
Abstract
Although there are currently more than 30 antibody-drug conjugates (ADC) in clinical development for the treatment of blood cancers and solid tumors, comparison of their clinical pharmacokinetics (PK) is challenging because of the large number of, and differences between, the targets, ADC constructs, dosing regimens, and patient populations. In this review, we standardized the evaluation, using non-compartmental PK data reported at Cycle 1, i.e., following the first drug administration of what is usually a repeated-dose treatment, in monotherapy. We report ADC clinical PK properties, dosing regimen, determination of doses ranges and associated maximum tolerated doses. We also evaluated the effect of structural characteristics and target types (hematological vs. solid tumors) on PK. In addition, we discuss how integration of PK/pharmacodynamics approaches on top of classical dose escalation in first-in-human studies may improve dosing regimen determination for subsequent phases of clinical development.
Introduction
Antibody–drug conjugates (ADCs) consist of a monoclonal antibody (mAb) and a cytotoxic drug (small molecule drug) associated through a linker. The target antigen should be expressed at high density on malignant cells and have limited expression on cells of normal tissues. The cytotoxic drug (most frequently auristatins, maytansinoids, and calicheamicins) must be highly potent to kill tumor cells at the intracellular concentrations that can be achieved with mAb-mediated delivery. They are designed to induce tumor cell death by causing irreversible DNA damage or interfering with the mechanism of cell division. ADCs are designed with linkers that release biologically-active drug following antigen-specific internalization and trafficking to lysosomes. The “cleavable” linkers rely on intracellular processes to release the cytotoxic drug, such as reduction of disulfide bonds mediated by glutathione (GSH) in the cytoplasm, exposure to acidic conditions (pH ~4) in the lysosome, or cleavage by specific proteases. Conversely, “non-cleavable” or “stable” linkers require catabolic degradation of the antibody to release the cytotoxic drug. Following administration in patients, ADCs consist of a sum of antibody species carrying different numbers of cytotoxic molecules, varying from zero (unconjugated) to ~7–8, for which the average value is the drug-to-antibody ratio (DAR). In principle, the distribution and elimination of the different entities varies, translating to decreasing average DAR during the course of the dosing interval.
Phase 1 studies of ADCs generally enroll patients with advanced cancer, whose disease is usually refractory to available treatment, in order to evaluate the safety and toxicity of new therapeutic agents; document the pharmacokinetic (PK) properties of those agents; determine the maximum tolerated dose (MTD), which is defined as the highest dose with a relatively low risk of dose-limiting toxicity (DLT); and to determine an appropriate dose level/regimen for phase 2 trials.
ADC PK information, which is generally retrieved from patients’ studies to document the time-course of the drug in the circulation, is a required element of the registration files submitted to regulatory authorities. Furthermore, the determination of dose-exposure effect relationships is now recognized to be a crucial part of the drug development process. Exposure-response is of particular importance because of the relatively narrow therapeutic index of ADCs, and, consequently, the need for dose and regimen optimization.
ADCs are administered as intravenous infusion, and, following in vivo processing, multiple analytes are detected in systemic circulation. The analytes commonly assessed for ADC bioanalysis are the conjugated antibody (antibody with DAR of at least 1), the total antibody (conjugated, partially deconjugated and fully deconjugated), the antibody-conjugated drug (the total small molecule drug conjugated to antibody), the unconjugated drug (small molecule drug not conjugated to antibody), and possibly metabolites of the small molecule drug including or not part of the linker, according to Gorovits et al.Citation1
There are currently around 30 ADC in clinical developmentCitation2 for the treatment of blood cancers and solid tumors and two ADC, brentuximab vedotin (Adcetris®) and ado-trastuzumab emtansine (Kadcyla®), are currently approved by the US Food and Drug Administration (FDA). However, given the large number of, and differences between, targets, ADC constructs, dosing regimens and patient populations, the comparison of ADC PK is challenging. We evaluated ADC PK in first-in-human (Phase 1) studies because study designs at this stage of development are relatively comparable and ADCs are frequently administered as monotherapy. This bibliography review covered the ADCs currently in development with reported information on PK parameters at multiple doses or information on dose range and dosing regimen. More specifically, a particular focus was provided on: (1) the starting doses, dose escalations and determination of the MTD; (2) the determination of the dosing regimen; and (3) the comparative PK of ADC according to structural characteristics (isotypes, linkers) and target types (hematological vs. solid tumors).
Results
Structural properties of ADC reviewed
The structural features, target, indication and dosing regimen information for 21 ADC evaluated in clinical studies are presented in . Isotype data was found for 19 of the ADCs; 3 were IgG4, 2 were IgG2, and the remainder were IgG1.The indications for the three ADC based on IgG4 backbone were hematological cancers. Most of the ADCs (18/21, 85%) had “cleavable” linkers. All ADCs had a similar average drug-to-antibody ratio (DAR), in the 3.5–4 range, except for CMC-544, which had a DAR of 6. The cytotoxic drugs were maytansinoids (DM1 and DM4), auristatins (MMAE and MMAF) or calicheamicin.
Table 1. Properties of ADC and dosing regimen in first-in-man studies
PK protocols of first-in-human studies with ADC
All ADCs were administered by intravenous infusion. The reported PK data had been analyzed by non-compartmental analysis (NCA) and most of the PK parameters were determined at Cycle 1, i.e., following the first ADC administration, over a limited time course of usually 21 d. The parameters most frequently reported for ADC were the AUC, area under the concentration-time curve as a measure of drug exposure, observed concentrations including the maximum concentration (Cmax, or end-of-infusion), or trough (Ctrough) concentrations at end of dosing interval, CL (clearance), Vss (an estimate of volume of distribution at steady-state) and t½ (elimination half-life). To provide a comparative view of PK properties of ADC administered at different doses and dosing regimen, only the parameters determined at MTD or nearest dose level were selected and are presented in .
Table 2. Comparative PK parameters of ADCs at MTD or nearest dose level, at Cycle 1
Regarding unconjugated drug, Cmax and AUC were reported for a limited number of therapeutic agents. Other PK parameters were generally not calculated since only a limited number of samples were associated with quantifiable levels during the first 21-d cycle.
Extensive sampling was the rule. Nevertheless, the actual number and distribution of samples and the total number of patients available for the PK analysis was only reported in peer-reviewed articles. The number of patient per dose group could be as low as one.
Although immunogenicity is a safety endpoint defined as a primary or secondary criterion in protocols, the incidence of anti-drug antibodies, time of onset and persistence were not reported. Incidence of immunogenicity for a limited number of ADC was reported to be low, between zero and five percent (), but the number of dosing cycles was not provided.
Table 3. Incidence of patients with persistent anti-therapeutic antibody response, following ADC administration in Phase 1
Dose ranges and maximum tolerated doses
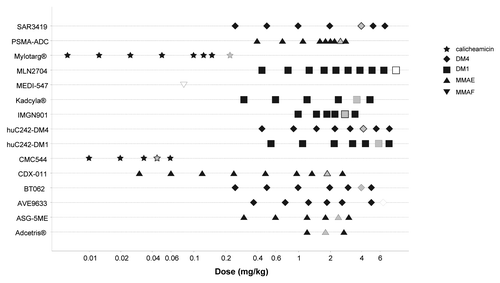
The detailed range of doses administered in first-in-human studies and the associated maximum tolerated doses (MTD) were available for 15 ADC. Data were plotted on a log-dose scale to accommodate the graph with the very large range of doses ().
Figure 1. ADC dose ranges and maximum tolerated doses in first-in-humans studies. Gray symbol: MTD; open symbol: MTD not reached.
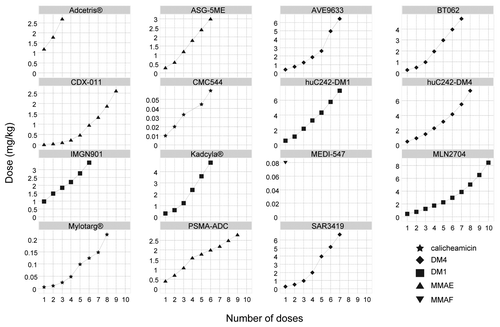
Typically, dose escalation methods were based on 3+3 or accelerated titration designs and the escalation algorithms followed a modified Fibonacci sequence, as shown in .
Figure 2. ADC dose escalation profiles in first-in-human studies.
Calicheamicin-containing ADC were evaluated in a much lower dose range (0.005 ∼0.2 mg/kg), about 2 orders of magnitude lower than auristatin- or maytansinoid-containing ADC. The latter have been evaluated in a similar dose range (0.3 ∼7 mg/kg), but maytansinoid ADC, conjugated with either DM1 or DM4, showed a trend to higher MTD. AVE9633 and MLN2704 were reported to have been well-tolerated and MTD was not reached at the end of the dose escalation (>∼7 mg/kg).
A median number of six dose-level escalations were necessary from the first dose level to reach MTD. However, evidence of potential therapeutic activity was rapidly observed for brentuximab vedotin and only three dose levels were investigated in phase I before registration trials were conducted. On the opposite, only one dose of MEDI-547 was evaluated because dose escalation was halted at the first dose level due to unexpected safety issues.
ADC pharmacokinetics
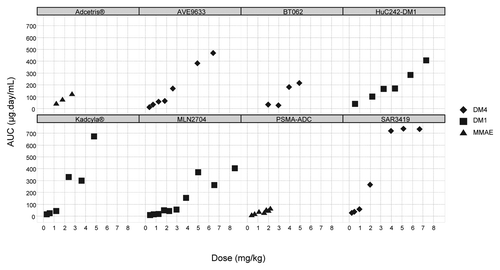
Summary PK parameters of ADC at MTD or nearest dose, determined by non-compartmental analysis at Cycle 1, are provided in . Calicheamicin ADCs exhibited much higher clearance than the other classes of ADC. The MTD of calicheamicin ADCs were significantly lower than those of other classes of ADC and showed much lower exposure. MTD of maytansinoid ADCs were ~2-fold higher than those of auristatin ADCs, and these higher doses resulted in higher ADC exposures. Dose-exposure relationship (expressed as conjugated antibody) for ADCs administered in the same q3w regimen is presented in . Overall, plasma AUC increased supra dose proportionally at the highest doses.
Figure 3. Dose-exposure relationship for ADC administered every 3 wk.
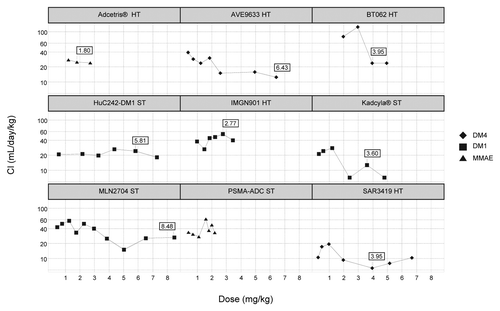
The analysis of dose/clearance relationship could be performed with 9 ADCs with complete PK information reported and revealed two distinct patterns (). On the one hand, a decrease in clearance with increasing doses was observed with AVE9633, BT062, MLN2704, and ado-trastuzumab emtansine. On the other hand, huC242-DM1, IMGN901, and PSMA-ADC showed no dose-dependent change of clearance. Brentuximab vedotin was administered in a narrow range of three doses; therefore, the effect of dose on clearance could not be evidenced. Overall, the dose-dependence of clearance could not clearly be attributed to the site of targeted antigen, i.e., solid tumor or hematological tumor. For ADC showing dose-dependence of clearance, the MTD were within the doses associated with linear clearance.
Figure 4. Effect of dose on ADC clearance, by target antigen type (HT, hematological tumor; ST, solid tumors. (Boxes indicate maximum tolerated dose in mg/kg)
Elimination half-life at MTD varied in large range, from 0.7 to 6.9 d. Vss (estimate of volume of distribution at steady-state) was limited and equivalent to the blood volume.
ADC containing IgG2 isotypes were not clearly associated with different PK parameters, as compared with IgG1. IgG4 ADC contained calicheamicin and displayed higher clearance than IgG1-based ADC.
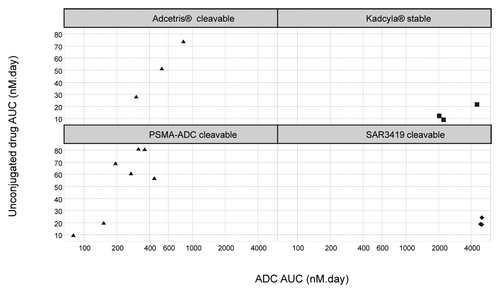
The body of literature on the time-course of unconjugated drug plasma concentration was very limited. The maximum concentration of unconjugated drug and related metabolites could be reached either just after intravenous infusion or a few days later. The unconjugated drug plasma exposure was proportional to the ADC exposure for brentuximab vedotin and PSMA-ADC (expressed as conjugated antibody). The unconjugated drug AUCs were ~200-fold less than that of their respective ADC, after adjustment for molecular weights (). However, unconjugated drug AUC was independent of ADC exposure for ado-trastuzumab emtansine, which has a “stable” SMCC linker, and for SAR3419, which has a “cleavable” linker.
Figure 5. Unconjugated drug plasma exposure vs. ADC plasma exposure.
Dosing regimen
In this review, the majority of the ADCs had clinical indication in solid tumors cancers (13/21, 62%) and the others were indicated in hematological cancers. Several dosing regimen for ADCs have been evaluated by their sponsors during the Phase 1 studies reviewed in this analysis ().
The most frequently reported dosing approaches in these studies were the q3w administration (18/21) and a comparison of q1w vs. q3w dosing regimen (8/21). The other two regimens reported, q2w or q4w, have more rarely been evaluated.
Dosing of ADC was based equally on bodyweight (11/21) or body surface area (10/21); no ADC was administered using fixed dose. Maytansinoid ADC were administered using both approaches, whereas auristatin ADC were administered using the bodyweight approach only.
Discussion
Earlier in the history of ADC clinical development, attempts were made to determine the plasma concentrations of each component of the investigational ADC, including the conjugated antibody, the total or free unconjugated drug, the total antibody, and the unconjugated antibody, because it was not obvious which analytes, or subset of analytes, may provide crucial information to understand safety and efficacy. Indeed, unlike small molecule drug or therapeutic proteins, it is possible that one species might account for efficacy, whereas another might account for toxicity.
As the field has evolved, and after decades of experience, it appeared that measuring all of these components may not be feasible or essential for characterization of the disposition of the ADC. In particular, no single assessment from the plasma is able to capture all aspects of the complex in vivo disposition of these large molecules, such as the rate of drug loss from an ADC (i.e., linker stability), the specific and non-specific cellular processing, and ultimately the exposure-response (E-R) relationship.
Following the publication of a white paperCitation1 from the American Association of Pharmaceutical Scientists and FDA, it is now generally considered that the most important and useful parameters can be derived from three key analytes in systemic circulation: intact conjugated antibody, total antibody, and unconjugated drug, as well as determination of anti-therapeutic antibodies (ATA). These multiple assays require the use of both small and large molecules bioanalytical techniques, but may also include additional technologies for the determination of DAR, to better define the composition of intact conjugated antibody during the dosing interval.
Comparative assessment of total antibody PK with conjugated antibody PK provides information on the rate of drug loss from the ADC. A greater separation of the conjugated antibody PK curve from the total antibody PK curve indicates a more rapid loss of drug from the ADC. Unconjugated antibody concentration is rarely measured because it is expected to have little or no biological activity. However, the unconjugated antibody clearance should be lower than that of the ADC, and it could accumulate following repeated administration of the ADC, especially for dosing regimen consisting of closer drug infusions.Citation3,Citation4 The concentration of unconjugated antibody may be approximated by mathematical modeling of the total antibody and conjugated antibody profilesCitation5 or can be directly quantified from plasma samples.Citation6 Unconjugated drug concentrations are used to infer the systemic exposure to the cytotoxic drug released from the ADC, or resulting of processing of the ADC by antigen-positive cells, followed by release of the cytotoxic drug. The cytotoxic-related products are also important to determine the potential drug-drug interaction potential associated with the cytotoxic moiety.
In this review, the comparison of ADC PK parameters in Phase 1 studies required some data transformation for harmonization of both dosing units and parameters. However, given the very low number of patients per dose-level, which was as low as one, the large inter-individual variability of exposure at low doses for ADC exhibiting target-mediated drug disposition (TMDD), the effect of the bioanalytical assay format and their accuracy, as well as the limited number of plasma samples that can be collected in cancer patients, the accuracy of ADC PK parameters in these first-in-human Phase 1 studies is inherently moderate.
Immunogenicity data are highly dependent on the sensitivity and specificity of the test methods used. The observed incidence of a positive result in a test method may be influenced by several factors, including sample handling, timing of sample collection, drug interference, concomitant medication and the underlying disease. Although direct comparison of the incidence may be misleading, this survey showed that ADC immunogenicity in Phase 1 was reported as generally low and the incidence was less than 5%. However, the treatment duration and number of doses administered are too low in the context of first-in-human studies to draw definitive conclusions regarding ADC immunogenicity.
Peak ADC plasma concentrations typically occurred at the end of infusion and declined in a biphasic manner. Vss varied in a limited range and were compatible with values usually reported for mAbs.Citation7 For ADC, mAbs, and other large therapeutic proteins, the reported volume of distribution after intravenous administration is close to the plasma volume, suggesting limited distribution into tissues. However, determination of Vss of antibody-based drugs using non-compartmental analysis is based on the assumption that the site of antibody elimination is in rapid equilibrium with plasma (i.e., it is assumed that all elimination is from the “central” compartment).Citation8 This assumption is likely partially incorrect for ADC that bind to and internalize within cells in tissue sites.
The influence of the IgG isotypes on ADC PK could not be evidenced. In our PK data set, IgG4 isotypes were associated with calicheamicin, and both gemtuzumab ozogamicin (Mylotarg®) and CMC-544 displayed higher clearance than other ADC. However, there are multiple confounding factors since the higher clearance could be associated with their much lower MTD susceptible to target-mediated clearance or with higher CD33 antigen turnover for gemtuzumab ozogamicin or with their higher DAR values. The Mylotarg® drug substance consists of a mixture of unconjugated and conjugated antibody, with significant amounts of unconjugated antibody.Citation9 The actual DAR value for the conjugated antibody fraction of gemtuzumab ozogamicin is therefore higher than the reported mean DAR of 2.5.
No general rule regarding the effect of “stable” vs. “cleavable” linkers on ADC exposure could be derived because PK parameters were available for only one ADC with stable linker (ado-trastuzumab emtansine). However, similar molar AUC were observed with ado-trastuzumab emtansine (stable SMCC linker) and SAR3419 (cleavable SPDB linker) at similar dose ranges. In addition, for both drugs, the unconjugated drug AUC was not proportional to the AUC of ADC. On the other hand, brentuximab vedotin and PSMA-ADC using the same protease-cleavable linker showed similar profiles, as their unconjugated drug AUC were proportional to the AUC of ADC. This observation adds another level of complexity for the interpretation of ADC analytes PK as their relationships may or may not be correlated.
ADC clinical PK data are rather limited and, consequently, the relationships between all variables of interest (e.g., linker, payload) and PK may not be addressed directly with clinical data because there are no data available that compare clinical investigations of the same mAb and payload with different linkers, or the same mAb and linker with different payloads. Nonclinical structure-activity data are generated to support lead optimization by companies developing ADCs, but limited results are publicly available.
Although the disposition of ADCs is conditioned by their antibody backbone, the elimination half-life of ADCs in this review were shorter than expected for Ig1 or IgG4 antibodiesCitation10 and were generally around 2–3 d. However, the terminal half-life for ADCs at Cycle 1 is likely an underestimate because of both limited sampling duration (<5 half-lives) and number of collection samples over usually 21 d. MAbs are thought to be eliminated via target-mediated and non-specific uptake into cells, followed by proteolytic degradation. Non-specific degradation, which involves FcRn-mediated recycling, predominantly occurs in endothelial cells and mononuclear phagocytes; target-mediated degradation, which involves both receptor-mediated internalization and intracellular lysosomal degradation, primarily occurs in target-expressing cells. In addition to these catabolic processes, ADC elimination may also occur by deconjugation which includes release of cytotoxic drug-containing products from the ADC via enzymatic or chemical processes. This dual elimination pathway may explain the higher clearance, determined in population PK studies, for ado-trastuzumab emtansine vs. trastuzumab, which are 0.68 L/dayCitation11 and 0.23 L/day,Citation12 respectively, although both drugs are based on the same IgG1 backbone.
Dose-dependent decrease of clearance was observed for AVE9633, BT062, MLN2704, and ado-trastuzumab emtansine, which suggested target-mediated disposition (TMD). Upon increasing dose levels, the curve reaches a plateau when the target-mediated, saturable clearance becomes negligible compared with the normal, linear IgG clearance.Citation13 These profiles were irrespective of the ADC target location, i.e., hematologic cancer or solid tumors. TMD was not observed for the other ADCs of our data set, but overall interpretation of these results is limited because all drugs were not administered in similar dose ranges. It is unclear whether this pattern might be observed at higher doses since dose escalations were limited by the safety profiles.
A dose-dependent PK profile may suggest high internalization by the tumor, or the presence of an antigen sink in tissues. This is particularly important for ADCs which exhibit therapeutic windows that are dependent upon the difference of the level and distribution of target between normal tissues and tumors. More detailed analysis should be performed in the context of the characteristics of the targets, such as receptor copy number, heterogeneity, and specificity of expression, internalization rate, and intracellular trafficking; however these data from patients are not published. The prediction of these effects in the clinic from in vitro studies may be biased by other factors such as cytokinetics, cytogenetics, multidrug resistance, and other prognostic factors. The influence of target expression and turnover on ADC PK may also be evaluated by comparing PK parameters between first cycle of administration and several cycles of therapy. In four studies, repeated dosing of ado-trastuzumab emtansine at 3.6 mg/kg q3w did not result in any noticeable accumulation.Citation11 On the other hand, increased concentrations of gemtuzumab ozogamicin were observed after the second dose and are believed to be due to a decrease in clearance by CD33-positive blast cells, a result of the reduced tumor burden following the first dose.Citation14
Until recently, no clear E-R relationships for safety and efficacy were available. No relationships were observed between the PK parameters of gemtuzumab ozogamicin and the maximum remission achieved by patients.Citation14 The E-R for brentuximab vedotin was more complex, since the probability of objective response rate increased with increasing average ADC Ctrough,ss, whereas it showed a decreasing trend with increasing MMAE Ctrough,ss. There was no clear E-R between thrombocytopenia and either ADC or MMAE concentration.Citation15 In a pooled analysis of four ado-trastuzumab emtansine studies (one Phase 1 and three Phase 2 studies), where PK parameters were calculated using standard non-compartmental approaches, ADC exposure did not correlate with clinical responses or key adverse events.Citation16 However, a recent Kaplan-Meier analysis of the pivotal ado-trastuzumab emtansine registration studies suggests a significant difference in survival for patient groups, divided according to quartiles of Cmin,C1D21. This PK endpoint is a population PK-predicted ADC trough concentration on Day 21 in Cycle 1.Citation11 Overall, the E-R analysis for efficacy suggested that, in the range investigated, higher ADC exposures correlated with greater overall survival or progression-free survival improvement. Since the tumor burden is expected to affect the PK, this may be interpreted in two ways: either higher exposure may be more effective or higher exposure at the same dose level may be related to a lower tumor burden that has a better prognosis.
Early optimization of dosing regimen is one of most important objectives in drug development. Phase 1 clinical trials are an essential step in the development of anticancer drugs, as their main goal is to establish the recommended dose or schedule for Phase 2 trials. To safely assess new drugs, cancer patients in initial cohorts of Phase 1 oncology studies receive low drug doses that are successively increased until the maximum tolerated dose (MTD) is determined. A recent surveyCitation17 on dose-levels and signs of efficacy in oncology Phase 1 trials showed that a median number of 5 dose-levels were tested to determine MTD with molecular-targeted agents (n = 74) and conventional cytotoxic agents (n = 201), whereas 4 dose-levels were tested to determine MTD with combination of both (n = 42). Our analysis on this limited ADC data set revealed that MTD was reached after a median number of six dose levels. Possible explanation for this higher number of dose-levels could be the choice of low starting doses, derived from current guidelines, or the use of conservative dose-escalation rules.Citation18 Toxicity of ADCs may be related to on-target or off-target binding. The present analysis shows little differences in MTD for ADCs with the same cytotoxic, although they are specific to different antigens types. This suggests that the MTD would be more dependent on the drug than on the target on the ADC.
ADC dose selection generally results from conservative approaches based on toxicological data in the non-human primate, regardless of whether or not the ADC binds to the animal target, and may use the same criteria as for traditional small molecule cytotoxics, e.g., selecting 1/6th HNSTD based on body surface area, 1/10th NOAEL based on bodyweight. Moreover, the classical dose escalation rules using “3+3” design and Fibonacci sequence are frequently applied. On the other hand, clinical experience with MEDI-547 showed unexpected safety issues from the lowest dose administered in patients, which were not predicted by nonclinical toxicology studies.Citation19
It is expected that the recent PK/pharmacodynamics (PD) modeling framework of translation of ADC efficacy from preclinical species to the clinic should provide a more rational basis for selecting the first-in-human dosing regimen.Citation20 These approaches may be useful to define a starting dose that has the potential for antitumor activity, but provides an acceptable toxicity profile based on the risk/benefit for the patient.
The most frequent dosing regimen selected at the end of the Phase 1 studies reviewed herein was the q3w administration. Given the relatively short elimination half-life of ADC, this decision is usually not based on PK principles alone, but is a combination of safety and efficacy endpoints as well as ease of ADC combination with standards of care in many cases.
The dose escalation methods have the advantage of rapidly providing some data on PK interpatient variability and exposure/response information. For example, early PK information during SAR3419 Phase 1 program allowed testing an optimized schedule based on the occurrence of late/cumulative adverse events at the recommended dose, supported by data showing ADC plasma accumulation after 4 weekly doses. Reversible corneal deposits were dose limiting in the q3w schedule, whereas the q1w schedule was well tolerated and active. The optimized schedule consisted of 4 weekly doses followed by 4 bi-weekly doses at the recommended dose. It showed an improved safety profile compared with schedules previously tested, and the clinical efficacy was preserved. The optimized schedule is being assessed in two Phase 2 studies.Citation21
Another example of dosing schedule optimization supported by PK/PD is provided by gemtuzumab ozogamicin. In 2000, gemtuzumab ozogamicin was granted accelerated approval by the US FDA based on promising Phase 2 data in relapsed older adults with acute myeloid leukemia (AML). The recommended treatment course was a total of two 9 mg/m2 doses with 14 d between the doses. The drug was voluntarily withdrawn in 2010 when efficacy could not be demonstrated and toxicity appeared excessive. To minimize toxicity, a new dosing regimen based on the repetition of lower dose of 3 mg/m2 on days 1, 4, and 7 was evaluated. The new protocol was reported to allow the safe delivery of higher cumulative doses and significantly improved outcomes in patients with AML.Citation22 The rationale for lower doses was based on the responses to doses of 1–4 mg/m2 in the initial Phase 1 study and a saturation of more than 80% of the CD33 sites after dosing with 4 mg/m2 or 6 mg/m2. In addition, recent dataCitation23 indicated high receptor occupancy (median ∼80%) at doses as low as 2 mg/m2. The rationale for administration of fractionated doses was based on the rapid re-expression of CD33 molecules on the cell surface after a first exposure. Indeed, a previous Phase 1 study had shown an increase in ADC AUC from first to second dose (q2w). Peripheral blast counts decreased after the first dose, and so did the clearance of gemtuzumab ozogamicin due to reduced internalization by blasts.Citation14 Since then, four randomized studies with low and fractionated doses have been completed that overall support its efficacy in newly diagnosed AML, with acceptable toxicity.Citation4
The dosing approaches for ADC in this review were either defined upon body weight or body surface area (BSA). The determination of the first-in-human dose should typically be done on a body weight basis because high molecular weight ADC often scale across species better with the circulating volume than body surface area.Citation24 As binding of the ADC to a cell-surface target and internalization may be a major clearance pathway, the disposition of the ADC may be target antigen-dependent so that animal species lacking the target antigen could have a different toxicological profile than humans. Extrapolation of the toxicity data to humans should be done cautiously in such situations and the traditional methods for establishing the first-in-human dose for cytotoxic agents can be appropriate for ADCs.Citation18 The choice of a BSA-based approach may be based on the observation that the anti-tumor effect is related to the cytotoxic drug dose, leading to customary use of BSA in dose calculations. Interestingly, ADCs containing the DM1 cytotoxic were dosed either on a mg/kg basis for ado-trastuzumab emtansine or a mg/m2 basis for MLN2704, IMGN901, and huC242-DM1. Indeed, comparable cross-reactivity of T-DM1 was observed for human and monkey tissues, whereas the targets were not expressed in monkeys for the above mentioned ADC. The ado-trastuzumab emtansine body weight-based dose of 3.6 mg/kg every 3 wk was considered acceptable upon review of the submission dossier since baseline body weight was identified as the significant covariate affecting the ADC’s steady-state AUC and Cmax. In addition, considering that the ado-trastuzumab emtansine MTD was established based on body weight-based dosing and that it had been used in clinical trials, the body weight-based dosing of ado-trastuzumab emtansine appeared acceptable to FDA.Citation11
Conclusion
This review, although limited by the relatively small number of published reports on complete ADC clinical PK, provides new insights regarding comparative ADC PK during early clinical development. Although early Phase 1 identification of E-R relationships of potentially multiple clinically relevant analytes remains a challenge, early ascending dose and repeat-dose PK studies with ADCs rapidly provide critical data on interpatient PK variability, drug accumulation, target-mediated drug disposition, and correlation to translational efficacy index, such as tumor static concentrations, which overall aid in the design of subsequent optimal dosing regimen.
The ability to project a starting dose for the first-in-human clinical studies may be difficult since the disposition of the ADC may be target antigen-dependent, so that animal species having a different pattern of target antigen expression could have a different toxicological profile than humans. It is expected that new modeling framework of translation of ADC efficacy from preclinical species to the clinic will provide a more rational basis for selecting the first-in-human dosing regimen.
Bioanalytical assessments of multiple component of ADCs are needed to describe the complex in vivo disposition of these large molecules. However, the incorporation of the DAR dynamics with these PK metrics would be useful to better characterize the time course of active entities. Since the DAR value changes dynamically, knowledge of time course of drug potency would help in dosing regimen optimization.
After many years of clinical experience, new dosing regimen strategies are emerging. Dose fractionation studies using more frequent administration of lower doses with calicheamicin-based ADC suggest improved efficacy and reduced toxicity. This approach incorporating efficacy and safety, but also PK/PD and target turnover/expression, is challenging the conventional process of ADC dosing regimen selection, as the recommended Phase 2 dose are usually defined through a classical dose escalation up to MTD.
Material and Methods
For the purpose of this review, databases were built, using data from either peer-reviewed journals, biologics license applications from FDA (Clinical Pharmacology section) or conferences posters, as of October 2013 (see refs. Citation3,Citation11,Citation14,Citation15,Citation16,Citation19,Citation21 and Citation25–Citation34). First, a PK parameters database was built for ADCs with non-compartmental PK analysis reported at Cycle 1, i.e., following the first drug administration of usually a repeated-dose treatment. A second database was built to compile information on ADC structural features, as well as range of Phase 1 doses, dosing regimen and maximum tolerated doses.
Since doses and PK parameters units were not reported in a consistent way, data were standardized to a body weight of 70 kg and body surface area of 1.73m2 and numbers were rounded to 3 significant figures. ADC molecular weights were assumed to be comparable, ~150 kDa, allowing for direct dose comparisons. ADC concentrations were reported in µg/mL units, whereas the cytotoxics concentrations were reported in ng/mL units. Because of data transformation, the PK parameters presented in this analysis may slightly differ from the original reported values. Graphical analysis was performed using TIBCO Spotfire ® v4.0.2.
Sznol M., Hamid O., Hwu P., Kluger H., Hawthorne T., Crowley E., et al. Pharmacokinetics (PK) of CR011-vcMMAE, an antibody-drug conjugate (ADC), in a phase (Ph) I study of patients (pts) with advanced melanoma. J Clin Oncol 2009; 27:Suppl.1 9063
Disclosure of Potential Conflicts of Interest
The author is full-time employee of Sanofi.
Acknowledgments
The author thanks the following individuals for their review and feedback on this manuscript: F. Donat, N. Fagniez, G. Sanderink, A. Bousseau, and C. Ribard.
References
- Gorovits B, Alley SC, Bilic S, Booth B, Kaur S, Oldfield P, Purushothama S, Rao C, Shord S, Siguenza P. Bioanalysis of antibody-drug conjugates: American Association of Pharmaceutical Scientists Antibody-Drug Conjugate Working Group position paper. Bioanalysis 2013; 5:997 - 1006; http://dx.doi.org/10.4155/bio.13.38; PMID: 23641692
- Mullard A. Maturing antibody-drug conjugate pipeline hits 30. Nat Rev Drug Discov 2013; 12:329 - 32; http://dx.doi.org/10.1038/nrd4009; PMID: 23629491
- Lapusan S, Vidriales MB, Thomas X, de Botton S, Vekhoff A, Tang R, Dumontet C, Morariu-Zamfir R, Lambert JM, Ozoux ML, et al. Phase I studies of AVE9633, an anti-CD33 antibody-maytansinoid conjugate, in adult patients with relapsed/refractory acute myeloid leukemia. Invest New Drugs 2012; 30:1121 - 31; http://dx.doi.org/10.1007/s10637-011-9670-0; PMID: 21519855
- Rowe JM, Löwenberg B. Gemtuzumab ozogamicin in acute myeloid leukemia: a remarkable saga about an active drug. Blood 2013; 121:4838 - 41; http://dx.doi.org/10.1182/blood-2013-03-490482; PMID: 23591788
- Lu D, Joshi A, Wang B, Olsen S, Yi JH, Krop IE, Burris HA, Girish S. An integrated multiple-analyte pharmacokinetic model to characterize trastuzumab emtansine (T-DM1) clearance pathways and to evaluate reduced pharmacokinetic sampling in patients with HER2-positive metastatic breast cancer. Clin Pharmacokinet 2013; 52:657 - 72; http://dx.doi.org/10.1007/s40262-013-0060-y; PMID: 23553425
- Pascual MH, Verdier P, Malette P, Mnich J, Ozoux ML. Validation of an immunoassay to selectively quantify the naked antibody of a new Antibody Drug Conjugate--SAR566658--for pharmacokinetic interpretation improvement. J Immunol Methods 2013; 396:140 - 6; http://dx.doi.org/10.1016/j.jim.2013.06.012; PMID: 23892158
- Dirks NL, Meibohm B. Population pharmacokinetics of therapeutic monoclonal antibodies. Clin Pharmacokinet 2010; 49:633 - 59; http://dx.doi.org/10.2165/11535960-000000000-00000; PMID: 20818831
- Lobo ED, Hansen RJ, Balthasar JP. Antibody pharmacokinetics and pharmacodynamics. J Pharm Sci 2004; 93:2645 - 68; http://dx.doi.org/10.1002/jps.20178; PMID: 15389672
- EMA [Internet] Refusal assessment report for Mylotarg, Procedure No. EMEA/H/C/000705 2008 [Cited 2014 Feb 20] Available from http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/000705/WC500070677.pdf
- Keizer RJ, Huitema AD, Schellens JH, Beijnen JH. Clinical pharmacokinetics of therapeutic monoclonal antibodies. Clin Pharmacokinet 2010; 49:493 - 507; http://dx.doi.org/10.2165/11531280-000000000-00000; PMID: 20608753
- Drugs@FDA:Kadcyla [Internet] Application Number: 125427orig1s000, Clinical Pharmacology and Biopharmaceutics Reviews [Cited 2014 Feb 20] Available from http://www.accessdata.fda.gov/drugsatfda_docs/nda/2013/125427Orig1s000ClinPharmR.pdf
- Bruno R, Washington CB, Lu JF, Lieberman G, Banken L, Klein P. Population pharmacokinetics of trastuzumab in patients with HER2+ metastatic breast cancer. Cancer Chemother Pharmacol 2005; 56:361 - 9; http://dx.doi.org/10.1007/s00280-005-1026-z; PMID: 15868146
- Tabrizi MA, Tseng CM, Roskos LK. Elimination mechanisms of therapeutic monoclonal antibodies. Drug Discov Today 2006; 11:81 - 8; http://dx.doi.org/10.1016/S1359-6446(05)03638-X; PMID: 16478695
- Dowell JA, Korth-Bradley J, Liu H, King SP, Berger MS. Pharmacokinetics of gemtuzumab ozogamicin, an antibody-targeted chemotherapy agent for the treatment of patients with acute myeloid leukemia in first relapse. J Clin Pharmacol 2001; 41:1206 - 14; http://dx.doi.org/10.1177/00912700122012751; PMID: 11697753
- Drugs@FDA:Adcetris [Internet] Application Number: 125388Orig1s000, Clinical Pharmacology and Biopharmaceutics Reviews [Cited 2014 Feb 20] Available from http://www.accessdata.fda.gov/drugsatfda_docs/nda/2011/125388Orig1s000ClinPharmR.pdf
- Girish S, Gupta M, Wang B, Lu D, Krop IE, Vogel CL, Burris Iii HA, LoRusso PM, Yi JH, Saad O, et al. Clinical pharmacology of trastuzumab emtansine (T-DM1): an antibody-drug conjugate in development for the treatment of HER2-positive cancer. Cancer Chemother Pharmacol 2012; 69:1229 - 40; http://dx.doi.org/10.1007/s00280-011-1817-3; PMID: 22271209
- Ferte C, Soria JC, Penel N. Dose-levels and first signs of efficacy in contemporary oncology Phase 1 clinical trials. PLoS One 2011; 6:e16633; http://dx.doi.org/10.1371/journal.pone.0016633; PMID: 21415927
- Roberts SA, Andrews PA, Blanset D, Flagella KM, Gorovits B, Lynch CM, Martin PL, Kramer-Stickland K, Thibault S, Warner G. Considerations for the nonclinical safety evaluation of antibody drug conjugates for oncology. Regul Toxicol Pharmacol 2013; 67:382 - 91; http://dx.doi.org/10.1016/j.yrtph.2013.08.017; PMID: 24012707
- Annunziata CM, Kohn EC, LoRusso P, Houston ND, Coleman RL, Buzoianu M, Robbie G, Lechleider R. Phase 1, open-label study of MEDI-547 in patients with relapsed or refractory solid tumors. Invest New Drugs 2013; 31:77 - 84; http://dx.doi.org/10.1007/s10637-012-9801-2; PMID: 22370972
- Haddish-Berhane N, Shah DK, Ma D, Leal M, Gerber HP, Sapra P, Barton HA, Betts AM. On translation of antibody drug conjugates efficacy from mouse experimental tumors to the clinic: a PK/PD approach. J Pharmacokinet Pharmacodyn 2013; 40:557 - 71; http://dx.doi.org/10.1007/s10928-013-9329-x; PMID: 23933716
- Coiffier B, Morschhauser F, Dupuis J, Haioun C, Laine F, Houot R, et al. Phase I study cohort evaluating an optimized administration schedule of SAR3419, an anti-CD19 DM4-loaded antibody drug conjugate (ADC), in patients (pts) with CD19 positive relapsed/refractory b-cell non-Hodgkin's lymphoma. J Clin Oncol 2012; 30:Suppl. 1 8057
- Castaigne S, Pautas C, Terré C, Raffoux E, Bordessoule D, Bastie JN, Legrand O, Thomas X, Turlure P, Reman O, et al, Acute Leukemia French Association. Effect of gemtuzumab ozogamicin on survival of adult patients with de-novo acute myeloid leukaemia (ALFA-0701): a randomised, open-label, phase 3 study. Lancet 2012; 379:1508 - 16; http://dx.doi.org/10.1016/S0140-6736(12)60485-1; PMID: 22482940
- Sapra P, Betts A, Boni J. Preclinical and clinical pharmacokinetic/pharmacodynamic considerations for antibody-drug conjugates. Expert Rev Clin Pharmacol 2013; 6:541 - 55; http://dx.doi.org/10.1586/17512433.2013.827405; PMID: 23978126
- Food and Drug Administration Guidance for Industry: Estimating the Maximum Safe Starting Dose in Initial Clinical Trials for Therapeutics in Adult Healthy Volunteers, U.S. Department of Health and Human Services,, Center for Drug Evaluation and Research (CDER), July 2005
- Advani A, Coiffier B, Czuczman MS, Dreyling M, Foran J, Gine E, Gisselbrecht C, Ketterer N, Nasta S, Rohatiner A, et al. Safety, pharmacokinetics, and preliminary clinical activity of inotuzumab ozogamicin, a novel immunoconjugate for the treatment of B-cell non-Hodgkin’s lymphoma: results of a phase I study. J Clin Oncol 2010; 28:2085 - 93; http://dx.doi.org/10.1200/JCO.2009.25.1900; PMID: 20308665
- Chanan-Khan A, Wolf JL, Garcia J, Gharibo M, Jagannath S, Manfredi D, et al. Efficacy analysis from phase I study of lorvotuzumab mertansine (IMGN901), used as monotherapy, in patients with heavily pre-treated CD56-positive multiple myeloma - A preliminary efficacy analysis. Blood 2010; 116:21
- Chanan-Khan AA, Jagannath S, Heffner LT, Avigan D, Lee KP, Lutz RJ, et al. Phase I study of BT062 given as repeated single dose once every 3 weeks in patients with relapsed or relapsed/refractory multiple myeloma. Blood 2009; 114:22
- Galsky MD, Eisenberger M, Moore-Cooper S, Kelly WK, Slovin SF, DeLaCruz A, Lee Y, Webb IJ, Scher HI. Phase I trial of the prostate-specific membrane antigen-directed immunoconjugate MLN2704 in patients with progressive metastatic castration-resistant prostate cancer. J Clin Oncol 2008; 26:2147 - 54; http://dx.doi.org/10.1200/JCO.2007.15.0532; PMID: 18362364
- Krop IE, Beeram M, Modi S, Jones SF, Holden SN, Yu W, Girish S, Tibbitts J, Yi JH, Sliwkowski MX, et al. Phase I study of trastuzumab-DM1, an HER2 antibody-drug conjugate, given every 3 weeks to patients with HER2-positive metastatic breast cancer. J Clin Oncol 2010; 28:2698 - 704; http://dx.doi.org/10.1200/JCO.2009.26.2071; PMID: 20421541
- Petrylak DP, Kantoff PW, Mega AE, Vogelzang NJ, Stephenson J, Fleming MT, Stambler N, Petrini M, Blattman S, Israel RJ. Prostate- specific membrane antigen antibody drug conjugate (PSMA ADC): A phase I trial in metastatic castration-resistant prostate cancer (mCRPC) previously treated with a taxane. J Clin Oncol 2013; 31 Suppl. 1
- Sauter A, Kloft C, Gronau S, Bogeschdorfer F, Erhardt T, Golze W, Schroen C, Staab A, Riechelmann H, Hoermann K. Pharmacokinetics, immunogenicity and safety of bivatuzumab mertansine, a novel CD44v6-targeting immunoconjugate, in patients with squamous cell carcinoma of the head and neck. Int J Oncol 2007; 30:927 - 35; PMID: 17332932
- Tolcher AW, Ochoa L, Hammond LA, Patnaik A, Edwards T, Takimoto C, Smith L, de Bono J, Schwartz G, Mays T, et al. Cantuzumab mertansine, a maytansinoid immunoconjugate directed to the CanAg antigen: a phase I, pharmacokinetic, and biologic correlative study. J Clin Oncol 2003; 21:211 - 22; http://dx.doi.org/10.1200/JCO.2003.05.137; PMID: 12525512
- Younes A, Kim S, Romaguera J, Copeland A, Farial SdeC, Kwak LW, Fayad L, Hagemeister F, Fanale M, Neelapu S, et al. Phase I multidose-escalation study of the anti-CD19 maytansinoid immunoconjugate SAR3419 administered by intravenous infusion every 3 weeks to patients with relapsed/refractory B-cell lymphoma. J Clin Oncol 2012; 30:2776 - 82; http://dx.doi.org/10.1200/JCO.2011.39.4403; PMID: 22753910