
Abstract
Because drug development is not a static process, a drug’s market authorisation may change over time. In many cases, the number of indications for which a drug is approved increases. Because this facet of drug development also comes at significant costs, a corresponding patent filing strategy is required to protect these investments. The strategy as applied to rituximab, which is approved for a variety of indications, is discussed in this review.
Keywords: :
Introduction
Antibodies are today’s most important class of therapeutic drugs. To enable exclusive commercialization of a new antibody for a given amount of time, patent protection is the method of choice. However, the lifetime of a patent is restricted to 20 y, with an effective lifetime of 21 y if a priority is claimed. While such a lifetime may be sufficient in other fields of technology, where the half-life of a product is often substantially less than 20 y, it is commonly too short for pharmaceutics because, once discovered, the clinical development and marketing authorisation periods for drugs often take a total of 8–10 y, thus reducing the time during which the drug, once approved, can be marketed under patent protection.
In major jurisdictions such as the US and Europe, this problem has been realized, and compensatory tools, “patent term extension” (PTE) in the US, and „supplementary protection certificate“ (SPC) in Europe, that effectively extend the exclusivity term for a given pharmaceutic in the case of a time-consuming authorisation procedure were developed. Drug manufacturers have also developed strategies to effectively extend the time during which their product is under protection by filing sequential patent applications that cover different stages of a drug’s lifetime.Citation1 The most important of these options are: (1) second medical indication patents; (2) drug formulation patents; (3) dosage regimen patents; and (4) combination therapy patents. Such a strategy of filing sequential patent applications is often described as „patent lifecycling“ (or as „patent evergreening“ by those who disagree with the approach).
Use of this strategy reflects the reality underlying drug development, i.e., it is a costly endeavor, with biologics being more expensive to develop than small molecular drugs. According to a study performed at Tufts University, the estimated average costs of developing a new biologic is 1.2 billion USD,Citation2 while development times are slightly longer than those reported for small molecular drugs.Citation3
Drug development, however, does not end with the first market authorisation. Oftentimes, a manufacturer makes findings and inventions related to a given pharmaceutic after it has been approved by the regulatory authorities. Quite understandably, sponsors may want to make these findings and inventions the subject of subsequent patent applications in order to obtain exclusivity and, at the same time, secure freedom to operate with respect to such secondary embodiments that still rely on the drug as such.
This strategy is discussed here using the example of rituximab, which is a chimeric anti-CD20 antibody marketed by Genentech/Biogen in the US under the brand name Rituxan®, and by Roche in Europe under the brand name MabThera®. This article focuses on the correlation between European patents and patent applications protecting rituximab, and the respective indications authorised in Europe; however, similar principles and findings apply to other regulated markets, like the US.
Research and Development History of Rituximab
The development of rituximab followed the discovery of CD20, which is an antigen widely expressed, in particular, on malignant B cells, from early pre-B cells to differentiated B cells. The discovery was accomplished by Lee Nadler from the Dana Farber Cancer Institute in 1980. Nadler also created murine antibodies against CD20 using the Köhler-Milstein techniqueCitation4 and administered them to lymphoma patients.Citation5
Later, the rights to one of these antibodies, called B1, were sold to Coulter Pharmaceuticals (now Glaxo Smith Kline, GSK), who used it to develop tositumomab (Bexxar®), which is a murine anti-CD20 antibody, and its radiolabelled analog, (131I) tositumomab. Marketing of Bexxar® was discontinued as of February 2014.
Rituximab, a chimeric antibody that was also know as IDEC-C2B8, was originally developed by IDEC Pharmaceuticals. It binds to amino acids 170–173 and 182–185 on CD20, which is a 297 amino acid tetra-transmembrane protein; the amino acids bound form a loop due to a disulfide bond between amino acids 167 and 183. Similar to tositumomab, a murine radiolabelled (90Y) version of rituximab, ibritumomab tiuxetan (Zevalin®), has been approved for marketing, too.
In August 1990, IDEC researchers began to immunize mice with a human B cell line. In January 1991, a hybridoma (2B8) was identified that recognized CD20. Based on the respective murine antibody, a chimeric antibody (C2B8) was then engineered. The first quantities of rituximab were generated by heterologous expression from a Chinese hamster ovary (CHO) cell in hollow fiber reactors in spring 1992.Citation6
In malignant B cells, rituximab causes a polarization upon binding, involving a reorganization of CD20, intercellular adhesion molecule 1, and moesin, and orientation of the microtubule organizing center. Accordingly, the polarization of B cells induced by rituximab augments its therapeutic role in triggering antibody-dependent cell-mediated cytotoxicity by effector cells.Citation7
Approval History of Rituximab
In December 1992, Biogen filed an investigational new drug (IND) application with the US Food and Drug Adminstration (FDA), which was only about two and a half years after the first immunization of mice with CD20 (in August 1990), and only about one and a half year after the first quantities of rituximab were produced in a CHO cell line.
The IND resulted in the first approval, which was for the treatment of relapsed/refractory CD20-positive B-cell non-Hodgkin lymphoma (NHL), in November 1997. Hence, the entire developent of rituximab through its first approval took only seven years. It appears that one reason for this rapid development was the fact that rituximab was granted an orphan drug designation for the indication of the first approval (i.e., a status assigned to a rare disease under which some regulatory requirements are reduced), which facilitated approval due to the reduced reglulatory requirements for drugs with this designation, e.g., the underlying pivotal trial only required 166 individuals.Citation6
Soon thereafter, in June 1998, the European Commission issued the first European approval for the treatment of grade III-IV follicular lymphoma patients who are chemoresistant or are in their second or subsequent relapse after chemotherapy. In both jurisdictions, further approvals followed quickly (). Most recently, a marketing application for a subcutaneous formulation comprising 1400 mg rituximab to be administered over approximately five minutes was approved by the European Commission in March 2014. The underlying data come from the Phase 3 SABRINA study in which a new formulation that includes recombinant human hyaluronidase was tested.
Table 1a. Approval history of rituximab in the United States and Europe
shows a feature analysis of the indications underlying the different approvals in the European Union, together with information regarding a potential orphan status of a given indication in the European Union (Art 3 (1) a) of Regulation (EC) No 141/2000, according to which the disease must not affect more than five in 10000 persons in the European Union).
Table 1b. Feature analysis of the indications underlying the different approvals in the European Union
Collaborations and Mergers
In 1995, IDEC entered into a collaboration with Genentech, based in South San Francisco, in order to accelerate the development of rituximab. Genentech financed the development costs and obtained the right to co-market rituximab in the United States. In 2003, IDEC merged with Cambridge-based firm Biogen, with rituximab as IDEC’s dowry. At that time, Genentech was already partly-owned by Swiss drugmaker Roche, who had acquired a first share of Genentech in 1990. In 2009, Roche completed the acquisition of Genentech and took over the remaining 44% of the shares.
Global Sales of Rituximab
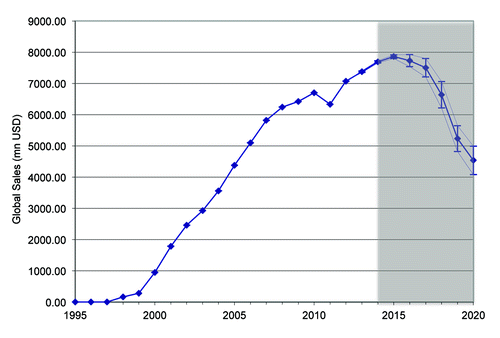
As mentioned already, rituximab’s initial approval was by the FDA in 1997. In the same year, global sales achieved 5.5 million USD. From that date on, the number of approved indications, as well as global sales, rose steadily. With global sales of 7.072 billion USD in 2012, rituximab is considered a blockbuster drug. shows global sales of rituximab between 1997 and 2020 (sales data from DrugAnalyst Ltd.)
Figure 1. Global sales of rituximab between 1997 and 2020. Data from information provider Drug Analyst. Note that figures from 2014 – 2020 are estimated. Error bars indicate upper and lower limits as used in the underlying data model.
Off-label Use
In addition to the approved indications, rituximab has been prescribed frequently for the treatment of dieases where no approval exists, i.e., so called „off-label use“. Indications encompass primary thrombocytopenia, immune thrombocytopenic purpura, macroglobulinemia, autoimmune hemolytic anemia, Burkitt lymphoma, multiple sclerosis, Wegener granulomatosis, post-transplant lymphoproliferative disorder, bullous dermatoses and hypogammaglobulinemia.Citation8 Other off-label indications include pemphigus, systemic lupus erythematosus, and angioedema.Citation9 Notably, indications that were approved at a later stage (e.g., chronic lymphocytic leukemia (CLL) in 2009) had already been used off-label before that date.Citation6 Between 2005 to 2007, ~ 17.1% of all rituximab reimbursements in the US related to off-label use,Citation8 which comprised an important share of rituximab sales.
Although US doctors may legally prescribe approved drugs for non-approved indications, drug companies are generally barred from actively promoting off-label uses of their drugs. In a recent US lawsuit,Citation10 a whistleblower accused Genentech of encouraging oncologists and other physicians to bill Medicare and other reimbursement programs for off-label uses of rituximb, thereby making the use the result of an independent medical judgment. The case was settled in November 2011 upon payment of 20 million USD, but, as part of the whistleblower settlement, Genentech did not admit guilt to the charges.
Only some off-label indications were made the subject of patent applications. The treatment of pemphigus is, for example, subject to newly filed US application US20130330332, which has a priority date of May 7, 1999. Only two European counterparts exist in this family, i.e., EP1176981 (), which was revoked in opposition, and is now in appeal, and EP1649870 (), which was refused in prosecution. For this reason, no further divisional applications can be filed in Europe, although pemphigus is disclosed, as a suitable indication, in the original specification filed in 1999 underlying the entire patent family.
Table 2. Non-exhaustive list of selected patent families assigned to Idec, Biogen, Genentech or Roche, which have been filed to protect rituximab, variants thereof, or the use thereof
The treatment of Waldenström’s macroglobulinemia (WM) was initially claimed in EP1946775 (), but was then deleted from the claims, and the application was later withdrawn. Because a pending European application in the respective family (EP2275136, ) that discloses the treatment of WM exists, it may still be possible to file a further divisional to again prosecute Waldenström’s macroglobulinemia, given that, in April 2013, the European Patent Office reinstated the former divisional rules, according to which a divisional can be filed from any pending application, without any time limits.
The treatment of systemic lupus erythematosus (SLE) is claimed in a dependent claim of EP2062916 (), which is still pending. The independent claim of EP2062916 is directed to a method for treating an autoimmune disease in a mammal who experiences an inadequate response to a tumor necrosis factor (TNF) inhibitor. Regarding the latter disease, rituximab seemed to be a promising candidate in early trials, whereas it failed in a pair of Phase 2/3 trials investigating its use in lupus nephritis, which is an inflammation of the kidney caused by SLE, so that no approval was obtained.Citation11 Probably due to the approaching patent expiry, Genentech and Roche refrained from investing in further registrational trials. It appears, however, that rituximab is still prescribed off-label on a regular basisCitation11 for SLE. Such development may encourage drug manufacturers to put a stronger focus on less formal tracks of clinical development, i.e., so-called “nonregistrational studies“.Citation12
Generally, it may seem both difficult and economically unsound to make off-label indications subject to a patent application because, for most of these indications, only insufficient data exist to support the requirements to enablement, written description and non-obviousness/inventive step. If existent, these data have oftentimes not been raised by the owners of the earlier patents. Further, some off-label indications discussed above relate to orphan diseases, which, under some circumstances, may not justify the expenses related with a patent application. It must be mentioned, however, that, with the exception of rheumatoid arthritis, most indications for which rituximab is approved qualify as orphan diseases, at least in Europe (), but the smaller patient pool that coincides with orphan status does not automatically mean that drugs addressing such indications are commercially unrewarding.Citation13
Patent Prosecution vs. Approval Procedure
While both a drug patent application and a drug approval application are subject to substantive examination, the respective bars are markedly different. To pass the test for non-obviousness/inventive step in a patent application, as for example set forth in Art. 52 of the European Patent Convention (EPC), non-clinical data that render it plausible that the claimed drug, or the alleged new medical use, formulation, dosage or combination thereof, exhibits some surprising effect, may be sufficient. It is not always clear, however, as to whether such effect provides useful in the clinical practice, let alone whether the drug, medical use, formulation dosage or combination will be eventually approved.
In contrast thereto, marketing applications submitted to regulators require clinical data that prove sufficient quality, safety and efficacy of a drug for which approval is sought, as for example set forth in Art 26 of the European Directive relating to medicinal products for human use (Directive 2001/83/EC). The respective bars appear much higher than those to be taken to meet the ”surprising effect” bar of the non-obviousness/inventive step test. A comparison between the requirements for market authorisation and patentability in Europe is shown in .
Table 1c. Comparison between the requirements for market authorization and patentability in Europe
For regulatory approval, however, no novelty requirement similar to patent examination exists. Thus, even if a patent application is rejected or revoked for lack of novelty or inventive step over the pertinent prior art, the claimed drug, medical use, formulation, dosage or combination can still receive regulatory approval. Hence, a drug or the alleged new medical use, formulation, dosage or combination thereof can receive approval even if no patent protection could be obtained, and vice versa.
Patent History of Rituximab
To date, public patent databases (e.g., www.orbit.com, as of Jan 1, 2014) contain 1659 patent families that have, in their claims, the terms “CD20” and “antibody”, out of which 236 are assigned to IDEC, Biogen, Genentech or Roche. Some of the early patent families from this list are devoted to methods for classifying white blood cells in a patient sample, in which method an anti-CD20 antibody is used (e.g., US5234816 assigned to Becton Dickinson, claiming a priority of July 12, 1992, or EP0472522 assigned to Coulter Corp, claiming a priority of December 16, 1988).
The earliest patent that is related to anti-CD20-based therapy is US patent US6652852 assigned to Xoma, which has a priority date of October 27, 1986. Xoma was already working on an anti-CD20 monoclonal antibody in the late 1980, but never got a respective product approved. The patent claims a method for treating a B-cell disorder with an antibody comprising a variable region having specificity for a CD20 antigen. The antibody is 2H7, which is a murine antibody produced by a murine hybridoma cell line deposited as HB9303 with the American Type Culture Collection (ATCC). The antibody was initially created by Ingene, which in turn was acquired by Xoma in August 1989. Though Xoma has never marketed said antibody, their early filing date put them into a position to negotiate a royalty agreement with Genentech, which gave rise, eventually, to a humanized antibody that is now developed by Biogen and Genentech, as discussed below.
Another early patent is US4987084 assigned to Dana Farber, which has a priority date of February 21, 1989. The patent claims a method of testing the effect of an agonist or an antagonist to B lymphocyte cell surface protein CD20 on B lymphocyte function, wherein optionally said agonist or antagonist comprises an antibody to B lymphocyte cell surface protein CD20.
shows a non-exhaustive list of selected patent families assigned to IDEC, Biogen, Genentech or Roche, which have been filed to protect rituximab, variants thereof, or the use thereof. The patents from family 1 have a priority date of July 24, 1992 and mark IDEC’s first CD20-related patents.European patent EP0605442 claims a chimeric anti-CD20 antibody that has a constant region from human or chimpanzee, while the antigen binding region is from an Old World Monkey, and does, as such, not protect rituximab (in which the variable regions are of murine origin). The other family members also relate to fully primate antibodies. This family will therefore no longer be discussed herein.
The patents from family 2 have a priority date of November 13, 1992, and mark IDEC’s first patents that provide compound protection for rituximab. They will thus herein be considered as the “first-generation patent family.” The different patents of this family derive from divisional applications that rely on the parent application EP0669836, and specify, in the claims, the hybridoma (EP0669836), the heavy chain (HC) and light chain (LC) sequence (EP2000149), the use and dosage in NHL (EP1005870), and the combination of rituximab with a radiolabelled anti-CD20 antibody (EP0752248).
It is, in this context, important that the claims of EP1005870 are not restricted to rituximab, i.e., their scope of protection also encompasses other anti-CD20 antibodies. Likewise, the remaining three patents also encompass ibritumomab tiuxetan because the latter is made with the same hybridoma and has the same HC and LC sequences.
Biogen IDEC has filed requests for SPCs for two members of family 2, namely for EP0669836, with ibritumomab tiuxetan as the drug for which supplementary protection is sought, and for EP2000149 with rituximab. Requests were filed in different European countries, including Germany, the UK and Ireland. While both requests are still pending in Germany, the request for EP0669836 has been granted in the UK and Ireland, already extending the protection for ibritumomab tiuxetan by five years until November 11, 2018. The request for EP2000149 is still pending in the UK, but has been rejected in Ireland. It is thus still uncertain whether the November 2013 date is really the date when compound protection for rituximab expires in Europe.
In the second-generation patents (i.e., patent family 3 and higher), second medical uses (e.g., EP2062916), combinations with other drugs (e.g., EP1176981), dosage regimen (e.g., EP1616572), formulations (e.g., EP2475353) or hybrids thereof are protected. While, with the exception of EP1112084, which protects the use of ibritumomab tiuxetan, all active patents from these families are either pending, or in opposition, they still represent a significant threat to competitors, because they either create insecurity with respect to future investments, or are, at least therorectially, enforceable although currently in opposition.
In families 2 – 10, five patents were, or still are, the subject of post-grant oppositions. EP1112084 (which relates to the use of a radiolabelled anti-CD20 antibody) was maintained in amended form. EP1613350 was finally revoked after appeal proceedings, because the main request and some auxiliary requests contained added subject matter that was not diclosed in the specification, while the 4th auxiliary request lacked novelty. EP1616572 was revoked in the first instance because during prosecution a dosage regimen was introduced into claim 1 that was not ipsis verbis disclosed in the application. The case is now in appeal. EP1176981 was revoked in the first instance for lack of inventive step. The case is now in appeal. In the opposition against EP1974747, the patentee has declared, recently, that he no longer approves the text in which the patent was granted, which equals a request for revocation.
It is thus quite surprising that out of 19 patents or patent applications in patent families 3 - 10, only one is now fully enforceable without any restrictions, i.e., it is (1) granted, (2) not yet expired, and (3) not the subject of a pending opposition. Ironically, this patent is EP1112084, which relates to the use of a radiolabeled anti-CD20 antibody, e.g., ibritumomab tiuxetan or tositumomab, not to the use of rituximab, and will for this reason no longer be discussed herein.
How the Patent Filing Strategy Reflects Rituximab’s Approval History
To demonstrate the relationship between rituximab’s approval history and its patent filing strategy, a feature analysis was first been performed, in which the different features of the different indications approved in the European Union () and the independent claims of the European members from patent families 2–10 () were distributed into particular feature categories (Disease, Stratification, Patient history, Combination with other drugs, Therapy modalities and Dosage), and type numbers were assigned. Results are shown in . These features were then correlated by means of a three-dimensional cluster analysis to demonstrate which patent or patent application reflects which authorisation. Results are shown in .
Table 3. Feature analysis of the indications approved in the European Union and the independent claims of the patent families 2 – 10
Table 4. Three dimensional cluster analysis to demonstrate which patent or patent application reflects which authorization
shows time bars reflecting the history of the European members from patent families 2 – 10. Flags indicate the date the corresponding authorisation was obtained in the European Union.
Figure 2. History of the European members from patent families 2–10. Flags indicate the date the corresponding authorization was obtained in the European Union. Patents or patent applications which have expired their maximum lifetime, or are revoked, rejected or withdrawn, are marked in italics. In some families selected withdrawn members are not shown.

Because clinical trials can represent novelty destroying prior art, at least in Europe,Citation14 patent applications are usually filed before a clinical trial is launched. Thus, a patent application that is meant to protect a given indication, dosage, formulation or drug combination is usually drafted at a time when the exact particulars of the corresponding authorisation are not yet known. This bears the risk, in case characteristics of the authorisation change during the approval process, that the resulting authorisation can have features not been disclosed in the specification.
Such a thing may have happened in EP1616572 (see above), which was revoked in the first instance because the dosage regimen introduced into claim 1 during patent prosecution was not ipsis verbis disclosed in the application. The latter disclosed weekly administration of an escalated dosage regimen, but the authorisation does not have the restriction to weekly administration.. To ensure that the patent protection covers the approved indication, the patentee thus simply omitted this restriction, which eventually gave rise to the revocation in the first instance due to inadmissible amendments.
further demonstrates that, whenever a patent was about to be granted in a given family, timely filing of a divisional occurred, because, under European law, a divisional application can only be filed relating to a European patent application that is still pending (Rule 36 EPC).
Patent Disputes
Not surprisingly, rituximab was the subject of various patent disputes, some of which relied on patents protecting enablement technologies, while others relied on patents protecting compounds, e.g., an anti-CD20 antibody.
Enablement Technology Patents
As regards the former, Biogen IDEC and Genentech were engaged in several lawsuits related to the alleged infringement of patents protecting enablement technologies that were used, allegedly, for the generation or production of rituximab.
In 2003, Genentech was involved, together with other biotechnology firms, in a lawsuit with Columbia UniversityCitation15 for the validity of Columbia’s Axel patent estate, which is related to gene expression systems that were said to be used in the generation of rituximab, and for which Genentech has paid royalties. The lawsuit was settled eventually.
In 1999, GlaxoWellcome (now GSK) sued Genentech for the infringement of four of their patents that covered stabilized immunoglobulin compositions and antibodies carrying a particular glycosylation pattern,Citation16 asking for a royalty payment on sales of rituximab. The claim was dismissed for invalidity of the underlying patents.
Quite notably, furthermore, are the different disputes related to the Cabilly family of patents, which is assigend to Genentech, and which covers key steps of bicistronic antibody expression. The patents family not only protects the production of rituximab, but many other therapeutic antibodies, and is thus subject to a large number of license contracts, and has furthermore gained a reputation for its long lifetime. The history and relevance of the Cabilly family of patents were discussed in a previous review.Citation17
Shortly thereafter, in September 2010, GSK sued Genentech for violation of patents RE 40,070 and RE 41,555. GSK claimed that the production of trastuzumab (Herceptin®) infringes the said patents, which cover the purification of IgG with hydrophobic interaction chromatography.Citation18 On the same day, Genentech responded by filing an action for declaratory judgement of non-infringement and invalidity of the two patents. Allegedly, both parties settled after the discovery process in 2012.
Compound Patents
Biogen IDEC and Genentech were likewise engaged in several lawsuits related to the alleged infringement of patents protecting rituximab, or its competitors, as a compund.
As regards compound patents, litigation took place between IDEC and Corixa (now GSK) over their anti-CD20 antibodies ibritumomab tiuxetan (Zevalin ®) and tositumomab (Bexxar ®). Citation19 IDEC claimed that four of Corixa’s patents protecting tositumomab were unenforceable. While the US District Court for the Southern District of California first granted IDEC’s motion for summary judgment in October 2003, and thus ruled that Corixa cannot use four of their patents to block sales of IDEC’s ibritumomab tiuxetan, that decision was revoked by the same court in January 2004, based on new evidence. Eventually, the parties settled their dispute and engaged in a cross-licensing agreement that encompassed ibritumomab tiuxetan and tositumomab, under which IDEC made royalty payments on their sales of ibritumomab tiuxetan to Corixa.
As discussed already, the Californian biotechnology company Xoma was alreading working on an anti-CD20 monoclonal antibody in the late 1980, called 2H7, which came into Xoma’s portfolio with the acquisition of Ingene. Xoma put this project on hold, but retained the respective patents, which covered the therapeutic use of chimeric chimeric IgG1 antibodies specific for the CD20 antigen on the surface of human B cells (among others, US5500362). These patents claim a priority of January 1987 and thus predate Biogen/IDECs own portfolio, the eariest priority of which is November 1992 (family 2 in ). On May 15, 1996, Xoma granted an exclusive license to Genentech and IDEC with respect to these patents, for which Genentech payed, and still pays, a royalty. Interestingly, one other result of this agreement seems to be the humanization of 2H7, then called hu2H7, which was the basis for the development of ocrelizumab (see below).
As part of an almost epic battle between Genentech and GSK, Genentech and Biogen sued GSK and Genmab on March 24, 2010 for infringement of US Patent US7682612 at the US District Court for the Southern District of California.Citation20 The patent is from the same family as EP1616572 and covers the treatment of CLL with a non-radiolabeled anti-CD20 antibody. Genentech claimed that GSK's anti-CD20 mAb ofatumumab (Arzerra®, see below), developed together with Genmab, violates said patent.
Although both ofatumumab and rituximab target CD20, ofatumumab binds a different epitope of the latter than rituximab, and with a different affinity. Genentech, who is the licensee of US7682612, advocated that ofatumumab infringes the patent because its claim language was not per se restricted to a particular epitope of CD20. However, in order to overcome an office objection related to lack of enablement, Biogen had, during the patent prosecution, stated that the term “anti-CD20 antibody” shall mean “antibodies having similar affinity and specifity as rituximab.”
Based on this prosecution history, the court construed the patent claims as being restricted to anti-CD20 antibodies having similar affinity and specifity as rituximab. The Court thus concluded that ofatumumab does not fall under the scope of said patent. Further, and without recoursing to prosecution history again, the court also construed the terms “does not include treatment with a radiolabeled anti-CD20 antibody” and “radiation is not used” as to exclude the use of a radiolabeled anti-CD20 antibody or the administration of a separate radiolabeled anti-CD20 antibody. Thereby, the court has signaled that the combination use of ofatumumab with a radiolabeled antibody, like GSK's tositumomab and radiolabelled I131 tositumomab, does not qualify as an infringement of the patent either.
Genentech and Biogen appealed the decision to the US Court of Appeals for the Federal Circuit, who confirmed the decision in April 2013.Citation21 The decision again makes clear how dangerous it can be to make conceding statements during patent prosecution. Such statements can strike back eventually because a US court may use them for a restrictive claim construction, in particular if advised thereof by a competitor.Citation22 gives an overview of some selected patents on which the above cases were based.
Table 5. Selected patents that were subject to litigation related to rituximab in the United States
The Advent of Biosimilars
Not surprisingly, the tremendous success of rituximab has triggered the development of follow-on biologicals, also called biosimilars. The first biosimilar to rituximab, Reditux, from India’s Dr. Reddy’s, has already been introduced to selected emerging markets, starting with India in 2007. According to the equity research firm FirstWord Pharma, 22 rituximab biosimilars were subject to clinical or preclinical trials in 2013 (see also www.clinicaltrialsregister.eu), making it the most attractive branded biologic for biosimilar manufacturers (though the first biosimilar antibody recently approved in the EU is a biosimilar version of anti-TNF antibody infliximab).Citation23
So far, no rituximab biosimilar has yet been approved in the US or Europe, despite the fact that, in Europe, the basic patent family protecting rituximab as a compound expired in November 2013 (, family 2). The reason for this delay are manifold. First, as discussed above, Biogen IDEC has filed requests for supplementary protection certificates (SPCs) for two members of the 2nd family (), that marked compound protection for rituximab. The requests are still pending. It is thus still unsure whether the oft-cited November 2013 date is really the date when compound protection for rituximab expires in Europe. Second, the second-generation patents still in force (i.e., patent families 3 and higher) represent an effective obstacle for market entry. This means that, even though, theoretically, competitors could enter the market with their biosimilars upon expiry of the first-generation patent (and, if applicable, the corresponding SPCs), second-generation patents may have to be considered, e.g., because competitors may not be allowed to advertise the respective indication, dosage, combination or formulation, or write it into the product label.
Regarding the latter, European regulatory law provides a so-called “carve-out”-option under which biosimilar manufacturers are entitled to leave away from the label (i.e., the summary of product characteristics and the patient information leaflet) any references to indications or dosage forms that are protected by patents in force. The respective permission is subject to national law, e.g., § 11e of the German Medicinal Products Act, as is the decision whether a statement must be added why certain therapeutic indications or dosage forms that are subject of the underlying authorisation are missing. In addition, it seems that the requirements set by the European Medicines Agency to provide evidence for safety and efficacy of a biosimilar antibody are higher than what was expected,Citation24 thus prolonging development times for biosimilar manufacturers.
The Quest Goes On: Biobetters
In the recent years, our understanding of the mechanism of action of rituximab, and anti-CD20 antibodies in general, has significantly increased.Citation25 This process resulted in the development of a number of second-generation anti-CD20 antibodies (sometimes also called “biobetters”), which have been characterized into two subtypes based on their ability to redistribute CD20 in membrane lipid rafts.
Type I “rituximab-like” anti-CD20 antibodies redistribute CD20 into membrane lipid rafts and potently activate complement,Citation26 whereas type II anti-CD20 antibodies weakly activate complement but more potently evoke direct programmed cell death. Both subtypes show equal ability in activating FcγR-bearing immune effector cells.
Second- or third-generation anti-CD20 antibodies are currently in the pipeline, some of which are developed by Biogen, Roche or Genentech, who have joined their forces to commercialize rituximab. gives an overview of some candidate molecules. Data were taken from information provided by the respective sponsors.
Table 6. Biobetters to rituximab that are approved or in clinical development
The market entry of these alternatives is not only subject to the respective authorisation, but also to the existence of third-party patents. While a thorough analysis of the patent situation is thus necessary to determine whether there is freedom to operate in a given market, it is noteworthy to mention that those patents from claiming rituximab or its use would not be relevant in this regard, while those patents claiming a mere anti-CD20 antibody could probably be relevant. shows some biobetter candidates that are already approved or still in the research and development pipeline. It remains questionable, however, whether the anti-CD20 market is big enough for that many successors – an outlook which stands, symbolically, for development in personalized medicine, where the number of drugs increases, while patient cohort sizes shrink.
Conclusion
Both the patent filing history and the market authorisation history mark the continued development of a drug that has already obtained its first authorisation. It is important, for biopharmaceutical companies, to protect 2nd or higher authorisations by corresponding patents, to block competitors from offering follow-on versions of the original drug for the respective particulars that are subject of the respective authorisations, once the patent protecting the basic compound has expired.
It can be challenging to synchronize the two strategies, mainly because a patent application is oftentimes filed years before the respective authorisation has been obtained, so that there may be a delta between what has been disclosed in the patent application and what has made it into the authorisation, respectively. At the same time, the standards of examination in patent prosecution and regulatory authorisation are markedly different, so that it is not unlikely that, e.g., for a given indication, a patent was awarded, but no market authorisation, and vice versa.
| Abbreviations: | ||
| ADCC | = | antibody-dependent cell-mediated cytotoxicity |
| ANCA | = | anti-neutrophil cytoplasmic antibodies |
| ATCC | = | American Type Culture Collection |
| C | = | constant region |
| Ca | = | calcium |
| CDC | = | complement-dependent cytotoxicity |
| CDR | = | complementarity-determining region |
| CHO | = | Chinese hamster ovary |
| CHOP | = | cyclophosphamide, doxorubicin, oncovin, and prednisone or prednisolone |
| CLL | = | chronic lymphocytic leukemia |
| Cu | = | copper |
| CVP | = | cyclophosphamide, vincristine, and prednisone or prednisolone |
| DIV | = | divisional application |
| DLCL | = | diffuse large cell lymphoma |
| EC | = | European Commission |
| EPC | = | European Patent Convention |
| FDA | = | Food and Drug Administration |
| Fc | = | fragment, crystallizable |
| FC | = | fludarabine and cyclophosphamide |
| GPA | = | granulomatosis with polyangiitis |
| HC | = | heavy chain |
| Ig | = | immunoglobulin |
| IND | = | investigational new drug application |
| iv | = | intravenous |
| LC | = | light chain |
| MCL | = | mantle cell lymphoma |
| MPA | = | microscopic polyangiitis |
| MTX | = | methotrexate |
| NHL | = | non-Hodgkin lymphoma |
| PCD | = | programmed cell death |
| RA | = | rheumatoid arthritis |
| sc | = | subcutaneous |
| SLE | = | systemic lupus erythematosus |
| TNF | = | tumor necrosis factor |
| V | = | variable region |
| WG | = | Wegener’s granulomatosis |
| WM | = | Waldenström’s macroglobulinemia |
Disclosure of Potential Conflicts of Interest
The author is involved in pending oppositions against some of the patents mentioned herein.
Disclaimer
The information provided herein reflect the personal views and considerations of the author. They do not represent legal counsel and should not be attributed to Michalski • Hüttermann and Partners Patent Attorneys or to any of its clients. Patent numbers and patent lifetimes have been verified with utmost care, but no liability is taken for their correctness.
References
- Storz U. IP Issues of Therapeutic Antibodies. In: Storz (ed), SpringerBriefs in Biotech Patents: “Intellectual Property Issues: Therapeutics, Vaccines and Molecular Diagnostics”, 2012, 1-16.
- DiMasi JA, Grabowski HG. The cost of biopharmaceutical R&D: Is biotech different?. Manage Decis Econ 2007; 28:469 - 79; http://dx.doi.org/10.1002/mde.1360
- Grabowski H, Cockburn I, Long G. The market for follow-on biologics: how will it evolve?. Health Aff (Millwood) 2006; 25:1291 - 301; http://dx.doi.org/10.1377/hlthaff.25.5.1291; PMID: 16966725
- Köhler G, Milstein C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 1975; 256:495 - 7; http://dx.doi.org/10.1038/256495a0; PMID: 1172191
- Nadler LM, Stashenko P, Hardy R, Kaplan WD, Button LN, Kufe DW, Antman KH, Schlossman SF. Serotherapy of a patient with a monoclonal antibody directed against a human lymphoma-associated antigen. Cancer Res 1980; 40:3147 - 54; PMID: 7427932
- Reff ME. The Discovery of Rituxan, in: Smith, C. G., O'Donnell, J. (Eds.), The process of new drug discovery and development. 2nd edition ed. Informa Healthcare, New York, 2006, 565-584.
- Rudnicka D, Oszmiana A, Finch DK, Strickland I, Schofield DJ, Lowe DC, Sleeman MA, Davis DM. Rituximab causes a polarization of B cells that augments its therapeutic function in NK-cell-mediated antibody-dependent cellular cytotoxicity. Blood 2013; 121:4694 - 702; http://dx.doi.org/10.1182/blood-2013-02-482570; PMID: 23613524
- Van Allen EM, Miyake T, Gunn N, Behler CM, Kohlwes J. Off-label use of rituximab in a multipayer insurance system. J Oncol Pract 2011; 7:76 - 9; http://dx.doi.org/10.1200/JOP.2010.000042; PMID: 21731512
- Bomm L, Fracaroli TS, Sodré JL, Bressan A, Gripp AC. Off-label use of rituximab in dermatology: pemphigus treatment. An Bras Dermatol 2013; 88:676 - 8; http://dx.doi.org/10.1590/abd1806-4841.20131905; PMID: 24068154
- ex. rel. Underwood vs Genentech, Civil Action No. 03-3983, District Court for the Eastern District of Pennsylvania.
- Eisenstein M. Approval on a knife edge. Nat Biotechnol 2012; 30:26 - 9; http://dx.doi.org/10.1038/nbt.2084; PMID: 22231087
- Beach & Strehlow, in: Steiner, Clinical Research Law And Compliance Handbook, Jones and Bartlett Publishers, 2006, 265-299.
- Meekings KN, Williams CS, Arrowsmith JE. Orphan drug development: an economically viable strategy for biopharma R&D. Drug Discov Today 2012; 17:660 - 4; http://dx.doi.org/10.1016/j.drudis.2012.02.005; PMID: 22366309
- EPA Technical Board of Appeal decision T0007/07.
- Genentech vs. Columbia, N.D. Cal. 2003, Case No 3:03-cv-01603.
- Genentech vs. GlaxoWellcome, D. Delaware, Case No. 99-335-RRM.
- Storz U. The Cabilly patents: Status quo and relevance for antibody companies. MAbs 2012; 4:274 - 80; http://dx.doi.org/10.4161/mabs.4.2.19253; PMID: 22453097
- GSK vs. Genentech, ND. California, Case No 5:10-cv-04255-PVT.
- IDEC vs. Corixa, SD. California, Case No P-01-1637 (RFC)
- Biogen vs. GSK, Case no 3:2010cv00608.
- Appeal No. 2012-1120 Fed. Cir. Apr. 16, 2013.
- Sandercock CG, Storz U. Antibody specification beyond the target: claiming a later-generation therapeutic antibody by its target epitope. Nat Biotechnol 2012; 30:615 - 8; http://dx.doi.org/10.1038/nbt.2291; PMID: 22781681
- Beck A, Reichert JM. Approval of the first biosimilar antibodies in Europe: a major landmark for the biopharmaceutical industry. MAbs 2013; 5:621 - 3; http://dx.doi.org/10.4161/mabs.25864; PMID: 23924791
- Reichert JM. Next generation and biosimilar monoclonal antibodies: essential considerations towards regulatory acceptance in Europe. February 3-4, 2011, Freiburg, Germany. MAbs 2011; 3:223 - 40; http://dx.doi.org/10.4161/mabs.3.3.15475; PMID: 21487235
- Cang S, Mukhi N, Wang K, Liu D. Novel CD20 monoclonal antibodies for lymphoma therapy. J Hematol Oncol 2012; 5:64 - 72; http://dx.doi.org/10.1186/1756-8722-5-64; PMID: 23057966
- Cragg MS, Morgan SM, Chan HT, Morgan BP, Filatov AV, Johnson PW, French RR, Glennie MJ. Complement-mediated lysis by anti-CD20 mAb correlates with segregation into lipid rafts. Blood 2003; 101:1045 - 52; http://dx.doi.org/10.1182/blood-2002-06-1761; PMID: 12393541