 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
CD81 is an essential receptor for hepatitis C virus (HCV). K21 is a novel high affinity anti-CD81 antibody with potent broad spectrum anti-HCV activity in vitro. The pharmacokinetics (PK), pharmacodynamics and liver distribution of K21 were characterized in cynomolgus monkeys after intravenous (i.v.) administration of K21. Characteristic target-mediated drug disposition (TMDD) was shown based on the PK profile of K21 and a semi-mechanistic TMDD model was used to analyze the data. From the TMDD model, the estimated size of the total target pool at baseline (Vc • Rbase) is 16 nmol/kg and the estimated apparent Michaelis-Menten constant (KM) is 4.01 nM. A simulation using estimated TMDD parameters indicated that the number of free receptors remains below 1% for at least 3 h after an i.v. bolus of 7 mg/kg. Experimentally, the availability of free CD81 on peripheral lymphocytes was measured by immunostaining with anti-CD81 antibody JS81. After K21 administration, a dose- and time-dependent reduction in free CD81 on peripheral lymphocytes was observed. Fewer than 3% of B cells could bind JS81 3 h after a 7 mg/kg dose. High concentrations of K21 were found in liver homogenates, and the liver/serum ratio of K21 increased time-dependently and reached ~160 at 168 h post-administration. The presence of K21 bound to hepatocytes was confirmed by immunohistochemistry. The fast serum clearance of K21 and accumulation in the liver are consistent with TMDD. The TMDD-driven liver accumulation of the anti-CD81 antibody K21 supports the further investigation of K21 as a therapeutic inhibitor of HCV entry.
Introduction
Hepatitis C virus (HCV) is a major cause of chronic hepatitis, liver cirrhosis, and hepatocellular carcinoma worldwide.Citation1,Citation2 HCV infection begins with the entry of virus into hepatocytes; prevention of HCV entry is therefore an attractive concept for preventing viral infection, reinfection of transplanted liver, or spread of infection. Effective inhibitors of HCV entry may provide substantial medical benefit in monotherapy for the protection of liver transplants or in combination therapy for the treatment of HCV infection.
HCV enters hepatocytes via internalization and fusion of viral envelope with cell membrane, which is mediated by the interaction between its envelope proteins E1 and E2 and cellular receptors. The host proteins CD81, scavenger receptor BI (SR-BI), claudin I and occludin are required for HCV entry.Citation3-Citation7 CD81, a member of the tetraspanin protein family, is a 26 kDa transmembrane protein, consisting of four hydrophobic transmembrane helices and two extracellular loops ECL1 and ECL2.Citation6,Citation8,Citation9 CD81 is widely expressed across hematopoietic and non-hematopoietic cells.Citation10-Citation12 Among hematopoietic cells, CD81 is expressed by B and T cells, macrophages, dendritic cells, natural killer (NK) cells and eosinophils, but not by neutrophils, platelets and erythrocytes. In addition to hepatocytes, it is also expressed by endothelial and epithelial cells in spleen, liver, kidney, testis, skin, gastrointestinal tract, brain, and skeletal muscle.
CD81 was the first host factor identified as critical for HCV entry.Citation6 The potential of CD81 as a therapeutic target was demonstrated by the efficient blockage of HCV infection in cell culture and in mouse models by anti-CD81 antibodies.Citation13-Citation16 In the human liver urokinase-type plasminogen activator-severe combined immunodeficiency (uPA-SCID) mouse HCV infection model, we (unpublished results) and othersCitation14 have shown that anti-human CD81 antibodies could fully protect these humanized mice from HCV challenge.
To enable the evaluation of CD81 as a potential target for HCV infection prevention and treatment, we generated several anti-CD81 monoclonal antibodies (mAb) that show high binding affinity to human and cynomolgus monkey CD81 and potent inhibitory activity on HCV entry. The medical use of anti-CD81 antibodies for therapeutic intervention of HCV infection will require an understanding of the relative distribution of the antibodies to hepatic target cells compared with non-hepatic cells, and the kinetics of target occupancy. MAbs against cell membrane-associated antigens are usually subject to target-mediated drug disposition (TMDD).Citation17,Citation18 Target binding of anti-CD81 antibodies was therefore expected to significantly influence the anti-CD81 antibody distribution and elimination kinetics. The understanding of anti-CD81 antibody TMDD would help determine what drug concentration and dosing schedule is required for antiviral efficacy.
The main objectives of this exploratory study were to 1) evaluate the pharmacokinetic (PK) and pharmacodynamics (PD) profiles and liver distribution of one of the newly identified anti-human CD81 mAb K21 after single dose administration in healthy cynomolgus monkeys; and 2) build a TMDD model to describe the PK, target binding, target saturation and their mutual relationship after single and repeat administration of K21 in monkeys.
Results
K21 mAb binds to human and monkey CD81 with similar affinity
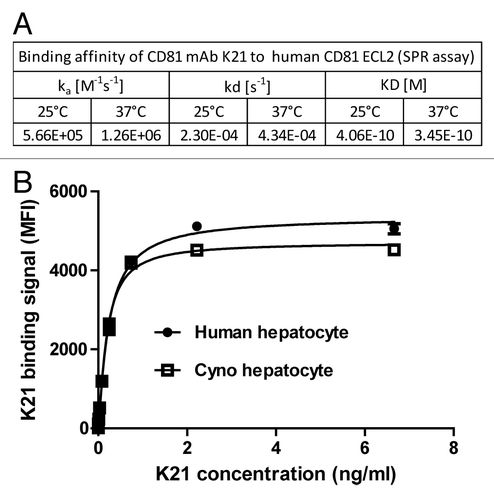
Through hybridoma screening, several murine mAbs that show high binding affinity to human and monkey CD81 and potent antiviral activity were identified. One of the new anti-CD81 mAb (named K21) showed high binding affinity to the large extracellular domain of human CD81 as measured by surface plasmon resonance (SPR), with a KD value of 0.35 nM at 37°C (). K21 was humanized to human IgG4 isotype for further development. To assess whether monkey would be an appropriate animal model for preclinical evaluations of K21, we determined the cross-reactivity of K21 to monkey CD81. As shown in , the apparent K21 binding affinity to native human and cynomolgus monkey CD81 on primary human or monkey hepatocytes was similar. Mean fluorescence intensities (MFI) observed by FACS were similar after K21 mAb binding to human and monkey hepatocytes, suggesting similar surface CD81 densities on hepatocytes of both species (). Therefore, the cynomolgus monkey was considered to be a relevant nonclinical model to evaluate the PK, PD, and liver distribution of the humanized K21 antibody.
Figure 1. Humanized K21 antibody binds to human and cynomolgus monkey CD81 with similar affinity. (A) Binding affinity measurement of anti-CD81 mAb K21 to recombinant human CD81 ECL2 protein using SPR assay. K21 antibody was immobilized using rabbit anti-mouse antibody. The analyte CD81 ECL2 was injected at 100 µl/min for three minute association time. The dissociation was monitored for 10 min. Data are from one representative experiment. (B) Human and cynomolgus monkey primary hepatocytes were incubated with serially diluted K21 mAb and PE- labeled anti-human IgG1 antibody, and analyzed by FACS. Data are from 3 independent experiments showing mean and standard deviation.
PK in cynomolgus monkeys
After a single intravenous (i.v.) bolus administration of K21 to cynomolgus monkeys at doses in 0.3–7 mg/kg range, the serum concentrations were monitored up to 672 h post-dose and the result from initial simple PK analysis is shown in . A follow-up repeat dose pilot PK study was conducted in the spirit of 3R’s (i.e., replacing animal tests where possible, reducing the number of required animals and refining existing scientific practices while still maintaining comparable results) using one animal per dose level, for the purpose of completing the understanding of target engagement by K21. The design of that study was assisted by the predictions of a TMDD PK model built on the data from the single dose study to optimize the information gained. The combined data were then used to build a refined model (see below) for future study design in non-human primates and human. The K21 serum concentration-time profiles up to 96 h from both single dose and repeat dose administration are shown in . The PK results from all dose groups are summarized in Table S1.
Table 1. Pharmacokinetic parameters of K21 from non-compartmental analysis following a single intravenous administration in cynomolgus monkeys
Figure 2. Serum concentrations of K21 vs. time plot following a single or repeat intravenous administration of K21 in cynomolgus monkeys. The symbols represent observed data and lines represent simulated data. The time scale for this figure is set for 96 h for illustration of the dynamics of early time points. The full data set can be found in Supplemental material.
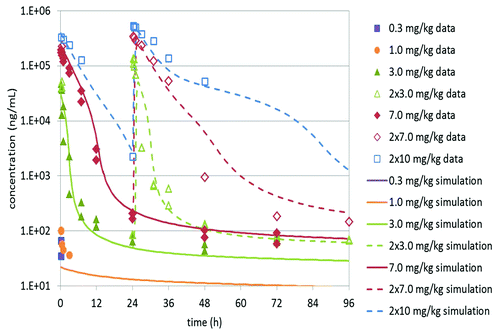
Nonlinear kinetics of K21 was apparent from the data shown in and over the dose range tested. At the lower doses, K21 was detected in serum only at the first measured time point of 15 min from the 0.3 mg/kg group and up to the 3 h post-dose time point from the 1 mg/kg group, and therefore very limited PK parameters could be obtained. At 3 and 7 mg/kg doses, a biphasic PK profile was observed, starting from very high initial serum concentrations with a rapid several log-fold decline in serum concentrations within 24 h followed by a slow decline at time points up to 504 h. The extrapolated initial concentrations (C0) from the α phase were used to calculate the volume of the central compartment (Vc). Due to limited time points available for the two lower dose levels, most PK parameters could not be determined. At doses 3 and 7 mg/kg, the calculated Vc values were 41 and 35 mL/kg, respectively. These volumes are comparable to the reported plasma volume in monkeys (~45 ml/kg),Citation19 and to the reported volume of distribution for mAbs that are primarily distributed intravascularly (33 – 71 mL/kgCitation20). Based on non-compartmental analysis (NCA), the volumes of distribution at steady-state (Vss) were 4810 mL/kg and 300 mL/kg, at the 3 and 7 mg/kg dose, respectively, and the apparent serum clearances (CL) at 3 and 7 mg/kg doses were 40 and 9.6 mL/hr/kg. These values are clearly dose-dependent, suggesting strong TMDD, and thus a TMDD PK model is needed to describe the PK properties of K21. On the other hand, the terminal half-life (T1/2) of 229–355 h (10–15 d) observed at both 3 and 7 mg/kg was long and consistent with typical serum half-lives of IgG mAbs.
PK modeling and target-mediated drug disposition
All collected PK data were fitted to a model that was previously described for TMDD.Citation21 The observed and model-fit profiles for the single and repeat-dose data are shown in . Parameter estimation converged rapidly, with most parameters having reasonable levels of relative standard errors (r.s.e.). A complete table of estimated parameters and their r.s.e. is displayed in the supplementary appendix. Figures for individual fits and diagnostic plots can be found in Figure S2–S4.
The size of the total target pool per kg body weight, expressed by was estimated to be 16 nmol/kg (7% r.s.e.). This number needs to be put into context with the direct measurement of 500,000 CD81 copies per cell on primary human hepatocytes (Table S2): assuming a liver cellularity of 100 million cells per gCitation22 and a liver volume of 150 ml for a 5 kg monkey,Citation19 the liver alone is estimated to contribute approximately 2.5 nmol/kg to the total target load. This model therefore predicts a significant volume of CD81 outside of the liver, consistent with CD81 expression analysis showing high CD81 levels in non-hepatic cells.Citation10-Citation12
The value for KD estimated from the TMDD modeling was found to be 0.21 nM (16% r.s.e.) and thus close to the value measured in vitro. The dissociation rate of antibody-target complexes could not be determined well from the available data (r.s.e. > 100%), and was estimated to be 5.2 h−1 (ie, 0.0014 sec−1). This lack of precision may be due to the rapid decline in concentrations during early hours post-dose and very limited measurement at lower doses, which is not sufficiently resolved with the available data in the study. The target was estimated to have slow turnover with kdeg of 0.061 h−1, i.e., 11 h of half-life (16% r.s.e.). On the time-scale of the study, antibody-target complex internalization appears to be nearly insignificant based on estimated kint of 0.0032 h−1 (36% r.s.e.) or is superseded by other processes.
While the full TMDD model is needed to calculate the time-course of receptor saturation, the above parameters can be used to project receptor saturation under steady-state conditions. This is done in a very similar way to the standard calculation of receptor occupancy except that the constant of dissociation KD needs to be replaced by the constant KM as a result of internalization and target turnover:Citation23
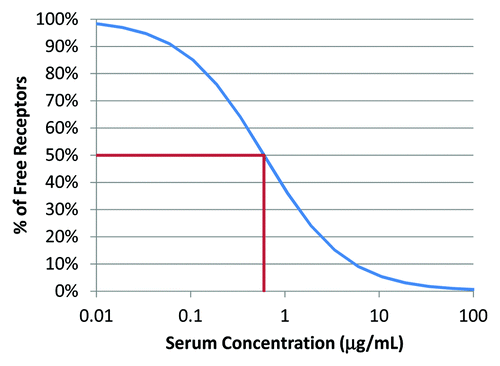
The KM value calculated from the above parameters for K21 in monkey is 4.0 nM, which predicts that a concentration of 60 μg/mL would be needed to maintain the number of free receptors at or below 1%. This is illustrated in .
Figure 5. Relationship between percentage of free targets and serum concentration simulated based on KM value estimated from the TMDD model.
The linear clearance (CL) from the central compartment was estimated to 0.193 mL/h/kg (r.s.e. > 100%) similar to values observed with other antibodies.Citation20 The central volume (Vc) of 19 mL/kg (11% r.s.e.), was found to be ~2-fold smaller than the physiological plasma volume (45 mL/kg)Citation19 and also smaller than the one by simple extrapolation from initial time points in the α phase after single dose of 3 or 7 mg/kg.
Pharmacodynamics of anti-CD81 antibody K21 in cynomolgus monkeys
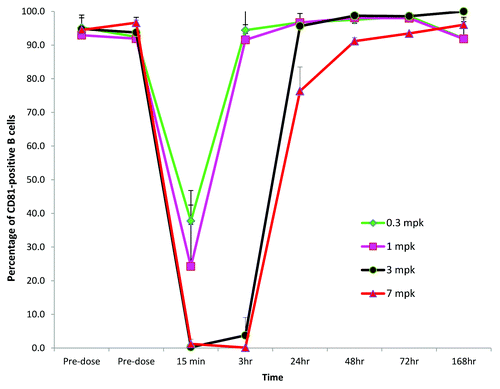
We assessed the CD81 antigen expression on peripheral lymphocytes using immunostaining with the previously described anti-CD81 mAb JS81.Citation24 Phycoerythrin (PE)-labeled JS81 competes with K21 for CD81 binding, indicative of binding to an overlapping epitope and binding in a mutually exclusive manner. As shown in Figure S1, pre-incubation of CD81 expressing cells with K21 completely prevented JS81 binding to the cell surface. This assay therefore can be used to detect the expression of free CD81 (unoccupied by K21) on the cell surface. Among blood cells, CD81 staining was observed on monocytes, NK, T and B cells. Monocytes and B cells demonstrated the highest staining with JS81 with > 95% cells being CD81-positive. After K21 administration, the percentage of NK, T and B cells and monocytes that could be stained with JS81 antibody changed in a dose- and time-dependent manner. shows the kinetics of unoccupied CD81 on B cells after administration of K21 to cynomolgus monkeys. Similar kinetics were observed for the other CD81-positive blood cell types investigated (NK, T cells, monocytes). Within 15 min after K21 administration, the percentage of B cells that could be stained with JS81 antibody was reduced in a K21 dose-dependent manner reaching levels below 3% after single dose at 3 or 7 mg/kg of K21. We did not determined whether the reduction of binding of JS81 to B cells was due to the occupancy of CD81 with K21 or CD81 internalization after K21 binding. The recovery kinetics of JS81 binding to B cells was also K21 dose- and time-dependent. At 0.3 and 1 mg/kg doses of K21, CD81 on B cells available for JS81 binding restored to baseline level by 3 h post K21 administration. In contrast, in the animals dosed with 3 and 7 mg/kg of K21, free CD81 recovered to baseline level by 24 and 48 h, respectively. No changes in B, T, NK and monocyte cell counts were observed after K21 administration (data not shown). Based on JS81 staining data, we selected the 7 mg/kg dose to assess the time course of K21 distribution in the liver in the next part of this study.
Figure 3. Mean (± SD) percentage of CD81-positive peripheral B cells over time after a single administration of K21 to cynomolgus monkeys. The percentage of CD81-positive cells was determined by staining the whole blood ex vivo with anti-CD81 antibody JS81.
Liver distribution of K21
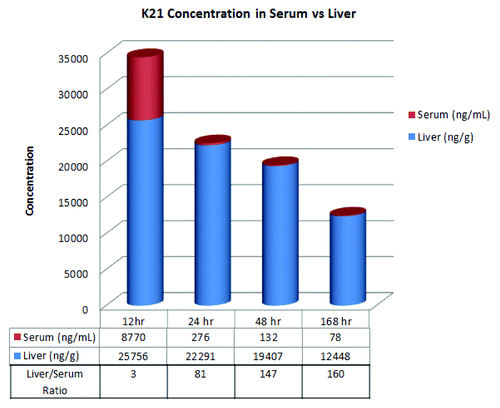
The time-course of K21 distribution in the liver was evaluated by two different methods. First, K21 levels were analyzed by ELISA in liver homogenates from liver biopsies collected at different time points after a single 7 mg/kg i.v. bolus. The time courses of K21 detected in the serum and in the liver using this method are shown in . In the liver homogenates, high levels of K21 were detected at all time points up to 168 h after administration. The liver concentrations of K21 were reduced to approximately half comparing the 12 h and 168 h time points. The liver/serum (L/S) ratio of K21 concentrations was ~3 at 12 h and reached 160 at 168 h post-administration ().
Figure 4. K21 concentrations in serum and liver and liver/serum ratio following a 7 mg/kg intravenous dose to cynomolgus monkeys.
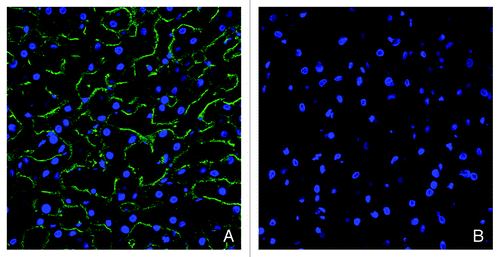
In addition to the measurement of K21 levels in liver homogenates, we also analyzed K21 presence in the tissue by immunohistochemistry. Double immunolabeling was performed to detect K21 and hepatocytes in cynomolgus monkey liver by staining with anti-human-IgG antibody and with an anti-cytokeratin 8 antibody. Using immunofluorescence, the presence of K21 on hepatocytes could be detected up to 48 h after dosing of the mAb, as determined by detectable binding of an anti-human IgG antibody and detection of fluorescent green signal within tissues (). Green fluorescence was maximal at 12 and 24 h post-dosing, and was present in both endothelial sinusoidal areas (green fluorescence alone), and co-localized with the hepatocyte marker (orange fluorescence). After 168 h, no significant difference from vehicle-treated tissues was observed. To confirm the lack of CD81 binding sites on hepatocytes after K21 treatment, we evaluated tissue staining with JS81. While hepatocytes could be stained with JS81 antibody in the vehicle-treated animal, the JS81 signal was absent after K21 administration (). These results are consistent with the lack of free CD81-binding sites on liver hepatocytes due to the bound K21 after a single administration of 7 mg/kg; however, the results of neither the staining nor the ELISA allowed us to distinguish between mAb bound extracellularly and internalized mAb.
Figure 6. Immunofluorescence detection of K21 in liver. K21 in vehicle- (A) and in K21- treated monkeys after 12 (B), 24 (C), 48 (D) and 168 h (E) was detected using anti-human IgG antibody (green fluorescent detection signal). Hepatocytes were labeled with an anti-cytokeratin-8 antibody (red fluorescent signal). K21 was detected on hepatocytes up to 48 h after dosing. K21 was observed in both sinusoidal areas (single-label green fluorescence) and in hepatocytes (orange signal, corresponding to co-localization with cytokeratin-8 fluorescence). Nuclei are stained with DAPI (blue).
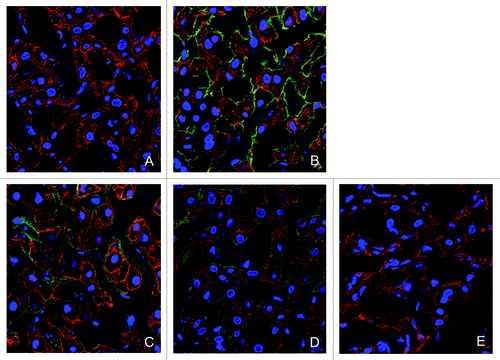
Figure 7. Immunofluorescence detection of cell-surface CD81 in the liver with anti-CD81 mAb JS81. Green fluorescent signal indicative of JS81 binding could be observed in the liver from vehicle-treated monkey (A), while no signal could be detected in the liver of K21-treated monkey at 12 h after a single 7 mg/kg dose (B). Liver nuclei are stained with DAPI (blue).
Discussion
The current study evaluated PK, PD and liver distribution of a novel humanized anti-CD81 antibody K21 in cynomolgus monkeys after single and repeat dose i.v. administration. Consistent with widespread expression of CD81, K21 exhibited fast target-mediated clearance from serum over a wide range of doses, resulting in a rapid decline of K21 serum levels and a sustained target-mediated accumulation in the liver. Our data demonstrate that K21 specifically accumulates in the liver and is detectable in the liver at times when the antibody has already been largely cleared from the circulation.
There are many factors that can influence the PK and tissue distribution of mAbs, including target biology, antibody structure, FcRn binding and proteolytic catabolism.Citation25,Citation26 In general, antibodies can be eliminated from systemic circulation by non-specific (non-target-mediated) and by specific target-mediated clearance mechanisms.Citation18 Non-specific clearance occurs primarily through the cells of the reticuloendothelial system (RES) via proteolytic catabolism and is regulated through the interaction with various Fc receptors.Citation17 Antibodies against soluble antigens with low endogenous levels (e.g., TNFα, IL-4, IL-5) often demonstrate non-specific dose-independent linear clearance.Citation27 In contrast, the clearance of antibodies targeting cell-surface antigens can be greatly accelerated through a target-mediated process and result in dose-dependent nonlinear kinetics.Citation28 In the case of the anti-CD81 antibody K21, we observed steep nonlinear PK ( and ). Thus, a simple PK analysis cannot describe the complete properties of K21 and a TMDD PK model was built in our second analysis.
The PK profiles suggest that, immediately after dosing, K21 is very rapidly removed from the circulation due to binding to cell-surface CD81 antigen that is highly expressed in the vascular compartment and in the liver. Clear signs of saturation of targets were seen at doses around 3 and 7 mg/kg, as indicated by the steep increase in serum exposure at these doses compared with the lower doses. This hypothesis is supported by the prolonged residence time of K21 in the liver due to CD81 binding on hepatocytes and by over ~16-fold reduction in Vss value when a single dose increased from 3 to 7 mg/kg (). To test this hypothesis, a repeat dose pilot PK was conducted in which the second dose of K21 was administered 24 h after the first dose at 3, 7, or 10 mg/kg (Table S1). Compared with the serum concentrations at the same time points after the first dose, the concentrations after the second dose were much higher than what would be expected based on “carry-over” from the pre-dose level, suggesting that the targets were still occupied when the second dose was administered. A TMDD model was built using the complete PK data describing the PK semi-mechanistically as a combination of a linear disposition and nonlinear processes involving the target. This model fits the data well and offers a plausible explanation to the observed PK and PD. Several related models were tested to see whether some of the remaining inconsistencies could be removed (such as 2-fold lower value for Vc); however, none of these could justify the complexity introduced by the added mechanisms. For the nonlinear target-mediated disposition, the TMDD model estimates the size of the total target pool at baseline is 16 nmol/kg. This indicates that the target pool is able to bind up to 2.4 mg of K21 per kg of body weight, consistent with our hypothesis based on early PK analysis. The efficacious concentration can be aimed by blocking receptors using estimated Km (4.0 nM in this case) from the TMDD model.Citation34 At steady-state, we simulated that the percentage of free receptors would be 50% for a serum concentration of 0.6 μg/mL and 1% for a serum concentration of 60 μg/mL (). After a single i.v. dose of 7 mg/kg, the serum concentrations of K21 were > 60 ug/mL for at least 3 h (Table S1), which should result in < 1% of free CD81 receptors. The rates of internalization of CD81 and cell-surface presentation of newly synthesized CD81 have not been determined experimentally, but the TMDD model predicts that both target turnover and internalization rate are slow. Further investigations are desirable to determine how long the free targets need to be blocked and what percentage of target occupancy is required for clinically effective inhibition of HCV entry by the anti-CD81 antibody K21. Such information could be valuable in the selection of dosing regimens for clinical studies of K21.
The liver is the target for HCV and thus our main objective was to investigate antibody distribution in the liver and target blockage after a single administration. MAbs can freely travel through the large (~100 nm) sinusoidal clefts in the liverCitation29 and thus readily access target proteins on the hepatocyte cell surface. We used a serial design and evaluated K21 distribution in the liver at several time points by measuring K21 level in the tissue and in the serum by ELISA and by immunostaining of K21 bound to hepatocytes. Both methodologies clearly demonstrated continued liver exposure to K21, while the observed disparity between ELISA readouts and immunolabeling at later time points could be due to the differences in assay sensitivity. The liver to serum (L/S) ratio increased time-dependently – it reached 150 by 48 h post-administration and 160 by 168 h (). In the absence of a strong antigen sink, the typical tissue/serum (T/S) ratios for mAbs in monkeys are below 0.1.Citation26 A T/S ratio > 30 was previously observed in the case of anti-Fas antibody, which also binds to a target in a highly perfused tissue.Citation30 The high L/S ratio observed in our study is consistent with specific and tight binding of K21 to liver hepatocytes. We have confirmed the sustained presence of K21 bound to CD81 on liver hepatocytes by double immunostaining for K21 and hepatocyte specific marker cytokeratin 8 () and by the lack of immunostaining with a competitive anti-CD81 antibody JS81 ().
Taken together, our data demonstrate that anti-CD81 mAb K21 can effectively block CD81 targets in the liver after a single i.v. administration at 7 mg/kg. These results support further investigation of K21 as a therapeutic inhibitor of HCV entry.
Material and Methods
Generation of anti-human CD81 mAb clone K21
Anti-CD81 mAbs were generated by conventional hybridoma technology.Citation31 Eight to 12 weeks old Balb/c and NMRI mice (Charles River, Sandhofer Weg 7, D 97633 Sulzfeld, Germany) were immunized with CHO CD81 cells. 107 CHO CD81 cells were mixed with CFA and administered intraperitoneally (i.p.) followed by i.p. administration of 107 cells in IFA and a final i.v. boost of 107 cells in phosphate buffered saline (PBS) with no adjuvant. Fusions of spleenocytes with P3X63 Ag8.653 were performed. Hybridomas that produced the antibodies showing positive binding for human and monkey CD81, but not CD9, were tested in the HCVpp con1 assay and VSVpp assay as a counter screen control in Huh 7 cells. Hybridomas that showed the most potent inhibitory activity in the HCVpp assay and no inhibitory activity in the VSVpp assay were then cloned. The murine CD81 antibody K21 was humanized using the complementarity-determining regions (CDRs) grafting method, i.e., by keeping the six loops that recognize the antigen intact and exchanging the murine framework for a human IgG1.Citation32 The CDRs were identified according to the Kabat nomenclature, whereas several frameworks for both heavy and light chains were chosen from the human germline IMGT databaseCitation33 and the human IgG4-SPLE framework was used.Citation34 Humanized CD81 antibody K21 was transiently expressed in HEK293 cells by transfection of the light and heavy chain plasmids via lipofectin according to the manufacturer (Life Technologies, Carlsbad, USA). Supernatant was collected 7 d after transfection and the antibody was purified by using MabSelect SuRe (GE Healthcare Bio-Sciences AB, Uppsala, Sweden). Peak fractions were pooled and underwent analytical size exclusion chromatography (10/30 Superdex 200, GE Healthcare Life Sciences) to achieve > 95% monomer content. Concentrations were determined by UV280, and were adjusted to 1 mg/ml and aliquots were stored at -80°C. Pools were analyzed further by SDS-PAGE and analytical SEC; identity was confirmed by mass spectroscopy.
Determination of anti-CD81 mAb binding affinity to human CD81 ECL2 by surface plasmon resonance
Binding of the murine anti-CD81 mAbsto recombinant CD81 extracellular loop 2 (ECL2) protein was measured using a BIAcore T100 instrument (GE Healthcare). A CM5 sensor was preconditioned by a 1 min injection at 100 µl/min of 0.1% SDS, 50 mM NaOH, 10 mM HCl and 100 mM H3PO4. The instrument was run in 10 mM HEPES pH 7.4, 150 mM NaCl, 1 mM EDTA, 0.05% (w/v) P20. Thirty µg/ml polyclonal rabbit anti-mouse (RAMIgG) antibody (Biacore GE healthcare) in 10 mM sodium acetate buffer at pH 5.0 were immobilized at 8000 RU in all flow cells using standard EDC/NHS chemistry according to the manufacturer’s instructions. The sensor was saturated by a 7 min injection at 5 µl/min with a 1 M ethanolamine pH 8.0 solution. Antibody capturing was done at 20 µl/min by a 15 sec injection of 60 nM antibody at 25°C and 40 nM antibody at 37°C. The analyte CD81 ECL2 was injected at 100 µl/min for three minute association time in a concentration series starting from 0 nM (buffer), 1.1 nM, 3.3 nM, 10 nM, 30 nM, 60 nM and 180 nM. The dissociation was monitored for 10 min. After each concentration series, the RAMFcy capture sensor was washed 15 sec with 100 mM HEPES pH 7.4, 1.5 M NaCl, 30 mM EDTA, 0.1% (w/v) TWEEN 20, 2% DMSO, injected at 20 µl/min and was subsequently regenerated by a three min injection of 10 mM glycine buffer pH 1.7 at 10 µl/min over all flow cells. The sensorgrams were evaluated using the Biacore T100 evaluation software Ver.1.0. Kinetic rates, ka (1/Ms), kd (1/s) and affinity KD (M) were determined according to a Langmuir model with RMAX local.
Evaluation of anti-CD81 mAb binding affinity to human and monkey hepatocytes by fluorescence-activated cell sorting (FACS)
Cryopreserved primary human and monkey hepatocytes were purchased from Celsis In Vitro Technologies (Baltimore, MD). Hepatocytes (1 × 105) were incubated with serially-diluted humanized K21 antibody in FACS buffer (PBS containing 0.5% FBS) for 45 min on ice. After washing, the cells were incubated with a labeled anti-human IgG secondary antibody (BD Biosciences, San Diego, CA) for 30 min on ice. After three washes, the stained cells were analyzed with a Calibur flow cytometer (BD Biosciences, San Diego, CA). Mean fluorescence intensity (MFI) values were graphed against antibody concentrations using the one-site specific binding regression fitting in GraphPad Prism™ software (Intuitive Software for Science, San Diego, CA).
Determination CD81 levels on cell surface by FACS
Antibodies bound per cell (ABC) were analyzed with flow cytometry using Becton Dickinson QuantiBRITE PE beads (Cat# 340495, BD, San Jose, CA) following manufacturer's instructions. CD81 mAb JS81-PE conjugate (1:1) specific was custom ordered from BD PharMingen (BD, La Jolla, CA). The number of CD81 molecule per cell was calculated using the QuantiBRITE beads as standards according to manufacturer’s instructions.
PK and PD study in cynomolgus monkeys
The cynomolgus monkey PK and PD study was approved by the institutional animal care and use committee (IACUC) at Charles River Laboratories (Reno, NV). The test article was administered to the experimentally naïve cynomolgus monkeys (Macaca fascicularis) aged 3–6 y via a slow bolus i.v. injection by a single dose or two doses, given approximately 24 h apart. Monkeys were administered K21 once at dose levels of 0.3, 1, 3, and 7 mg/kg (n = 2). Blood was collected at predose, and 0.25, 0.5, 1, 3, 7, 12, 24, 48, 72, 120, 168, 336, 504, and 672 h after dosing. Monkeys were also administered K21 twice at dose levels of 3 mg/kg (n = 2), 7 mg/kg (n = 1), and 10 mg/kg (n = 1). Blood was collected at predose, 0.5 and 24 h after the first dose (day 1) and at 0.25, 0.5, 1, 3, 7, 12, 24, 48, 72, 120, 168, 336, 504, and 672 h after the second dose (day 2) of either 3 or 7 mg/kg. The monkey that received 10 mg/kg twice was sampled at predose, and 0.25, 1, 3, 7, and 24 h after the first dose (day 1), and at 0.25, 0.5, 1, 3, 7, 12, 24, 96, 168, 336, 504, and 672 h after the second dose (day 2).
A single dose liver distribution study in cynomolgus monkeys
The study was approved by the IACUC at Hoffmann-La Roche (Nutley, NJ). A single 7 mg/kg dose was administered to the experimentally naïve male cynomolgus monkeys (Macaca fascicularis) aged 4–8 y via a slow bolus i.v. injection. Blood samples were collected for serum for the determination of serum K21 concentrations and for the determination of PD activity. At specified time points (n = 1 per time point), necropsies were conducted and the liver was collected to assess the level of K21 and CD81 expression in the tissue. A single dose of vehicle (20 mM histidine, 140 mM NaCl, and 0.02% Tween 20, with a pH of 6.) was administered to a control animal (n = 1), necropsy was conducted at 168 h time point and the liver was collected to assess CD81 expression in the tissue.
Pharmacodynamic analysis
Whole blood samples were analyzed by flow cytometry for CD81 protein expression on lymphocyte subpopulations. Blood samples were collected into sodium heparin vacutainers and stored at ambient temperatures of 18–22°C. One hundred µL of blood was added to a 2.2 mL/well 96-well plate that contained 1.8 mL/well of stain buffer (BD Biosciences, San Jose, CA) and 100 µL/well of antibody cocktail. The plate was incubated for 10 min at room temperature in the dark and then lysed with 1.8 mL of 1X BD FACS Lyse solution and centrifuged at room temperature for 8 min at 300 x g. The supernatant was removed and the cells were fixed with 600 ml/well of stain buffer and analyzed immediately. The samples were analyzed by FACS Canto II flow cytometer (BD Biosciences, San Jose, CA). The flow cytometry data were analyzed using DIVA® 6.0 software.
The following antibody panel was used for the analysis of CD81 expression on peripheral CD4 and CD8 T cell and B cell – CD4-FITC, CD81-PE, CD8-PE-Cy7, CD45-PerCP, CD20-APC, CD3-APC-Cy7. CD81 expression on peripheral monocyte and NK cells was monitored by staining with CD14-FITC, CD81-PE, CD45-PerCP, CD16-APC, CD3-APC-Cy7. Anti-CD81 antibody clone JS81 that competes with K21 for CD81 binding was used for this analysis. All antibodies used for flow cytometry analysis were obtained from BD Bioscience.
Absolute cell counts
BD TruCount™ tubes (BD Biosciences, San Jose, CA) were used in combination with CD45 labeling for real-time quantification of B, CD3, CD4, and CD8 lymphocytes, NK cells and monocytes absolute counts in peripheral blood. The samples were analyzed by FACS Canto II flow cytometer (BD Biosciences, San Jose, CA).
Measurement of K21 in cynomolgus monkey serum
The concentrations of the chimeric human IgG1 antibody drug were determined by using an enzyme-linked immune-sorbent assay (ELISA). A streptavidin-coated 96-well Nunc Maxisorp microtiter plate was coated with a capture reagent, biotin-labeled CD81 ligand, at 0.211 μg/mL in PBS containing 0.5% bovine serum albumin (BSA) and incubated for one hour at room temperature with non-vigorous shaking. The plate was washed three times with washing buffer (PBS containing 0.05% Tween 20). K21 drug was used as a reference standard, and known amounts of the drug was spiked into 100% naïve monkey serum pool at different concentrations for the preparation of standards and quality control samples (QCs). The standards were used for the generation of a calibration curve and the QCs were used to monitor the performance of the assay. The lower limit of quantitation (LLOQ) in serum was 30 ng/mL. All study serum samples, standards, and QCs were diluted at 1:20 in PBS with 0.5% BSA. One hundred μL of diluted standards, QCs, or study samples were added to each well of the plate and incubated for one hour with non-vigorous shaking. After three washes, 100 μL of 0.030 μg/mL digoxigenin-conjugated mouse monoclonal anti-human IgG antibody was added to each well and incubated for one hour with non-vigorous shaking. The plate was washed three times, and 100 μL of 25 mU/mL horseradish peroxidase-conjugated anti-digoxigenin Fab fragments was added to each well and incubated for one hour with non-vigorous shaking. The plate was washed three times, and 100 μL of TMB substrate was added to each well and incubated for approximately 5 min. Color development was photometrically determined at 450 nm (with reference wavelength at 650 nm), and was proportional to the analyte concentration in the serum sample. Unknown sample assay responses were plotted against the standard curve to measure concentration of drug in samples. Any experimental samples below LLOQ were annotated as BLQ (below limit of quantitation). The precision (% CV of replicates) and accuracy (% relative error) of the assay were within 20% as calculated from the quality control samples that were assayed on each plate. Any samples above the limit of quantitation of the assay (800 ng/mL) were diluted with blank monkey serum to bring the samples within the assay range prior to analysis.
Measurement of K21 in the liver
Liver solid tissue samples were washed and weighed. Homogenization buffer (Tissue Extraction Reagent I, Invitrogen, catalog no. FNN0071) and protease inhibitor cocktail (Sigma-Aldrich, catalog no. P8340) was then added to the samples such that the homogenization buffer is 2 volumes of the tissue weight. The tissues were homogenized (2010 Geno/Grinder, SPEX SamplePrep LLC) and the resulting homogenate was centrifuged for approximately 30 min at 4°C at 3500 rpm. The tissue supernatants were transferred to fresh tubes and stored at approximately -70°C until analysis. The ELISA assay described for measurement of K21 concentrations in serum was modified for the measurement of antibody concentrations in liver tissues as follows. A standard calibration curve was generated by using serial dilutions of drug in blank monkey liver tissue supernatant. The LLOQ in the tissue supernatant was 300 ng/mL. All study tissue supernatant samples, standards, and QCs were diluted at 1:4000 in sample dilution buffer (10% homogenization buffer with 2% BSA). One hundred μL of diluted standards, QCs or study samples were added to the plate and incubated for one hour with gentle shaking. After three washes, 100 μL of 0.090 μg/mL digoxigenin-conjugated mouse monoclonal anti-human IgG antibody was added to each well, and the plate was incubated for one hour with non-vigorous shaking. The plate was washed three times, and 100 μL of horseradish peroxidase conjugated anti-digoxigenin Fab fragments was added to each well at 75 mU/mL final concentration. The rest of the experimental steps are as described for the serum assay. Any samples above the limit of quantitation of the assay (8000 ng/mL) were diluted with blank monkey liver tissue supernatant to bring the samples within the assay range prior to analysis.
Liver to serum concentration ratios (L/S) at a given time point were calculated using the ratio of K21 concentration on the liver (ng/g) to that in serum (ng/mL).
Pharmacokinetic analysis
Initial PK assessment of the single dose data up to 7 mg/kg was performed using Phoenix™ WinNonlin® (Pharsight Inc., St. Louis, MO), where parameters were calculated using non-compartmental analysis for half-life (T1/2), area under the serum concentration-time curve up to the last measurable time point (AUClast), CL and Vss. Sampling times were reported as nominal time and concentrations below the limit of quantitation were considered as zero. The AUCs were calculated using the linear trapezoidal rule. The C15 min values were at first sampling time post dose taken directly from measured serum concentrations. The extrapolated serum concentrations immediately after dosing (C0) was estimated by extrapolating from the α phase only, and the extrapolated Vc values were then estimated based on the given i.v. dose and C0.
PK modeling and target-mediated drug disposition
The TMDD model described by Mager and Jusko,Citation21 which empirically describes the PK as a combination of a linear disposition and nonlinear processes involving the target, was selected for this analysis. Briefly, the linear disposition is described by two compartments (volumes and
) with first-order exchange between them (
) and first-order elimination from the central compartment (
). Antibodies in the central compartment are binding to free target molecules by law of mass action and involving the constant of dissociation
. Before dosing, the target is at a dynamic equilibrium between constant synthesis and first-order degradation at a rate
, in such a way that it is maintained at a concentration
.Citation21 Antibody-target complexes can either dissociate at a rate
or are irreversibly internalized at a rate
. All equations are displayed in the Supplementary Information (Appendix).
Data of all treated animals were fitted simultaneously using nonlinear mixed effects modeling allowing estimation of a single set of typical parameter values shared by all animals (“fixed effects”). Small deviations from these typical values (“random effects”) were allowed for the parameters central volume (), baseline size of the target pool (
), and target turnover (
); however, due to the limited number of animals, and to avoid bias by dose group, we fixed them to a small value (ω = 0.1). A residual error model (describing the remaining differences between observed data and model-simulated profiles) that combines additive and concentration-proportional contributions was used. The analysis was performed using MONOLIX 4.1.3. The full data set used for this analysis is shown in Figures S3, S4, and S5. Measurements that are below the limit of quantitation (30 ng/mL) are included as “left-censored data” in this analysis.
Liver immunohistochemistry
Fresh liver tissues (left lateral, right lateral and left median lobes) of vehicle- (168 h) and K21-treated (12, 24, 48 and 168 h) cynomolgus monkeys were embedded in OCT compound (Tissue Tek), frozen, and stored at -70°C until sectioning. For specific detection of K21 in the tissue, we used a proprietary biotin-labeled mouse anti-human mAb, which binds to human immunoglobulins while showing no cross-reactivity with cynomolgus monkey IgG. For the immunolabeling of hepatocytes, anti-human cytokeratin-8 mouse mAb (Millipore, Billerica, MA) was used as a primary antibody. Also, a commercially-available anti-CD81 antibody (clone JS81, BD Biosciences, San Jose, CA) was used in selected experiments.
Briefly, frozen liver sections were cut in a cryostat at 5 um thickness, and immunofluorescence labeling was performed in an Ultra automated stainer (Ventana Medical Systems, Tucson, AZ). Sections were incubated with primary antibody (biotin-mouse anti-human IgG), followed by streptavidin-Alexa 488 (Invitrogen, Carlsbad, CA). For double immunolabeling, sections were then incubated with cytokeratin-8 primary antibody followed by goat anti-mouse IgG-Alexa 568 (Invitrogen, Carlsbad, CA). Similarly, for detection of clone JS81, primary antibody incubation was followed by detection with anti-mouse-AlexaFluor-488 antibody (Invitrogen, Carlsbad, CA). The immunolabeled sections were mounted in Prolong Gold plus antifade mounting media with DAPI.
Immunofluorescence confocal microscopy was performed using Zeiss LSM 510 laser scanning microscope. The fluorescence of individual fluorochrome was captured separately after optimization to reduce bleed-through between channels. All the images were taken at the same settings and magnification. Two sections of each liver lobe were used for immunolabeling and six images of each liver lobe (3 images per section) were captured for evaluation.
For quantification of JS81 liver staining, image analysis was performed on the captured images from all different liver lobes as described above, using Definiens Tissue Studio image analysis software. Total intensity of the fluorescent signal was measured, and results were normalized to the number of nuclei in the image field.
| Abbreviations: | ||
| PK | = | pharmacokinetics |
| PD | = | pharmacodynamics |
| mAb | = | monoclonal antibody |
| TMDD | = | target-mediated drug disposition |
| HCV | = | hepatitis C virus |
| L/S | = | liver to serum ratio |
| C15 min | = | serum concentration at 15 min post dose |
| AUClast | = | area under the curve up to the last measurable concentration |
| C0 | = | initial concentrations |
| Vc | = | volume of central compartment |
| Vss | = | volume of steady state distribution |
| T1/2 | = | elimination half-life |
| CL | = | clearance |
| mpk | = | mg/kg |
| iv | = | intravenous |
Additional material
Download Zip (420.9 KB)Acknowledgments
The authors would like to thank our colleagues, Johannes Auer, Georg Fertig, and Silke Hansen for the generation of humanized anti-CD81 mAb K21 and technical assistance, as well as Roland F Staack for the development of the original bioanalytical assay which enabled quantification of K21 in serum and in liver.
Disclosure of Potential Conflicts of Interest
No potential conflict of interest was disclosed.
References
- Lavanchy D. The global burden of hepatitis C. Liver Int 2009; 29:Suppl 1 74 - 81; http://dx.doi.org/10.1111/j.1478-3231.2008.01934.x; PMID: 19207969
- Brown RS. Hepatitis C and liver transplantation. Nature 2005; 436:973 - 8; http://dx.doi.org/10.1038/nature04083; PMID: 16107838
- Dorner M, Horwitz JA, Robbins JB, Barry WT, Feng Q, Mu K, et al. A genetically humanized mouse model for hepatitis C virus infection. Nature 2011; 474:208 - 11; http://dx.doi.org/10.1038/nature10168; PMID: 21654804
- Evans MJ, von Hahn T, Tscherne DM, Syder AJ, Panis M, Wölk B, et al. Claudin-1 is a hepatitis C virus co-receptor required for a late step in entry. Nature 2007; 446:801 - 5; http://dx.doi.org/10.1038/nature05654; PMID: 17325668
- Meredith LW, Wilson GK, Fletcher NF, McKeating JA. Hepatitis C virus entry: beyond receptors. Rev Med Virol 2012; 22:182 - 93; http://dx.doi.org/10.1002/rmv.723; PMID: 22392805
- Pileri P, Uematsu Y, Campagnoli S, Galli G, Falugi F, Petracca R, et al. Binding of hepatitis C virus to CD81. Science 1998; 282:938 - 41; http://dx.doi.org/10.1126/science.282.5390.938; PMID: 9794763
- Scarselli E, Ansuini H, Cerino R, Roccasecca RM, Acali S, Filocamo G, et al. The human scavenger receptor class B type I is a novel candidate receptor for the hepatitis C virus. EMBO J 2002; 21:5017 - 25; http://dx.doi.org/10.1093/emboj/cdf529; PMID: 12356718
- Kitadokoro K, Bordo D, Galli G, Petracca R, Falugi F, Abrignani S, et al. CD81 extracellular domain 3D structure: insight into the tetraspanin superfamily structural motifs. EMBO J 2001; 20:12 - 8; http://dx.doi.org/10.1093/emboj/20.1.12; PMID: 11226150
- Levy S, Nguyen VQ, Andria ML, Takahashi S. Structure and membrane topology of TAPA-1. J Biol Chem 1991; 266:14597 - 602; PMID: 1860863
- Oren R, Takahashi S, Doss C, Levy R, Levy S. TAPA-1, the target of an antiproliferative antibody, defines a new family of transmembrane proteins. Mol Cell Biol 1990; 10:4007 - 15; PMID: 1695320
- Maecker HT, Todd SC, Levy S. The tetraspanin superfamily: molecular facilitators. FASEB J 1997; 11:428 - 42; PMID: 9194523
- Yáñez-Mó M, Alfranca A, Cabañas C, Marazuela M, Tejedor R, Ursa MA, et al. Regulation of endothelial cell motility by complexes of tetraspan molecules CD81/TAPA-1 and CD151/PETA-3 with alpha3 beta1 integrin localized at endothelial lateral junctions. J Cell Biol 1998; 141:791 - 804; http://dx.doi.org/10.1083/jcb.141.3.791; PMID: 9566977
- Lindenbach BD, Evans MJ, Syder AJ, Wölk B, Tellinghuisen TL, Liu CC, et al. Complete replication of hepatitis C virus in cell culture. Science 2005; 309:623 - 6; http://dx.doi.org/10.1126/science.1114016; PMID: 15947137
- Meuleman P, Hesselgesser J, Paulson M, Vanwolleghem T, Desombere I, Reiser H, et al. Anti-CD81 antibodies can prevent a hepatitis C virus infection in vivo. Hepatology 2008; 48:1761 - 8; http://dx.doi.org/10.1002/hep.22547; PMID: 19030166
- Bartosch B, Dubuisson J, Cosset FL. Infectious hepatitis C virus pseudo-particles containing functional E1-E2 envelope protein complexes. J Exp Med 2003; 197:633 - 42; http://dx.doi.org/10.1084/jem.20021756; PMID: 12615904
- Wakita T, Pietschmann T, Kato T, Date T, Miyamoto M, Zhao Z, et al. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat Med 2005; 11:791 - 6; http://dx.doi.org/10.1038/nm1268; PMID: 15951748
- Lobo ED, Hansen RJ, Balthasar JP. Antibody pharmacokinetics and pharmacodynamics. J Pharm Sci 2004; 93:2645 - 68; http://dx.doi.org/10.1002/jps.20178; PMID: 15389672
- Tabrizi MA, Tseng CM, Roskos LK. Elimination mechanisms of therapeutic monoclonal antibodies. Drug Discov Today 2006; 11:81 - 8; http://dx.doi.org/10.1016/S1359-6446(05)03638-X; PMID: 16478695
- Davies B, Morris T. Physiological parameters in laboratory animals and humans. Pharm Res 1993; 10:1093 - 5; http://dx.doi.org/10.1023/A:1018943613122; PMID: 8378254
- Dong JQ, Salinger DH, Endres CJ, Gibbs JP, Hsu CP, Stouch BJ, et al. Quantitative prediction of human pharmacokinetics for monoclonal antibodies: retrospective analysis of monkey as a single species for first-in-human prediction. Clin Pharmacokinet 2011; 50:131 - 42; http://dx.doi.org/10.2165/11537430-000000000-00000; PMID: 21241072
- Mager DE, Jusko WJ. General pharmacokinetic model for drugs exhibiting target-mediated drug disposition. J Pharmacokinet Pharmacodyn 2001; 28:507 - 32; http://dx.doi.org/10.1023/A:1014414520282; PMID: 11999290
- Barter ZE, Bayliss MK, Beaune PH, Boobis AR, Carlile DJ, Edwards RJ, et al. Scaling factors for the extrapolation of in vivo metabolic drug clearance from in vitro data: reaching a consensus on values of human microsomal protein and hepatocellularity per gram of liver. Curr Drug Metab 2007; 8:33 - 45; http://dx.doi.org/10.2174/138920007779315053; PMID: 17266522
- Grimm HP. Gaining insights into the consequences of target-mediated drug disposition of monoclonal antibodies using quasi-steady-state approximations. J Pharmacokinet Pharmacodyn 2009; 36:407 - 20; http://dx.doi.org/10.1007/s10928-009-9129-5; PMID: 19728050
- Ziegler S, Kronenberger B, Albrecht BA, Kaul A, Gamer AL, Klein CD, et al. Development and evaluation of a FACS-based medium-throughput assay for HCV entry inhibitors. J Biomol Screen 2009; 14:620 - 6; http://dx.doi.org/10.1177/1087057109337161; PMID: 19531666
- Bumbaca D, Boswell CA, Fielder PJ, Khawli LA. Physiochemical and biochemical factors influencing the pharmacokinetics of antibody therapeutics. AAPS J 2012; 14:554 - 8; http://dx.doi.org/10.1208/s12248-012-9369-y; PMID: 22610647
- Tabrizi M, Bornstein GG, Suria H. Biodistribution mechanisms of therapeutic monoclonal antibodies in health and disease. AAPS J 2010; 12:33 - 43; http://dx.doi.org/10.1208/s12248-009-9157-5; PMID: 19924542
- Deng R, Jin F, Prabhu S, Iyer S. Monoclonal antibodies: what are the pharmacokinetic and pharmacodynamic considerations for drug development?. Expert Opin Drug Metab Toxicol 2012; 8:141 - 60; http://dx.doi.org/10.1517/17425255.2012.643868; PMID: 22248267
- Ng CM, Stefanich E, Anand BS, Fielder PJ, Vaickus L. Pharmacokinetics/pharmacodynamics of nondepleting anti-CD4 monoclonal antibody (TRX1) in healthy human volunteers. Pharm Res 2006; 23:95 - 103; http://dx.doi.org/10.1007/s11095-005-8814-3; PMID: 16308668
- Weinstein JN, van Osdol W. The macroscopic and microscopic pharmacology of monoclonal antibodies. Int J Immunopharmacol 1992; 14:457 - 63; http://dx.doi.org/10.1016/0192-0561(92)90176-L; PMID: 1618598
- Saito M, Yoshigae Y, Nakayama J, Ogawa Y, Ohtsuki M, Kurihara A, et al. Tissue distribution of humanized anti-human Fas monoclonal antibody (R-125224) based on fas antigen-antibody reaction in collagen-induced arthritis monkeys. Life Sci 2007; 80:2005 - 14; http://dx.doi.org/10.1016/j.lfs.2007.02.043; PMID: 17477938
- Köhler G, Milstein C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 1975; 256:495 - 7; http://dx.doi.org/10.1038/256495a0; PMID: 1172191
- Lo BK. Antibody humanization by CDR grafting. Methods Mol Biol 2004; 248:135 - 59; PMID: 14970494
- Lefranc MP. IMGT, the international ImMunoGeneTics database. Nucleic Acids Res 2003; 31:307 - 10; http://dx.doi.org/10.1093/nar/gkg085; PMID: 12520009
- Stubenrauch K, Wessels U, Regula JT, Kettenberger H, Schleypen J, Kohnert U. Impact of molecular processing in the hinge region of therapeutic IgG4 antibodies on disposition profiles in cynomolgus monkeys. Drug Metab Dispos 2010; 38:84 - 91; http://dx.doi.org/10.1124/dmd.109.029751; PMID: 19850673