleavage peptides and some inconsistently recovered tryptic peptides (e.g., one very hydrophobic peptide, peptides ≤ 3 residues eluting very early in the chromatogram) were not considered for structural footprinting assessments because of the complexities in quantitation.
Ultimately, it is the rate constants of the tryptic peptides that reflect the susceptibility of different regions in the IgG1 to oxidative labeling. These values provide the basis for inferring structural data; a ratio between the dimer rate constant (dimer k) and the monomer rate constant (monomer k) for each tryptic peptide is the comparator value used for assigning regions of protection or relative accessibility differences. The individual rate constants for the tryptic peptides of the dimer and monomer are shown in the Tables S4A and S4B. The resulting uncorrected rate constant ratios between the dimer rate constants and the monomer rate constants are shown in and . Comparison of the rate constant ratios between dimer and monomer, however, could not be performed without first performing a normalization step due to the differences in their starting protein concentrations and small differences in their formulation excipient concentrations. The starting concentration of the monomer was ~3× greater than the dimer, and there were likely differences in the amounts of radical-scavenging formulation excipients (e.g., surfactants, sugars) between samples due to variability of the centrifugal filter buffer-exchange step. These differences were maintained even after the 10× dilution with a phosphate buffer performed just prior to synchrotron irradiation. The effects of these factors were noted in the preliminary fluorophore dose response analysis, in which the effective dose for the monomer was observed to be 3-4 times less than the dimer (Fig. S2). Previous oxidative footprinting studies have noted that differences in protein concentration and levels of radical quenchers affect the labeling reaction, and must be taken into account.Citation45,Citation46 For this data set, the non-normalized rate constant ratios have a mean value of 2.7, and a median value of 2.5. Using a strategy similar to one used in microarray analysis or metabolomics to correct for non-biological variations between samples (e.g., division by central tendency, mean scaling),Citation47,Citation48 we used the average of the mean and the median, 2.6, to normalize the ratios to a value of 1. A normalization factor of 2.6 also appears to be in line with the 3–4× difference in effective dose observed between the dimer and monomer. The rate constant ratios were thus divided by 2.6 to produce the normalized, or corrected, ratios between the dimer and monomer, with mean and median values of 1.0 ( and ).
Once the normalized rate constant ratios were calculated, an assessment of relative protection or accessibility differences between the dimer and monomer could be made. A normalized rate constant ratio of 1 was considered to be the nominal value signifying no protection differences between the dimer and monomer. Tryptic peptides with corrected ratios that deviate by 50% or greater from the value of 1 (≤ 0.5 or ≥1.5), were designated as regions that showed differences in relative protection levels in the dimer relative to the monomer. Ratios ≤ 0.5 were assigned as peptides with more protection (or slower rates of oxidation) in the dimer, whereas ratios ≥1.5 were assigned as peptides with less protection (or faster rates of oxidation) in the dimer. Generation of additional data sets in the future will eventually allow for more granular assessment, such that gradients of relative protection level differences can be established (e.g., distinguishing between very small and very large differences). For the present study, we applied a very conservative 50% deviation from the normalized rate constant ratio of 1 for assigning protection level differences. The observed variability in rate constants in previous footprinting studies would support this conservative approach.Citation49,Citation50 A recent study by Kiselar and ChanceCitation49 noted a ~3% error in rate constants using similar data-dependent acquisition methods we employed; the rate constant ratios in our study with large deviations from the ratio of 1 (≤ 0.5 or ≥ 1.5) would not be significantly changed with such levels of variability in the individual rate constants of the dimer and monomer (Table S4A and Table S4B).
The normalized rate constant ratios and assignments of more or less protection in the dimer relative to the monomer are summarized in and . A color-coded bar graph representation of the normalized rate constant ratios for the heavy and light chain peptides is shown in . Peptides that showed no difference are uncolored bars, green bars represent peptides with more protection in the dimer, and yellow bars represent peptides with less protection in the dimer. Based on these assignments, it became apparent that trends of increased protection (slower rate of oxidation) are present in several light chain tryptic peptides (). Four out of the ten light chain tryptic peptides oxidized by hydroxyl radicals, encompassing residues 1-42 and 109-142, showed increased protection in the dimer (, green bars). One tryptic peptide showing increased protection, LC 19-42, encompasses CDR 1 (complimentary determining region) of the light chain. These areas of increased protection are thus presumed to be involved in the interface region of the dimer.
Figure 5. Bar graphs of corrected rate constant ratios (dimer:monomer). (A) Corrected rate constant (k) ratios of heavy chain tryptic peptides (). (B) Corrected rate constant (k) ratios of light chain tryptic peptides (). For both panels, yellow bars indicate peptides with decreased protection in the dimer. Green bars indicate peptides with increased protection in the dimer. Uncolored bars indicate peptides that show no relative difference in protection levels between the dimer and monomer.
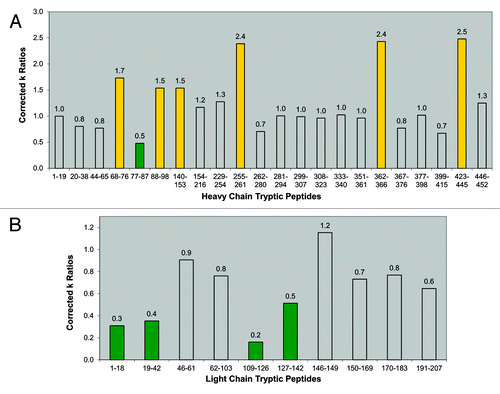
A single peptide (HC 77-87) in the heavy chain showed increased protection in the dimer (, green bar). Although no consistent trends or extended protected regions were observed as in the light chain, it is possible that this small region of the heavy chain in the Fab domain is also involved in the dimeric interface. There appeared to be some regions in the heavy chain (HC 88-98 and HC 140-153) that displayed decreased protection (faster rates of oxidation). Also, three peptides (HC 255-261, HC 362-366, and HC 423-445) in the dimer displayed apparent large increases in their rates of oxidation, with corrected rate constant ratios approximately 150% greater than the nominal corrected ratio value of 1 ( and ). These peptides also showed significant amounts of pre-existing oxidized forms in the dimer, prior to synchrotron irradiation. These three heavy chain peptides contained Fc methionine residues (Met258, Met364, and Met434) that are known to be susceptible to oxidation in therapeutic antibodies,Citation51,Citation52 and upon synchrotron irradiation, they oxidized much faster than all other peptides of the dimer. In a sense, these peptides in the dimer appeared to be already “pre-disposed” to accelerated hydroxyl radical oxidation. Previous studies have shown a correlation of oxidation to aggregation in therapeutic mAbs,Citation53 although whether oxidation increases the propensity for aggregation, or vice versa, has not been established. The accelerated rate of oxidation of heavy chain peptides in the dimer reflects regions of increased solvent exposure, and may be indicative of partially unfolded local conformations in the dimer.
Taking into account the observed peptides displaying increased protection, we can determine that the interface region between the monomeric subunits of the dimer occurs primarily between their Fab domains. This conclusion is consistent with the results of the lower resolution fragment-SEC methods described earlier. The higher resolution afforded by the hydroxyl radical footprinting method allowed for more precise structural mapping, wherein the probable dimeric interface points were further localized to specific peptidic regions in the light and heavy chains. The larger regions of increased protection in the light chain (compared to just a small patch in the heavy chain) suggest its more prominent involvement in the interface.
Mapping of oxidative footprinting data onto IgG1 homology structural models
The regions of more or less protection were mapped onto a homology crystal structure model of an IgG1 antibody (). The IgG1 model was constructed from representative IgG1 Fab and IgG1 Fc crystal structures, and the regions of more or less protection were mapped onto the model according to the described color-coding scheme. We considered two factors in building a proposed dimeric orientation model: (1) the oxidative footprinting data implicating a dimeric interface involving the Fab domains; and (2) the native orientation/position of the two light chains and heavy chains in the Fab domains of our IgG1 model structure. A dimeric interface region occurring primarily between the Fab domains can be visualized by two possible models: a single arm-bound “Fab-to-Fab” dimer orientation, or a double arm-bound “Fab-to-Fab,” or more appropriately, a “Fabʹ2-to-Fabʹ2” orientation.
Figure 6. IgG1 homology structure with mapped protection levels. (A) Model of IgG1 constructed using 1BJ1 and 1IGY crystal structures. Colored according to the relative protection levels derived from oxidative labeling. Yellow indicates regions with decreased protection in the dimer. Green indicates regions with increased protection in the dimer. Uncolored areas indicate regions which show no relative difference in protection levels between the dimer and monomer. The blue region represents the Asn-linked Fc glycans typically found in IgG1 therapeutic mAbs. (B) Space-filled cartoon model of the IgG1 with the heavy chains colored purple and light chains colored red. The light chains of an IgG1 are on opposite planes with respect with each other. The heavy chains in the Fab region are also on opposite planes.
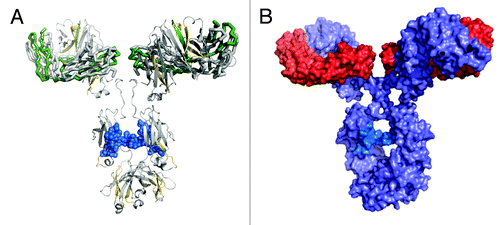
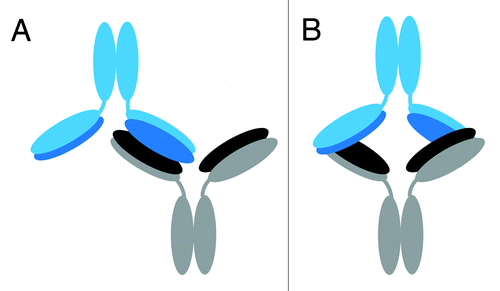
The two possible models were further defined as “head-to-head” configurations due to the three-dimensional orientation of the light chains and heavy chains in the Fab domain of an IgG1. The representative IgG1 model structure shows that the two light chains and two heavy chains in the Fab regions in a mAb are arranged in such a way that they are on opposite planes relative to each other (). In the case of either the “Fab-to-Fab” or “Fabʹ2-to-Fabʹ2” dimeric model, for the light chains/heavy chains of one mAb to interface with the light chains/heavy chains of another mAb, the two mAb units must be in a “head-to-head” orientation in which their respective Fab or Fabʹ2 regions are aligned to each other. This orientation allows for the light chains/heavy chains in the Fab regions to be involved in the interface. Cartoon models of the head-to head, single arm-bound Fab-to-Fab and the head-to head, double arm-bound Fabʹ2-to-Fabʹ2 orientations are shown in ; these represent the two possible structures of our representative IgG1 dimer that can be deduced from the oxidative footprinting data. Either orientation could account for the observed regions of increased protection observed in the dimer’s Fab domain. Either orientation may also be expected to produce the observed drop in potency for the dimer relative to the monomer in the cell killing assay described earlier, as some of CDRs in the dimer would be at least partially blocked.
Figure 7. Proposed possible orientations of the IgG1 dimer. Cartoon representations of proposed possible dimer orientations based on the hydroxyl radical footprinting data. The bottom mAb has heavy chains in gray and light chains in black. The top mAb has heavy chains in light blue and light chains in dark blue. Head-to-head orientation allows for the light and heavy chains of the dimer to associate with each other in the interface region. (A) Cartoon depiction of a head-to head, a single-arm bound Fab-to-Fab dimer. (B) Cartoon depiction of a head-to head, double arm-bound Fabʹ2-to-Fabʹ2 dimer.
Discussion
In this study, hydroxyl radical footprinting was used, for the first time, to study the structure of a therapeutic IgG1 mAb dimer. The differential rates of oxidation of the amino acid side chains, quantified at the tryptic peptide level, were used to delineate the probable regions of the dimeric interface, and allowed for the modeling of proposed dimer orientations. Regions of increased protection (or reduced rates of oxidation) in the dimer’s Fab domain led to its assignment as the primary interface region. The high resolution oxidative footprinting data further localized the likely interface points to more extended tryptic peptide regions in the light chain (LC 1-42 and LC 62-103), and a short tryptic peptide region in the heavy chain (HC 77-87), and allowed us to model two possible orientations: a head-to head, a single-arm bound Fab-to-Fab dimer, and a head-to head, double arm-bound Fabʹ2-to-Fabʹ2 dimer (). Our lower resolution fragment-SEC analysis using papain and FabRICATOR® enzymes provided complimentary evidence to support the Fab/Fab interface of our IgG1 dimer. These orientations, in which some CDRs would be at least partially blocked, could also help to explain the dimer’s reduced biological activity.
Previous structural studies on mAbs and other proteins have also implicated the Fab domain as a likely dimeric interface region. Moore et al. postulated the involvement of the Fab domain in the interface of a therapeutic IgG1 dissociable dimer based on competitive binding studies between the dimer and monomer.Citation54 Kanai and colleagues used viscosity measurements to determine that self-association of a therapeutic IgG1 mAb was due to Fab/Fab interactions.Citation55 The previously cited studies from Amgen suggested Fab domain participation in their mAb dimers.Citation36,Citation37 Wang et al. used computational methods to predict aggregation prone regions in commercial therapeutic mAbs.Citation56 According to their predictive tools, aggregation prone motifs in the variable domains are primarily found in or adjacent to the CDRs. As our oxidative footprinting data showed, a tryptic peptide encompassing one CDR in the light chain showed relatively increased protection, and was implicated as part of the interface region of our IgG1 dimer ( and ). Light chains, specifically, have been cited in other protein studies as being involved in aggregation interface regions. A study on myeloma-derived light chains (Bence-Jones proteins) revealed their propensity for aggregation or dimerization,Citation57 while a molecular modeling study proposed dimerization through light chains as the first step in amyloid fibril self-association.Citation58
More detailed structural elucidation of therapeutic mAb dimers has been attempted using other techniques, including H/D exchange and Transmission Electron Microscopy (TEM). Zhang et al. used H/D exchange to probe the structure of therapeutic IgG1 dimers induced by freeze-thaw and thermal stress conditions.Citation59 Their freeze-thaw dimer was composed of “native-like” monomers, as determined by Trp fluorescence and ANS binding data. The H/D data for this dimer showed only subtle changes in protection or solvent accessibility in the heavy chains, and no distinct regions could be identified as the interface region of the dimer. The H/D data for their thermally-induced, non-native dimer did reveal three short pepsin peptides in the CDR (one light chain and two heavy chain peptides) that showed increased protection, thus implicating the involvement of the Fab region in the dimer interface. Although the H/D data pointed to some Fab/Fab interaction for a forced stress-induced dimer, there was still probably not enough structural information to allow for modeling of its dimeric orientation.
More recently, Paul et al. used Transmission Electron Microscopy (TEM) to visualize the structures of stress-induced dimers.Citation60 Their “process stress” dimer, which is analogous to the native dimer in our study, appeared to have a “bone-like” orientation. This “bone-like” dimer was depicted with a single arm Fab/Fab interface region, and approximates the possible head-to-head, single-arm bound Fab-to-Fab model we hypothesized from our oxidative footprinting data. In addition, their TEM data for a pH stress-induced dimer appeared to show a head-to-head Fabʹ2-to-Fabʹ2 orientation similar to the other possible, footprinting-derived structure we proposed for our dimer. TEM, however, does not provide detailed structural information at the peptide or residue level, and it is thus not possible to pinpoint more specific interaction sites or areas of local conformational changes (in this sense, TEM can perhaps be considered a medium resolution technique). The correlation between our proposed model dimer structures and the structures observed by Paul et al. suggests that hydroxyl radical footprinting and TEM can be used together in a complimentary fashion for future structural studies.
Although we propose two possible structures for our IgG1 dimer, the specific orientation and interface regions of dimers from different therapeutic mAbs will likely vary. Data from Wang and co-workersCitation56 suggested that sites prone to aggregation can be sequence specific, while TEM data from Paul et al.,Citation60 and from Roux and Tankersely,Citation61 visually show that different dimeric orientations involving the Fab arms exist. Roux also showed that the inherent flexibility of an IgG molecule allows different non-covalent dimeric orientations to exist in equilibrium.Citation62 Other factors such as concentration, formulation, and the forces driving the non-covalent associations may also play a role in a dimer’s structure. Our IgG1 dimeric association appears to be driven by hydrophobic interactions, as demonstrated by the in-situ dimerization of its papain-generated Fabs persisting in the high salt conditions of the SEC analysis.
The hydroxyl radical footprinting data seems to provide a level of sensitivity and structural detail for a therapeutic mAb native dimer that has not previously been described. The oxidative footprinting data delineated specific areas of protection or solvent accessibility differences in the dimer compared to the monomer control. The peptide-level solvent accessibility differences we present here provided sufficient data to identify the likely dimeric interface regions in the Fab domain. The residue-level solvent accessibility differences in the dimer were also captured with this permanent covalent labeling (CL) technique, and can be more readily mined for further structural information in the future as software packages designed for these analyses continue to be developed. While the Fab domain of mAbs have been implicated in past studies as interaction sites for dimers, identification of the discrete regions (and residues) involved in those interactions are made possible with this higher resolution technique. In addition to identifying an interface region, other areas of conformational changes were detected in our dimer. Several heavy chain peptides displayed decreases in protection (faster rates of oxidation), and may thus indicate localized regions of partial unfolding in the dimer. This may be supportive of the idea that aggregates occurring under non-stress conditions are formed from partially unfolded molecules that are part of the native state population.Citation32 It is possible that a partial unfolding of the Fab domains in our IgG1 is part of the pathway that eventually leads to its dimerization through those regions.
We demonstrated here the utility of hydroxyl radical footprinting for probing the higher order structure of a therapeutic IgG1 mAb dimer. The high resolution information offered by this covalent labeling (CL) technique can provide orthogonal or complimentary data to those provided by other analytical methods, such as H/D exchange, TEM, or fragment-SEC analysis, and may help provide further insights into the structure and function of mAb aggregates. With its fast reaction times, apparent sensitivity, and high potential for structural resolution, hydroxyl radical footprinting holds promise beyond structural studies of dimers and aggregates, and future work will hopefully show its applicability to other aspects of therapeutic mAb higher order structure characterization.
Materials and Methods
Materials
A therapeutic IgG1 mAb produced in Chinese hamster ovary cells was chosen as a model mAb for the hydroxyl radical footprinting study. Approximately 3 g of the formulated drug substance of the mAb was obtained for dimer and monomer enrichment.
Enrichement of IgG1 mAb dimer and monomer samples via size exclusion chromatography (SEC)
For automated fraction collection, 100 μL of undiluted mAb drug substance at 25 mg/mL were applied per injection onto a Tosoh-Bioscience size exclusion chromatography (SEC) TSKgel G3000SWxl (7.8 × 300mm, 5 μm) column equilibrated with the mobile phase of 0.2 M K2HPO4, 0.25 M KCl, pH 6.2. The runtime was 30 minutes, the flow rate was at 0.5 mL/min, and the column was kept at ambient temperature. The elution profile was monitored at 280 nm. An Agilent 1100 HPLC system equipped with an automated fraction collector was used to collect the peaks corresponding to the dimer and monomer components into a temperature-controlled sample compartment. Fractionated dimer and monomer samples were pooled and concentrated with iCONTM Protein Concentrators (Pierce). Samples were also buffer-exchanged into its formulation buffer at the final concentration step. The final concentrations of the enriched dimer and monomer were ~10 mg/mL and 30 mg/mL, respectively. For confirmation of enriched dimer and monomer purities, 25 µL of each sample, diluted to 1 mg/mL with mobile phase buffer, was applied to the TSKgel G3000SWxl column for analytical-scale analysis. Sample aliquots were kept frozen until immediately needed for experimental use.
Size-exclusion chromatography with multi-angle laser light scattering (SEC-MALLS)
To measure the average molar mass of the various sized species, 100 μg per sample were injected by Agilent 1100 HPLC and separated by a Tosoh-Bioscience size exclusion chromatography (SEC) TSKgel G3000SWxl (7.8 × 300mm, 5 μm) column at 0.5mL/min, in series with a multi-angle laser light scattering instrument (MALLS) (Dawn HELEOS, Wyatt Technology). Protein concentration was determined by Agilent HPLC UV absorbance at 280 nm or by refractive index at 658 nm (Optilab rex, Wyatt Technology). The refractive index increment (dn/dc) is 0.185. Results were analyzed with Debye plot using Wyatt Astra software (version 5.3.4).
Hydroxy radical oxidation via synchrotron irradiation
Synchrotron irradiation was performed by NeoProteomics, Inc., using beamline X28C of the National Light Synchrotron Light Source (NSLS) of the Brookhaven National. Details of the beamline setup and standard irradiation procedures have been described in detail previously.Citation18,Citation27,Citation28,Citation63 Briefly, exposure conditions were predetermined by following the dose-dependent degradation of the fluorescent compound Alexa 488 in a dilution buffer of 20 mM sodium phosphate, pH 6.5. The mirror was set to 5.5 mrad and 0 mm focus for dose response analysis. IgG1 dimer and monomer (shipped frozen from Genentech) were thawed and diluted approximately 10× with 50 mM sodium phosphate, pH 6.2 immediately prior to irradiation. For sample radiolysis, the mirror was set to 5.5 mrad and 6 mm focus, and the beam current was approximately 200 mA. A steady state concentration of approximately 1 µM of hydroxyl radicals is generated by synchrotron irradiation. Two-hundred μL of sample was used for each exposure time interval, which varied from 0 ms to 17.5 ms. Irradiated samples were collected in Eppendorf tubes with methionine-NH2 quench further hydroxyl radical activity. Samples were then frozen and shipped back to Genentech.
Tryptic peptide mapping and RP-UHPLC-MS analysis
Synchrotron-irradiated dimer and monomer samples were received from Brookhaven National Laboratory, and thawed prior to digestion. Samples were initially buffer-exchanged into 100 mM Tris-HCl pH 8.0, 7.2 M GnCl (guanidine hydrochloride), 20 mM EDTA using iCON Protein Concentrators (Pierce). Subsequently, samples were incubated at 37°C for one hour after addition of DTT to a final concentration of 10 mM, then at ambient temperature in the dark for 30 minutes after addition of sodium iodoacetate to a final concentration of 25 mM, and finally for 15 minutes in the dark after addition of DTT to final concentration of 50 mM. S-carboxymethylated samples were loaded onto NAP-5 columns (GE Healthcare) and eluted with 0.5 mL of 50 mM Tris, pH 8.0, 20 mM CaCl2. Samples were digested at 37°C with 3% trypsin (w/w, Roche recombinant) for four hours. TFA was added to a final concentration of 0.2% to stop the digest.
Tryptic peptides were separated using an Agilent 1200 SL UHPLC with an Agilent ZORBAX Rapid Resolution HT SB-C8, 2.1 × 150 mm, 1.8 µm column equilibrated with solvent A (Water, 0.1% TFA). Initial conditions were held for 4 minutes, then the chromatogram was developed with a gradient of solvent B (ACN, 0.1% TFA) of 0.75% solvent B/min for 60 minutes. The column was washed with 95% solvent B for 5 minutes before initial conditions were restored. Flow rate was at 0.5 mL/min, and temperature was at 70°C. Mass spectrometric analysis of chromatographic peaks observed at 214 nm was performed with a Thermo Fisher Orbitrap XL operating in the positive ion mode. Data analysis was performed with ProtMap MS or Thermo Excalibur software.
IgG1 antibody homology modeling
Two copies of an IgG1 Fab (PDB ID = 1BJ1, Muller 1998) were aligned on to the full-length structure of Mus musculus IgG1 antibody (PDB ID=1IGY, Harris JMB 1998) using Pymol (DeLano, WL, www.pymol.org). The root mean square deviation was 2.1 Angstroms when aligning the Fab onto the IgG1 structure. The model was then orientated manually in Pymol to match the protected regions on the light chain of one antibody with another antibody model.
LC-MS operating in a two-dimensional format
The LC-MS method used for the study has been described in detail by Alvarez at al.Citation38 Various sized species separated by a Tosoh-Bioscience size exclusion chromatography (SEC) TSKgel G3000SWxl (7.8 × 300mm, 5 μm) column at 0.5 mL/min were characterized on-line by LC-MS using a two-dimensional approach. SEC separation was carried out on a Dionex Ultimate 3000, dual gradient pump HPLC equipped with two temperature-controlled column compartments equipped with a set of built-in divert valves. The system was configured to run two independent gradients, thus making it suitable for on-line two-dimensional chromatographic applications, and the second column compartment was equipped with a set of 7-port, 6-position valves, thus being able to accommodate multiple reverse-phase (RP) trap-cartridges. During the elution of a peak of interest, the SEC flow was diverted onto one of the available trap cartridges for peak trapping. The process was repeated until all peaks of interest were trapped as discrete fractions. Trapped fractions were then desalted for a period of 5 minutes with 95% solvent A containing 0.1% formic acid in water. Afterwards, fractions were sequentially step-eluted by ramping the RP gradient to 65% solvent B containing 0.1% formic acid in acetonitrile at a reduced flow rate of 100uL/min for efficient desolvation and diverting the flow to each trap for a period of 5 minutes per trap. Effluents were directed onto a QTOF-Premier mass spectrometer operating in a positive ionization mode by electrospray for detection. Deconvolution of multiply-charged species was performed using MaxEnt 1 program supplied with the Waters MassLynx software package. Fraction segments and trapping times were determined by an initial injection of the sample prior to on-line LC-MS analysis.
Papain and FabRICATOR® Digestion for Fragment-SEC analysis
Samples were incubated with papain at an enzyme to protein weight ratio of 1:100 in a buffer consisting of 100 mM Tris, 1 mM cysteine, 4 mM EDTA, pH 7.4 at 37°C for 2 hours. Antipain was added to a final concentration of 0.4 μM to stop the digest. For digest time course experiments, incubation times at 37°C were varied from 30 minutes to 4 hours. Samples were incubated with FabRICATOR® (Bulldog Bio) at an enzyme unit to protein ratio of 2.5:1 in a buffer consisting of 50 mM sodium phosphate, 150 mM NaCl, pH 6.6 at 37°C for 3 hours.
Fragment species generated by either papain or FabRICATOR® were separated using the TSKgel G3000SWxl column and size-exclusion chromatography method described above. Peaks separated by size-exclusion chromatography were analyzed by the described MALLS and LC-MS methods.
Potency assay
The Human Umbilical Vein Endothelial Cell (HUVEC) anti-proliferation bioassay, which has been described previously,Citation60 is based upon the ability of the IgG1 antibody to inhibit antigen-induced HUVEC proliferation. The assay was performed in 96-well tissue culture microtiter plates. In this assay, varying concentrations of the antibody samples were mixed with antigen (30 ng/mL final concentration) for 30–90 minutes at ambient temperature. Fifty μL of the antigen/antibody sample mixtures were added to the microtiter plates, followed by addition of 50 μL of HUVEC suspension (10,000 cells/well). The plates were incubated at 37°C, 5% CO2 in a humidified incubator for four days, after which 25 μL of the redox dye, AlamarBlue was added. The plates were incubated for six to seven hours at 37°C, 5% CO2 in a humidified incubator, and the relative number of viable cells were quantified indirectly by reading the fluorescence using a 96-well fluorometer with excitation at 530 nm and emission at 590 nm. AlamarBlue is blue and non-fluorescent in its oxidized state, but is reduced by the intracellular environment into a pink form, which is highly fluorescent. The changes in color and fluorescence were proportional to the number of viable cells. The results, expressed in relative fluorescence units (RFU), were plotted against antibody concentrations, and a parallel line bioassay (PLB) computer program was used to estimate the inhibitory activity of antibody samples relative to the unfractionated starting drug substance used a reference material. Inhibition of HUVEC proliferation is proportional to antibody concentrations.
Non-reduced CE-SDS
The method has been described in detail previously.Citation64 Briefly, samples were labeled with fluorogenic [3-(2-furoyl)quinoline-2-carboxaldeyde (FQ)] dye according to the following procedure. IgG1 dimer and monomer samples were exchanged into sodium phosphate reaction buffer using NAP-5 columns (GE Healthcare) to remove potentially competing formulation constituents. Aliquots of desalted samples were mixed with N-ethylmaleimide dissolved in 2% SDS and incubated for 5 min at 70°C to control disulfide reshuffling under denaturing conditions. FQ and potassium cyanide (KCN) reagents were added to the SDS-IgG1 solutions, and the final solutions were incubated for 10 min at 50°C before diluted with 1% SDS to quench the reaction.
Separation of IgG1 species was performed with 31.2 cm (20 cm effective length) bare fused-silica capillaries (Polymicro Technologies) encased in 40°C thermal-controlled cartridges. A Beckman PA800+ system (Beckman Coulter) was used with 32 Karat versions 9.0 to control the instrument and the LIF detector. Fluorescence was excited by a 3.5 mW, 488 nm laser and filtered with a 600 ± 20 nm bandpass filter and 488 nm notch filter. Voltage was applied in the negative mode. Sample solutions were introduced electrokinetically at 5 kV for 40 s and separated at 15 kV.
| Abbreviations: | ||
| AUC | = | analytical ultracentrifugation |
| CD | = | circular dichroism |
| CDR | = | complimentary determining region |
| CE-SDS | = | capillary electrophoresis sodium dodecyl sulfate |
| CL | = | covalent labeling |
| Da | = | Dalton |
| EIC | = | extracted ion chromatograms |
| FPOP | = | fast photochemical oxidation of proteins |
| FTIR | = | Fourier-transform infrared spectroscopy |
| HC | = | heavy chain |
| H/D | = | hydrogen deuterium |
| HUVEC | = | Human Umbilical Vein Endothelial Cells |
| LC | = | light chain |
| LC-MS | = | liquid chromatography-mass spectrometry |
| mAb | = | monoclonal antibody |
| MALLS | = | multi-angle laser light scattering |
| MS | = | mass spectrometry |
| MW | = | molecular weight |
| NMR | = | nuclear magnetic resonance |
| NSLS | = | National Synchrotron Light Source |
| RP-UHPLC | = | reverse-phase ultra-high pressure liquid chromatography |
| SEC | = | size-exclusion chromatography |
| TEM | = | transmission electron microscopy |
Additional material
Download Zip (961.7 KB)Acknowledgements
We would like to graciously acknowledge Dr. Mark Chance for all his consultations and advice on the hydroxyl radical footprinting technique and data analysis. This work would not be possible without his groundbreaking work and continued support. We would also like to thank the NeoProteomics team of John Schenkel, Parminder Kaur, and Sayan Gupta, and Rhijuta D’Mello for their assistance in the synchrotron experiments and software-related items. At Genentech, we would like to thank Charlie Eigenbrot, Adelle Lohse, Mary Nguyen, Lisa Vampola, Marian Eng, David A. Michels, Monica Parker, Linda Chan, Kathy Kosewic, Helene Gazzano-Santoro, and John Stults for their valuable assistance, input, and support.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Related Research Data
References
- Harris M. Monoclonal antibodies as therapeutic agents for cancer. Lancet Oncol 2004; 5:292 - 302; http://dx.doi.org/10.1016/S1470-2045(04)01467-6; PMID: 15120666
- Reichert JM. Monoclonal antibodies as innovative therapeutics. Curr Pharm Biotechnol 2008; 9:423 - 30; http://dx.doi.org/10.2174/138920108786786358; PMID: 19075682
- Chan AC, Carter PJ. Therapeutic antibodies for autoimmunity and inflammation. Nat Rev Immunol 2010; 10:301 - 16; http://dx.doi.org/10.1038/nri2761; PMID: 20414204
- Kaltashov IA, Bobst CE, Abzalimov RR, Wang G, Baykal B, Wang S. Advances and challenges in analytical characterization of biotechnology products: mass spectrometry-based approaches to study properties and behavior of protein therapeutics. Biotechnol Adv 2012; 30:210 - 22; http://dx.doi.org/10.1016/j.biotechadv.2011.05.006; PMID: 21619926
- Berkowitz SA, Engen JR, Mazzeo JR, Jones GB. Analytical tools for characterizing biopharmaceuticals and the implications for biosimilars. Nat Rev Drug Discov 2012; 11:527 - 40; http://dx.doi.org/10.1038/nrd3746; PMID: 22743980
- Pelton JT, McLean LR. Spectroscopic methods for analysis of protein secondary structure. Anal Biochem 2000; 277:167 - 76; http://dx.doi.org/10.1006/abio.1999.4320; PMID: 10625503
- Garidel P, Hegyi M, Bassarab S, Weichel M. A rapid, sensitive and economical assessment of monoclonal antibody conformational stability by intrinsic tryptophan fluorescence spectroscopy. Biotechnol J 2008; 3:1201 - 11; http://dx.doi.org/10.1002/biot.200800091; PMID: 18702089
- Bertucci C, Pistolozzi M, De Simone A. Structural characterization of recombinant therapeutic proteins by circular dichroism. Curr Pharm Biotechnol 2011; 12:1508 - 16; http://dx.doi.org/10.2174/138920111798357276; PMID: 21542799
- Jiang Y, Li C, Nguyen X, Muzammil S, Towers E, Gabrielson J, et al. Qualification of FTIR spectroscopic method for protein secondary structural analysis. J Pharm Sci 2011; 100:4631 - 41; http://dx.doi.org/10.1002/jps.22686; PMID: 21713773
- Houde D, Berkowitz SA, Engen JR. The utility of hydrogen/deuterium exchange mass spectrometry in biopharmaceutical comparability studies. J Pharm Sci 2011; 100:2071 - 86; http://dx.doi.org/10.1002/jps.22432; PMID: 21491437
- Houde D, Arndt J, Domeier W, Berkowitz S, Engen JR. Characterization of IgG1 conformation and conformational dynamics by hydrogen/deuterium exchange mass spectrometry. Anal Chem 2009; 81:2644 - 51; http://dx.doi.org/10.1021/ac802575y; PMID: 19265386
- Engen JR. Analysis of protein conformation and dynamics by hydrogen/deuterium exchange MS. Anal Chem 2009; 81:7870 - 5; http://dx.doi.org/10.1021/ac901154s; PMID: 19788312
- Hoofnagle AN, Resing KA, Ahn NG. Protein analysis by hydrogen exchange mass spectrometry. Annu Rev Biophys Biomol Struct 2003; 32:1 - 25; http://dx.doi.org/10.1146/annurev.biophys.32.110601.142417; PMID: 12598366
- Mendoza VL, Vachet RW. Probing protein structure by amino acid-specific covalent labeling and mass spectrometry. Mass Spectrom Rev 2009; 28:785 - 815; http://dx.doi.org/10.1002/mas.20203; PMID: 19016300
- Mendoza VL, Vachet RW. Protein surface mapping using diethylpyrocarbonate with mass spectrometric detection. Anal Chem 2008; 80:2895 - 904; http://dx.doi.org/10.1021/ac701999b; PMID: 18338903
- Gómez GE, Mundo MR, Craig PO, Delfino JM. Probing protein surface with a solvent mimetic carbene coupled to detection by mass spectrometry. J Am Soc Mass Spectrom 2012; 23:30 - 42; http://dx.doi.org/10.1007/s13361-011-0266-x; PMID: 22006407
- Zhou X, Lu Y, Wang W, Borhan B, Reid GE. ‘Fixed charge’ chemical derivatization and data dependant multistage tandem mass spectrometry for mapping protein surface residue accessibility. J Am Soc Mass Spectrom 2010; 21:1339 - 51; http://dx.doi.org/10.1016/j.jasms.2010.03.047; PMID: 20452239
- Maleknia SD, Ralston CY, Brenowitz MD, Downard KM, Chance MR. Determination of macromolecular folding and structure by synchrotron x-ray radiolysis techniques. Anal Biochem 2001; 289:103 - 15; http://dx.doi.org/10.1006/abio.2000.4910; PMID: 11161303
- Xu G, Chance MR. Radiolytic modification and reactivity of amino acid residues serving as structural probes for protein footprinting. Anal Chem 2005; 77:4549 - 55; http://dx.doi.org/10.1021/ac050299+; PMID: 16013872
- Xu G, Chance MR. Hydroxyl radical-mediated modification of proteins as probes for structural proteomics. Chem Rev 2007; 107:3514 - 43; http://dx.doi.org/10.1021/cr0682047; PMID: 17683160
- Guan JQ, Chance MR. Structural proteomics of macromolecular assemblies using oxidative footprinting and mass spectrometry. Trends Biochem Sci 2005; 30:583 - 92; http://dx.doi.org/10.1016/j.tibs.2005.08.007; PMID: 16126388
- Hambly DM, Gross ML. Laser flash photolysis of hydrogen peroxide to oxidize protein solvent-accessible residues on the microsecond timescale. J Am Soc Mass Spectrom 2005; 16:2057 - 63; http://dx.doi.org/10.1016/j.jasms.2005.09.008; PMID: 16263307
- Hambly D, Gross M. Laser flash photochemical oxidation to locate heme binding and conformational changes in myoglobin. Int J Mass Spectrom 2007; 259:124 - 9; http://dx.doi.org/10.1016/j.ijms.2006.08.018
- Gau BC, Sharp JS, Rempel DL, Gross ML. Fast photochemical oxidation of protein footprints faster than protein unfolding. Anal Chem 2009; 81:6563 - 71; http://dx.doi.org/10.1021/ac901054w; PMID: 20337372
- Watson C, Sharp JS. Conformational analysis of therapeutic proteins by hydroxyl radical protein footprinting. AAPS J 2012; 14:206 - 17; http://dx.doi.org/10.1208/s12248-012-9336-7; PMID: 22382679
- Chance MR, Sclavi B, Woodson SA, Brenowitz M. Examining the conformational dynamics of macromolecules with time-resolved synchrotron X-ray ‘footprinting’. Structure 1997; 5:865 - 9; http://dx.doi.org/10.1016/S0969-2126(97)00241-4; PMID: 9261085
- Chance MR. Unfolding of apomyoglobin examined by synchrotron footprinting. Biochem Biophys Res Commun 2001; 287:614 - 21; http://dx.doi.org/10.1006/bbrc.2001.5628; PMID: 11563839
- Kiselar JG, Maleknia SD, Sullivan M, Downard KM, Chance MR. Hydroxyl radical probe of protein surfaces using synchrotron X-ray radiolysis and mass spectrometry. Int J Radiat Biol 2002; 78:101 - 14; http://dx.doi.org/10.1080/09553000110094805; PMID: 11779360
- Takamoto K, Chance MR. Radiolytic protein footprinting with mass spectrometry to probe the structure of macromolecular complexes. Annu Rev Biophys Biomol Struct 2006; 35:251 - 76; http://dx.doi.org/10.1146/annurev.biophys.35.040405.102050; PMID: 16689636
- Maleknia SD, Wong JW, Downard KM. Photochemical and electrophysical production of radicals on millisecond timescales to probe the structure, dynamics and interactions of proteins. Photochem Photobiol Sci 2004; 3:741 - 8; http://dx.doi.org/10.1039/b315904c; PMID: 15295629
- Cromwell ME, Hilario E, Jacobson F. Protein aggregation and bioprocessing. AAPS J 2006; 8:E572 - 9; http://dx.doi.org/10.1208/aapsj080366; PMID: 17025275
- Mahler HC, Friess W, Grauschopf U, Kiese S. Protein aggregation: pathways, induction factors and analysis. J Pharm Sci 2009; 98:2909 - 34; http://dx.doi.org/10.1002/jps.21566; PMID: 18823031
- Hawe A, Kasper JC, Friess W, Jiskoot W. Structural properties of monoclonal antibody aggregates induced by freeze-thawing and thermal stress. Eur J Pharm Sci 2009; 38:79 - 87; http://dx.doi.org/10.1016/j.ejps.2009.06.001; PMID: 19540340
- Hermeling S, Crommelin DJ, Schellekens H, Jiskoot W. Structure-immunogenicity relationships of therapeutic proteins. Pharm Res 2004; 21:897 - 903; http://dx.doi.org/10.1023/B:PHAM.0000029275.41323.a6; PMID: 15212151
- den Engelsman J, Garidel P, Smulders R, Koll H, Smith B, Bassarab S, et al. Strategies for the assessment of protein aggregates in pharmaceutical biotech product development. Pharm Res 2011; 28:920 - 33; http://dx.doi.org/10.1007/s11095-010-0297-1; PMID: 20972611
- Remmele RL Jr., Callahan WJ, Krishnan S, Zhou L, Bondarenko PV, Nichols AC, et al. Active dimer of Epratuzumab provides insight into the complex nature of an antibody aggregate. J Pharm Sci 2006; 95:126 - 45; http://dx.doi.org/10.1002/jps.20515; PMID: 16315222
- Lau H, Pace D, Yan B, McGrath T, Smallwood S, Pater K, et al. Investigation of degradation processes in IgG1 monoclonal antibodies by limited proteolysis couples with weak cation-exchange HPLC. J Chrom B 2010; 878:868 - 76; http://dx.doi.org/10.1016/j.jchromb.2010.02.003
- Alvarez M, Tremintin G, Wang J, Eng M, Kao YH, Jeong J, et al. On-line characterization of monoclonal antibody variants by liquid chromatography-mass spectrometry operating in a two-dimensional format. Anal Biochem 2011; 419:17 - 25; http://dx.doi.org/10.1016/j.ab.2011.07.033; PMID: 21867674
- von Pawel-Rammingen U, Johansson BP, Björck L. IdeS, a novel streptococcal cysteine proteinase with unique specificity for immunoglobulin G. EMBO J 2002; 21:1607 - 15; http://dx.doi.org/10.1093/emboj/21.7.1607; PMID: 11927545
- Gsponer J, Vendruscolo M. Theoretical approaches to protein aggregation. Protein Pept Lett 2006; 13:287 - 93; http://dx.doi.org/10.2174/092986606775338407; PMID: 16515457
- Gupta S, Sullivan M, Toomey J, Kiselar J, Chance MR. J. The Beamline X28C of the Center for Synchrotron Biosciences: a national resource for biomolecular structure and dynamics experiments using synchrotron footprinting. Synchrotron Radiat. 2007; 14:233 - 43; http://dx.doi.org/10.1107/S0909049507013118
- Sullivan MR, Rekhi S, Bohon J, Gupta S, Abel D, Toomey J, et al. Installation and testing of a focusing mirror at beamline X28C for high flux x-ray radiolysis of biological macromolecules. Rev Sci Instrum 2008; 79:025101; http://dx.doi.org/10.1063/1.2839027; PMID: 18315323
- Xu G, Kiselar J, He Q, Chance MR. Secondary reactions and strategies to improve quantitative protein footprinting. Anal Chem 2005; 77:3029 - 37; http://dx.doi.org/10.1021/ac048282z; PMID: 15889890
- Kaur P, Kiselar JG, Chance MR. Integrated algorithms for high-throughput examination of covalently labeled biomolecules by structural mass spectrometry. Anal Chem 2009; 81:8141 - 9; http://dx.doi.org/10.1021/ac9013644; PMID: 19788317
- Tong X, Wren JC, Konermann L. Effects of protein concentration on the extent of gamma-ray-mediated oxidative labeling studied by electrospray mass spectrometry. Anal Chem 2007; 79:6376 - 82; http://dx.doi.org/10.1021/ac070724u; PMID: 17628115
- Bohon J, Jennings LD, Phillips CM, Licht S, Chance MR. Synchrotron protein footprinting supports substrate translocation by ClpA via ATP-induced movements of the D2 loop. Structure 2008; 16:1157 - 65; http://dx.doi.org/10.1016/j.str.2008.04.016; PMID: 18682217
- Rao Y, Lee Y, Jarjoura D, Ruppert AS, Liu CG, Hsu JC, et al. A comparison of normalization techniques for microRNA microarray data. Stat Appl Genet Mol Biol 2008; 7:e22; http://dx.doi.org/10.2202/1544-6115.1287; PMID: 18673291
- van den Berg RA, Hoefsloot HC, Westerhuis JA, Smilde AK, van der Werf MJ. Centering, scaling, and transformations: improving the biological information content of metabolomics data. BMC Genomics 2006; 7:142; http://dx.doi.org/10.1186/1471-2164-7-142; PMID: 16762068
- Kiselar JG, Chance MR. Future directions of structural mass spectrometry using hydroxyl radical footprinting. J Mass Spectrom 2010; 45:1373 - 82; http://dx.doi.org/10.1002/jms.1808; PMID: 20812376
- Kiselar JG, Datt M, Chance MR, Weiss MA. Structural analysis of proinsulin hexamer assembly by hydroxyl radical footprinting and computational modeling. J Biol Chem 2011; 286:43710 - 6; http://dx.doi.org/10.1074/jbc.M111.297853; PMID: 22033917
- Bertolotti-Ciarlet A, Wang W, Lownes R, Pristatsky P, Fang Y, McKelvey T, et al. Impact of methionine oxidation on the binding of human IgG1 to Fc Rn and Fc γ receptors. Mol Immunol 2009; 46:1878 - 82; http://dx.doi.org/10.1016/j.molimm.2009.02.002; PMID: 19269032
- Liu D, Ren D, Huang H, Dankberg J, Rosenfeld R, Cocco MJ, et al. Structure and stability changes of human IgG1 Fc as a consequence of methionine oxidation. Biochemistry 2008; 47:5088 - 100; http://dx.doi.org/10.1021/bi702238b; PMID: 18407665
- Luo Q, Joubert MK, Stevenson R, Ketchem RR, Narhi LO, Wypych J. Chemical modifications in therapeutic protein aggregates generated under different stress conditions. J Biol Chem 2011; 286:25134 - 44; http://dx.doi.org/10.1074/jbc.M110.160440; PMID: 21518762
- Moore JM, Patapoff TW, Cromwell ME. Kinetics and thermodynamics of dimer formation and dissociation for a recombinant humanized monoclonal antibody to vascular endothelial growth factor. Biochemistry 1999; 38:13960 - 7; http://dx.doi.org/10.1021/bi9905516; PMID: 10529242
- Kanai S, Liu J, Patapoff TW, Shire SJ. Reversible self-association of a concentrated monoclonal antibody solution mediated by Fab-Fab interaction that impacts solution viscosity. J Pharm Sci 2008; 97:4219 - 27; http://dx.doi.org/10.1002/jps.21322; PMID: 18240303
- Wang X, Das TK, Singh SK, Kumar S. Potential aggregation prone regions in biotherapeutics: A survey of commercial monoclonal antibodies. MAbs 2009; 1:254 - 67; http://dx.doi.org/10.4161/mabs.1.3.8035; PMID: 20065649
- Stevens F, Myatt E. Polymerization of immunoglobulin domains: a model system for the development of facilitated macromolecular assembly. Nanotechnology 1991; 2:206 - 13; http://dx.doi.org/10.1088/0957-4484/2/4/007
- Stevens FJ, Myatt EA, Chang CH, Westholm FA, Eulitz M, Weiss DT, et al. A molecular model for self-assembly of amyloid fibrils: immunoglobulin light chains. Biochemistry 1995; 34:10697 - 702; http://dx.doi.org/10.1021/bi00034a001; PMID: 7662653
- Zhang A, Singh SK, Shirts MR, Kumar S, Fernandez EJ. Distinct aggregation mechanisms of monoclonal antibody under thermal and freeze-thaw stresses revealed by hydrogen exchange. Pharm Res 2012; 29:236 - 50; http://dx.doi.org/10.1007/s11095-011-0538-y; PMID: 21805212
- Paul R, Graff-Meyer A, Stahlberg H, Lauer ME, Rufer AC, Beck H, et al. Structure and function of purified monoclonal antibody dimers induced by different stress conditions. Pharm Res 2012; 29:2047 - 59; http://dx.doi.org/10.1007/s11095-012-0732-6; PMID: 22477068
- Roux KH, Tankersley DL. A view of the human idiotypic repertoire. Electron microscopic and immunologic analyses of spontaneous idiotype-anti-idiotype dimers in pooled human IgG. J Immunol 1990; 144:1387 - 95; PMID: 2303712
- Roux K. Immunoglobulin structure and function as revealed by electron microscopy. In Arch Allergy Immunol 1999; 129:85-99.
- Adilakshmi T, Soper SF, Woodson SA. Structural analysis of RNA in living cells by in vivo synchrotron X-ray footprinting. Methods Enzymol 2009; 468:239 - 58; http://dx.doi.org/10.1016/S0076-6879(09)68012-5; PMID: 20946773
- Michels DA, Parker M, Salas-Solano O. Quantitative impurity analysis of monoclonal antibody size heterogeneity by CE-LIF: example of development and validation through a quality-by-design framework. Electrophoresis 2012; 33:815 - 26; http://dx.doi.org/10.1002/elps.201100528; PMID: 22430180