
Abstract
Monoclonal antibody (mAb) products are extraordinarily heterogeneous due to the presence of a variety of enzymatic and chemical modifications, such as deamidation, isomerization, oxidation, glycosylation, glycation, and terminal cyclization. The modifications in different domains of the antibody molecule can result in different biological consequences. Therefore, characterization and routine monitoring of domain-specific modifications are essential to ensure the quality of the therapeutic antibody products. For this purpose, a rapid and informative methodology was developed to examine the heterogeneity of individual domains in mAb products. A recently discovered endopeptidase, IdeS, cleaves heavy chains below the hinge region, producing F(ab')2 and Fc fragments. Following reduction of disulfide bonds, three antibody domains (LC, Fd, and Fc/2) can be released for further characterization. Subsequent analyses by liquid chromatography/mass spectrometry, capillary isoelectric focusing, and glycan mapping enable domain-specific profiling of oxidation, charge heterogeneity, and glycoform distribution. When coupled with reversed phase chromatography, the unique chromatographic profile of each molecule offers a simple strategy for an identity test, which is an important formal test for biopharmaceutical quality control purposes. This methodology is demonstrated for a number of IgGs of different subclasses (IgG1, IgG2, IgG4), as well as an Fc fusion protein. The presented technique provides a convenient platform approach for scientific and formal therapeutic mAb product characterization. It can also be applied in regulated drug substance batch release and stability testing of antibody and Fc fusion protein products, in particular for identity and routine monitoring of domain-specific modifications.
Introduction
The recombinant monoclonal antibody (mAb) is a proven therapeutic modality, with more than 30 products approved for marketing and more than three hundred candidates under clinical development in the pharmaceutical industry.Citation1 Marketed therapeutic mAbs are predominantly immunoglobulin G (IgG)1 molecules, with only a few IgG2s and IgG4s. IgG3 antibodies have not been developed for therapeutic purposes because of their shorter half-life, susceptibility to proteolysis in the hinge region and extensive allotypic polymorphism.Citation2 Structurally, IgGs are composed of two identical heavy chains (HCs) and two identical light chains (LCs). Functionally, IgGs consist of one crystallizable fragment (Fc) and two identical antigen binding fragments (Fabs) connected by a flexible hinge region. Each Fab consists of one LC and the N-terminal half of one heavy chain (Fd). The complementarity-determining regions (CDRs) that reside in the variable regions of LC and Fd are responsible for the antigen binding specificity. The Fc domain interacts with the neonatal Fc receptor (FcRn), which protects the IgG from degradation and contribute to its long half-life. The Fc domains of IgG1 and IgG3 are also responsible for engaging immune-mediated effector functions via interactions with various Fcγ receptors and immune molecules such as complement proteins.Citation3
Therapeutic antibody products are inherently heterogeneous due to a wide spectrum of enzymatic and chemical modifications.Citation4,Citation5 Natural cellular processing and degradation events during production and storage can lead to product variants generated by oxidation, deamidation, isomerization, glycosylation, glycation, terminal processing and cyclization. Although human IgGs are highly homologous and share more than 95% sequence homology,Citation6 each mAb drug product displays different heterogeneity due to differences in variable regions, manufacturing processes, and storage conditions. Besides IgGs, fusion proteins, which consist of the IgG Fc domain and a fusion partner protein, are also emerging as therapeutic constructs that provide improved pharmacokinetic profile compared with the fusion partner proteins alone. IgG and Fc fusion proteins are complex molecules that contain multiple domains. In biopharmaceutical development, characterization of domain-specific modifications is essential because of their potential effects on the product’s efficacy and safety. From the product control perspective, simple, fast, and robust analytical methods are required to monitor such modifications.
Peptide mapping, i.e., proteolytic digestion of protein molecules into small peptides followed by LC-MS analysis, is generally the method of choice for characterization of protein modifications. Although analysis of small peptides provides high site specificity, complicated sample preparation, lengthy chromatographic separation, and time-consuming data processing often prevent it from being used in routine monitoring of product quality. For the purpose of product characterization, results from peptide mapping need to be interpreted with caution as artifacts during lengthy digestion could occur and lead to incorrect conclusions.Citation7-Citation11 Another important issue to consider is that any correlation between modifications of different sites can be lost after the protein is cleaved into small peptides.
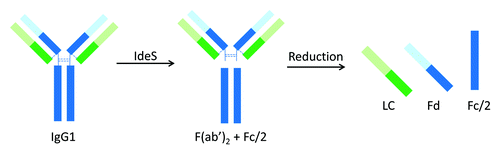
Limited proteolysis of IgG molecules is an alternative approach that can overcome those limitations of conventional peptide mapping. Because the hinge region of IgG molecules is flexible and solvent-exposed, Fc, Fab, and F(ab’)2 fragments can be readily generated by enzymatic cleavage under native condition. Reduction of these large fragments results in three antibody domains of ~25 kDa, including LC, monomeric Fc (Fc/2), and Fd. Domain-specific characterization can then be achieved by subsequent analyses. This approach has been demonstrated using proteolytic enzymes such as papain and Lys-C.Citation12,Citation13 Pepsin is another enzyme that can generate F(ab’)2 fragment; however, Fc is degraded into smaller peptides upon pepsin digestion. When using papain and Lys-C to generate antibody fragments, the digestion condition needs to be carefully controlled to achieve high yields of the desired cleavage products and avoid over-digestion. So far, limited proteolysis of antibody by Lys-C has only been reported for IgG1 molecules. Papain digestion requires enzyme activation and method optimization for IgGs of different subclasses.Citation14 Effective papain digestion of IgG2 antibodies can only be achieved following reduction of disulfide bonds in the hinge region.Citation15 However, partial reduction could occur, leading to a mixture of Fd and Fd dimer, and complete digestion of IgG2 antibodies by papain remains challenging.Citation15,Citation16
IdeS (immunoglobulin-degrading enzyme of Streptococcus pyogenes) is a recently discovered cysteine protease that cleaves at the hinge region of all IgG subclasses. IdeS digestion of an IgG1 molecule is illustrated in . Compared with the aforementioned proteases, IdeS provides a few advantages, including high site specificity, simple and robust digestion procedure, and high yield.Citation17-Citation19 Application of IdeS has been demonstrated in many literature reports on IgG1 and IgG2 molecules,Citation20-Citation28 as well as Fc fusion proteins.Citation29,Citation30 Almost all the reports to date used primarily LC-MS techniques in subsequent analysis of the resulting antibody fragments. This is because the size of the antibody fragments produced by IdeS is well-suited for so-called middle-up or middle-down structural characterization.Citation31,Citation32 Detection of antibody deamidation with a mass change of only 1 Da, however, remains challenging. Unlike HPLC and CE-based analytical techniques, MS is rarely used in a routine quality testing environment. IdeS has also been successfully applied to characterize domain-specific glycosylation at the released N-glycan level, where N-glycans released from Fc and F(ab’)2 were analyzed separately using either MS or HPLC technique.Citation33,Citation34 For example, in the characterization of intravenous immunoglobulin (IVIG, a pool of all IgG subclasses), detailed profiling of Fab and Fc glycans was achieved using HPLC-based glycan mapping method.Citation34 In that study, F(ab’)2 and Fc fragments from IdeS digestion were isolated by size exclusion chromatography (SEC).
Figure 1. Limited proteolysis of IgG1 by IdeS.
In the present study, the utility of IdeS in domain-specific characterization is expanded to the IgG subclasses of therapeutic value (IgG1, IgG2, and IgG4), as well as Fc fusion proteins. The combinations of IdeS proteolysis with HPLC, CE, and MS methods, which are the three major biochemical techniques available for therapeutic antibodies, are demonstrated. LC-MS is used to explicitly identify the IdeS cleavage site in all IgG subclasses and characterize their oxidations in individual domains. Reversed phase (RP) HPLC is used to establish the identity of each molecule studied. Capillary isoelectric focusing (cIEF) is employed to evaluate domain-specific charge heterogeneity and monitor charge profile changes caused by deamidation. HPLC-based glycan mapping is performed on N-linked glycans released from different domains of an Fc fusion protein, where the individual domains produced by IdeS cleavage can be rapidly separated using a protein A spin column without the need for HPLC fractionation.
Results
Production of IgG and Fc fusion protein domains by IdeS proteolysis
Ten IgG molecules, including five IgG1s, three IgG2s, and two IgG4s, and one Fc fusion protein were digested using IdeS and reduced by dithiothreitol (DTT). The reduced IdeS digests were analyzed by RP HPLC and the resulting chromatograms are shown in . Three major chromatographic peaks were observed for each IgG molecule, representing the three released domains – LC, Fd, and Fc/2. The reduced IdeS digest of the Fc fusion protein, namely F1, showed two chromatographic peaks corresponding to Fc/2 and its fusion partner. All peaks were identified by online MS analysis and the results are summarized in . The mass of the F1 receptor domain is not listed because it contains multiple glycosylation sites and displays a very heterogeneous mass distribution. A single cleavage site can be identified for each molecule based on the measured mass of the Fc/2 and Fd domains. A summary of the identified cleavage sites is shown in .
Figure 2. RP HPLC of IdeS digested IgG1 (A1-A5), IgG2 (B1-B3), IgG4 (C1-C2), and Fc fusion protein (F1) molecules.
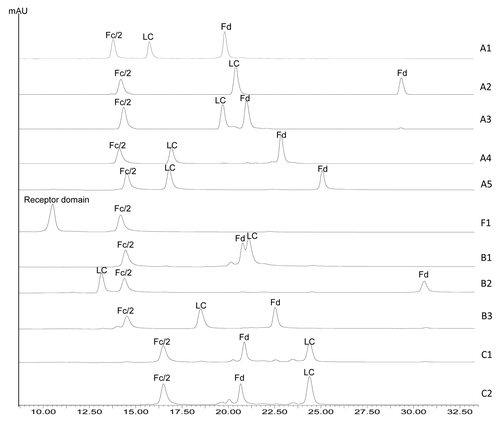
Table 1. Identification of IgG and Fc fusion protein domains by RP LC-MS
Figure 3. IgG hinge region sequence comparison. IdeS cleavage sites identified by LC-MS are indicated by vertical lines.

Identification assay for IgG and Fc fusion proteins
The individual domains of each IgG or Fc fusion protein are well resolved by RP separation except for mAb B1 (). The Fc/2 domains of all IgG1 and IgG2 molecules elute at ~13.6–14.5 min, whereas those of IgG4 molecules elute at ~16.5 min. The retention times of Fd and LC domains vary significantly regardless of the IgG subclass. The retention times of the three domain peaks are shown in . For any given pair of molecules in this study, there was at least one peak having a retention time difference of 1 min or greater. The only pair of molecules that showed comparable retention times for all three domain peaks is mAbs C1 and C2. mAb C2 is a wild type IgG4 and mAb C1 has identical amino acid sequence to mAb C2 except for a S228P mutation in the hinge region. The engineering of S228 to P228 is an effective approach to prevent Fab arm exchange commonly observed for IgG4 antibodies.Citation35,Citation36 MAbs C2 and C1 thus represent the wild type and the commonly adopted core-hinge mutation constructs of IgG4 therapeutic candidates. Although there is only a single amino acid difference between mAbs C1 and C2, a slight difference in the Fd retention time can be noticed in , demonstrating the differentiating power of this method. In conclusion, the method described here can be potentially used as an identity test for antibody or Fc fusion protein therapeutics.
Table 2. Retention times of the three major peaks in HPLC and corresponding identified domains
Domain-specific methionine oxidation
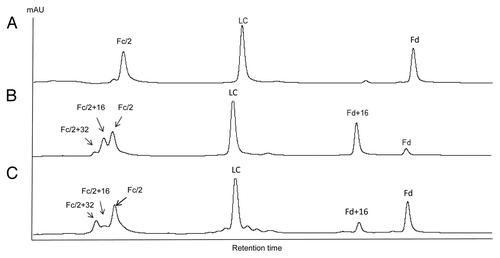
The susceptibility of methionine residues to oxidation was evaluated by forced oxidation of IgG antibodies. Following peroxide or light exposure, oxidized IgG molecules were digested with IdeS, reduced by DTT, and analyzed by RP LC-MS. The resulting RP chromatograms of mAb A2 before and after tert-butylhydroperoxide (tBHP) treatment are shown in and . All peaks were identified by online MS. It is known that tBHP oxidizes methionine residues specifically.Citation37-Citation39 For tBHP-treated A2, two peaks eluting immediately before Fc/2 were observed. Those two peaks had molecular weight increases of 16 Da and 32 Da, indicating oxidation of one and two methionine residues, respectively. There was no oxidized LC detected. The Fd peak decreased significantly, with concurrent increase of a singly oxidized Fd peak. mAb A2 contains one methionine in LC (M4), three methionines in Fd (M34, M83, and M101), and three methionines in Fc (M257, M363, and M433). The results indicated that M4 on LC was not susceptible to oxidation and at least two of the three methionine residues on Fc could be readily oxidized. Although the majority of Fd was converted to singly oxidized Fd, no double or triple oxidations were detected following tBHP treatment. This observation indicated that one of the three methionines on Fd had high susceptibility to oxidation, whereas the other two methionines were almost completely resistant to oxidation upon tBHP treatment.
Figure 4. IdeS domain mapping of mAb A2 by RP LC-MS. A, untreated; B, tBHP treated; C, exposed to 0.5X ICH light exposure level.
Using the approach outlined above for mAb A2, the susceptibility to oxidation of methionine residues in Fc/2, Fd, and LC domains was compared between antibodies of different IgG subclasses. For ease of comparison, methionine residues are numbered based on EU numbering systemCitation40 in the discussions.
The Fc/2 oxidation profiles of five IgG1, two IgG2, and one IgG4 molecules are compared. The chromatographic region where Fc/2 and its oxidized forms elute is shown in . mAb B2 is not included because oxidized Fc/2 peaks are only partially resolved from the LC peak. mAb C2 is not included because it has identical amino acid sequence to mAb C1 except for a S228P mutation. The oxidation profile of mAb C2 is virtually identical to that of mAb C1. The %Peak Area of Fc/2 (0 oxidation), Fc/2+16 Da (1 oxidation), and Fc/2 + 32 Da (2 oxidations) is shown in .
Figure 5. Comparison of tBHP induced oxidation on Fc. RP chromatographic regions of Fc/2 related peaks are presented. Highly conserved Fc methionine residues are denoted by black dots and the non-conserved methionine residue is denoted by gray dot.
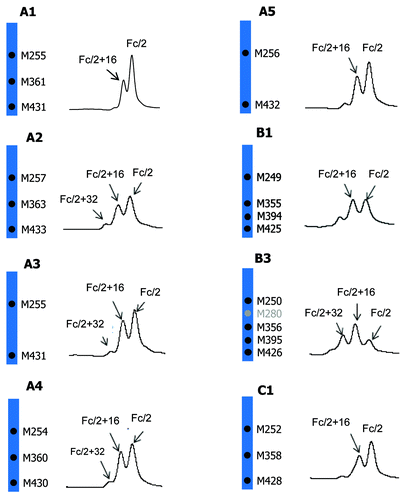
Table 3. Percent oxidation of Fc/2 by HPLC peak area
As shown in and , all IgG1 antibodies (A1, A2, A3, A4, and A5) showed significant increases in singly oxidized Fc/2, accompanied with small increases in doubly oxidized Fc/2. The strictly conserved Fc methionine residues, M252 and M428, are present in all five IgG1 antibodies whereas methionine M358 is present in A1, A2, and A4 only (). All five IgG1 antibodies showed comparable increases in singly and doubly oxidized Fc/2 peaks. These results suggested that M358 did not oxidize to any appreciable level under the stress condition used. This finding is consistent with literature indicating that the propensity to oxidation of Fc methionine residues follows the order of M252 > M428 > M358 when treated with tBHP.Citation41 The IgG4 antibody, mAb C1, showed a similar oxidation level to the IgG1 antibodies. In contrast, the oxidation levels were noticeably higher in IgG2 antibodies B1 and B3. Besides the three methionine residues present in IgG1 and IgG4 (M252, M358, and M428), IgG2 antibodies have one additional conserved methionine residue at M397. mAb B3, which showed the highest oxidation levels, has one more methionine in the Fc domain. The higher oxidation levels in IgG2 antibodies B1 and B3 were likely due to the oxidation of the additional methionine residue(s).
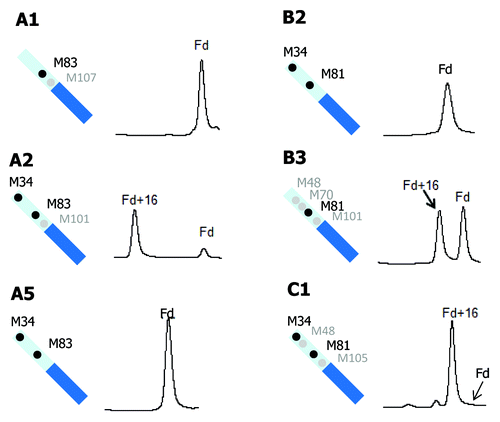
Methionine oxidation in the Fd domain was also compared for three IgG1 (A1, A2, and A5), two IgG2 (B2 and B3), and one IgG4 (C1) antibodies (). MAbs A3 and B1 are not included because the Fd or the oxidized Fd peaks are only partially resolved from the LC peaks. mAb A4 is not included because three of its four Fd methionine residues do not align with those of any other molecules studied. Of the six antibodies shown in , mAbs A1, A5, and B2 showed no detectable oxidation in their Fd domain. MAbs A5 and B2 both have two methionine residues in their Fd domain, M34 and M81. These two methionine residues appeared to have very low tendency to oxidation. Following tBHP treatment, the Fd domain in mAb A2 was almost completely converted to its singly oxidized form. Compared with mAbs A5 and B2, the Fd domain of mAb A2 has one additional methionine residue at M101. The methionine M101 in mAb A2 is located in HCDR3 and likely solvent exposed, which can readily explain the drastic increase in oxidation. Despite the very high single oxidation level of Fd in mAb A2, there was no detectable doubly oxidized form (see for full chromatogram). This observation confirmed that M34 and M81 were largely resistant to oxidation, and the oxidation of Fd in mAb A2 was predominantly caused by M101 oxidation. MAbs B3 and C1 also have a methionine residue in their HCDR3 region, M101 and M105, respectively. Like mAb A2, both mAbs B3 and C1 showed great increases in singly oxidized Fd without detectable double oxidation. It is thus reasonable to conclude that M101 or M105 in HCDR3 have high propensity to oxidation and the other methionine residues in Fd (M34, M48, M70, and M81) are far less prone to oxidation. Peptide mapping of tBHP-stressed A2, B3, and C1 confirmed that M101 or M105 is the dominant oxidation site, and the other Fd methionine residues in those molecules are not oxidized to any appreciable level (data not shown). mAb A1 also has one methionine residue (M107) in the HCDR3 region. In contrast to mAbs A2, B3, and C1, there was no detectable Fd oxidation in mAb A2. The crystal structure of mAb A1 indicates that M107 is buried in the Fab interface, which will be discussed in detail later.
Figure 6. Comparison of tBHP induced oxidation on Fd. RP chromatographic regions of Fd related peaks are presented. Highly conserved Fd methionine residues are denoted by black dots and non-conserved methionine residues are denoted by gray dots.
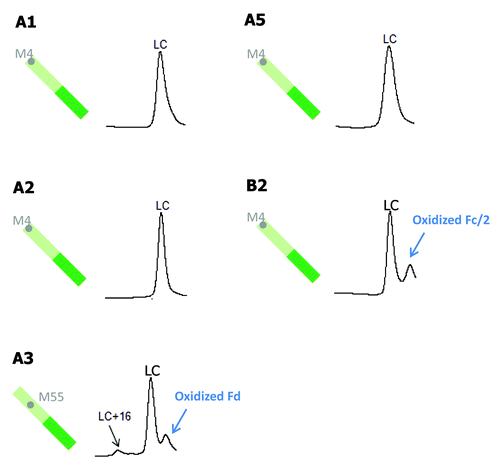
In , LC methionine oxidation following tBHP treatment is compared for five antibodies (A1, A2, A3, A5, and B2). MAbs A4, B1, B3, C1, and C2 do not have any methionine residues on their LC. Of the five antibodies shown in , mAbs A1, A2, A5, and B2 all have one LC methionine at the same position, M4. There was no detectable LC oxidation for those four antibodies, indicating that M4 is likely buried in the high order structure. The only antibody that showed a slight increase in LC oxidation is mAb A3, which has one LC methionine residue (M55) in LCDR2.
Figure 7. Comparison of tBHP induced oxidation on LC. RP chromatographic regions of LC related peaks are presented. Grey dots denote non-conserved methionine residues on LC.
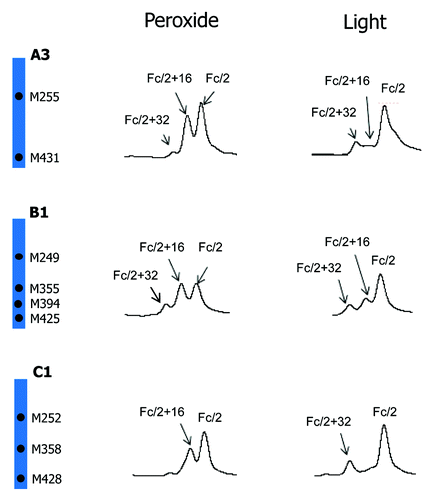
Besides peroxide, protein oxidation can also be induced by light exposure. Oxidation profiles of mAb A2 induced by light and tBHP is compared in . A noticeable difference between peroxide and light-induced oxidation profiles is the relative distribution of singly and doubly oxidized Fc/2. Whereas tBHP treatment led to mainly singly oxidized Fc/2, light stress induced mainly doubly oxidized Fc/2. Because light stress may also cause oxidation of amino acid residues other than methionine, such as tryptophan, peptide mapping was performed to examine the sites of oxidation. It was found that, following light exposure at 0.5X ICH level, all detectable Fc oxidation occurred on methionine residues (26% on M257, 5% on M363, and 19% on M433). These results suggested that Fc methionine oxidations induced by tBHP treatment are mainly independent events and those induced by light exposure were more correlated. A similar conclusion has been made about a different IgG1 antibody.Citation42 To examine if a similar phenomenon occurs on IgG of other subclasses, light stress studies were performed on another IgG1 (A3), one IgG2 (B1), and one IgG4 (C1). The comparison of their oxidation profiles induced by tBHP and light is presented in . Similar to mAb A2, both IgG1 A3 and IgG4 C1 antibodies showed greater increases of doubly oxidized Fc/2 than singly oxidized Fc/2 following light stress. Although doubly oxidized Fc/2 of IgG2 antibody B1 was only comparable to or slightly lower than that of singly oxidized Fc/2, the relative abundance of doubly oxidized Fc/2 induced by light was noticeably higher than that induced by tBHP treatment.
Figure 8. Comparison of tBHP (left) and light (right, 1X ICH light exposure level) induced oxidation on Fc. RP chromatographic regions of Fc/2 related peaks are presented. Highly conserved Fc methionine residues are denoted by black dots.
Domain-specific charge profiling
Asparagine deamidation is a common degradation pathway that introduces charge heterogeneity in protein molecules. Asparagine deamidation can be accelerated at high pH or high temperature. In this study, cIEF analysis of an IgG1 antibody (A1) was used as an example to demonstrate domain-specific charge profiling by IdeS digestion or partial reduction of the antibody molecule.
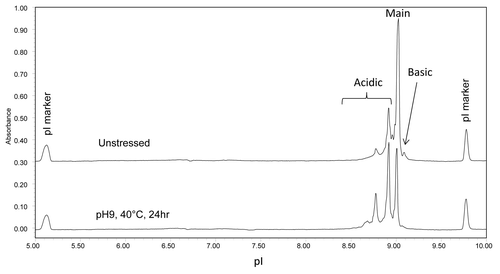
mAb A1 has identical amino acid sequence to trastuzumab (Herceptin®) and contains potential deamidation sites at N30 on LC, N55 on Fd, and N318, N387, N392 on Fc. To investigate the relative susceptibility of the asparagine residues in different domains to deamidation, mAb A1 was stressed at pH 9 and 40 °C for 24 h. shows the cIEF electropherograms of mAb A1 before and after high pH stress. As seen in , high pH stressed mAb A1 showed a significant increase in acidic variants, suggesting extensive deamidation.
Figure 9. cIEF electropherogram overlay of unstressed (top) and high pH stressed (bottom) mAb A1.
The mAb A1 samples were then partially reduced by DTT under native conditions that are known to reduce almost all inter-chain disulfide bonds without reducing intra-chain disulfide bonds.Citation43 Because protein isoelectric point can be affected by the number of free sulfhydryl groups and its higher order structure, the partial reduction condition is adopted to minimize additional charge groups and conformational changes. The cIEF electropherograms of partially reduced mAb A1 is shown in . Because of the strong Fab interface interactions, partially reduced mAb A1 appeared as intact antibody molecule in the electropherogram under native cIEF condition (0 M urea). To separate LC and HC peaks, increasing amounts of urea were added to the cIEF running buffer. As shown in , separated LC and HC peaks became visible when urea concentration was increased to 5 M. Complete separation of LC and HC peaks was achieved with 7 M urea in the running buffer. Further increase of urea concentration to 8 M did not reduce the number of peaks, but caused peak distortion.
Figure 10. Effect of urea concentration on cIEF analysis of partially reduced mAb A1.
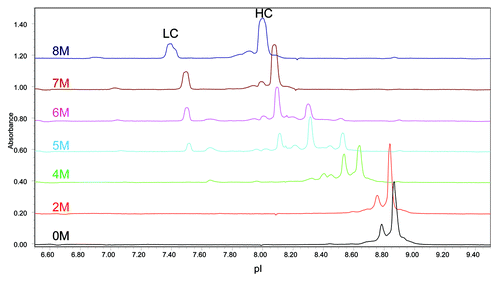
Domain-specific charge profiles of mAb A1 were determined for LC () after partial reduction, Fc () after IdeS digestion, and Fd () after IdeS digestion and partial reduction. To separate LC from HC or Fd, 7 M urea was added to the running buffer to generate the electropherograms shown in . When analyzing the IdeS digest by cIEF in the presence of 7 M urea, the acidic peak of Fc split into two peaks and moved closer to the native Fc peak (data not shown). Therefore, the two unresolved peaks at ~7.6 in were assigned as acidic variant peaks of Fc and the peak at ~7.4 was assigned as acidic variant peak of LC. The increase in %Acidic species of individual domains is summarized in . As can be seen in and , most of the acidic species induced by high pH stress came from LC. There was a moderate increase in acidic species of Fc and only a slight increase in acidic species of Fd. Despite the great increase in LC %Acidic, there was no double deamidation peak detected for LC (). This observation was consistent with the single deamidation site (N30) on LC.
Figure 11. cIEF electropherogram overlays of unstressed (top) and high pH stressed (bottom) mAb A1 following (A) partial reduction, (B) IdeS digestion, and (C) IdeS digestion and partial reduction. Acidic species are denoted by “-A.”
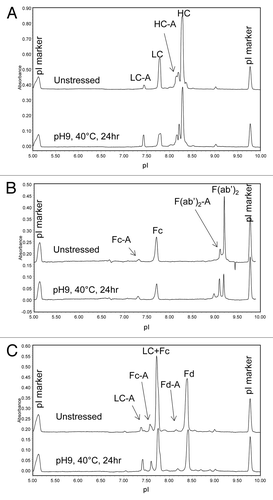
Table 4. Percent Acidic species of mAb A1 domains by cIEF analysis
The results indicate that the propensity of different domains to deamidation in mAb A1 follows the order of LC >> Fc > Fd. In an earlier study, CEX peaks of trastuzumab were characterized in detail.Citation44 The majority of the CEX acidic species were assigned to deamidation on LC N30. Deamidation of Fd N55 was also detected, but at much lower abundance. Information on Fc deamidation of trastuzumab was not provided in that study.
Domain-specific N-Glycan profiling
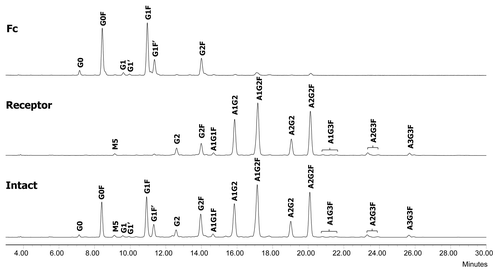
The F1 molecule is a dimeric Fc fusion protein composed of the Fc domain of human IgG1 and the soluble domain of a receptor protein. The fusion protein F1 contains six N-glycosylation sites, three on each half-molecule. In addition to the conserved N-glycosylation site in the Fc domain, there are two N-glycosylation sites in the receptor domain. In order to characterize and monitor domain-specific glycosylation, the fusion protein F1 was digested by IdeS. The resulting Fc/2 and receptor domains were then separated by a protein A spin column. The receptor domain was collected in the flow throw fraction and the Fc/2 domain was collected in the eluate (). Both fractions were at high purity as determined by SEC (>97%, data not shown). The glycosylation profiles of isolated receptor and Fc/2 domains were determined by N-glycan mapping. As shown in , there was a clear segregation of neutral and charged N-glycans released from the Fc/2 and the receptor domains. The majority of the Fc N-glycans are neutral biantennary N-glycans such as G0F, G1F, and G2F. In great contrast, the receptor domain possesses primarily sialylated N-glycans. Except for G2F, all glycoforms are present primarily in only one of the two domains. Because the Fc glyans are partially sequestered within the CH2 domains, it is as expected that they are less completely processed than those in the more exposed receptor domain.
Figure 12. Illustration of the experimental procedure for Receptor and Fc domain-specific N-glycan mapping of the Fc fusion protein F1.
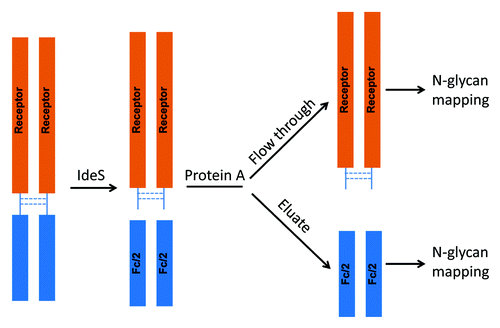
Figure 13. N-glycan profiles of the Fc fusion protein F1 (bottom) and its receptor (middle) and Fc (top) domains. G, non-reducing end galactose; F, core fucose; A, sialic acid; M, high mannose.
Discussion
The IdeS cleavage site on IgG1 molecules has been determined experimentally by N-terminal sequencing and LC-MS, and found to be at the GG bond of the hinge sequence PKSCDKTHTC PPCPAPELLG GP.Citation17,Citation45 For IgG2 molecules, putative cleavage sites on the AG and GP bonds of the hinge sequence RKCCVECPPC PAPPVAGP have both been proposed.Citation18,Citation19 IdeS cleavage at the AG bond of IgG2 molecule is more likely based on structural modeling and sequence alignment with IgG1. Similarly, the IdeS cleavage site on IgG4 molecules is likely at the GG bond of the hinge sequence SKYGPPCPSC PAPEFLGGP. There is, however, no published sequencing or MS data in literature confirming the cleavage sites on IgG2 and IgG4 molecules. In the current work, a number of IgG antibodies of different subclasses and an IgG1 Fc fusion protein were subjected to IdeS proteolysis followed by reduction and online LC-MS analysis. The results from this study showed that the IdeS cleaves IgG1, IgG2, and IgG4 molecules at a single site at the GG, AG, and GG bonds in the hinge region, respectively. The IgG4 hinge region S228P substitution does not affect IdeS cleavage. This substitution is a common strategy to prevent Fab arm exchange of IgG4 in vivo.Citation35,Citation36 Although IdeS demonstrated strict substrate specificity to IgG and IgG Fc domain with a precise cleavage site (GG or AG bond) at the lower hinge region, it appeared to be able to tolerate certain variations in the hinge region. For example, there appeared to be no effect on IdeS digestion when the hinge region cysteines were replaced with serine or conjugated to small molecules.Citation25,Citation30 It is thus not surprising that engineering of wild type IgG4 CPSC segment to that of IgG1 and IgG2 (CPPC) does not affect the cleavage, as shown in the current study.
Domain mapping of IgG molecules by IdeS proteolysis provides a useful alternative to peptide mapping for antibody characterization. The simplicity in sample preparation and data analysis is better suited for routine monitoring of antibody product quality. In comparison with other means of generating antibody domains, IdeS has the advantages of high site specificity, a robust digestion procedure readily applicable to all IgG subclasses, and digestion at native conditions that allow subsequent functional studies. The Fc/2, Fd and LC domains released from each molecule by IdeS proteolysis and subsequent DTT reduction presented unique chromatographic patterns in RP separation. Except for the two IgG4 antibodies differing in only one amino acid in the hinge region, all molecules tested in this study can be unambiguously distinguished from each other. The procedure outlined here can therefore be used as an identification test of IgG and Fc fusion protein products.
Methionine oxidation is one of the most common post-translational modifications of protein molecules, and it is the subject of extensive studies in literature. Oxidation of methionine residues can be induced under thermal, peroxide, or light stress conditions leading to methionine sulfoxide. Further oxidation to methionine sulfone is rare, and requires much harsher oxidative stress. Methionine oxidation in Fc domain of mAbs can alter the higher order structure and affect stability,Citation46 binding to Protein A and Protein G,Citation41,Citation47 and affinity to FcRn.Citation41,Citation48,Citation49 Although Fc methionine oxidation does not appear to affect binding to FcγRs to any appreciable extent,Citation48 oxidation of methionine residues in CDRs could affect binding of the antibody to its ligand. Therefore, methionine oxidation in antibody therapeutics could adversely affect their stability, purification, serum half-life, and biological activity.
Oxidation of methionine side chain to sulfoxide results in decreased hydrophobicity, and proteins containing oxidized methionine can be separated from their non-oxidized forms by RP chromatography. The mass addition of 16 Da in a methionine-containing antibody or antibody domain is characteristic of methionine sulfoxide. In this study, a number of IgG antibodies of different subclasses were exposed to chemical oxidation stress using tBHP. It has been found that tBHP oxidize methionine residues specifically,Citation37-Citation39 and site susceptibility to oxidation upon tBHP treatment correlates with that under thermal stress.Citation50 Chemically-stressed IgG molecules were analyzed by IdeS domain mapping with online MS detection. The results indicate that, among the highly conserved Fc methionine residues, M252 and M428 have higher susceptibility to chemically-induced oxidation than M358. The additional methionine conserved in IgG2 antibodies, M397, also showed a noticeable propensity to oxidation upon tBHP treatment.
Of the 10 IgGs tested, nine showed no detectable oxidation on LC, including four IgGs containing LC M4. Only the IgG molecule containing LC M55 in LCDR2 showed a slight increase in oxidation under the chemical oxidation stress condition.
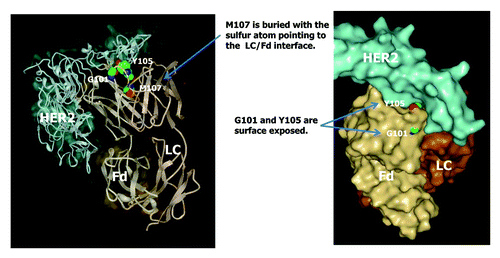
The common Fd methionine residues M34, M48, and M81 are not prone to oxidation even though M34 is located in HCDR1. Methionines in HCDR3 showed drastically different susceptibility to chemically-induced oxidation. Whereas M101 or M105 in three of the IgG antibodies studied (mAbs A2, B3, and C1) showed very high propensity to oxidation, M107 in mAb A1 showed no detectable oxidation following tBHP treatment. In order to investigate the different susceptibility of HCDR3 methionine residues to oxidation, the crystal structure of a commercial IgG1 antibody therapeutics, trastuzumab, is closely examined. mAb A1 has identical amino acid sequence to trastuzumab and is expected to have the same crystal structure. The crystal structure of the HER2 ligand and the Fab domain of trastuzumab is shown in . As can be seen in , M107 in trastuzumab is buried with the sulfur atom pointing to the LC/Fd interface. Assuming similar crystal structures of mAbs A2, B3, and C1 in the variable domain, the M101 and M105 residues (corresponding to G101 and Y105 in trastuzumab) are likely surface exposed. Therefore, the difference in the propensity to oxidation of HCDR3 methionine residues can be explained by the degree of solvent exposure. Further examination of trastuzumab crystal structure revealed that amino acids in the G101-Y105 region are all surface exposed, G100 and A106 are partially exposed, and the rest of the amino acids in HCDR3 (W99, M107 to Y109) are completely buried. Based on the results from this study, it can be speculated that methionine residues in alignment within the G101-Y105 region of trastuzumab are likely to have high susceptibility to oxidation regardless of IgG subclass.
Figure 14. Crystal structure of trastuzumab (Herceptin®) Fab in complex with the extracellular domain of human HER2 (pdb code: 1N8Z). Sphere presentations are shown for Fd amino acid residues G101, Y105, and M107.
Besides chemical stress by peroxide, light exposure of protein molecules can also induce methionine oxidation via a few possible mechanisms. The protein molecule may dissipate the absorbed light energy causing an increase in temperature and consequent methionine oxidation. Alternatively, primary photo-oxidation of photo-sensitive residues such as tryptophan, tyrosine, and phenylalanine may lead to indirect oxidation of methionine via formation of reactive oxygen species such as peroxy radicals.Citation51 It has also been suggested that methionine could be oxidized by singlet oxygen generated by the reaction between molecular oxygen and photo-activated polysorbate in protein formulation.Citation52 Oxidation of an IgG1 antibody was studied under different stress conditions to examine the susceptibility of M252 and M428 in the Fc region to oxidation.Citation42,Citation50 It was found that M252 and M428 were oxidized randomly when treated with tBHP, but were oxidized preferentially on the same HC following thermal and light stresses. Results from the current study indicate that light-induced M252 and M428 oxidation is correlated in IgG2 and IgG4 as well. Because of the close proximity of M252 and M428 in the crystal structure,Citation41 it is possible that a light-induced oxidizing intermediate such as persulfoxide on one methionine residue could oxidize the other methionine, forming two methionine sulfoxides.Citation53 In contrast, methionine oxidations by peroxide appeared to be independent events in all three IgG subclasses (IgG1, IgG2, and IgG4) studied here. Oxidation of methionine by peroxide may proceed via an acid-catalyzedCitation53 or water-mediated mechanism,Citation54 where oxygen is transferred from peroxide to the nucleophilic sulfide on methionine side chain through a nucleophilic substitution.
Charge variants are commonly observed in IgG antibodies, including therapeutic mAbs.Citation55,Citation56 Many of the post-translational modifications could lead to charge variants either directly by changing the net charge on the molecule or indirectly by altering protein structure, and thus pKa of charged residues. Deamidation is one of the common modifications that introduce charge heterogeneity in mAbs. Despite the advancement in MS instrumentation and methodology, characterization of deamidation on intact antibody or large antibody fragments remains challenging due to the small mass increment post-deamidation (+1 Da). Separate charge profiles of Fab and Fc domains have been obtained using limited Lys-C or papain proteolysis in combination with CEX separation.Citation57-Citation61 It is known that the variable regions of both LC and HC could contribute to the overall charge heterogeneity of the antibody product,Citation55,Citation56 and deamidation of LC and HC CDR asparagine residues could cause loss in bioactivity.Citation44,Citation62 There is, however, no previous report on domain-specific charge profiling of LC and Fd domains. In this study, charge heterogeneity in individual LC, Fd, and Fc domains of an IgG1 antibody, mAb A1, were studied by cIEF analysis following IdeS proteolysis or partial reduction. Because of the strong Fab interface interactions, charge profiling of individual LC and Fd domains can only be achieved under somewhat denaturing conditions. Addition of 7 M urea in the cIEF running buffer was found to be sufficient to disrupt the non-covalent interactions between LC and HC or Fd. When applied in high pH stress studies, IdeS proteolysis in combination with denaturing cIEF provides a convenient approach to rank the susceptibility of different domains to deamidation. In comparison with CEX chromatography, cIEF separation of charge variants is faster and does not require method development for individual molecules. Therefore, the methodology developed here is readily applicable to other antibody products, either for assessment of domain sensitivity to deamidation or for routine monitoring of domain-specific charge profiles.
Another common post-translational modification studied here is N-glycosylation. Human IgG antibodies possess a conserved N-glycosylation site at N297 in Fc. Approximately 15–20% of human IgG also contain N-linked glycans in the Fab region.Citation6 One therapeutic antibody, cetuximab (Erbitux®), bears an N-linked glycan at N88 of the Fd domain. Fc fusion proteins, such as etanercept (Enbrel®), can also have N-linked glycans on the Fc fusion partner. N-linked glycans can be highly heterogeneous and vary widely between different cell lines, clones, production conditions, as well as between different sites of the same molecule. Glycosylation of protein products has diverse roles in protein folding, stability, function, and PK behavior. N-glycan in different domains could have different safety and efficacy consequences. It is thus important to use quantitative and convenient approaches to obtain a comprehensive assessment of glycoform distribution on a routine basis.
Application of IdeS in glycan profiling has been reported in a number of publications.Citation20-Citation25,Citation30,Citation33,Citation34,Citation63 In most reported studies, antibody fragments were analyzed by LC-MS techniques directly to determine glycoform distributions. Whereas MS-based methods are superior in speed and sample consumption, the complexity in operating MS instruments and in data analysis often prevent them from being used in routine quality tests. More accurate relative quantitation of different glycoforms, including minor glycan species, is often achieved in analysis of released glycans using HPLC or CE-based techniques. In a study on IVIG, detailed profiling of Fab and Fc glycans was achieved on released and fluorescently labeled glycans.Citation34 In that study, F(ab’)2 and Fc fragments were isolated by SEC.
We present here a simpler approach to provide detailed domain-specific glycan profiling. The Fc and the receptor domains of an Fc fusion protein produced by IdeS proteolysis was conveniently isolated by a protein A spin column in bind-and-elute mode. Glycans were then released from the isolated Fc and receptor domain fractions and the fluorescently labeled glycans were separated by HPLC. The complex glycoforms in this Fc fusion protein were clearly segregated in the two domains. The Fc domain possesses primarily neutral fucosylated bianntenary complex glycans and the receptor domain contains mostly sialylated glycans with or without core fucosylation. The same approach may be applied to IgG antibodies as well, where the F(ab’)2 and Fc domains can be isolated in the protein A flow-through and eluate fractions, respectively.
IdeS proteolysis is shown to be a convenient tool in the characterization of IgG and Fc fusion protein domain-specific modifications. IgG domain mapping by IdeS proteolysis provides advantages over the conventional peptide mapping methods, including simple sample preparation, ease of data interpretation, fewer method artifacts, and information on site correlation. Subsequent analysis of antibody domains produced by IdeS proteolysis are demonstrated using analytical techniques, including liquid chromatography such as RP HPLC, electrophoresis such as cIEF, and glycan mapping following simple domain isolation by protein A affinity spin column. The combination of IdeS proteolysis with those QC-compatible methods allows routine testing of product identity and domain-specific modifications. When coupled with forced degradation studies such as peroxide, light, pH, and thermal stresses, susceptibility of domain-specific sequence hot-spots to different degradation pathways can be rapidly screened for a large number of therapeutic candidates. It is expected that IdeS proteolysis will have broader application in product characterization, purity and identity tests in release and stability studies, developability assessment, characterization of biosimilar mAbs and Fc fusion proteins,Citation64 and other areas of biopharmaceutical development.
Materials and Methods
IdeS (FabRICATOR® Genovis), dithiothreitol (DTT), 2-mercaptoethanol (BME), isopropanol (IPA), acetonitrile (ACN), trifluoroacetic acid (TFA) were purchased from Fisher Scientific. TRIS-HCl (1 M, pH 8.0), ammonium hydroxide, methylcellulose (MC), and sodium dodecyl sulfate (SDS) were purchased from Sigma-Aldrich. NAb Protein A plus spin column, Zeba spin desalting column (7 K MWCO), formic acid, 8 M guanidine hydrochloride (GuHCl), and NP-40 were purchased from Thermo Scientific. Peptide N glycosidase F (PNGase-F) and LudgerTag™ 2-AB (2-aminobenzamide) Glycan Labeling Kit were purchased from QA-Bio, LLC. tBHP (70%) was purchased from Acros Organics. All other chemicals were analytical reagent grade or higher. All IgGs and the Fc fusion protein used in this study were expressed in Chinese hamster ovary (CHO) cells and purified by chromatographic steps at Merck.
IdeS digestion
One unit of IdeS (67 unit/µL) was added to each µg of protein. The protein/enzyme mixture was then diluted with a dilution buffer (50 mM sodium phosphate, 150 mM NaCl, pH 6.6) to a final protein concentration of 10 mg/mL and incubated at 37 °C for 60 min.
Reduction
For RP HPLC analysis, the IdeS digested sample was reduced by 20 mM DTT in 50 mM TRIS-HCl (pH 8.0) and 6M GuHCl at 56 °C for 30 min. For cIEF analysis, DTT reduction was conducted in TRIS-HCl (pH8.0) buffer without GuHCl and incubated at room temperature in dark for 10 min.
RP HPLC
The RP HPLC separation of IdeS digested and DTT reduced samples was performed on a Halo C8 column (150 × 4.6 mm, 2.7µm, MAC-MOD Analytical, Inc.). The column was held initially at 90% mobile phase A (0.1% TFA in 10% IPA) and 10% mobile phase B (0.1% TFA in 10% IPA and 90% ACN) for 1 min, followed by a 5 min gradient from 10% B to 25% B. Separation was achieved with a linear gradient from 25% B to 40% B in 30 min. The column temperature and flow rate were maintained at 75 °C and 0.25 mL/min, respectively. Typically, 10 µg of sample was injected onto the column for analysis.
LC-MS
Online MS analysis was performed in positive ion mode on a Waters Xevo Q-TOF mass spectrometer equipped with an ESI source. The capillary and cone voltages were set at 3.2 keV and 35 V, respectively. The desolvation and source temperatures were set at 370 °C and 120 °C, respectively. All of the other voltages were optimized to provide maximal signal intensity. The instrument was calibrated in an m/z range of 100–3000. All raw data were processed using Waters MassLynx MaxEnt 1 software to obtain the deconvoluted mass.
cIEF analysis
Profiling of charged variants was performed using imaging cIEF technique on an iCE3 analyzer (ProteinSimple). The separation was conducted on a 50 mm long silica capillary (100 µm i.d. and 200 µm o.d.) inner coated with fluorocarbon. The catholyte was 0.1 M NaOH in 0.1% MC and the anolyte was 0.08 M phosphoric acid in 0.1% MC. Samples were focused at 1500 V for 1 min and 3000 V for 10 min. The absorbance of the focused proteins was monitored at 280 nm. The resulting electropherograms were integrated using Empower software. Typically, 30 µL of sample at 2 mg/mL was added to 170 µL of cIEF mixing buffer containing 70 µL of 1% MC, 9 µL of pharmalyte 3–10, 1 µL of high pI marker, 1 µL of low pI marker, and 89 µL of water. For partially reduced samples, varying amount of urea was added to the cIEF mixing buffer and the volume of water was reduced accordingly to achieve the same concentrations of all other components. For example, 84 mg of urea and 26 µL of water were added to the cIEF mixing buffer to achieve final urea concentration of 7 M. CIEF mixing buffer containing different concentration of urea was prepared by adjusting urea weight and water volume accordingly.
Domain isolation by protein A spin column
NAb Protein A plus spin column was used to separate the Fc/2 and (Receptor)2 domains from an IgG1 Fc fusion protein (F1). Briefly, the IdeS digested sample was mixed with the binding buffer provided in the NAb kit and loaded onto a Protein A spin column pre-conditioned with the binding buffer. The column was capped at both ends and placed on a shaker to shake at ~400 rpm at ambient temperature for 10 min. After removing the bottom cap and loosening the top cap, the column was centrifuged at 5000 × g for 1 min. The flow through fraction containing (Receptor)2 was collected. The column was washed with binding buffer three times. The Fc/2 fraction was eluted from the column using IgG elution buffer provided in the kit. The Fc/2 fraction was neutralized to ~pH 6 using the neutralization buffer provided in the kit.
Enzymatic N-Glycan release and labeling
The (Receptor)2 and Fc/2 fractions isolated from protein A spin column were brought to complete dryness in a speedVac. The dried samples were reconstituted in 45 µL PBS (0.02 M phosphate buffer, 0.0054 M potassium chloride and 0.274 M sodium chloride, pH7.4) and incubated with 1 µL 10% BME and 1.5 µL 5% SDS at 37 °C for 10 min. The sample was digested with 10 µL PNGaseF and 5 µL NP40 at 37 °C overnight. Cold ethanol was then added to precipitate protein content. The supernatant containing N-glycans was brought to complete dryness. N-glycans were labeled with 10 µL of 2-AB labeling reagent provided in the labeling kit at 65 °C for 3 h. The excess dye was removed using a lysate plate (Promega Cat# A2241) with ACN. The N-glycans were eluted with water. After drying in a speedVac, the labeled N-glycan was reconstituted with 70% ACN.
N-Glycan profiling
Fluorescently labeled N-glycans were separated on an Acquity UPLC BEH glycan column (100 × 2.1 mm, 1.7 μm) using a Waters Acquity UPLC instrument. Five μL of sample was injected. The column was held initially at 25% mobile phase A (100 mM ammonium formate, pH4.5) and 75% mobile phase B (100% ACN). The separation was achieved using a linear gradient of 25−40% mobile phase A at 0.4 mL/min in 30 min followed by increase to 100% mobile phase A in 0.1 min and held for 4.9 min. Samples were kept at 12 °C in the autosampler prior to injection, and the column temperature was 60 °C. The fluorescence detection wavelengths were λex = 330 nm and λem = 420 nm with PMT gain of 10.
Stress conditions
Chemically-induced oxidation was conducted in 0.1% tBHP and incubated at 25 °C for 24 h. Residual tBHP was removed by buffer exchange using Zeba spin desalting column with a MW cut-off of 7 kDa.
Light induced oxidation was conducted at 25 °C using a Caron photostability chamber (Caron 4565–2). Light exposure of 0.5 X or 1 X ICH level (1X ICH level = 1200 klux-hours of white light and 200 W-hours/m2 of UV light) was applied to the studied mAbs.
High pH stress of mAb A1 was conducted by adjusting sample pH to 9.0 with 0.5N NaOH and incubated at 40 °C for 24 h. High pH stressed mAb A1 was buffer exchanged to pH 6.0 using Zeba spin desalting column prior to analysis.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Reichert JM. Marketed therapeutic antibodies compendium. MAbs 2012; 4:413 - 5; http://dx.doi.org/10.4161/mabs.19931; PMID: 22531442
- Salfeld JG. Isotype selection in antibody engineering. Nat Biotechnol 2007; 25:1369 - 72; http://dx.doi.org/10.1038/nbt1207-1369; PMID: 18066027
- Jiang X-R, Song A, Bergelson S, Arroll T, Parekh B, May K, Chung S, Strouse R, Mire-Sluis A, Schenerman M. Advances in the assessment and control of the effector functions of therapeutic antibodies. Nat Rev Drug Discov 2011; 10:101 - 11; http://dx.doi.org/10.1038/nrd3365; PMID: 21283105
- Liu H, Gaza-Bulseco G, Faldu D, Chumsae C, Sun J. Heterogeneity of monoclonal antibodies. J Pharm Sci 2008; 97:2426 - 47; http://dx.doi.org/10.1002/jps.21180; PMID: 17828757
- Beck A, Wagner-Rousset E, Ayoub D, Van Dorsselaer A, Sanglier-Cianférani S. Characterization of therapeutic antibodies and related products. Anal Chem 2013; 85:715 - 36; http://dx.doi.org/10.1021/ac3032355; PMID: 23134362
- Jefferis R. Glycosylation of recombinant antibody therapeutics. Biotechnol Prog 2005; 21:11 - 6; http://dx.doi.org/10.1021/bp040016j; PMID: 15903235
- Dick LW Jr., Mahon D, Qiu D, Cheng K-C. Peptide mapping of therapeutic monoclonal antibodies: improvements for increased speed and fewer artifacts. J Chromatogr B Analyt Technol Biomed Life Sci 2009; 877:230 - 6; http://dx.doi.org/10.1016/j.jchromb.2008.12.009; PMID: 19112052
- Lippincott J, Apostol I. Carbamylation of cysteine: a potential artifact in peptide mapping of hemoglobins in the presence of urea. Anal Biochem 1999; 267:57 - 64; http://dx.doi.org/10.1006/abio.1998.2970; PMID: 9918655
- Xiao G, Bondarenko PV, Jacob J, Chu GC, Chelius D. 18O labeling method for identification and quantification of succinimide in proteins. Anal Chem 2007; 79:2714 - 21; http://dx.doi.org/10.1021/ac0617870; PMID: 17313184
- Fodor S, Zhang Z. Rearrangement of terminal amino acid residues in peptides by protease-catalyzed intramolecular transpeptidation. Anal Biochem 2006; 356:282 - 90; http://dx.doi.org/10.1016/j.ab.2006.06.023; PMID: 16859627
- Zang L, Carlage T, Murphy D, Frenkel R, Bryngelson P, Madsen M, Lyubarskaya Y. Residual metals cause variability in methionine oxidation measurements in protein pharmaceuticals using LC-UV/MS peptide mapping. J Chromatogr B Analyt Technol Biomed Life Sci 2012; 895-896:71 - 6; http://dx.doi.org/10.1016/j.jchromb.2012.03.016; PMID: 22483985
- Yan B, Valliere-Douglass J, Brady L, Steen S, Han M, Pace D, Elliott S, Yates Z, Han Y, Balland A, et al. Analysis of post-translational modifications in recombinant monoclonal antibody IgG1 by reversed-phase liquid chromatography/mass spectrometry. J Chromatogr A 2007; 1164:153 - 61; http://dx.doi.org/10.1016/j.chroma.2007.06.063; PMID: 17640657
- Gadgil HS, Bondarenko PV, Pipes GD, Dillon TM, Banks D, Abel J, Kleemann GR, Treuheit MJ. Identification of cysteinylation of a free cysteine in the Fab region of a recombinant monoclonal IgG1 antibody using Lys-C limited proteolysis coupled with LC/MS analysis. Anal Biochem 2006; 355:165 - 74; http://dx.doi.org/10.1016/j.ab.2006.05.037; PMID: 16828048
- Adamczyk M, Gebler JC, Wu J. Papain digestion of different mouse IgG subclasses as studied by electrospray mass spectrometry. J Immunol Methods 2000; 237:95 - 104; http://dx.doi.org/10.1016/S0022-1759(00)00135-6; PMID: 10725455
- Yan B, Eris T, Yates Z, Hong RW, Steen S, Kleemann G, Wang W, Liu JL. Analysis of human antibody IgG2 domains by reversed-phase liquid chromatography and mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci 2009; 877:1613 - 20; http://dx.doi.org/10.1016/j.jchromb.2009.03.043; PMID: 19411197
- Chen S, Lau H, Brodsky Y, Kleemann GR, Latypov RF. The use of native cation-exchange chromatography to study aggregation and phase separation of monoclonal antibodies. Protein Sci 2010; 19:1191 - 204; http://dx.doi.org/10.1002/pro.396; PMID: 20512972
- von Pawel-Rammingen U, Johansson BP, Björck L. IdeS, a novel streptococcal cysteine proteinase with unique specificity for immunoglobulin G. EMBO J 2002; 21:1607 - 15; http://dx.doi.org/10.1093/emboj/21.7.1607; PMID: 11927545
- Vincents B, von Pawel-Rammingen U, Björck L, Abrahamson M. Enzymatic characterization of the streptococcal endopeptidase, IdeS, reveals that it is a cysteine protease with strict specificity for IgG cleavage due to exosite binding. Biochemistry 2004; 43:15540 - 9; http://dx.doi.org/10.1021/bi048284d; PMID: 15581366
- Wenig K, Chatwell L, von Pawel-Rammingen U, Björck L, Huber R, Sondermann P. Structure of the streptococcal endopeptidase IdeS, a cysteine proteinase with strict specificity for IgG. Proc Natl Acad Sci U S A 2004; 101:17371 - 6; http://dx.doi.org/10.1073/pnas.0407965101; PMID: 15574492
- Chevreux G, Tilly N, Bihoreau N. Fast analysis of recombinant monoclonal antibodies using IdeS proteolytic digestion and electrospray mass spectrometry. Anal Biochem 2011; 415:212 - 4; http://dx.doi.org/10.1016/j.ab.2011.04.030; PMID: 21596014
- Goetze AM, Zhang Z, Liu L, Jacobsen FW, Flynn GC. Rapid LC-MS screening for IgG Fc modifications and allelic variants in blood. Mol Immunol 2011; 49:338 - 52; http://dx.doi.org/10.1016/j.molimm.2011.09.002; PMID: 21945018
- Jäger C, Ferrara C, Umaña P, Zeck A, Regula JT, Koll H. Determination of antibody glycosylation by mass spectrometry. Methods Mol Biol 2012; 901:195 - 208; http://dx.doi.org/10.1007/978-1-61779-931-0_13; PMID: 22723103
- Wang B, Gucinski AC, Keire DA, Buhse LF, Boyne MT 2nd. Structural comparison of two anti-CD20 monoclonal antibody drug products using middle-down mass spectrometry. Analyst 2013; 138:3058 - 65; http://dx.doi.org/10.1039/c3an36524g; PMID: 23579346
- Ayoub D, Jabs W, Resemann A, Evers W, Evans C, Main L, Baessmann C, Wagner-Rousset E, Suckau D, Beck A. Correct primary structure assessment and extensive glyco-profiling of cetuximab by a combination of intact, middle-up, middle-down and bottom-up ESI and MALDI mass spectrometry techniques. MAbs 2013; 5:699 - 710; http://dx.doi.org/10.4161/mabs.25423; PMID: 23924801
- Wagner-Rousset E, Janin-Bussat M-C, Colas O, Excoffier M, Ayoub D, Haeuw J-F, Rilatt I, Perez M. Corvaȉa N, Beck A. Antibody-drug conjugate model fast characterization by LC-MS following IdeS proteolytic digestion. MAbs 2014; 6:1 - 12; http://dx.doi.org/10.4161/mabs.26773
- Goetze AM, Liu YD, Arroll T, Chu L, Flynn GC. Rates and impact of human antibody glycation in vivo. Glycobiology 2012; 22:221 - 34; http://dx.doi.org/10.1093/glycob/cwr141; PMID: 21930650
- Zhou S, Mozziconacci O, Kerwin BA, Schöneich C. Fluorogenic tagging methodology applied to characterize oxidized tyrosine and phenylalanine in an immunoglobulin monoclonal antibody. Pharm Res 2013; 30:1311 - 27; http://dx.doi.org/10.1007/s11095-012-0970-7; PMID: 23412915
- Woodard J, Lau H, Latypov RF. Nondenaturing size-exclusion chromatography-mass spectrometry to measure stress-induced aggregation in a complex mixture of monoclonal antibodies. Anal Chem 2013; 85:6429 - 36; http://dx.doi.org/10.1021/ac401455f; PMID: 23742703
- Strand J, Huang C-T, Xu J. Characterization of Fc-fusion protein aggregates derived from extracellular domain disulfide bond rearrangements. J Pharm Sci 2013; 102:441 - 53; http://dx.doi.org/10.1002/jps.23421; PMID: 23242781
- Lynaugh H, Li H, Gong B. Rapid Fc glycosylation analysis of Fc fusions with IdeS and liquid chromatography mass spectrometry. MAbs 2013; 5:641 - 5; http://dx.doi.org/10.4161/mabs.25302; PMID: 23839239
- Zhang Z, Pan H, Chen X. Mass spectrometry for structural characterization of therapeutic antibodies. Mass Spectrom Rev 2009; 28:147 - 76; http://dx.doi.org/10.1002/mas.20190; PMID: 18720354
- Fornelli L, Ayoub D, Aizikov K, Beck A, Tsybin YO. Middle-down analysis of monoclonal antibodies with electron transfer dissociation orbitrap fourier transform mass spectrometry. Anal Chem 2014; 86:3005 - 12; http://dx.doi.org/10.1021/ac4036857; PMID: 24588056
- Bondt A, Rombouts Y, Hazes JMW, Wuhrer M, Dolhain RJEM. European Workshop for Rheumatology Research, 20–22 February 2014, Lisbon, Portugal. Abstracts. Ann Rheum Dis 2014; 73:Suppl 1 A1; http://dx.doi.org/10.1136/annrheumdis-2013-205124.2; PMID: 24489135
- Anumula KR. Quantitative glycan profiling of normal human plasma derived immunoglobulin and its fragments Fab and Fc. J Immunol Methods 2012; 382:167 - 76; http://dx.doi.org/10.1016/j.jim.2012.05.022; PMID: 22683540
- Angal S, King DJ, Bodmer MW, Turner A, Lawson ADG, Roberts G, Pedley B, Adair JR. A single amino acid substitution abolishes the heterogeneity of chimeric mouse/human (IgG4) antibody. Mol Immunol 1993; 30:105 - 8; http://dx.doi.org/10.1016/0161-5890(93)90432-B; PMID: 8417368
- Labrijn AF, Buijsse AO, van den Bremer ETJ, Verwilligen AYW, Bleeker WK, Thorpe SJ, Killestein J, Polman CH, Aalberse RC, Schuurman J, et al. Therapeutic IgG4 antibodies engage in Fab-arm exchange with endogenous human IgG4 in vivo. Nat Biotechnol 2009; 27:767 - 71; http://dx.doi.org/10.1038/nbt.1553; PMID: 19620983
- Shen FJ, Kwong MY, Keck RG, Harris RJ. The application of tert-butylhydroperoxide oxidation to study sites of potential methionine oxidation in a recombinant antibody. Techniques in Protein Chemistry 1996; VII:275 - 84; http://dx.doi.org/10.1016/S1080-8914(96)80031-7
- Keck RG. The use of t-butyl hydroperoxide as a probe for methionine oxidation in proteins. Anal Biochem 1996; 236:56 - 62; http://dx.doi.org/10.1006/abio.1996.0131; PMID: 8619496
- Ji JA, Zhang B, Cheng W, Wang YJ. Methionine, tryptophan, and histidine oxidation in a model protein, PTH: mechanisms and stabilization. J Pharm Sci 2009; 98:4485 - 500; http://dx.doi.org/10.1002/jps.21746; PMID: 19455640
- Edelman GM, Cunningham BA, Gall WE, Gottlieb PD, Rutishauser U, Waxdal MJ. The covalent structure of an entire gammaG immunoglobulin molecule. Proc Natl Acad Sci U S A 1969; 63:78 - 85; http://dx.doi.org/10.1073/pnas.63.1.78; PMID: 5257969
- Pan H, Chen K, Chu L, Kinderman F, Apostol I, Huang G. Methionine oxidation in human IgG2 Fc decreases binding affinities to protein A and FcRn. Protein Sci 2009; 18:424 - 33; http://dx.doi.org/10.1002/pro.45; PMID: 19165723
- Liu H, Gaza-Bulseco G, Zhou L. Mass spectrometry analysis of photo-induced methionine oxidation of a recombinant human monoclonal antibody. J Am Soc Mass Spectrom 2009; 20:525 - 8; http://dx.doi.org/10.1016/j.jasms.2008.11.011; PMID: 19103498
- Liu H, Chumsae C, Gaza-Bulseco G, Hurkmans K, Radziejewski CH. Ranking the susceptibility of disulfide bonds in human IgG1 antibodies by reduction, differential alkylation, and LC-MS analysis. Anal Chem 2010; 82:5219 - 26; http://dx.doi.org/10.1021/ac100575n; PMID: 20491447
- Harris RJ, Kabakoff B, Macchi FD, Shen FJ, Kwong M, Andya JD, Shire SJ, Bjork N, Totpal K, Chen AB. Identification of multiple sources of charge heterogeneity in a recombinant antibody. J Chromatogr B Biomed Sci Appl 2001; 752:233 - 45; http://dx.doi.org/10.1016/S0378-4347(00)00548-X; PMID: 11270864
- Ryan MH, Petrone D, Nemeth JF, Barnathan E, Björck L, Jordan RE. Proteolysis of purified IgGs by human and bacterial enzymes in vitro and the detection of specific proteolytic fragments of endogenous IgG in rheumatoid synovial fluid. Mol Immunol 2008; 45:1837 - 46; http://dx.doi.org/10.1016/j.molimm.2007.10.043; PMID: 18157932
- Liu D, Ren D, Huang H, Dankberg J, Rosenfeld R, Cocco MJ, Li L, Brems DN, Remmele RL Jr.. Structure and stability changes of human IgG1 Fc as a consequence of methionine oxidation. Biochemistry 2008; 47:5088 - 100; http://dx.doi.org/10.1021/bi702238b; PMID: 18407665
- Gaza-Bulseco G, Faldu S, Hurkmans K, Chumsae C, Liu H. Effect of methionine oxidation of a recombinant monoclonal antibody on the binding affinity to protein A and protein G. J Chromatogr B Analyt Technol Biomed Life Sci 2008; 870:55 - 62; http://dx.doi.org/10.1016/j.jchromb.2008.05.045; PMID: 18567545
- Bertolotti-Ciarlet A, Wang W, Lownes R, Pristatsky P, Fang Y, McKelvey T, Li Y, Li Y, Drummond J, Prueksaritanont T, et al. Impact of methionine oxidation on the binding of human IgG1 to Fc Rn and Fc γ receptors. Mol Immunol 2009; 46:1878 - 82; http://dx.doi.org/10.1016/j.molimm.2009.02.002; PMID: 19269032
- Wang W, Vlasak J, Li Y, Pristatsky P, Fang Y, Pittman T, Roman J, Wang Y, Prueksaritanont T, Ionescu R. Impact of methionine oxidation in human IgG1 Fc on serum half-life of monoclonal antibodies. Mol Immunol 2011; 48:860 - 6; http://dx.doi.org/10.1016/j.molimm.2010.12.009; PMID: 21256596
- Chumsae C, Gaza-Bulseco G, Sun J, Liu H. Comparison of methionine oxidation in thermal stability and chemically stressed samples of a fully human monoclonal antibody. J Chromatogr B Analyt Technol Biomed Life Sci 2007; 850:285 - 94; http://dx.doi.org/10.1016/j.jchromb.2006.11.050; PMID: 17182291
- Kerwin BA, Remmele RL Jr.. Protect from light: photodegradation and protein biologics. J Pharm Sci 2007; 96:1468 - 79; http://dx.doi.org/10.1002/jps.20815; PMID: 17230445
- Lam XM, Yang JY, Cleland JL. Antioxidants for prevention of methionine oxidation in recombinant monoclonal antibody HER2. J Pharm Sci 1997; 86:1250 - 5; http://dx.doi.org/10.1021/js970143s; PMID: 9383735
- Li S, Schöneich C, Borchardt RT. Chemical instability of protein pharmaceuticals: Mechanisms of oxidation and strategies for stabilization. Biotechnol Bioeng 1995; 48:490 - 500; http://dx.doi.org/10.1002/bit.260480511; PMID: 18623513
- Chu J-W, Yin J, Brooks BR, Wang DIC, Ricci MS, Brems DN, Trout BL. A comprehensive picture of non-site specific oxidation of methionine residues by peroxides in protein pharmaceuticals. J Pharm Sci 2004; 93:3096 - 102; http://dx.doi.org/10.1002/jps.20207; PMID: 15514984
- Vlasak J, Ionescu R. Heterogeneity of monoclonal antibodies revealed by charge-sensitive methods. Curr Pharm Biotechnol 2008; 9:468 - 81; http://dx.doi.org/10.2174/138920108786786402; PMID: 19075686
- Du Y, Walsh A, Ehrick R, Xu W, May K, Liu H. Chromatographic analysis of the acidic and basic species of recombinant monoclonal antibodies. MAbs 2012; 4:578 - 85; http://dx.doi.org/10.4161/mabs.21328; PMID: 22820257
- Moorhouse KG, Nashabeh W, Deveney J, Bjork NS, Mulkerrin MG, Ryskamp T. Validation of an HPLC method for the analysis of the charge heterogeneity of the recombinant monoclonal antibody IDEC-C2B8 after papain digestion. J Pharm Biomed Anal 1997; 16:593 - 603; http://dx.doi.org/10.1016/S0731-7085(97)00178-7; PMID: 9502155
- Vlasak J, Bussat MC, Wang S, Wagner-Rousset E, Schaefer M, Klinguer-Hamour C, Kirchmeier M, Corvaïa N, Ionescu R, Beck A. Identification and characterization of asparagine deamidation in the light chain CDR1 of a humanized IgG1 antibody. Anal Biochem 2009; 392:145 - 54; http://dx.doi.org/10.1016/j.ab.2009.05.043; PMID: 19497295
- Lau H, Pace D, Yan B, McGrath T, Smallwood S, Patel K, Park J, Park SS, Latypov RF. Investigation of degradation processes in IgG1 monoclonal antibodies by limited proteolysis coupled with weak cation-exchange HPLC. J Chromatogr B Analyt Technol Biomed Life Sci 2010; 878:868 - 76; http://dx.doi.org/10.1016/j.jchromb.2010.02.003; PMID: 20206584
- Kim J, Jones L, Taylor L, Kannan G, Jackson F, Lau H, Latypov RF, Bailey B. Characterization of a unique IgG1 mAb CEX profile by limited Lys-C proteolysis/CEX separation coupled with mass spectrometry and structural analysis. J Chromatogr B Analyt Technol Biomed Life Sci 2010; 878:1973 - 81; http://dx.doi.org/10.1016/j.jchromb.2010.05.032; PMID: 20554483
- Pace AL, Wong RL, Zhang YT, Kao Y-H, Wang YJ. Asparagine deamidation dependence on buffer type, pH, and temperature. J Pharm Sci 2013; 102:1712 - 23; http://dx.doi.org/10.1002/jps.23529; PMID: 23568760
- Huang L, Lu J, Wroblewski VJ, Beals JM, Riggin RM. In vivo deamidation characterization of monoclonal antibody by LC/MS/MS. Anal Chem 2005; 77:1432 - 9; http://dx.doi.org/10.1021/ac0494174; PMID: 15732928
- Janin-Bussat M-C, Tonini L, Huillet C, Colas O, Klinguer-Hamour C, Corvaïa N, Beck A. Cetuximab Fab and Fc N-glycan fast characterization using IdeS digestion and liquid chromatography coupled to electrospray ionization mass spectrometry. Methods Mol Biol 2013; 988:93 - 113; http://dx.doi.org/10.1007/978-1-62703-327-5_7; PMID: 23475716
- Beck A, Diemer H, Ayoub D, Debaene F, Wagner-Rousset E, Carapito C, Van Dorsselaer A, Sanglier-Cianférani S. Analytical characterization of biosimilar antibodies and Fc-fusion proteins. Trends Analyt Chem 2013; 48:81 - 95; http://dx.doi.org/10.1016/j.trac.2013.02.014