Abstract
The European Medicines Agency received recently the first marketing authorization application for a biosimilar monoclonal antibody (mAb) and adopted the final guidelines on biosimilar mAbs and Fc-fusion proteins. The agency requires high similarity between biosimilar and reference products for approval. Specifically, the amino acid sequences must be identical. The glycosylation pattern of the antibody is also often considered to be a very important quality attribute due to its strong effect on quality, safety, immunogenicity, pharmacokinetics and potency. Here, we describe a case study of cetuximab, which has been marketed since 2004. Biosimilar versions of the product are now in the pipelines of numerous therapeutic antibody biosimilar developers. We applied a combination of intact, middle-down, middle-up and bottom-up electrospray ionization and matrix assisted laser desorption ionization mass spectrometry techniques to characterize the amino acid sequence and major post-translational modifications of the marketed cetuximab product, with special emphasis on glycosylation. Our results revealed a sequence error in the reported sequence of the light chain in databases and in publications, thus highlighting the potency of mass spectrometry to establish correct antibody sequences. We were also able to achieve a comprehensive identification of cetuximab’s glycoforms and glycosylation profile assessment on both Fab and Fc domains. Taken together, the reported approaches and data form a solid framework for the comparability of antibodies and their biosimilar candidates that could be further applied to routine structural assessments of these and other antibody-based products.
Introduction
With more than 40 products currently approved and ~30 molecules investigated in advanced clinical trials, monoclonal antibodies (mAbs) and derivatives constitute the most important and the fastest growing class of human therapeutics.Citation1 These large proteins can be used for a variety of indications such as inflammatory diseases and cancer. Most of the first generation approved molecules such as rituximab, trastuzumab, infliximab, cetuximab, adalimumab and bevacizumab will be off patent soon.Citation2 This will open the way for the approval of biosimilar mAbs in the European Union (EU) and in the United States (US). Biosimilar antibodies are “generic” versions of marketed antibodies produced through different manufacturing processes and from different clones.Citation3 The marketed antibodies are referred to as originator or reference products when compared with the biosimilar version. Due to the inherent variability of bioproduction and the large number of parameters that influence it, it is difficult, or rather impossible, to produce exact copies of large biomolecules such as antibodies because of their inherent structural complexity. This is in sharp contrast with the relative ease of manufacture of low-cost generic versions of small pharmaceutical molecules. As a consequence, different variants such as glycosylation variants and other microvariations, like charge variants, may occur in biosimilar mAbs. These could affect the final quality, safety and potency,Citation4 and that is why the term biogeneric should be avoided.Citation2 Nevertheless, it is now possible to produce proteins and glycoproteins that are highly similar to reference products owing to the tremendous progress that has been achieved in the last few years. This implies the need for a new regulatory framework for the approval of biosimilar products based on comparability with the reference molecule. The European Medicines Agency (EMA) was the first to initiate the regulatory pathways for biosimilar products in 2005, which has to date led to marketing authorization in Europe for 14 recombinant drugs encompassing 3 product classes (human growth hormone, granulocyte colony-stimulating factor and erythropoietins).Citation5 Specific guidelines are also available in Europe for biosimilar insulin, interferon and low molecular weight heparins.Citation6 Outside Europe, biosimilar antibodies have already been approved in India, South Korea and China. These biosimilar products are copies of current important therapeutic antibodies such as rituximab and abciximab.Citation7 Additional biosimilar candidates currently in development include copies of infliximab (Remicade®; Janssen Biotech, Merck), trastuzumab (Herceptin®; Genentech/Roche), cetuximab (Erbitux®; ImClone/Lilly, Merck-Serono), bevacizumab (Avastin®; Genentech/Roche) and etanercept, an Fc-fusion protein (Enbrel®; Amgen, Pfizer, Takeda).
At the end of 2012 the EMA released the final version of the guidelines on similar medicinal products containing mAbs.Citation8 These guidelines discuss relevant animal model, non-clinical and clinical studies that are recommended to establish the similarity and the safety of a biosimilar compared with a reference mAb approved in the EU. For the approval of biosimilar products, the EMA requires high similarity to the reference product in terms of physico-chemical characteristics, functional properties and clinical efficiency. The primary amino acid sequence should be the same for the biosimilar and the reference product. If appropriately justified with regard to its potential effects on safety or pharmacokinetic (PK) and pharmacodynamic (PD) properties, small differences in the micro-heterogeneity pattern of the molecule may be acceptable. First-generation approved originator mAbs sequences, however, are not explicitly published in patents and other official documents. Some patents only contain the complementary determining region and the variable domain sequences. The International Immunogenetics Information System (IMGT) is one of the main sources of structural and biological information on immunoglobulins (including monoclonal antibodies), T cell receptors, major histocompatibility of human and other vertebrate species and other. The IMGT provides a common access to sequence, genome and structure immunogenetics data. These data are based on the scientific information available in other databases or published in the scientific literature and patents and, as such, are an extremely useful and helpful resource in the development of biosimilar products. However, sequence errors do exist in the scientific data and databases. Sequence errors have been reported for trastuzumab,Citation9 rituximabCitation10 and etanercept.Citation11 Another major quality attribute of mAbs is their N-glycosylation profile,Citation12 which has important effects on effector functions such as antibody-dependent cell-mediated cytotoxicity (ADCC) and complement dependent cytotoxicity (CDC).Citation13,Citation14 The N-glycosylations of mAbs can also affect safety and PK/PD. It is evident that extensive structural and functional comparison of the biosimilar and the reference product is the foundation of biosimilar development and that assessment of the amino acid sequences and N-glycosylation patterns are among the most important criteria. Mass spectrometry (MS) is a key technique that plays a primary role in the assessment and the structural comparison of biosimilar and reference product. Throughout all stages of mAbs development and production, mass spectrometry based methodologies are used to provide essential information on the primary structures including amino-acid sequences, glycosylation and other post-translational modifications.Citation15 Here we chose cetuximab for a case study because it is one of the therapeutic mAbs that will be off-patent soon.
Cetuximab is a chimeric mouse-human IgG1 targeting epidermal growth factor receptor (EGFR). It is approved for use in the EU and US as a treatment for colorectal cancer and squamous cell carcinoma of the head and neck. The amino acid sequence for both the light and heavy chains of cetuximab are reported in the IMGT database (www.imgt.org) and the drug bank (www.drugbank.ca). The crystal structure of the antigen binding fragment (Fab) has been reported by Li et al.Citation16 and is referenced in the RCSB Protein Data Bank. Cetuximab’s primary sequence has also been reported by Dubois et al. who used liquid chromatography (LC)-MS/MS and matrix assisted laser desorption ionization-time of flight-MS (MALDI-TOF-MS) for PK studies.Citation17
Cetuximab is produced by SP2/0 murine myeloma cells and is N-glycosylated both in the Fc and the Fab domains. A high prevalence of hypersensitivity reactions to cetuximab has been reported in some areas of the US. Some of the glycoforms were demonstrated to be responsible for these hypersensitivity reactions and anaphylaxis.Citation18 Here, we used the latest generation of high resolution electrospray ionization (ESI) and MALDI mass spectrometry instruments and methods for the detailed and rapid structural characterization of the EMA-approved version and formulation of cetuximab. Intact molecular weight (MW) measurements, middle-up and middle-down, i.e., MW determination and direct mass spectrometric sequencing on the domain level, as well as bottom-up techniques were used to assess the glycosylation variants and the amino acid sequence. Detailed sequence information of the antibodies subunits were then determined using MALDI N- and C-terminal top-down sequencing (TDS) analysis.Citation19,Citation20 LC-MS/MS peptide mapping experiments on tryptic and GluC digests enabled post-translational modifications and sequence variants to be further localized. Using a proprietary search engine to query carbohydrate structure databases, glycopeptide and glycan identifications and profiles were automatically generated. From the LC-ESI-MS mass spectra of cetuximab subunits (middle-up) we derived glycosylation site-specific accurate masses of the various antibody glycoforms. Our results confirmed an unexpected modification revealing a sequence error in the light chain terminal region. The methodologies we describe here form a solid framework for routine biosimilar structure verification.
Results and Discussion
Mass measurement of intact cetuximab
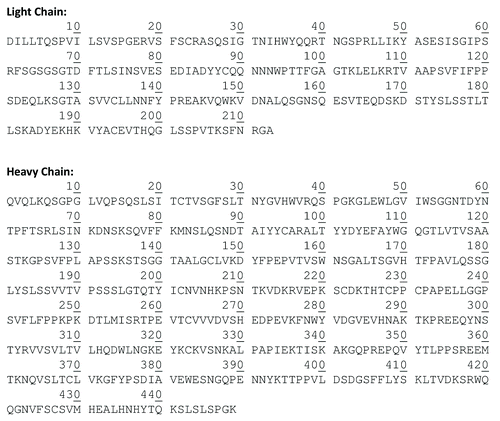
The intact LC-ESI-TOF-MS mass measurement of cetuximab indicates a strong heterogeneity of the antibody (). This heterogeneity is not chromatographically resolved by the simple LC method that was applied. The observed mass peaks in the deconvoluted spectrum result from an overlap of different isoforms due to post translational modifications (PTMs). The most important PTMs of cetuximab are the complex glycosylations resulting from four glycosylation sites and incomplete lysine clipping of the two heavy chain C-termini.Citation21-Citation23 To interpret the intact measurements, the observed heterogeneity needs to be reduced and the theoretical MWs of the expected isoforms calculated. The sequences of the light and heavy chain of cetuximab reported in the IMGT database and the previously cited referencesCitation16,Citation17 are shown in .
Figure 1. Deconvoluted ESI-Q-TOF mass spectrum of intact cetuximab. The calculated MW for the G0F/G0F; G2FGal2/G2FGal2 isoform with pyroglutamic acid formation at the N-termini of the heavy chains is 152235 Da, which is 116 Da lower than the experimental MW at 152351 Da.
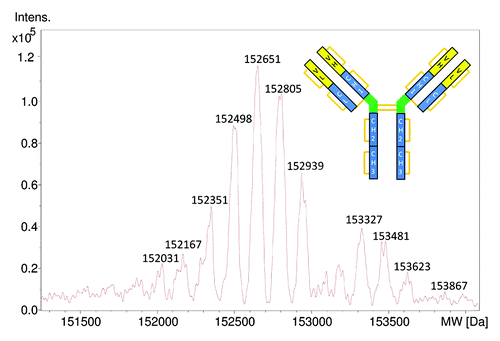
Figure 2. Sequences of the light and heavy chains of cetuximab as reported in the IMGT and in the literature.Citation16,Citation17 The N-glycosylation sites are indicated in bold character.
Intact IgG1 contain 32 cysteines and thus 16 disulfide bonds (-32 Da when calculating the intact antibody mass). It is noticeable by looking at the sequence of the light chain of cetuximab that it lacks the C-terminal cysteine found in IgG1. The thiol group of this particular cysteine usually links the light chain to the heavy chain by a disulfide bond. To calculate the theoretical MW, this missing cysteine was added to the C-terminal sequence and an average MW of 23368.7 Da for the light chain and 49371.7 Da for the heavy chain is obtained. N-terminal glutamines are usually converted to pyroglutamic acid, which results in a mass decrease of 17 Da per heavy chain. As already mentioned another major modification occurring in mAbs sequences is the clipping of the C-terminal lysine of heavy chains resulting in a mass decrease of 128 Da per heavy chain. Taking into consideration these modifications, the theoretical average MW of the aglycosylated form of intact cetuximab would be 145 158 Da. It should be noted that calculated average MWs depend on the source of the average atoms’ MWs used. In fact, the isotopic abundance of elements depends on their source. When calculating the theoretical masses of natural proteins, the atomic weights from organic sources are preferred.Citation24 We have observed that different MW calculators, open source and proprietary, would calculate different average MWs for the same sequence depending on the source of the atomic weights they use. These added to rounding errors can induce up to 1 Da calculated MW difference for an intact antibody, the equivalent of a 7 ppm difference.
Cetuximab bears two glycosylation sites on each heavy chain, one in the conserved site in the CH2 domain and one in the Fd domain. Glycoforms of the CH2 domain are mainly of the complex glycan type, with the most commonly occurring forms being G0, G0F, G1F and G2F with 1299.2, 1445.3, 1607.5 and 1769.6 Da mass increments, respectively. The Fd domain glycans can be more complex with tri-antennary and tetra-antennary glycans and differing degrees of sialylation.
If we consider the G0F/G0F; G2FGal2/G2FGal2 isoform, the calculated theoretical MW would be 152 236 Da. This MW is not consistent with any of the measured MW of the intact cetuximab and is -115 Da off the closest isoform 152 351 Da in the deconvoluted mass spectrum (). The possible reasons for this inconsistency were explored in the subsequent analyses.
Middle-up analysis
To further investigate this MW difference, we performed middle-up analysis of cetuximab by IdeS proteolysis followed by complete reduction of all disulfide bonds. Middle-up refers to the mass measurements of large fragments (subunits) of proteins after limited proteolysis.Citation24 IdeS or Immunoglobulin degrading enzyme of Streptococcus pyogenes has the advantage of being more specific than the other described IgG hinge cleaving enzymes.Citation25 IdeS cleavage followed by the reduction of disulfide bonds results in three subunits (light chain, Fc/2 domain and Fd domain) each with a MW of approximately 25 kDa. It permits easier and more straightforward analysis by LC-MS in a short time (less than 2 h for the whole analysis including digestion and LC-MS). The LC-MS chromatogram obtained shows three main peaks (). The first peak corresponds to the Fc/2 fragment with different isoforms. The theoretical MW of the G0F isoform of Fc/2 with clipped C-terminal lysine and reduced cysteines is 25 236.04 Da (average) and 25 220.463 Da (monoisotopic). The utilized ultra-high resolution Q-TOF provides isotopic resolution of the analyzed fragments and enables the determination of their monoisotopic masses. The monoisotopic mass is an intrinsic property of the molecule that is not dependent on the isotopic abundance of elements that make up the molecule, as is the case for the average mass.Citation24 The isotopic abundance of the elements in a biologic depends on the feeding source for the producing cell line;Citation26 therefore, if accessible, the monoisotopic mass is a better reference in accurate mass measurements.
Figure 3. Total ion chromatogram of IdeS cleaved and reduced cetuximab separating the three major subunits. For each of the Fc/2 and Fd subunits, two peaks were observed. Fc/2 shows lysine clipping at the heavy chain’s C-terminus, Fd exhibits glycosylation heterogeneity (presence of sialic acid).
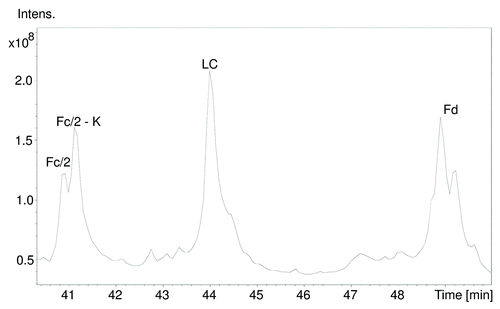
As shown in , the measured monoisotopic mass of the Fc/2-K G0F glycoform was determined to be 25220.462 Da, which corresponds to a MW error of -0.06 ppm. The measured MWs are consistent with different glycoforms with the cleaved/non-cleaved lysine heterogeneity mostly with sub ppm MW errors ().
Figure 4. Deconvoluted spectra (red) of the main isoforms of the subunits after IdeS digestion and reduction, (A) Fc/2, (B) LC and (C) Fd. The monoisotopic MWs were calculated from the baseline resolved isotopic peak patterns using the SNAP peak picking algorithm. The isotopic patterns calculated from the sequence of the antibody as specified in the IMGT database were also displayed for the three subunits (blue). The position of the average mass is marked with an arrow. For Fc/2, the isotopic pattern was calculated taking into account lysine clipping at the C-terminus and G0F glycosylation. For the Fd subunit N-terminal pyroglutamic acid formation and G2FGal2 glycosylation was assumed. While the agreement between the measured and expected monoisotopic mass is well below 1 ppm for the Fc/2 and Fd fragments [-1.5 mDa for Fc/2 (A) and 0.4 mDa for Fd (C)], a significant MW difference of 58.006 Da was observed for the LC (B) clearly indicating a sequence or structural variation.
![Figure 4. Deconvoluted spectra (red) of the main isoforms of the subunits after IdeS digestion and reduction, (A) Fc/2, (B) LC and (C) Fd. The monoisotopic MWs were calculated from the baseline resolved isotopic peak patterns using the SNAP peak picking algorithm. The isotopic patterns calculated from the sequence of the antibody as specified in the IMGT database were also displayed for the three subunits (blue). The position of the average mass is marked with an arrow. For Fc/2, the isotopic pattern was calculated taking into account lysine clipping at the C-terminus and G0F glycosylation. For the Fd subunit N-terminal pyroglutamic acid formation and G2FGal2 glycosylation was assumed. While the agreement between the measured and expected monoisotopic mass is well below 1 ppm for the Fc/2 and Fd fragments [-1.5 mDa for Fc/2 (A) and 0.4 mDa for Fd (C)], a significant MW difference of 58.006 Da was observed for the LC (B) clearly indicating a sequence or structural variation.](/cms/asset/c5c86971-c59e-49cd-9972-16d5dfacb1a2/kmab_a_10925423_f0004.gif)
Table 1. Theoretical and measured monoisotopical masses of the Fc/2 subunit identified glycoforms. ¤ Structure confirmed by GlycoQuest, * manually corrected due to overlapping
The second chromatographic peak corresponds to the light chain (). The light chain MW according to the IMGT sequence after adding the C-terminal cysteine is 23 368.69 Da (average) and 23 354.512 Da (monoisotopic). The measured monoisotopic mass of the light chain is 23 412.518 Da, which corresponds to a +58.006 Da mass difference (). Thus, we can speculate that there is an unidentified modification/variation in the light chain sequence.
The third chromatographic peak corresponds to the Fd fragment. The monoisotopic MW derived from the IMGT sequence of the most abundant Fd glycoform, G2FGal2, is 27530.3150 Da and corresponds to the measured MW of 27 530.3154 Da () with a 0.02 ppm mass error. The measured molecular weights are consistent with different glycoforms typically at the 1 ppm MW error level ().
Table 2. Theoretical and measured monoisotopical masses of the Fd subunit identified glycoforms. ¤ Structure confirmed by GlycoQuest, * manually corrected due to overlapping
Middle-down MALDI-in source decay (ISD) analysis
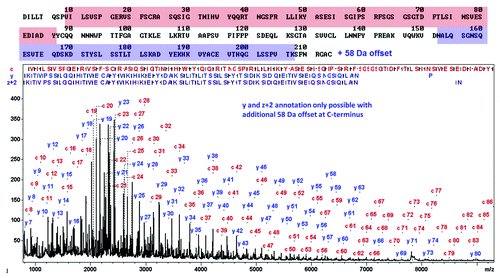
The middle-up results described above revealed that the light chain presents an unexpected modification. Middle-down MS, by analogy to top-down MS refers to the MS/MS sequencing of limited proteolysis generated subunits of a protein.Citation15,Citation24 Misuse of the top-down and middle-down terms is common in the literature when referring to the molecular weight measurement of intact IgG or limited proteolysis generated subunits without MS/MS. MALDI-ISD allows fast, straight-forward top-down sequence analysis of undigested proteins based on fragmentation of the entire protein chain caused by hydrogen radical transfer from the MALDI-ISD matrix.Citation27,Citation28 The data quality is high (mass accuracy approx. Ten ppm in reflector mode and typical sequence readout length is 60–90 residues from N- and C-terminus) and even permits de novo sequencing of medium-sized proteins.Citation29 Middle-down MALDI-ISD enables fast sequencing of terminal domains of mAbs in a targeted way. The cetuximab fragments after IdeS digest and chromatographic separation were collected for further MALDI-ISD analysis. The results confirm the N- and C-terminal sequences of the heavy chain, including the heterogeneities discussed above (N-terminal pyroglutamic acid presence/absence of the C-terminal lysine) (Fig. S1). The ISD sequencing of the light chain confirmed the first 86 N-terminal residues of the IMGT database sequence. The C-terminal y- and z+2 fragments match the light chain sequence only under the condition of a +58 Da modification C-terminal to residue 207. This offset is present in the smallest C-terminal fragment y7 which indicates that the modification is present within the 6 C-terminal residues. Taking this offset into account provides for a good match of the C-terminal domain of the light chain from residues 162‒207 (). The IMGT sequence of the 6 C-terminal residues is FNRGAC. Considering that no post-translational modifications are described for these residues in mAbs, we can speculate that it might be a substitution of one or more amino-acids (Fig. S2). In fact, the substitution of A by an E or of G by a D in that sequence would cause a +58 Da mass shift. To confirm this putative sequence modification, we performed a bottom-up analysis of cetuximab using trypsin and endoprotease GluC digestion.
Figure 5. LC-MALDI-ISD middle-down spectrum of cetuximab’s light chain after chromatographic separation, using sDHB as matrix. The N-terminal sequence of the first 86 residues matches the spectrum precisely (top) while the C-terminal sequence displays a +58 Da offset. After assuming an A to E exchange (+58 Da), the C-terminal sequence is in accordance with the ISD-spectrum.
Bottom-up analysis
The trypsin and the GluC digests were subjected to LC-MS/MS analysis. The combined results of the two digestions provided 100% sequence coverage for both the light (Fig. S3) and heavy chains. The C-terminal tryptic peptide is very short and was not detected by LC-MS/MS. However, the MS/MS spectrum of the C-terminal GluC peptide () allowed the unambiguous assignment of an A213E substitution in the cetuximab light chain, which is in agreement with the middle-down sequencing result and the intact MW of the light chain. The alanine-to-glutamic acid substitution results in an expected MW shift of 58.005 Da, in very good agreement with the observed +58.006 Da mass difference between the light chain MW determination shown in and the calculated MW based on the IMGT sequence. The mass error between the corrected theoretical and the experimental intact light chain data are only 0.04 ppm.
Figure 6. LC-MALDI-MS/MS mass spectrum of the gluC C-terminal peptide of cetuximab’s light chain confirming E213 instead of A.
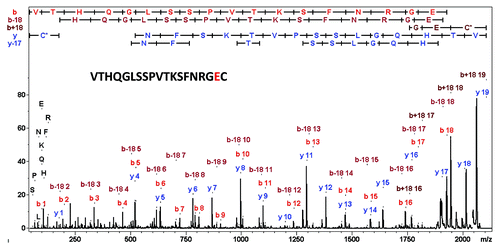
These results show that the IMGT sequence of the light chain of cetuximab contains 2 errors at the C-terminus: Cys214 is missing and Glu213 replaces Ala213. These sequence errors were not previously described. Dubois at al. used trypsin digestion in their bottom-up analysis of cetuximab and achieved 88% sequence coverage missing the three C-terminal residues.Citation17 Wang at al. used publically-available high-resolution crystal structures of cetuximab to determine potential aggregation-prone regions.Citation30 When performing the sequence alignment of different mAbs, this group used the IMGT sequence. The unique capabilities of the strategy adopted in this work highlight that MALDI-ISD middle-down sequence analysis provides reliable information for C-terminal sequences, rapidly—a capability that was not directly available using Edman sequencing, the classical method of protein terminal analysis that only accesses the N-terminus. Subsequently, the classical bottom-up strategy can then be used in a targeted way to confirm the findings and hypotheses from the top-down sequencing. The intact MW or subdomain MWs (middle-up approaches) provide a third, orthogonal dimension to confirming the overall assigned biosimilar structure.
Glycosylation assessment
Whereas the analysis of glycans released by endoglycosidases provides the averaged glycan profile of the mAb, the glycopeptide-centric approach employed in this study allows for determination of glycan heterogeneity at each of the two glycosylation sites of cetuximab separately (Fig. S4–6). In this approach, MS/MS spectra of glycopeptides are acquired during standard bottom-up LC-MS/MS experiments and classified as glycopeptide spectra. For this purpose, the ProteinScape software we used searches for specific fragmentation patterns and oxonium ions to classify the potential glycopeptide spectra (Fig. S7). In the case of MALDI-TOF/TOF-MS/MS data, the fragmentation pattern described by Wuhrer el al.Citation31 and Rapp et al.Citation32 is used for classification. From the fragmentation pattern of the glycopeptide (Fig. S8A), ProteinScape determines the m/z value of the peptide and the glycan moieties. These are used for their identification through database searches. Glycans were identified through the search engine GlycoQuest (integrated in ProteinScape) and peptides through Mascot (Matrix Science, USA) (Fig. S8). Initially, GlycoQuest searches public or user-defined databases for glycan structures that match each experimental parent MW within a given tolerance. From the candidate glycan structures, fragments are calculated and matched with the respective MS/MS spectra. As a result of this search, a list of glycan structures is obtained, which is ranked by a score. The score is based on the number of identified fragments and the intensity coverage of the MS/MS spectrum, i.e., the fraction of the sum of the intensities of the peaks assigned to a glycan structure vs. the sum of the intensities of all MS/MS fragment ions. A spectrum viewer provides the annotation of both glycan and peptide fragments for the interactive validation of GlycoQuest and Mascot database search results (Fig. S9–10). Glycan identification with the described mass spectrometric methods relates to the assignment of glycan composition and to some extent of the glycan structure. These methods, however, do not allow inferring any further reaching structural assignments, such as the definition of linkage. Such full structural assignments require dedicated structural methods, e.g., NMR or mass spectrometric analysis of permethylated and released glycans, which were not the objective in this study.
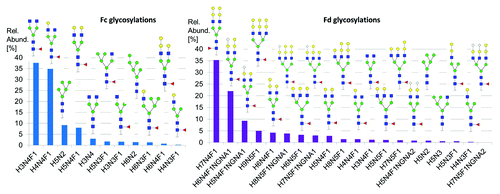
Cetuximab glycans have been extensively characterized by Qian et al. who identified 21 different glycans.Citation23 However, the method used did not allow differentiation between the Fc N-glycosylation (of the CH2 domain) and the Fd N-glycosylation since the authors released the glycans before analyzing them. Janin-Bussat et al. used a middle-up approach similar to the one described here and identified 6 different glycoforms for the conserved CH2 N-glycosylation and 11 different Fd N-glycosylations.Citation22 In our work, we could identify 11 glycans for the CH2 N-glycosylation and 20 different for the Fd N-glycosylation with a total of 24 different glycosylations ( and and ). We ranked the different glycoforms according to their relative abundances as derived from the middle-up MS glycoprofiling (). It is, to our knowledge, the most comprehensive identification of cetuximab’s glycoforms. In the Fd glycosylations, 41% contained N-glycolyl neuraminic acid (NGNA) and 78% contained Gal-α-1,3-gal in their structures. These glycosylations has been shown to be responsible for immunogenic responses.Citation18 Batch-to-batch variations in the glycosylation profiles of cetuximab have also been reported by Sundaram et al.Citation21
Figure 7. LC-ESI-MS spectra of (A) Fc/2 subunit and (B) Fd subunit. Only 19 out of 20 are displayed, the lowest abundant species is out of the mass range shown. The peaks were annotated with the correspondent glycoform.
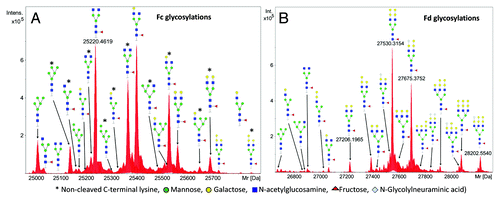
Figure 8.(A) Relative abundance of glycoforms attached to the N299 glycosylation site (Fc) as derived from middle-up glyco-profiling (Left). Eleven glycoforms were assigned to this glycosylation site. (B) Relative abundance of glycoforms of glycosylation site N88 (Fd) as derived from middle-up glyco-profiling (Right). Twenty glycoforms could be assigned to this glycosylation site.
Conclusion
To meet the expectations of regulatory agencies, marketing applications for biosimilar antibodies must include data from reliable comparability methodologies applied to the biosimilar and reference molecules. The state-of-the art mass spectrometry-based methods presented here constitute a solid framework for both amino acid sequence and glycosylation assessment. Starting with an intact antibody mass measurement and going through middle-up and middle-down and then bottom-up MS approaches, we were able to detect rapidly and unambiguously both an expected and an unexpected sequence error near the light chain C-terminus and to correct them. It is clear that biosimilar developers should start with such a comprehensive structural assessment and comparability exercise prior to other pre-clinical studies. In addition, we provided the most comprehensive glycoform identification and glycoprofiling of cetuximab to date using a new automated glycopeptides-based identification strategy, which also substantially reduced the time required for the overall analysis and data interpretation compared with traditional manual approaches. Taken together, the reported workflow and mass spectrometric techniques clearly demonstrated the ability to gain in-depth structural insights that will help biosimilar developers in the structural assessment of reference mAbs and in comparability studies.
Material and Methods
The cetuximab used in this study is the EMA-approved version and formulation.
Middle-up and middle-down sample preparation
Cetuximab was cleaved in the hinge region using limited proteolysis by IdeS (Immunoglobulin-degrading enzyme of Streptococcus pyogenes) (FabRICATOR, Genovis, Lund). After cleavage, 6 M of guanidine-HCl and 30 mM TCEP were added to perform reduction (30 min, RT) before adding 1% trifluoracetic acid (TFA) yielding the Fc/2, Fd and light chain subunits of cetuximab.
LC-MS of intact cetuximab and middle-up analysis
LC-MS analyses of intact cetuximab and of IdeS digested fragments were performed on an Acquity UPLC H-Class system (Waters, MA, USA) coupled to a maXis 4G high resolution Q-TOF type mass spectrometer (Bruker Daltonik, Bremen, Germany). For middle-up MW analysis of the cetuximab subunits, a modified maXis 4G with a novel collision cell design was used, which permits mass resolution of approx. 80 000 for the deconvoluted spectra of the antibody fragments, thus well-resolving the isotopic patterns of the cetuximab subunits.
For intact mAb analysis, 3 µg of cetuximab were loaded at a 0.3 mL/min flow rate of 0.1% formic acid in water (solvent A) on a BEH300 C4 2.1 × 100 mm column (Waters). The antibody was eluted using a linear gradient of 5‒95% of solvent B (0.1% formic acid in 60% acetonitrile and 40% isopropanol) in 8 min followed by a 3 min 93% solvent B wash stage before reconditioning of the column at 5% solvent B.
For IdeS-generated cetuximab subunits, 2 µg of the cetuximab digest were loaded on the column and eluted using a flow rate of 0.4 mL/min and 23 min at 5% solvent B followed by a linear gradient of 5 to 15% solvent B in 3 min then 15 to 45% in 30 min and 45 to 80% in 1 min followed by 5 min at 80% of solvent B and reconditioning of the column at 5% solvent B. The whole LC-MS system and analysis was controlled by BioPharma Compass 1.1 (Bruker Daltonik).
Monoisotopic molecular weights (MWs) of the IdeS-generated cetuximab subunits were determined by the SNAP algorithm. The SNAP algorithm fits a theoretical peak pattern derived from the average atomic composition found in proteinsCitation33 to the measured isotopic peak patterns.Citation34,Citation35 Raw data were inspected and sporadic peak mis-assignments were manually corrected.
MALDI-ISD middle-down analysis of cetuximab
For middle-down analysis, the cetuximab Ides digest was desalted using Micro Spin G 25 columns (GE Healthcare) and LC-separated using Agilent 1200 and Zorbax 300SB-C8 column with water/0.1% TFA as solvent A and acetonitrile/0.1% TFA as solvent B. Fractions were spotted directly to MTP BigAnchor plates. The dry sample spots were then covered with sDHB (9+1 mixture of 2,5-dihydroxybenzoic acid and 2-hydroxy-5-methoxybenzoic acid, 25 g/l in 50% acetonitrile/water/0.1% TFA) and intact protein spectra and ISD spectra were acquired using WARP-LC 1.3 and compass 1.4 flexSeries. ISD spectra were acquired in reflector mode and externally calibrated using bovine ubiquitin ISD fragments. MALDI-ISD middle-down spectra were further analyzed with BioTools 3.2 SR4.
Bottom-up analysis of cetuximab
100 µg of cetuximab (20 µL) were mixed with 80 µL of guanidine-HCl (6M), reduced with DDT at 56°C for 2 h and alkylated with iodoacetamide at 37°C for 30 min then desalted using Microspin G 25 column. Trypsin (seq. grade) was obtained from Promega and Endoproteinase GluC from Roche. Tryptic digestion was performed for 24 h at 37°C, the GluC digest at 25°C. Peptide mapping nanoLC-MS/MS analysis was performed using a Dionex RSLC nano-chromatography system (Thermo Scientific, MA, USA) coupled to a maXis impact high resolution Q-TOF mass spectrometer (Bruker Daltonik) equipped with Captivespray nanosprayer technology (Bruker Daltonik). The analytical column is a Dionex C18 Pepmap 100, 2 µm, 0.075 × 250 mm. The digestion peptides were loaded on the enrichment column (Dionex C18 Pepmap 100, 5 µ, 0.1 × 20 mm) at a flow rate of 5 µL/min, elution was performed at 0.300 µL/min with a 5‒40% linear gradient of solvent B in 55 min followed by 15 min at 97% solvent B before reconditioning of the column at 5% solvent B.
The maXis impact was operated in the positive ion mode. The TOF was mass calibrated prior to analysis using sodium TFA in the m/z 430‒2100 range. For tandem MS, the system automatically switched between MS and MS/MS modes. MS spectra were recorded over an m/z range of 50‒2200 at 2 spectra/sec. The MS/MS accumulation time was regulated by the intensity of the selected peak on the MS spectrum. The 3 most abundant peptide ions, preferably doubly or triply charged, were selected in each MS spectrum for further isolation and collision induced dissociation using optimized collision energies depending on charge state and m/z of the ion. The analyzed peptides were subsequently excluded from re-selection for 60 sec. The acquisition process was controlled by the Compass software (Bruker Daltonik).
Mass spectra were processed and the resulting peak lists transferred to ProteinScape 3.1 (Bruker Daltonik). The Mascot search engine (Matrix Sciences) was used for MS/MS searches against a custom database containing the IMGT light and heavy chain sequences of cetuximab and common likely contaminants (trypsin, human keratins). Carbamidomethylation of cysteine residues was set as a fixed modification while methionine oxidation, N-terminal cyclization of glutamine, deamidation of glutamine and asparagine where set as variable modifications. The peptide mass tolerance was set at 7 ppm and fragment mass tolerance was set to 0.05 Da. Up to two trypsin or GluC mis-cleavages were allowed. Search results were compiled using ProteinScape (Bruker Daltonik).
MALDI Bottom-Up analysis for light chain C-terminal sequence confirmation was performed on the GluC digested cetuximab. The digested sample was separated using nano-Advance UHPLC (Bruker Daltonik) equipped with Acclaim PepMap100, C18, 5 µm, 100 Å, 100 µm i.d. × 2 cm (Trap column) and Acclaim PepMap RSLC, C18, 2 µm, 100 Å, 75 µm i.d. × 25 cm, nanoViper (Analytical column). Five pmol of digest were loaded and eluted using a linear gradient starting from 2% acetonitrile/ 0.1% TFA/water to 35% acetonitrile in 64 min. Fractions of 10 sec were spotted using Proteineer FcII (Bruker Daltonik) with a matrix sheath flow (α-cyano-4-hydroxycinnamic acid) providing co-crystallization of sample and matrix on the MALDI sample holder. MALDI-TOF/TOF-MS and MS/MS spectra were acquired using ultrafleXtreme (Bruker Daltonik) and further analyzed using the ProteinScape 3.1 software platform.
For glycan and peptide identification based on glycopeptide MS/MS spectra, an automated four-step workflow was executed in ProteinScape. In step 1, each MS/MS spectrum was screened for the presence of characteristic N-linked glycopeptide fragmentation patterns (Fig. S8A). In step 2, the peptide [M+H]+ was extracted from that pattern, yielding both the precise glycan and the peptide MW from a single glycopeptides MS/MS spectrum. In step 3, Bruker’s GlycoQuest glycan search engine in ProteinScape obtained glycan identifications via GlycomeDBCitation36 database searches using the glycan fragments of the glycopeptide MS/MS spectra. Several input parameters were specified for the search: the glycan type was restricted to N-glycan; taxonomy and composition were not restricted. Only singly charged, protonated ions and a fragmentation type containing b, c, y and z-ions were used. MS tolerance was set to 0.5 Da and MS/MS tolerance to 0.8 Da. For every glycopeptide spectrum, the peptide moiety mass previously determined during classification was used as modification of the glycan. In step 4, Mascot searches allowed identification of the peptide parts of the glycopeptides, yielding overall the glycan and peptide structures, including the location of the glycosylation site in the peptide sequence (Fig. S8B).
Additional material
Download Zip (1.2 MB)Acknowledgments
We thank Stephanie Kaspar and Peter Brechlin for help with the bottom-up measurements and Ulrike Schweiger-Hufnagel (all Bruker-Daltonik, Bremen) for critically reading the manuscript. We further thank Jason Rouse’s team (Pfizer, Andover, MA) and Fredrik Olssen (Genovis, Lund) for many helpful discussions on subunit analysis of antibodies.
Submitted
05/08/2013
Revised
06/13/2013
Accepted
06/15/2013
Disclosure of Potential Conflicts of Interest
No potential conflict of interest was disclosed.
Supplemental Materials
All supplemental materials may be found here: www.landesbioscience.com/journals/mabs/article/24323.
References
- Beck A, Wurch T, Bailly C, Corvaia N. Strategies and challenges for the next generation of therapeutic antibodies. Nat Rev Immunol 2010; 10:345 - 52; http://dx.doi.org/10.1038/nri2747; PMID: 20414207
- Beck A. Biosimilar, biobetter and next generation therapeutic antibodies. MAbs 2011; 3:107 - 10; http://dx.doi.org/10.4161/mabs.3.2.14785; PMID: 21285536
- Beck A, Sanglier-Cianférani S, Van Dorsselaer A. Biosimilar, biobetter, and next generation antibody characterization by mass spectrometry. Anal Chem 2012; 84:4637 - 46; http://dx.doi.org/10.1021/ac3002885; PMID: 22510259
- Khawli LA, Goswami S, Hutchinson R, Kwong ZW, Yang J, Wang X, et al. Charge variants in IgG1: Isolation, characterization, in vitro binding properties and pharmacokinetics in rats. MAbs 2010; 2:613 - 24; http://dx.doi.org/10.4161/mabs.2.6.13333; PMID: 20818176
- McCamish M, Woollett G. Worldwide experience with biosimilar development. MAbs 2011; 3:209 - 17; http://dx.doi.org/10.4161/mabs.3.2.15005; PMID: 21441787
- Kálmán-Szekeres Z, Olajos M, Ganzler K. Analytical aspects of biosimilarity issues of protein drugs. J Pharm Biomed Anal 2012; 69:185 - 95; http://dx.doi.org/10.1016/j.jpba.2012.04.037; PMID: 22633839
- Schneider CK, Kalinke U. Toward biosimilar monoclonal antibodies. Nat Biotechnol 2008; 26:985 - 90; http://dx.doi.org/10.1038/nbt0908-985; PMID: 18779806
- Weise M, Bielsky MC, De Smet K, Ehmann F, Ekman N, Giezen TJ, et al. Biosimilars: what clinicians should know. Blood 2012; 120:5111 - 7; http://dx.doi.org/10.1182/blood-2012-04-425744; PMID: 23093622
- Xie H, Chakraborty A, Ahn J, Yu YQ, Dakshinamoorthy DP, Gilar M, et al. Rapid comparison of a candidate biosimilar to an innovator monoclonal antibody with advanced liquid chromatography and mass spectrometry technologies. MAbs 2010; 2:2; PMID: 20458189
- Beck A, Diemer H, Ayoub D, Debaene F, Wagner-Rousset E, Carapito C, et al. Trends in biosimilar antibody and Fc-fusion protein analytical characterization. Trends Analyt Chem 2013; In press http://dx.doi.org/10.1016/j.trac.2013.02.014
- Tan Q, Guo Q, Fang C, Wang C, Li B, Wang H, et al. Characterization and comparison of commercially available TNF receptor 2-Fc fusion protein products. MAbs 2012; 4:761 - 74; http://dx.doi.org/10.4161/mabs.22276; PMID: 23032066
- Goetze AM, Schenauer MR, Flynn GC. Assessing monoclonal antibody product quality attribute criticality through clinical studies. MAbs 2010; 2:500 - 7; http://dx.doi.org/10.4161/mabs.2.5.12897; PMID: 20671426
- Jiang XR, Song A, Bergelson S, Arroll T, Parekh B, May K, et al. Advances in the assessment and control of the effector functions of therapeutic antibodies. Nat Rev Drug Discov 2011; 10:101 - 11; http://dx.doi.org/10.1038/nrd3365; PMID: 21283105
- Anthony RM, Wermeling F, Ravetch JV. Novel roles for the IgG Fc glycan. Ann N Y Acad Sci 2012; 1253:170 - 80; http://dx.doi.org/10.1111/j.1749-6632.2011.06305.x; PMID: 22288459
- Beck A, Wagner-Rousset E, Ayoub D, Van Dorsselaer A, Sanglier-Cianférani S. Characterization of therapeutic antibodies and related products. Anal Chem 2013; 85:715 - 36; http://dx.doi.org/10.1021/ac3032355; PMID: 23134362
- Li S, Schmitz KR, Jeffrey PD, Wiltzius JJ, Kussie P, Ferguson KM. Structural basis for inhibition of the epidermal growth factor receptor by cetuximab. Cancer Cell 2005; 7:301 - 11; http://dx.doi.org/10.1016/j.ccr.2005.03.003; PMID: 15837620
- Dubois M, Fenaille F, Clement G, Lechmann M, Tabet JC, Ezan E, et al. Immunopurification and mass spectrometric quantification of the active form of a chimeric therapeutic antibody in human serum. Anal Chem 2008; 80:1737 - 45; http://dx.doi.org/10.1021/ac7021234; PMID: 18225864
- Chung CH, Mirakhur B, Chan E, Le QT, Berlin J, Morse M, et al. Cetuximab-induced anaphylaxis and IgE specific for galactose-alpha-1,3-galactose. N Engl J Med 2008; 358:1109 - 17; http://dx.doi.org/10.1056/NEJMoa074943; PMID: 18337601
- Suckau D, Resemann A. T3-sequencing: targeted characterization of the N- and C-termini of undigested proteins by mass spectrometry. Anal Chem 2003; 75:5817 - 24; http://dx.doi.org/10.1021/ac034362b; PMID: 14588022
- Hardouin J. Protein sequence information by matrix-assisted laser desorption/ionization in-source decay mass spectrometry. Mass Spectrom Rev 2007; 26:672 - 82; http://dx.doi.org/10.1002/mas.20142; PMID: 17492750
- Sundaram S, Matathia A, Qian J, Zhang J, Hsieh MC, Liu T, et al. An innovative approach for the characterization of the isoforms of a monoclonal antibody product. MAbs 2011; 3:505 - 12; http://dx.doi.org/10.4161/mabs.3.6.18090; PMID: 22123057
- Janin-Bussat MC, Tonini L, Huillet C, Colas O, Klinguer-Hamour C, Corvaïa N, et al. Cetuximab Fab and Fc N-glycan fast characterization using IdeS digestion and liquid chromatography coupled to electrospray ionization mass spectrometry. Methods Mol Biol 2013; 988:93 - 113; http://dx.doi.org/10.1007/978-1-62703-327-5_7; PMID: 23475716
- Qian J, Liu T, Yang L, Daus A, Crowley R, Zhou Q. Structural characterization of N-linked oligosaccharides on monoclonal antibody cetuximab by the combination of orthogonal matrix-assisted laser desorption/ionization hybrid quadrupole-quadrupole time-of-flight tandem mass spectrometry and sequential enzymatic digestion. Anal Biochem 2007; 364:8 - 18; http://dx.doi.org/10.1016/j.ab.2007.01.023; PMID: 17362871
- Zhang Z, Pan H, Chen X. Mass spectrometry for structural characterization of therapeutic antibodies. Mass Spectrom Rev 2009; 28:147 - 76; http://dx.doi.org/10.1002/mas.20190; PMID: 18720354
- Chevreux G, Tilly N, Bihoreau N. Fast analysis of recombinant monoclonal antibodies using IdeS proteolytic digestion and electrospray mass spectrometry. Anal Biochem 2011; 415:212 - 4; http://dx.doi.org/10.1016/j.ab.2011.04.030; PMID: 21596014
- Beavis RC. Chemical mass of carbon in proteins. Anal Chem 1993; 65:496 - 7; http://dx.doi.org/10.1021/ac00052a030
- Takayam MN. Cα bond cleavage of the peptide backbone via hydrogen abstraction. J Am Soc Mass Spectrom 2001; 12:1044 - 9; http://dx.doi.org/10.1016/S1044-0305(01)00289-6
- Demeure K, Quinton L, Gabelica V, De Pauw E. Rational selection of the optimum MALDI matrix for top-down proteomics by in-source decay. Anal Chem 2007; 79:8678 - 85; http://dx.doi.org/10.1021/ac070849z; PMID: 17939742
- Resemann A, Wunderlich D, Rothbauer U, Warscheid B, Leonhardt H, Fuchser J, et al. Top-down de Novo protein sequencing of a 13.6 kDa camelid single heavy chain antibody by matrix-assisted laser desorption ionization-time-of-flight/time-of-flight mass spectrometry. Anal Chem 2010; 82:3283 - 92; http://dx.doi.org/10.1021/ac1000515; PMID: 20329751
- Wang X, Singh SK, Kumar S. Potential aggregation-prone regions in complementarity-determining regions of antibodies and their contribution towards antigen recognition: a computational analysis. Pharm Res 2010; 27:1512 - 29; http://dx.doi.org/10.1007/s11095-010-0143-5; PMID: 20422267
- Wuhrer M, Catalina MI, Deelder AM, Hokke CH. Glycoproteomics based on tandem mass spectrometry of glycopeptides. J Chromatogr B Analyt Technol Biomed Life Sci 2007; 849:115 - 28; http://dx.doi.org/10.1016/j.jchromb.2006.09.041; PMID: 17049937
- Rapp U, Resemann A, Mayer-Posner FJ, Schäfer W, Feichtinger K. The fragmentation behavior of glycopeptides using the PSD technique. In: Townsend A, Hotchkiss A, eds. Techniques in Glycobiology. New York: Marcel Dekker Inc., 1997:53-65.
- Senko M, Beu S, McLaffertycor F. Determination of monoisotopic masses and ion populations for large biomolecules from resolved isotopic distributions. J Am Soc Mass Spectrom 1995; 6:229 - 33; http://dx.doi.org/10.1016/1044-0305(95)00017-8
- Tsybin YO, Fornelli L, Stoermer C, Luebeck M, Parra J, Nallet S, et al. Structural analysis of intact monoclonal antibodies by electron transfer dissociation mass spectrometry. Anal Chem 2011; 83:8919 - 27; http://dx.doi.org/10.1021/ac201293m; PMID: 22017162
- Koster C. Mass spectrometry method for accurate mass determination of unknown ions. US Patent 6188064, 2001.
- Ranzinger R, Herget S, von der Lieth CW, Frank M. GlycomeDB--a unified database for carbohydrate structures. Nucleic Acids Res 2011; 39:Database issue D373 - 6; http://dx.doi.org/10.1093/nar/gkq1014; PMID: 21045056