
Abstract
Nerve growth factor (NGF) is indispensable during normal embryonic development and critical for the amplification of pain signals in adults. Intervention in NGF signaling holds promise for the alleviation of pain resulting from human diseases such as osteoarthritis, cancer and chronic lower back disorders. We developed a fast, high-fidelity method to convert a hybridoma-derived NGF-targeted mouse antibody into a clinical candidate. This method, termed Library Scanning Mutagenesis (LSM), resulted in the ultra-high affinity antibody tanezumab, a first-in-class anti-hyperalgesic specific for an NGF epitope. Functional and structural comparisons between tanezumab and the mouse 911 precursor antibody using neurotrophin-specific cell survival assays and X-ray crystal structures of both Fab-antigen complexes illustrated high fidelity retention of the NGF epitope. These results suggest the potential for wide applicability of the LSM method for optimization of well-characterized antibodies during humanization.
More than two million Americans and 1 in 5 surveyed Europeans suffer from chronic pain.Citation1 This pain is debilitating, and has substantial significant economic effects as US companies spend an estimated $90 billion annually for employee absenteeism, loss of productivity and medical treatments.Citation2 Current pain therapies rely on five classes of drugs: corticosteroids, non-steroidal anti-inflammatory drugs (NSAIDs), cyclooxygenase inhibitors (coxibs), opioids and cannabinoids.Citation3 However, these medications are not always effective due to the limited pain etiologies targeted, dose-limiting side effects, and the potential for addiction. Therefore, a need exists for additional therapies that treat severe chronic pain. Antagonism of nerve growth factor (NGF) is a promising approach for effective pain management in a variety of pain syndromes.Citation4 Accordingly, we developed an anti-NGF antibody, tanezumab, which is currently in clinical trials. Tanezumab has efficacy for pain associated with osteoarthritisCitation5 and the lower back, and has also shown promise in pain associated with cancer and rodent models of visceral and neuropathic pain.Citation6,Citation7 As an anti-NGF therapy, tanezumab and other drugs of this class utilize the first new mechanism of pain management in decades.
A fundamental challenge in developing anti-NGF therapeutics is the overlap in structure and function among the neurotrophin family members such as NGF, NT-3, NT-4/5 and brain-derived neurotrophic factor (BDNF), which have an overall amino acid sequence identity of 52%.Citation8 Expressed as pro-peptides, neurotrophins undergo proteolytic maturation to form the biologically active 26 kiloDalton homodimer. Each neurotrophin has an independent biological function that is mediated through activation of one or more of the tropomyosin-related tyrosine kinase receptors, TrkA, TrkB or TrkC. NGF binds specifically to the extracellular domain of TrkA, leading to receptor phosphorylation followed by activation of several intracellular signaling pathways.Citation9 This may occur in a 2:2 stochiometry as was observed in the crystal structure of NGF bound to a TrkA extracellular fragment.Citation10,Citation11 In addition to binding Trk receptors, all neurotrophins bind to the p75 neurotrophin receptor (p75NTR, also known as the low-affinity neurotrophin receptor),Citation12 which is a member of the tumor necrosis factor receptor super-family.Citation13 The biological effects of NGF interaction with p75NTR in different cell types both with and without Trk expression remains to be fully elucidated. However, it is known that the association of NGF and p75NTR into a signaling complex with a 2:2 stochiometry is dependent on glycosylation of p75NTR.Citation14,Citation15
Given the role of NGF in pain, we sought to engineer an antibody that would preferentially block the signaling of NGF over other neurotrophins. Our starting point was the hybridoma-derived mouse 911 antibody that demonstrated efficacy in animal pain modelsCitation7 and cross-reactivity with rodent, human and non-human primate NGF orthologs, and we sought to retain these characteristics during optimization. We developed a method to boost the affinity of the antibody for NGF and retain the exact NGF-specific epitope of 911, resulting in the clinical candidate tanezumab. This broadly applicable method, library scanning mutagenesis (LSM), employs small libraries containing few mutants, in contrast to most large library methods with recombinant bacteriophage, yeast and ribosome display technologies.Citation16-Citation19 Analyzing and engineering the complementarity-determining region 3s (CDR3s) with LSM involves application of relationships between framework and variable domains described by Foote and Poljak.Citation20-Citation23 To appreciate fully the epitope fidelity imparted on tanezumab by LSM, we determined X-ray crystal structures of NGF in complex with both 911-Fab and with tanezumab-Fab and compared their biochemical and biological activities.
Results
Conversion by optimization of CDR1 and CDR2
The starting point for molecular conversion to a clinical lead antibody was the mouse antibody clone 911,Citation6,Citation7,Citation24 which binds human and murine NGF with high affinity (KD ~10 nM) [, workflow (1)]. The goal of this humanization was to build the mouse paratope onto a human antibody framework while maintaining the specificity and integrity of binding. Sensitivity to NGF binding drove the conversion and guided all CDR targeted mutations and libraries. The human frameworks having light and heavy chain segments O8 /JK2 and VH4–59 /JH4, respectively and with the NCBI V gene nomenclature, were used to generate the template antibody 8L2–6D5. They were selected due to the highest human germline similarity to 911 based on ImMunoGeneTics information system® (IMGT; www.imgt.org) database using Igblast.Citation25 8L2–6D5 contains one framework mutation in the heavy chain framework.Citation20 This was the starting antibody for humanization and affinity maturation after E. coli and hamster codon optimization and sub-cloning into an E. coli Fab expression vector using standard techniques [, workflow (2)].Citation26,Citation27
Figure 1. Design and biophysical analysis of the humanization of tanezumab by LSM. (A) Schematic diagram depicting the development of tanezumab by cloning the mouse 911 hybridoma-derived antibody (1), identifying the CDRs and constructing a human framework template (2), humanizing and adjusting the template (3), employing library scanning mutagenesis (LSM) on the CDR3s (4) and merging mutations into the final antibody (5). The template affinity for NGF was KD 25 nM. Following CDR 1 and 2 optimizations, the intermediate affinity for NGF was 1 nM. LSM analysis was then combined on the intermediate for a final set of library screening to arrive at tanezumab (KD < 12 pM). (B) LSM histogram for each complete mutagenesis at each position in L3 except P95 and all residues in H3. Mutants were better off-rate than wild-type (w.t., red), same as w.t. (beige), worse than w.t. (light blue) or no binding (dark blue). The rectangular box around L3 K92 highlights a 100% permissive position. Asterisks (*) identify positions targeting for combination in the last round of LSM. (C) Tanezumab binding competition for NGF receptors, p75NTR (green) and TrkA (pink) by percent capture of NGF to biosensor chip coated with either receptor. (D) 911 binding competition for NGF receptors, p75NTR (green) and TrkA (pink) by percent capture of NGF to biosensor chip coated with either receptor. (E) Percent neuron survival as a function of increasing antibody concentration of either tanezumab (purple) or 911 (orange).
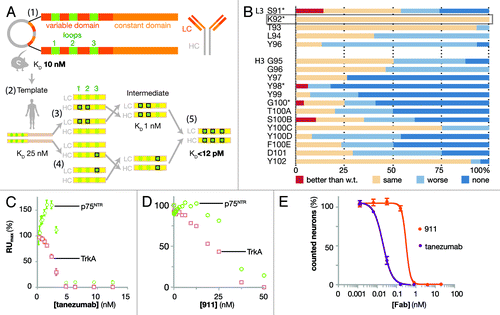
The amino acid sequences of 8L2–6D5 CDRs 1 and 2 were: L1 (24RASQDISNHLN34), L2 (50YISRFHS56), H1 (26GFSLIGYDIN35) and H2 (50MIWGDGTTDYNSAL63). The first variants were created within only these 4 CDRs of the 8L2–6D5 E. coli Fab format. Two criteria were used for directing mutations. Amino acid positions that were conserved in human CDRs were identified and replaced with the most commonly occurring amino acids at those few positions, and amino acids likely to be used during in vivo affinity maturation were left alone based on alignment of the selected germline with Igblast.Citation25,Citation28 These criteria were determined by taking the human germline frameworks selected and using them to query the IMGT database and protein data bank with Igblast to create a non-exhaustive framework alignment from the database. Focusing in CDRs, positions that were not conserved in the H1, H2, L1 and L2 CDRs across the alignment were identified; these are likely optimized for epitope recognition during affinity maturation. Conservation was observed at other positions. For example, when using the selected germline amino acid sequence as a probe of the entire antibody database, the human residue in L2 position 51 is frequently a small residue, while the mouse CDR is Ile, which is suggestive of a structural position not directly binding antigen in the paratope, rather than engaging in a critical interaction with the antigen. Therefore, we mutated conservativelyCitation29 CDR1 and CDR2 residues such as L2 51 to the human residue in order to match the framework around the paratope. Thus, a small number of mutations were made in these 4 CDRs with the goal of mimicking a human CDR sequence, and subsequently mutants were selected for further optimization based on measurement of a slower off-rate as compared with the starting sequence [, workflow (3); ]. The affinity of 8L2–6D5 for human NGF was 25 nM by surface plasmon resonance (SPR), with kon of 4 × 104 M−1s−1 and koff of 1 × 10−3 s−1. Mutants were tested unpurified from small-scale periplasmic E. coli expression, allowing for rapid, high-throughput, overnight analysis. Because most SPR measurements were performed on partially pure proteins, we ranked Fab mutants using measured values for koff and an 8L2–6D5 based, constant value of kon of 4 × 104 M−1s−1 to obtain estimates of the KD. Improved affinity clones (koff < 1 × 10−3 s−1) were sought among these conservative mutations. When screening the L1 and L2 mutants, the heavy chain was maintained as the template sequence and the light chain was likewise maintained for the screening of the H1 and H2 mutants [, workflow (3)]. This approach, which employed wobble codons,Citation29 was not exhaustive in the probing of amino acid sequence space, but sought to decrease the likelihood of a human immune response while maintaining the NGF epitope, minimizing chemical liabilities (oxidation, isomerization) and allowing for increased binding affinity. The intermediate clone optimized from this step, H19-L129, had a measured koff of 1.4 × 10−4, which represents a 7-fold improvement in koff (calculated KD of 1 nM), and consisted of CDR amino acid sequences: L1 (24RASQSISNNLN34), L2 (50YTSRFHS56), H1 (26GFSLIGYDLN35) and H2 (50IIWGDGTTDYNSAV63).
Table 1. Koff values determined during optimization of L1, L2, H1 and H2 for generation of the intermediate
Library Scanning Mutagenesis on CDR3: rapid optimization
The LSM method is a two-step process for rapid identification of mutations in L3 and H3 that maximize improvement in affinity for the antigen [, workflow (4)]. Screening a selected subset of amino acids to combine with the initially identified pivotal positions further optimizes the loop structure resulting in an increased affinity toward the target. For our anti-NGF antibody, mutants were screened by biosensor (SPR/BIAcore™) analysis in the context of the template antibody, 8L2–6D5. Using this lower affinity template antibody was important to allow increased sensitivity of the assay for improvements in binding as indicated by a slower off-rate (koff). Mutants identified by this first step in the LSM method were subsequently screened in the second step by incorporation into the CDR1 and CDR2 optimized intermediate, higher affinity H19-L129 and monitored for further improved off-rates (and higher binding affinities) to generate the final antibody [, workflow (5)].
For the first LSM step, each position in L3 and H3 was mutated to all 20 amino acids using the degenerate codon NNK (where N represented nucleotide abbreviated as A or T or G or C and K represented G or T). The total library size was kept small (32 codons per each oligonucleotide) by making a change in only one CDR position at a time. Each positional library was cloned and screened independently; for example, five independent libraries were generated from separate oligos each with one NNK for each L3 position and calculated as only 32+32+32+32+32 = 160 clones to cover L3. CDR L3 Pro95 was not altered due to a presumed structural role. Periplasmic E. coli Fab expression was followed by high-throughput off-rate analysis, while keeping track of phenotype with genotype. The L3 and H3 starting sequences were 91SKTLPY96 and 95GGYYYGTSYYFDY102, respectively. The single measurement values of koff were considered significantly improved when slower than 1 × 10−3 s−1, significantly worse when faster than 2 × 10−3 s−1, and unchanged otherwise [; Fig. S1). Amino acid CDR positions where most clones produced unchanged values of koff were considered to be “permissive,” and positions where most clones produced worse values of koff were considered to be “restrictive.” For instance, L3 residue K92 was permissive since all mutants had the same off-rate, while H3 residues Y98 and Y99 were considered to be restrictive sites since fewer than 7% of mutants retained binding, which is a narrow working space for binding in the paratope. Mutations at sites L3-S91, H3-Y98, H3-G100 and H3-S100B improved koff (). Since koff is determined from “single-shot” screening data (Fig. S1), some SPR curves with noise or artifacts produced misleading values of koff, which could easily be discarded upon inspection of the biosensor curves (e.g., H3-S100B was not improved).
Table 2. Koff values determined for a subset of mutants during LSM
For the second LSM step, four positions from CDRs L3 and H3 were selected for combinatorial library screening for affinity improvement in the context of the higher affinity intermediate, H19-L129. The intent of this small library was to exploit the positional and chemical information from the first round of off-rate screening with a minimum number of changes from the native sequence. Permissive sites were included to provide conformational flexibility and to allow for binding synergy. The positions selected for this micro-library were L3-S91, H3-Y98 and H3-G100 with addition of the completely permissive site L3-K92 (, * positions). Because of the limited number of positions (four) and analysis of the sequence information from the first NNK CDR3 libraries, sites were made into a library that combined mutations of L3 and H3 simultaneously and using wobble codonsCitation29 that included the improved amino acid plus a limited set of other mutants. H3 position 98 was changed to Y, W or C using codon TRS (R is A or G and S is C or G) and H3 position 100 was changed to A or P using codon SCC (S is C or G). L3 position 91 was changed to E using codon GAG and L3 position 92 was changed to all 20 amino acids using the degenerate codon NNK, since it was a permissive position and may allow unpredicted binding improvement. Based on off-rate screening of this library, two clones were dominant with improved values of koff. These clones had CDR L3 and H3 sequences 91EXTLPY96 and 95GGYWYATSYYFDY102, respectively, where X is H (clone ID 3E) or R (clone ID 3C), and apparent values of KD below 0.4 nM; sequence alignment of the constructs along the path from 911 to tanezumab highlight the differences generated during LSM (). Mammalian expression and purification as Fab and full-length IgG was performed for 3E and 3C to perform complete kinetic analysis. 3E has superior affinity for NGF and became the lead antibody—the clinical candidate, tanezumab—for functional analysis.
Table 3. Sequence alignment of CDRs from 911, 8L2–6D5 (template), H19-L129 (intermediate) and tanezumab
Analyzing receptor blocking by tanezumab and the 911 antibody
The ability of both tanezumab and 911 to block NGF binding to either TrkA or p75NTR was determined by equilibrating a range of concentrations of either Fab with NGF at either 2.5 nM or 5 nM. The receptors were soluble human Fc-fusion proteins amine-coupled to the biosensor chip at low density. Tanezumab blocked NGF binding to TrkA and p75NTR where the stoichiometry of 1:1 antibody binding sites to NGF binding sites was met (). The anomalous increase in p75NTR binding at low concentrations of antibody may have reflected detection of ternary complexes of p75NTR/(NGF)2/tanezumab, which would have increased the mass and therefore increased the measured signal. 911 also blocked NGF binding to both TrkA and p75NTR binding, but less potently than tanezumab due to its lower affinity for NGF ().
To ensure that tanezumab recognized an NGF epitope functionally equivalent to that of 911, neurotrophin-dependent survival assays were performed in the presence of competing Fabs (), where the NGF concentration is within range for viable neurons (Fig. S2A) and the Fabs have been shown to be equivalent to the full-length antibody (Fig. S2B). These cell-based assays used mouse embryonic day 13 trigeminal ganglion (TG) sensory neurons, which depend on NGF-TrkA signaling for survival. Tanezumab inhibited NGF-dependent survival with an IC50 of 15 pM where NGF was at a saturating concentration (15 pM). Under the same conditions, 911 inhibited NGF with an IC50 of 400 pM (). Due to the binary nature of the live/dead neuron counting as a function of antibody concentration, the transition data was rarely captured by more than 1 data point for either 911 or tanezumab, but was reproducible.
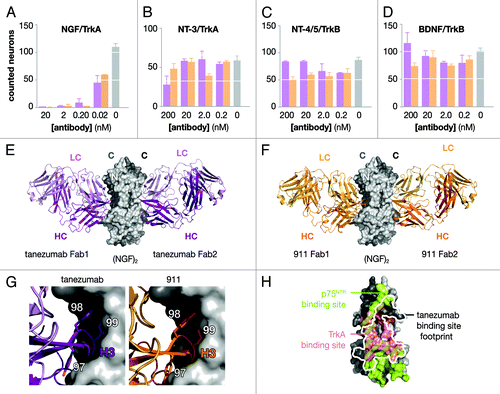
The extent of specificity of tanezumab and 911 for NGF over the other neurotrophins was evaluated by neuron survival assays specific for each of the neurotrophins. Using neurons, which specifically responded to neurotrophins via either TrkA or TrkB, cell survival assays were performed for each of the neurotrophins in competition with tanezumab or 911 (; Fig. S3). For competition with NGF and NT-3, embryonic day 18 trigeminal ganglion (TG) sensory neurons were used, which were highly responsive to NGF and marginally responsive to NT-3 via TrkA. For competition with BDNF and NT-4/5, embryonic day 17/18 nodose sensory neurons were used, which were responsive to BDNF and NT-4/5 via TrkB. With pre-incubation of neurons and within concentration ranges of 0.02–200 nM of either antibody, the most dramatic effect was observed for inhibition of NGF-dependent survival of TG neurons where 100% suppression of survival was obtained with 0.2 nM of antibody, while there was no significant effect on NT-3-dependent survival of TG neurons at 200 nM of antibody. Under the same assay conditions and using nodose neurons, neither tanezumab nor 911 affected either BDNF or NT-4/5 TrkB-dependent survival at concentrations up to 200 nM.
Figure 2. Functional and structural analysis of tanezumab and 911. Neuron survival assays (A–D) for decreasing concentrations of full-length antibody either tanezuamb (purple) or 911 (orange). The control (gray) represents neurotrophin without antibody added to the well. TG neurons express TrkA and survive in the presence of NGF (A) or NT-3 (B). Nodose neurons express TrkB and survive in the presence of NT-4/5 (c) or BDNF (d). X-ray crystal structures determined for two tanezuamb Fab (purple) bound to (NGF)2 (E) and two 911 Fab (orange) bound to (NGF)2 (F). Side-by-side comparison (G) tanezumab and 911 H3 residues 97–99. (H) Tanezumab-NGF binding site (white) overlaid “footprint” on the p75NTR (green) and TrkA (pink) binding sites on one side of (NGF)2.
Structural and functional epitope of tanezumab and 911
The molecular basis of tanezumab and 911 activities toward NGF was investigated by X-ray crystallography. Crystals of the NGF/Fab complexes were formed after purification by gel filtration. The structures were determined using molecular replacement methods, revealing the Fab2:(NGF)2 complexes (; ; Table S1; Figs. S4 and S5). The NGF monomers in complex with 911-Fab and tanezumab-Fab are essentially unchanged from the previously determined structures of free NGF. The N-terminal eight residues were disordered in the tanezumab complex and the N-terminal nine residues were disordered in the 911 complex, which was similar to the unbound NGF,Citation30 NGF/p75NTR complexCitation14 and other unbound neurotrophin structures.Citation31,Citation32 This N-terminal disorder differs from the NGF/TrkA complex structures,Citation10,Citation11 where those N-terminal residues were ordered and interacting with the TrkA receptor.
Table 4. Solvent-accessible surface areas for NGF/Fab complexes
Both tanezumab and 911 Fabs bound to the same epitope on NGF, at the interface between the NGF monomers (; Fig. S6A). The NGF epitope was composed of β-strand segments from both NGF monomers in both complexes. For each complex, 1 Fab at the (NGF)2 interface was analyzed because the structures were 2-fold symmetric either by crystallographic (911-Fab) or non-crystallographic (tanezumab-Fab) symmetry. In terms of the interactions with NGF, the tanezumab-Fab and 911-Fab are remarkably similar to each other. Both Fabs use all 6 CDRs to contact NGF, and the total size of the contact zones and of the individual variable domains are within experimental error. The single most buried Fab residue for both complexes is VH Y99, which accounts for about 14% of the buried Fab surface (Table S2).
Mutational Analysis of H3
Comparing the paratopes between tanezumab and 911 showed high structural similarity centered around H3 residue Y99. The Cα position for tanezumab H3 A100 was the same as 911 H3 G100. H3 Y100d is also in the same Cα position and side-chain conformation in both structures. These paratope features were used to design mutants to determine H3 features, which likely contribute to the higher affinity of tanezumab toward NGF compared with that of 911. Three H3 mutants were created initially to evaluate this hypothesis: a point mutant Y99A, a triple mutant W98A/Y99G/T100aG and a quadruple mutant W98A/Y99G/T100aG/Y100dA. Of these three mutants, W98A/Y99G/T100aG/Y100dA did not bind NGF at a concentration of 200 nM, whereas it bound a non-blocking anti-tanezumab antibody, 305.5A11 at 100 nM (Fig. S7). A subsequent set of either glycine or alanine mutants was designed to address the function of the H3 Cα conformational flexibility, where glycines mutations add conformational flexibility in additional to removing the side-chain interactions. Point mutants and double mutants retained NGF binding. However, the triple and quadruple mutants (W98G/Y99G/Y100dG plus T100aG, respectively) did not bind NGF but did bind 305.5A11, the non-blocking antibody. The analogous triple and quadruple Ala mutants had detectable NGF binding.
Discussion
The LSM method was developed to facilitate the high-throughput affinity maturation of antibodies in the highly diverse CDR3 region. During the preparation step, the starting antibody template 911 was humanized by first selecting the closest matching human germline sequence for each chain. Following this, a query of the antibody IMGT database allowed creation of an ad hoc alignment to guide the selection of minor mutations in CDRs 1 and 2 for further humanization, which is commonly performed.Citation28 This non-exhaustive preparatory step before LSM was completed in order to decrease the chance of immunogenicity and to improve the overall antibody structural fitness. For example, 911 L2 I51 was converted to a small amino acid in the human sequence alignment, where threonine was experimentally better than other small conserved residues, and this was retained in the optimized tanezumab.Citation25 The X-ray diffraction determined structures revealed that this residue was a good choice for structural optimization and humanization. Specifically, this CDR2 position has the same backbone conformation in both 911 and tanezumab paratopes and neither are within 6 Å of NGF, which is beyond any reasonable interaction. The humanized tanezumab amino acid, T51, is also preferred during our optimization based on 2-fold improvement in koff for both variants tested, which includes the I51T (). The additional intent of these preparative mutations was to remove potential liabilities, such as the H2 residue M50, which was an oxidation liability and was therefore replaced with several preferred amino acids including I50, where one combination with I50 also improved binding (). From this stage, very small LSM libraries were applied to L3 and H3, to identify critical or “restrictive” sites and additional “permissive” sites for improved affinity based on a high-throughput single-shot SPR assay, where improvements in off-rate were readily apparent (Fig. S1). In total, ten CDR mutations were combined during this process from 66 positions, which resulted in a 15% difference compared with the starting mouse CDRs and improved the affinity by more than 3 orders of magnitude while maintaining antibody specificity toward NGF over the closely related neurotrophins.
LSM has the potential to be applied to other antibody affinity maturation processes and could be expanded to include any CDR loop, e.g., the H2 loop, which sometimes plays an important role in binding in addition to L3 and H3.Citation33 Recent humanizations have used phage display, framework shuffling and affinity maturation from large libraries, which work well, but have the pitfalls of introducing errors of deletion or insertion or mutations that are unusual for traditional antibodies.Citation34 Other methods have used lower homology antibody frameworks with CDR grafting in combination with a softer mutagenesis technique called In Vitro Somatic Hypermutation.Citation28 Bowers and coworkers used this sophisticated and challenging method requiring mammalian HEK293 cells expressing activation-induced cytidine deaminase (AID) resulting in what is called somatic hypermutation mutagenesis (SHM). SHM mutations are directed based on in vivo sequence and phenotypic observations using flow cytometry and mammalian cell culture.Citation28 By comparison, LSM is likely to be of general applicability as a low-cost, minimal technology affinity maturation of antibodies that recognize a subset of highly homologous protein families, such as the fibroblast growth factors (FGFs), because it maintains fidelity of the binding epitope. Broader libraries, for example, risk the possibility of PCR errors, or dramatic epitope drifts in the individual CDR sequences or results in loss of a synergistic relationship between variable domain framework pairings. In the latter case, the possible adaptation of the light chain to recognize the antigen, which frequently has a minor role in binding, could degrade the binding site epitope fidelity due to the selective pressure of the binding optimization process. This could result in an undesirable light chain protrusion into the interacting epitope, which would alter the overall structural paratope and could result in a loss of fidelity toward highly homology targets such as neurotrophins.
Tanezumab and 911 blocked both TrkA and p75NTR binding to NGF and inhibited NGF-dependent neuron survival; the latter is a highly sensitive measure of function preferable to kinase signaling assays. Additionally, neither tanezumab nor 911 blocked neuron survival dependent upon the other neurotrophins: NT-3, NT-4/5 and BDNF (). This observation underscored the ability of LSM to maintain epitope fidelity for the highly redundant and identical neurotrophin family; blocking NGF over the other neurotrophins, which use the same receptors and have high homology in the p75NTR binding site,Citation15 was a high bar successfully cleared. The differences observed between tanezumab and 911 are likely due to differences in epitope affinity and may be magnified by the multi-valency of the system. For example, binding (NGF)2 results in two different binding sites that could have different affinities, which is consistent with the NGF-p75NTR and NT-3-p75NTR structures.Citation14,Citation15 Thus, one (NGF)2 could have partial occupancy at certain concentrations with a high-affinity, preferred interaction, such as the case for (NGF)2 and tanezumab, and show initially ‘sandwiching’ while blocking at saturating concentrations ().
The structures of the Fab-antigen complexes, tanezumab and 911 bound to NGF, are virtually identical and illustrate how both Fabs specifically block TrkA and p75NTR by blocking the binding site for these receptors on NGF (; Fig. S6A–C). Using the structural information combined with the results from the LSM analysis, a small number of additional mutations were designed to perturb tanezumab binding to NGF and to further delineate residues that contribute most strongly to the binding (Fig. S7). Based on the structural similarity of contact residues at the interface, we hypothesized that residues distant from the interface, in particular, the CDR3 heavy chain, along with backbone entropy were the largest contributors to the differences in the observed affinities of 911 and tanezumab toward NGF. In the LSM analysis (), H3 100A G→A improved binding without resulting in any additional interactions with NGF, suggesting that restricting the main chain conformation was beneficial to the binding affinity potentially by lowering the entropy cost upon binding to NGF. Thus, another benefit of LSM is the identification of sites that “pre-pay” for the conformational entropy of H3, thus improving affinity by having H3 restricted to bind NGF precisely and translating into binding improvements of as much as ~1,000–5,000 fold. Additionally, mutations to glycines in H3 (W98G/Y99G/Y100dG plus T100aG) resulted in a complete loss of binding to NGF at 200 nM concentration, while mutations to alanine at the same sites resulted in measurable, albeit weak, binding to NGF. This observation highlighted the role of backbone entropy on binding to antigen for H3, and suggests that backbone entropy should be considered and even exploited to improve future affinity maturation efforts where tailoring affinity is critical to function.
The mouse antibody 911 was successfully converted to the clinical candidate tanezumab using the LSM method. This high-throughput method resulted in an improvement in binding affinity for NGF of at least 2,000-fold while retaining fidelity and specificity of the epitope. Structural and mutational analysis of the two Fab:NGF complexes revealed high retention of the binding epitope and that conformational restriction of residues identified by LSM played a crucial role in the affinity improvement. Finally, biochemical and cellular assays showed a functional profile desirable in a clinical candidate. The analysis of the starting and final optimized clinical candidate tanezumab presented here and the general utility of the LSM method should be of help in guiding future efforts toward obtaining other high fidelity antibodies.
In summary, LSM has been shown to be a simple and yet powerful technology to affinity mature antibodies while maintaining high epitope fidelity during the process for developing therapeutic antibodies. Our technique significantly increased antibody affinity from ~25–50 nM to less than 10 pMCitation35 with decreased risk of epitope drift on the target, NGF, which is a valid concern when selecting clones from large, diverse Fab libraries, given their two chain format.Citation36,Citation37 LSM gave information rapidly about functional importance of amino acid positions and residues were deemed either permissive or restrictive for use in the subsequent, ultra-small library combinations for optimization. Side-by-side comparison of the tanezumab and 911 Fabs each bound to (NGF)2 () confirmed that epitope was maintained during LSM. Analysis of the tanezumab/(NGF)2 complex along with the p75NTR/(NGF)2 and TrkA/(NGF)2 complexes further illustrated why tanezumab blocks NGF signaling (Fig. S6). Further mutational analysis based upon the structures verified the role of tanezumab heavy chain CDR3 for binding NGF. Thus, by using LSM an ultra-high affinity antibody was generated that differentiated between NGF and highly similar neurotrophins, and maintained species cross-reactivity. The data presented here suggest possible wide applicability of this strategy for improvement of the affinity of lead antibodies with the goal of engineering superior human antibody therapeutics.
Materials and Methods
Antibody Fab vector and expression
The template vector used for Fab expression from E. coli periplasm was a pUC19-derived bis-cistronic vector employing the Lac-promoter, a periplasmic signal-peptide, the light chain (LC) followed by the heavy chain (HC) Hv and human IgG2a CH1 with a six-histidine purification tag and stop codon.
Fab library synthesis
Fab libraries were made using standard splicing by overlap extension PCR mutagenesisCitation38,Citation39 with doped nucleotidesCitation29 at the target positions and sub-cloned. LSM libraries were made using the same PCR method and incorporating degenerate oligonucleotides with the target position codon as NNK.
Fab preparation
Small-scale expression in 96-well plates was optimized for screening Fab libraries. Starting from E. coli transformed with Fab library, colonies were picked to inoculate both a master plate [agar LB + ampicillin (50 μg/mL) + 2% Glucose] and a working plate [2 ml/well, 96-well plate with each well containing 1.5 mL of LB and ampicillin (50 μg/mL) + 2% glucose]. Both plates were grown at 30 °C for 8–12 h. The master plate was stored at 4 °C and the cells from the working plate were pelletted at 5000 r.p.m. and re-suspended with 1 mL of LB+ampicillin (50 μg/mL) and 1 mM IPTG to induce expression of Fabs. Cells were harvested by centrifugation after 5 h expression time at 30 °C, and then re-suspended in 0.5 mL of buffer HBS-EP (10 mM HEPES buffer pH 7.4, 150 mM NaCl, 0.005% P20, 3 mM EDTA). Lysis of re-suspended cells was attained by one cycle of freezing (-80 °C) and thawing at 37 °C. Cell lysates were centrifuged at 5000 r.p.m. for 30 min to separate cellular debris from supernatants containing Fabs. The supernatants were then injected into the BIAcore 3000 to obtain kinetic parameters for ranking each Fab. Clones expressing Fabs were rescued from the master plate to sequence the DNA for future analysis.
BIAcore Assay
Affinities of anti-NGF Fabs and mAbs were determined using a BIAcore3000 ™ SPR system [GE Healthcare (formerly BIAcore, Inc.)]. CM5 chips were activated with EDC and NHS according to the supplier’s protocols. Human NGF was diluted into 10 mM sodium acetate pH 4.0 buffer and injected over the activated chip at a concentration of 0.005 mg/mL. Using variable flow time across the individual chip channels, two ranges of antigen density were achieved: 100–200 response units (RU) for detailed kinetic analyses and 500–600 RU for screening assays. The chip was blocked with ethanolamine. Regeneration studies showed that a mixture of Pierce elution buffer, (Pierce Chemicals) and 4 M NaCl (2:1 ratio) effectively removed the bound Fab while keeping the activity of hNGF on the chip for over 200 injections. HBS-EP buffer was used as the running buffer for all the BIAcore assays.
A screening assay was optimized to determine off-rate kinetics (koff) for Fab clones from the libraries (Fig. S1). Supernatants of the small culture lysates were injected at 50 uL/min for 2 min. Dissociation times of 10 to 15 min were used for determination of single-exponential dissociation rate (koff) using BIAevaluation software. Clones that showed koff values in the same range as the template (8L2–6D5, koff 1 × 10−3 s−1) were injected for confirmation and dissociation times of up to 45 min were allowed to obtain improved koff values. Clones showing improved koff values were expressed in large scale [200 mL culture volume and exploiting standard His-Tag affinity chromatography (Qiagen, Inc.)] and full kinetic parameters were determined on purified protein. The assay was capable of detecting difference in affinity that were approximately 2-fold or larger.
Neuron survival assays
The neuron survival assays were conducted on ganglia from embryos (at embryonic day 13 or 18 depending on the assay) from time-mated Swiss Webster female mouse. The trigeminal and nodose ganglia were dissected and cleaned. The ganglia were then treated with trypsin, mechanically dissociated and plated at a density of 100–300 cells per well in defined, serum-free medium in 96-well plates (Greiner Bio-One) coated with poly-L-ornithine and laminin. E13 and E18 trigeminal sensory neurons were grown either without added neurotrophins or in the presence of 0.4 ng/mL NGF (saturating concentrations for E13 and sub-saturating concentration for E18) or NT-3 (250 ng/mL, sub-saturating concentration). Triplicate cultures were set up in the presence of varying concentrations of tanezumab Fab and full-length antibodies. E18 nodose sensory neurons were grown either in the absence of neurotrophins, or with sub-saturating concentrations of NT-4/5 (0.4 ng/mL) or BDNF (0.4 ng/mL), again with and without the addition of antibodies in either previously mentioned forms. After culturing for 48 h, the total number of neurons surviving in each well under each condition was ascertained by immuno-staining with a neuron-specific antibody followed by automated-imaging and cell counting. The data represent the percentage of surviving neurons after 48 h in culture (± standard error of mean, n = 3 for each data point), relative to controls with no antibody defined as 100% (trigeminal neurons with NGF or nodose neurons with BDNF) ( and ; Figs. S2 and S3).
Measurement of antibody blocking and estimate of KDs
To test the ability of tanezumab and 911 to prevent the interaction of NGF with either of the receptors TrkA and p75, 5 nM of human NGF was pre-mixed and incubated with 0 to 50 nM of each antibody. After the incubation for 24 h, samples were injected at 10 μl/minute over a BIAcore CM5 chip with 260 RU of either amine-coupled p75NTR (channel 2) and 600 RU of TrkA (channel 3), and percent binding was determined (). Increased concentrations of either tanezumab or 911 blocked the interaction of NGF with both p75NTR and TrkA, as shown by decreased signal (measured in RU), indicating that tanezumab and 911 block the interaction of human NGF with both TrkA and p75NTR. When tanezumab concentration equaled NGF concentration (at about 5 nM NGF concentration), no NGF binding was observed (as shown by a signal of zero percent RUmax). No receptor binding was observed when concentration of NGF was equal to tanezumab concentration suggested that 5 nM NGF was at least 10-fold higher than the KD of tanezumab for NGF and at equilibrium. When the 911 concentration was much greater than the NGF concentration, although perhaps not 10-fold greater than the KD, 911 blocked both TrkA and p75NTR binding to NGF.
Structure determination and refinement for 911-Fab and tanezumab-Fab complexes
Tanezumab-Fab and 911-Fab each in complex with NGF were purified by size-exclusion chromatography (SEC) in buffer composed of 120 mM sodium chloride, 20 mM sodium acetate pH 5.5, concentrated to 10 mg/mL and subjected to sparse matrix crystallization trials. Optimized 911-Fab/NGF crystals grew from a 1:1 mixture with 20% w/v PEG 3350, 200 mM zinc acetate pH 6.3 and were preserved for cryo- data collection by boosting the PEG concentration to 30% and immersion into liquid nitrogen. Optimized tanezumab-Fab/NGF crystals grew from a 1:1 mixture with 10% w/v PEG 5000 monomethyl ether, 20% glycerol, 100 mM sodium cacodylate pH 6.8, and were preserved for cryo- data collection by using an additional 5% glycerol and immersion into liquid nitrogen. 911-Fab/NGF data was indexed and reduced in space group P1 with an NGF dimer and two 911-Fabs per asymmetric unit. Data resolution was extended to 2.5Å by using synchrotron radiation at the Stanford Synchrotron Radiation Laboratory (SSRL) beam line 11–1. Tanezumab-Fab/NGF data was indexed and reduced in space group C2221 with one NGF protomer and a single Fab in the asymmetric unit. Data resolution extending to 2.5Å resolution was collected at the Advanced Light Source (ALS) beam line 5.0.2. Data were reduced and scaled using HKL2000 and CCP4, respectively. Both tanezumab-Fab/NGF and 911-Fab/NGF were solved (PHASER) using a single NGF protomer (from pdb 1WWW) and an Fv and Fab constant region as search probes. The final refinements were performed using REFMAC5 and employed non-crystallographic restraints (where applicable) and TLS refinement (Table S1). Final coordinates and structure factors for 911-Fab and tanezumab-Fab complexes with NGF have been deposited in the Protein Data Bank (www.pdb.org) under accession codes 4EDX and 4EDW, respectively.
BIAcore mutant analysis
Surface preparation was the result of immobilization of mouse monoclonal anti-HIS onto CM5 sensor surface Anti-HIS surfaces were prepared by amine-coupling of a mouse monoclonal IgG1 anti-HIS tag antibody (R&D Systems; Catalog# MAB050) to a Biacore CM5 sensor chip surface at 25 °C on a Biacore 2000 SPR instrument. The running buffer for the immobilization procedure was HBS-T+ (10 mM HEPES, 150 mM NaCl, 0.05% Tween-20, pH 7.4). The following was performed on all flow cells simultaneously to result in all flow cells containing amine-coupled anti-HIS. The CM5 chip was activated by injecting a 1:1 (v/v) mixture of 400 mM EDC and 100 mM NHS for 7 min at a flow rate of 10 µL/min. Then, anti-HIS antibody was diluted to 50 µg/mL in 10 mM sodium acetate pH 5.0 and injected at 20 µL/min for 7 min. Then, 1 M ethanolamine pH 8.5 was injected for 7 min at 10 µL/min to block the surface. Three 30 s injections of 10 mM glycine, pH 1.7 were then performed to condition the surface. Subsequently, all binding assays were performed on a Biacore 2000 Surface Plasmon Resonance biosensor at 25 °C in a running buffer of HBS-T+ (10 mM HEPES, 150 mM NaCl, 0.05% Tween-20, pH 7.4). For a given analysis cycle, his-tagged recombinant Fab was captured onto the anti-HIS surface by injecting undiluted Fab supernatant over a given flow cell for 1 min at 10 µL/min. Different Fab samples were captured onto flow cells 2, 3 and 4 and flow cell 1 was used as a reference surface. After Fab capture, flow was initiated over all flow cells and running buffer was injected for 1 min at 10 µL/min. Then, analyte (running buffer, 200 nM rhNGF, 100 nM 305.5A11 mAb or 100 nM 305.1A12 Mab) was injected over all flow cells for 2 min at 10 µL/min. Three 30 s injections of 10 mM Glycine pH 1.7 were performed at 50 µL/min to regenerate the anti-HIS surface. Biosensor data was processed and analyzed using BIA Evaluation Software version 4.1.1. Reference subtracted response data were obtained by subtracting the response of flow cell 1 from the response of flow cells 2, 3 and 4. For a given Fab (on a given flow cell) the reference subtracted data was then double-referenced by subtracting the buffer analyte sensorgram from that of the protein analyte (mAb or rhNGF) sensorgram.
Additional material
Download Zip (2 MB)Disclosure of Potential Conflict of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We are grateful to C. Wiesmann, S. Hymowitz and P. Wu for assistance with X-ray data collection and to D. Reilly and D. Yansura for Fab expression. We thank Janet Finer-Moore at UCSF for feedback and discussions about protein interfaces, Heather Deacon (GoFigures) for figure development and Julie Lougheed for editing and revisions. Portions of this research were performed at the Stanford Synchrotron Radiation Lightsource operated for the US. Department of Energy Office of Science by Stanford University and the Berkeley Center for Structural Biology at the Advanced Light Source operated by the US. Department of Energy.
References
- Foley KM. Opioids and chronic neuropathic pain. N Engl J Med 2003; 348:1279 - 81; http://dx.doi.org/10.1056/NEJMe030014; PMID: 12660393
- Glickman-Simon R. Persistent Pain and Palliative Care. Medcsape Pharmacists 2006.
- Andersson DA, Gentry C, Alenmyr L, Killander D, Lewis SE, Andersson A, Bucher B, Galzi JL, Sterner O, Bevan S, et al. TRPA1 mediates spinal antinociception induced by acetaminophen and the cannabinoid Δ(9)-tetrahydrocannabiorcol. Nat Commun 2011; 2:551; http://dx.doi.org/10.1038/ncomms1559; PMID: 22109525
- Hefti FF, Rosenthal A, Walicke PA, Wyatt S, Vergara G, Shelton DL, Davies AM. Novel class of pain drugs based on antagonism of NGF. Trends Pharmacol Sci 2006; 27:85 - 91; http://dx.doi.org/10.1016/j.tips.2005.12.001; PMID: 16376998
- Lane NE, Schnitzer TJ, Birbara CA, Mokhtarani M, Shelton DL, Smith MD, Brown MT. Tanezumab for the treatment of pain from osteoarthritis of the knee. N Engl J Med 2010; 363:1521 - 31; http://dx.doi.org/10.1056/NEJMoa0901510; PMID: 20942668
- Shelton DL, Zeller J, Ho WH, Pons J, Rosenthal A. Nerve growth factor mediates hyperalgesia and cachexia in auto-immune arthritis. Pain 2005; 116:8 - 16; http://dx.doi.org/10.1016/j.pain.2005.03.039; PMID: 15927377
- Sevcik MA, Ghilardi JR, Peters CM, Lindsay TH, Halvorson KG, Jonas BM, Kubota K, Kuskowski MA, Boustany L, Shelton DL, et al. Anti-NGF therapy profoundly reduces bone cancer pain and the accompanying increase in markers of peripheral and central sensitization. Pain 2005; 115:128 - 41; http://dx.doi.org/10.1016/j.pain.2005.02.022; PMID: 15836976
- Pezet S, McMahon SB. Neurotrophins: mediators and modulators of pain. Annu Rev Neurosci 2006; 29:507 - 38; http://dx.doi.org/10.1146/annurev.neuro.29.051605.112929; PMID: 16776595
- Gerdin MJ, Eiden LE. Regulation of PC12 cell differentiation by cAMP signaling to ERK independent of PKA: do all the connections add up? Sci STKE 2007; 2007:pe15.
- Wiesmann C, Ultsch MH, Bass SH, de Vos AM. Crystal structure of nerve growth factor in complex with the ligand-binding domain of the TrkA receptor. Nature 1999; 401:184 - 8; http://dx.doi.org/10.1038/43705; PMID: 10490030
- Wehrman T, He X, Raab B, Dukipatti A, Blau H, Garcia KC. Structural and mechanistic insights into nerve growth factor interactions with the TrkA and p75 receptors. Neuron 2007; 53:25 - 38; http://dx.doi.org/10.1016/j.neuron.2006.09.034; PMID: 17196528
- Frade JM, Rodríguez-Tébar A, Barde YA. Induction of cell death by endogenous nerve growth factor through its p75 receptor. Nature 1996; 383:166 - 8; http://dx.doi.org/10.1038/383166a0; PMID: 8774880
- Nagata S. Apoptosis by death factor. Cell 1997; 88:355 - 65; http://dx.doi.org/10.1016/S0092-8674(00)81874-7; PMID: 9039262
- He XL, Garcia KC. Structure of nerve growth factor complexed with the shared neurotrophin receptor p75. Science (New York, NY 2004; 304:870-5.
- Gong Y, Cao P, Yu HJ, Jiang T. Crystal structure of the neurotrophin-3 and p75NTR symmetrical complex. Nature 2008; 454:789 - 93; PMID: 18596692
- Clackson T, Hoogenboom HR, Griffiths AD, Winter G. Making antibody fragments using phage display libraries. Nature 1991; 352:624 - 8; http://dx.doi.org/10.1038/352624a0; PMID: 1907718
- Boder ET, Wittrup KD. Yeast surface display for screening combinatorial polypeptide libraries. Nat Biotechnol 1997; 15:553 - 7; http://dx.doi.org/10.1038/nbt0697-553; PMID: 9181578
- Hanes J, Plückthun A. In vitro selection and evolution of functional proteins by using ribosome display. Proc Natl Acad Sci U S A 1997; 94:4937 - 42; http://dx.doi.org/10.1073/pnas.94.10.4937; PMID: 9144168
- Zahnd C, Amstutz P, Plückthun A. Ribosome display: selecting and evolving proteins in vitro that specifically bind to a target. Nat Methods 2007; 4:269 - 79; http://dx.doi.org/10.1038/nmeth1003; PMID: 17327848
- Holmes MA, Buss TN, Foote J. Structural effects of framework mutations on a humanized anti-lysozyme antibody. J Immunol 2001; 167:296 - 301; PMID: 11418663
- Holmes MA, Buss TN, Foote J. Conformational correction mechanisms aiding antigen recognition by a humanized antibody. J Exp Med 1998; 187:479 - 85; http://dx.doi.org/10.1084/jem.187.4.479; PMID: 9463398
- Amit AG, Mariuzza RA, Phillips SE, Poljak RJ. Three-dimensional structure of an antigen-antibody complex at 2.8 A resolution. Science (New York, NY 1986; 233:747-53.
- Bhat TN, Bentley GA, Fischmann TO, Boulot G, Poljak RJ. Small rearrangements in structures of Fv and Fab fragments of antibody D1.3 on antigen binding. Nature 1990; 347:483 - 5; http://dx.doi.org/10.1038/347483a0; PMID: 2215663
- Hongo JS, Laramee GR, Urfer R, Shelton DL, Restivo T, Sadick M, Galloway A, Chu H, Winslow JW. Antibody binding regions on human nerve growth factor identified by homolog- and alanine-scanning mutagenesis. Hybridoma 2000; 19:215 - 27; http://dx.doi.org/10.1089/02724570050109611; PMID: 10952410
- Ye J, Ma N, Madden TL, Ostell JM. IgBLAST: an immunoglobulin variable domain sequence analysis tool. Nucleic Acids Res 2013; 41:W34-40; http://dx.doi.org/10.1093/nar/gkt382; PMID: 23671333
- Prodromou C, Pearl LH. Recursive PCR: a novel technique for total gene synthesis. Protein Eng 1992; 5:827 - 9; http://dx.doi.org/10.1093/protein/5.8.827; PMID: 1287665
- Barbas CF. Phage display: a laboratory manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press, 2001.
- Bowers PM, Neben TY, Tomlinson GL, Dalton JL, Altobell L, Zhang X, Macomber JL, Wu BF, Toobian RM, McConnell AD, et al. Humanization of antibodies using heavy chain complementarity-determining region 3 grafting coupled with in vitro somatic hypermutation. J Biol Chem 2013; 288:7688 - 96; http://dx.doi.org/10.1074/jbc.M112.445502; PMID: 23355464
- Balint RF, Larrick JW. Antibody engineering by parsimonious mutagenesis. Gene 1993; 137:109 - 18; http://dx.doi.org/10.1016/0378-1119(93)90258-5; PMID: 7506686
- McDonald NQ, Lapatto R, Murray-Rust J, Gunning J, Wlodawer A, Blundell TL. New protein fold revealed by a 2.3-A resolution crystal structure of nerve growth factor. Nature 1991; 354:411 - 4; http://dx.doi.org/10.1038/354411a0; PMID: 1956407
- Butte MJ, Hwang PK, Mobley WC, Fletterick RJ. Crystal structure of neurotrophin-3 homodimer shows distinct regions are used to bind its receptors. Biochemistry 1998; 37:16846 - 52; http://dx.doi.org/10.1021/bi981254o; PMID: 9836577
- Robinson RC, Radziejewski C, Spraggon G, Greenwald J, Kostura MR, Burtnick LD, Stuart DI, Choe S, Jones EY. The structures of the neurotrophin 4 homodimer and the brain-derived neurotrophic factor/neurotrophin 4 heterodimer reveal a common Trk-binding site. Protein Sci 1999; 8:2589 - 97; http://dx.doi.org/10.1110/ps.8.12.2589; PMID: 10631974
- Kallewaard NL, McKinney BA, Gu Y, Chen A, Prasad BV, Crowe JE Jr.. Functional maturation of the human antibody response to rotavirus. J Immunol 2008; 180:3980 - 9; PMID: 18322207
- Fransson J, Teplyakov A, Raghunathan G, Chi E, Cordier W, Dinh T, Feng Y, Giles-Komar J, Gilliland G, Lollo B, et al. Human framework adaptation of a mouse anti-human IL-13 antibody. J Mol Biol 2010; 398:214 - 31; http://dx.doi.org/10.1016/j.jmb.2010.03.004; PMID: 20226193
- Abdiche YN, Malashock DS, Pinkerton A, Pons J. Exploring blocking assays using Octet, ProteOn, and Biacore biosensors. Anal Biochem 2009; 386:172 - 80; http://dx.doi.org/10.1016/j.ab.2008.11.038; PMID: 19111520
- Wang Z, Wang Y, Li Z, Li J, Dong Z. Humanization of a mouse monoclonal antibody neutralizing TNF-alpha by guided selection. J Immunol Methods 2000; 241:171 - 84; http://dx.doi.org/10.1016/S0022-1759(00)00203-9; PMID: 10915859
- Ohlin M, Owman H, Mach M, Borrebaeck CA. Light chain shuffling of a high affinity antibody results in a drift in epitope recognition. Mol Immunol 1996; 33:47 - 56; http://dx.doi.org/10.1016/0161-5890(95)00123-9; PMID: 8604223
- Horton RM, Cai ZL, Ho SN, Pease LR. Gene splicing by overlap extension: tailor-made genes using the polymerase chain reaction. Biotechniques 1990; 8:528 - 35; PMID: 2357375
- Horton RM. PCR-mediated recombination and mutagenesis. SOEing together tailor-made genes. Mol Biotechnol 1995; 3:93 - 9; http://dx.doi.org/10.1007/BF02789105; PMID: 7620981