Abstract
The epidermal growth factor receptor (EGFR) and the type I insulin-like growth factor receptor (IGF-1R) are two cell surface receptor tyrosine kinases known to cooperate to promote tumor progression and drug resistance. Combined blockade of EGFR and IGF-1R has shown improved anti-tumor activity in preclinical models. Here, we report the characterization of a stable IgG-like bispecific antibody (BsAb) dual-targeting EGFR and IGF-1R that was developed for cancer therapy. The BsAb molecule (EI-04), constructed with a stability-engineered single chain variable fragment (scFv) against IGF-1R attached to the carboxyl-terminus of an IgG against EGFR, displays favorable biophysical properties for biopharmaceutical development. Biochemically, EI-04 bound to human EGFR and IGF-1R with sub nanomolar affinity, co-engaged the two receptors simultaneously, and blocked the binding of their respective ligands with similar potency compared to the parental monoclonal antibodies (mAbs). In tumor cells, EI-04 effectively inhibited EGFR and IGF-1R phosphorylation, and concurrently blocked downstream AKT and ERK activation, resulting in greater inhibition of tumor cell growth and cell cycle progression than the single mAbs. EI-04, likely due to its tetravalent bispecific format, exhibited high avidity binding to BxPC3 tumor cells co-expressing EGFR and IGF-1R, and consequently improved potency at inhibiting IGF-driven cell growth over the mAb combination. Importantly, EI-04 demonstrated enhanced in vivo anti-tumor efficacy over the parental mAbs in two xenograft models, and even over the mAb combination in the BxPC3 model. Our data support the clinical investigation of EI-04 as a superior cancer therapeutic in treating EGFR and IGF-1R pathway responsive tumors.
Introduction
Both the epidermal growth factor receptor (EGFR) and the type I insulin-like growth factor receptor (IGF-1R) are commonly expressed in many types of human cancers. Upon activation by their respective ligands, they each stimulate multiple receptor downstream signaling transduction pathways, including the phosphatidylinositol-3-kinase (PI3K)/Akt and the mitogen-activated protein kinase/extracellular signal-regulated kinase (MAPK/ERK) cascades. Both receptors play important roles in cancer biology by regulating a variety of cellular processes involved in supporting tumor progression, such as cell proliferation, survival, transformation and migration.Citation1–Citation5
EGFR is a clinically validated cancer target with both monoclonal antibodies (mAbs; cetuximab and panitumumab) and small molecule tyrosine kinase inhibitors (TKIs; erlotinib and gefitinib) approved as treatments for multiple indications, e.g., metastatic colorectal cancer (mCRC), head and neck squamous cell carcinoma (HNSCC), non-small cell lung carcinoma (NSCLC) and pancreatic cancers.Citation6–Citation9 IGF-1R is a target of intense investigation with at least six mAbs and several small molecule inhibitors in different stages of clinical trials.Citation10,Citation11 The most advanced study with the anti-IGF-1R figitumumab in combination with chemotherapeutics paclitaxel and carboplatin in NSCLC was terminated early due to futility (ClinicalTrials.gov: NCT00596830). However, two other anti-IGF-1R mAbs, AMG 479 and dalotuzumab, recently demonstrated encouraging clinical responses in combination with other agents in pancreatic and breast cancers, respectively,Citation12,Citation13 supporting the continued development of IGF-1R-targeted cancer therapeutics.
EGFR and IGF-1R pathways can crosstalk with each other at different levels, and they often cooperate to promote tumor growth and progression.Citation14–Citation16 The interplay of these two receptor pathways may lead to resistance by the tumor to inhibition of one receptor via compensatory upregulation/activation of the reciprocal receptor, and dual inhibition of EGFR and IGF-1R has been shown to improve anti-tumor activity and overcome resistance to therapy against a single receptor in preclinical models.Citation17–Citation24 Moreover, co-expression of EGFR and IGF-1R has been reported in many human tumors, including lung, colorectal and pancreatic carcinoma,Citation25–Citation27 supporting dual targeting of these two receptors in these indications. Clinically, EGFR inhibitors are known to be efficacious in only a subpopulation of cancer patients, and intense research for molecular predictors of clinical outcomes to EGFR targeted therapies has identified K-Ras mutation as a predictive biomarker of resistance to EGFR mAbs treatment in colorectal cancer and EGFR gene mutation or high copy number as strong indicators of response to EGFR TKIs in lung cancer.Citation28–Citation30 Rational combination strategies may overcome tumor resistance to EGFR-targeted therapies and expand their target treatment populations. The safety and efficacy of combinations of EGFR and IGF-1R inhibitors are currently being evaluated in several clinical studies (ClinicalTrials.gov: NCT00845039, NCT00617734, NCT00788957).
Bispecific molecules such as bispecific antibodies (BsAbs) provide a means of simultaneously targeting multiple epitopes on the same molecular target or different targets with a single therapeutic agent. As cancer therapeutics, they have the potential to confer novel or more potent activities, lower the cost of goods and facilitate the development of new therapeutic regimens in contrast to a mixture of two mAbs.Citation31–Citation33 Recently, catumaxomab, a trifunctional bispecific antibody targeting human epithelial cell adhesion molecule (EpCAM) and CD3 has shown a clear clinical benefit in patients with peritoneal carcinomatosis of epithelial cancers,Citation34 and a bispecific T-cell engaging (BiTE) antibody with dual specificity for CD19 and CD3 has also demonstrated encouraging clinical activity in patients with CD19 expressing hematological malignancies.Citation35 Despite strong interest in the development of bispecific molecules as cancer therapeutics, technical challenges in the production of stable and active bispecific molecules have in the past hindered the clinical evaluation of most bispecific formats. Many engineered antibody formats, including an IgG-like bispecific antibody combining the variable regions of two antibodies targeting EGFR and IGF-1R, have compromised stability or solubility.Citation35–Citation37 Several strategies have been taken to increase the product quality and in vivo stability of bispecific molecules, including PEGylation, conjugation with human serum albumin and Fc engineering.Citation38,Citation39 MM-111, a bispecific molecule with dual binding arms specific for ErbB2 and ErbB3 fused to a modified human serum albumin for improved in vivo serum half-life has shown encouraging preclinical activity and is being tested in Phase 1/2 clinical trials.Citation40 An alternative approach using Adnectins™, which are composed of fibronectin domains genetically engineered to bind specific antigens, was employed to construct a bispecific agent targeting EGFR and IGF-1R. In this case, PEGylation was used to improve the solubility and serum half-life of this bispecific molecule, and the PEGylated bispecific Adnectin™ was shown to inhibit the growth of both EGFR and IGF-1R driven human tumor xenografts; however, it remains to be seen if this dual-targeting molecule is more efficacious compared to both EGFR and IGF-1R mAbs.Citation41
We previously reported a new and robust BsAb platform that utilizes stability-engineered scFvs appended to full length IgGs to target two tumor necrosis factor receptor family members or two distinct epitopes of IGF-1R.Citation42–Citation44 Both BsAbs displayed IgG-like pharmaceutical properties, dual specificity and improved anti-tumor activity. Here, we describe the development of a novel BsAb targeting EGFR and IGF-1R using this platform technology. The bispecific antibody provides a means of concurrent persistent blockade of two growth factor receptor pathways and offers greater anti-tumor efficacy than EGFR and IGF-1R mAbs, and thus has the potential to be a superior cancer therapeutic over the mAbs.
Results
Generation of bispecific antibody directed against EGFR and IGF-1R.
An inhibitory anti-EGFR Fab RR456, identified from panning a semi-synthetic human antibody phage library against EGFRvIII, a tumor specific mutant variant of EGFR resulting from an in-frame deletion of exons 2–7 deletion,Citation45,Citation46 was confirmed to bind to both wild-type and the mutant form of EGFR. M60-A02, an affinity-matured variant of RR456, had a ∼10-fold improvement in binding affinity to EGFR as measured by kinetic surface plasmon resonance and biolayer interferometry studies and showed a ∼32-fold improvement in binding activity over the parental RR456 in binding to human EGFR-expressing CHO cells, as measured by flow cytometry (data not shown). The stability-engineered BIIB5 scFv (C06 cFv), targeting an allosteric epitope on IGF-1R,Citation47 was selected for the anti-IGF-1R scFv portion of the BsAb. M60-A02 VH was attached to a chimeric aglycosylated IgG4.P/IgG1 constant domain (agly IgG4.P/IgG1), with the stability engineered anti-IGF-1R scFv appended to the carboxyl-terminus of the CH3 domain through a flexible (GGGGS)3 linker. The resulting bispecific antibody targeting EGFR and IGF-1R (EI-BsAb), denoted EI-04, is illustrated in .
The EI-04 bispecific construct was expressed well in CHO cells, with titers ranging from 100–200 mg/L in unamplified cell lines. Protein A chromatography eluates of EI-04 BsAb generally contained low levels (<10%) of aggregate; this aggregate was predominantly dimeric, and there was little or no higher order aggregate in the preparations. Such low levels of aggregate are consistent with those observed with traditional IgG constructs. Yields following a two-step purification schema consisting of protein A and ion exchange chromatography consistently resulted in greater than 1 g of highly purified bispecific antibody from 10–20 L of culture media. The product quality of the final purified bispecific antibody was generally very high with purity >97 % as assessed by SDS-PAGE and SEC ( and ). The thermal stability profile of purified EI-04 as assessed by differential scanning calorimetry (DSC), shown in , confirms the Tm of the stability-engineered BIIB5 scFv domain (∼68°C) within the context of the final bispecific antibody format.Citation44 Stability studies of purified EI-04 BsAb showed the material to remain monomeric, homogeneous and stable at 4°C over at least 2 months at 25 and 50 mg/ml in a citrate-based pre-formulation buffer (data not shown).
Surface plasmon resonance (SPR) was used to measure the binding kinetics of recombinant, soluble human EGFR ecto-domain to immobilized EI-04 BsAb () and the parental anti-EGFR mAb M60-A02 (). Rapid, saturable kinetics were observed for both the BsAb and the mAb. The intrinsic association constants for both antibodies were 3.6 × 106 M−1s−1, while the dissociation constants were 1.0 × 10−3 s−1, also for both molecules. From these kinetic rate constants, the affinities of both molecules for EGFR were determined to be 0.28 nM. In comparison, the clinically validated anti-EGFR cetuximab and panitumumab showed EGFR binding kinetics similar to that of EI-04 and M60-A02, with binding affinities measured at 0.58 nM and 0.27 nM, respectively (data not shown). In addition, the BsAb showed the expected target specificity in vitro—EI-04 bound only to EGFR, and not to recombinant human ErbB family members HER2, HER3 or HER4 as determined in a biolayer interferometry assay (Sup. Fig. S1).
Biolayer interferometry-based steady state/equilibrium binding analysis was used to determine the affinity of EI-04 BsAb binding to the IGF-1R ectodomain. Tight apparent affinities were measured for both the EI-04 BsAb and its parental anti-IGF-1R mAb C06 at 0.36 and 0.14 nM, respectively (). Thus, EI-04 binds tightly to the IGF-1R ectodomain, within 2- to 3-fold of the parental anti-IGF-1R antibody and the error of experimental measurement.
SPR analysis was also used to demonstrate that the EI-04 BsAb can bind simultaneously to both of its targets (). His-tagged IGF-1R ectodomain was captured by anti-His tag antibody immobilized onto a Biacore chip. EI-04, when flowed over the sensor chip, bound to the captured IGF-1R. Fc-tagged EGFR, when passed over the Biacore chip, bound specifically to the IGF-1R-bound BsAb. These results demonstrate that target binding to one end of the BsAb does not hinder binding to the other end, and that EI-04 can bind simultaneously to both EGFR and IGF-1R with similar activity compared to its original mAbs.
Inhibition of ligand binding and receptor activation by EI-04.
The ability of EI-04 bispecific antibody to block both EGF and IGF-1/IGF-2 binding to their cognate receptors was assessed by DELFIA and ELISA, respectively. EI-04 BsAb, the parental anti-EGFR mAb M60-A02, and anti-EGFR mAbs cetuximab and panitumumab were tested in a competitive binding DELFIA assay format with Europium-labeled EGF to plate-bound EGFR. All of the molecules showed fully competitive inhibition of labeled EGF binding (). The IC50 values for EI-04, M60-A02, cetuximab and panitumumab are 0.17, 0.16, 0.10 and 0.11 nM, respectively. Unlabeled EGF also fully inhibited Eu-EGF binding, with an IC50 of 6.3 nM, reflecting its intrinsically weaker affinity for the receptor (data not shown). Thus, EI-04 was as active as various anti-EGFR mAbs for inhibiting EGF binding to EGFR. As measured by ELISA, EI-04 blocked both IGF-1 and IGF-2 binding to IGF-1R () comparably to the parental anti-IGF-1R antibody C06. Neither EI-04 nor C06 brought IGF binding to baseline in the assay, consistent with the previously reported allosteric inhibitory mechanism of the parental anti-IGF-1R mAb.Citation47 The IC50 values were 1.5 nM (IGF-1) and 2.6 nM (IGF-2) for EI-04 and 1.0 nM (IGF-1) and 1.6 nM (IGF-2) for C06. The slight decrease in IC50 observed with EI-04 is within the error of the measurement. Thus, the stabilized scFv derived from C06 that resides at the C-terminus of EI-04 is capable of recapitulating the ligand-blocking properties of the parental anti-IGF-1R mAb.
The effect of EI-04 on ligand-induced activation of EGFR and IGF-1R was assessed in non-small cell lung carcinoma (NSCLC) H322M cells, which express similar high levels of both receptors (data not shown) and displayed robust response to EGF or IGF-1 stimulation in phosphorylation of EGFR and IGF-1R respectively as measured by western blot analysis ( and D). EI-04 was shown to block EGF-stimulated phosphorylation of EGFR as potently as its parental anti-EGFR mAb M60-A02 () and to inhibit IGF-1-induced phosphorylation of IGF-1R to a similar extent compared to the original anti-IGF-1R mAb C06 (). The data indicate that EI-04 recapitulated the combined inhibitory activities of M60-A02 and C06 on their respective target receptors.
Concurrent blockade of EGFR and IGF-1R signaling pathways and superior inhibition of tumor cell growth by EI-04.
In culture, tumor cell lines often express constitutively active EGFR and IGF-1R signaling components due to tumor-produced autocrine factors or serum EGF and IGFs. We evaluated the effects of EI-04 and single mAbs on EGFR and IGF-1R phosphorylation and downstream signaling events in head and neck squamous cell carcinoma (HNSCC) HN11 cells, which were shown to produce autocrine EGFR ligands and develop resistance to EGFR inhibition through upregulation of IGF-1R signaling,Citation19,Citation48 thus representing a model with both EGFR and IGF-1R signaling components. illustrates the result of western blot analysis of EGFR and IGF-1R phosphorylation status in HN11 cells grown in culture medium supplemented with 10% FBS after treatment with tested antibodies for 4 h. EI-04 significantly inhibited serum-dependent IGF-1R phosphorylation to a similar degree relative to the anti-IGF-1R mAb C06 alone or in combination with anti-EGFR mAbs, despite an apparent lack of IGF-1R downregulation induction by the BsAb. This indicates that the inhibitory effect of EI-04 on IGF-1R activation was mainly due to its ligand blocking activity and not induction of receptor internalization. Interestingly, M60-A02 alone exhibited little effect on EGFR phosphorylation, and only dual-targeting of EGFR and IGF-1R by the mAb combinations or EI-04 led to evident reduction of EGFR phosphorylation under the experimental conditions, likely due to receptor pathway cross-talk in this cell line. No detectable EGFR downregulation was observed with any antibody treatment in HN11 cells; however, both EI-04 and M60-A02 were capable of promoting EGFR downregulation to a similar extent, as shown in the GEO colon cancer cell line (Sup. Fig. S2).
To assess the effects of EI-04 and control mAbs on receptor downstream signaling, p-AKT and p-ERK levels in HN11 cells treated with various antibodies for 4 h were measured by MSD assay. As shown in , phosphorylation of AKT in HN11 cells was largely dependent on the IGF-1R pathway, whereas phosphorylation of ERK appeared to be entirely driven by the EGFR pathway. In this model, EI-04 demonstrated significantly greater inhibition of AKT phosphorylation than the anti-IGF-1R mAb C06 alone, and similar degree of inhibition compared with the combinations of C06 with anti-EGFR mAbs including M60-A02, cetuximab and panitumumab. EI-04 treatment also induced potent reduction in p-ERK levels, comparable to that of single anti-EGFR mAbs and the various EGFR/IGF-1R mAb combinations. In H322M cells, a similar pattern of preferential dependence of p-AKT on the IGF-1R pathway and p-ERK on the EGFR pathway was observed, and EI-04 demonstrated potent concurrent inhibition of both AKT and ERK pathways (data not shown).
The consequence of concomitantly inhibiting both EGFR and IGF-1R signaling by EI-04 in comparison to mAbs was evaluated in a 3-day cell viability assay. The dose response curves of EI-04 and control mAbs in inhibiting cell growth of HN11 cells are shown in . Anti-EGFR mAbs M60-A02, cetuximab and anti-IGF-1R mAb C06 displayed dose-dependent inhibition of tumor cell growth; M60-A02 appeared slightly more potent than cetuximab, while both anti-EGFR mAbs were much more effective than C06. EI-04 and the combinations of C06 with M60-A02 or cetuximab all demonstrated significantly superior inhibitory activity to single mAbs. EI-04 produced statistically similar level of inhibition to what was observed with the mAb combinations over the whole dose range tested, though the advantage of EI-04 was generally seen at the two higher antibody concentrations (100 nM and 300 nM).
The effect of EI-04 in comparison to mAbs on cell cycle progression was also investigated in HN11 cells. The cells were treated with all antibodies for 48 h before cell cycle analysis. As shown in , C06 had little effect, M60-A02 and cetuximab increased the percentage of cells arresting in the G1/G0 phase, while the combinations and the BsAb both further increased the cell population in G1/G0 phase and correspondingly reduced cells entering S and G2/M phase. EI-04 even showed a slight advantage compared to the combinations, consistent with the growth inhibition data, where EI-04 displayed modest improved inhibitory activity over the combination at 100 nM in these cells (). Interestingly, the sub-G1 population representative of apoptotic cells, although small, is only visible in cells treated with EI-04 or mAb combinations.
The inhibitory activity of EI-04 on tumor cell growth in comparison to mAbs was further assessed in a panel of tumor cell lines (thirty two) derived from different tissue origins (pancreas, lung, breast, colorectal, head and neck, etc.). The percent growth inhibition given by 300 nM of C06, M60-A02 or EI-04 on tumor cells grown in the absence of exogenous ligands in medium with 10% FBS was shown (Sup. Fig. S3). Compared to C06 and M60-A02 alone, EI-04 exhibited potent growth inhibition (≥30%) on an expanded spectrum of tumor cell lines (23 for EI-04, 4 for C06 and 13 for M60-A02), and consistently produced improved growth inhibitory activity in the majority of cell lines tested. In addition, the relative expression levels of EGFR and IGF-1R and the mutation status of several genes commonly altered in cancer, including K-Ras, B-Raf, PTEN, PI3K and p53, were analyzed relative to EI-04 activity across the cell line panel (Sup. Fig. S3). No obvious biomarkers for tumor response or resistance to EI-04 were identified from this analysis. Nonetheless, EI-04 showed potent growth inhibitory activity in multiple tumor cell lines carrying one or more gene mutations.
High avidity binding to tumor cells and synergistic inhibitory effect on ligand-stimulated cell growth by EI-04.
The binding characteristics of EI-04 to tumor cell lines expressing various levels of EGFR and IGF-1R, relative to its parental anti-EGFR mAb M60-A02 and anti-IGF-1R mAb C06, were analyzed by flow cytometry to define the potential targeting properties of bispecific antibodies on heterogeneous tumor populations. Dose titration binding curves of M60-A02, C06 and EI-04 to four tumor cell lines (epidermoid carcinoma line A431, pancreatic carcinoma line BxPC3, breast carcinoma line MCF7 and NSCLC-H322M) were generated (Sup. Fig. S4). Anti-EGFR mAb M60-A02 exhibited dose-dependent strong binding to BxPC3, A431 and H322M cells, and little binding to MCF7 cells. Anti-IGF-1R mAb C06 displayed dose-dependent significant binding to MCF7 and H322M cells, and low binding activity to BxPC3 and A431 cells. In comparison, EI-04 demonstrated significant overall binding to all four tumor cell lines, and particularly greater maximal binding than the single mAbs. These results demonstrate that EI-04 can bind to human cancer cells expressing EGFR and/or IGF-1R in a manner that is distinct from either monoclonal antibody, indicating its potential to target a broader range of tumor populations.
The binding characteristics of EI-04 in comparison to its parental mAbs to BxPC3 tumor cells, which express more EGFR than IGF-1R as measured by FACS, were further evaluated in competitive flow cytometry binding assays using Alexa 647-labeled parental mAb tracers. illustrates the titration curves of EI-04, C06 and M60-A02 competing with Alexa 647-C06 for binding to IGF-1R and with Alexa 647-M60-A02 for binding to EGFR, respectively. M60-A02 did not inhibit Alexa 647-C06 binding as expected, while C06 and EI-04 were able to block Alexa 647-C06 binding in a dose-dependent manner with IC50 values of 0.67 nM for C06 and 0.05 nM for EI-04. On the other hand, as anticipated, C06 did not inhibit Alexa 647-M60-A02 binding, while M60-A02 and EI-04 blocked Alexa 647-M60-A02 binding with IC50 values of 0.73 nM and 1.71 nM, respectively. The results indicate that in tumor cells where a higher number of EGFR than IGF-1R molecules are present, EI-04 could display significantly greater IGF-1R-binding activity than the parental IGF-1R mAb C06, likely due to an enhanced avidity effect mediated by the tetravalent EI-04 BsAb.
To better understand the functional consequences of the increased binding avidity of EI-04 to BxPC3 tumor cells, we evaluated the effect of EI-04 in comparison to mAbs on IGF-1 and EGF-driven cell growth. As shown in , EI-04 demonstrated a much more potent inhibitory activity than the parental anti-IGF-1R C06 or the C06/M60-A02 combination on IGF-1 stimulated cell growth, while anti-EGFR M60-A02 had little effect on IGF-1 stimulated growth. In comparison, the BxPC3 cells were less responsive to EGF stimulation and the anti-EGFR antibodies only demonstrated inhibition at relatively high antibody concentrations (>10 nM). EI-04 and the mAb combination showed similar activity to that of M60-A02 at inhibiting EGF-driven cell growth, while C06 had no effect. The improved potency of EI-04 in blocking IGF-1-driven growth is likely attributed to the increased IGF-1R-binding avidity of EI-04 mediated by the tetravalent format of the bispecific antibody.
Anchorage-independent growth is a hallmark of neoplastic transformation. To evaluate the ability of EI-04 and the parental mAbs to inhibit anchorage-independent growth, a soft agar colony formation assay was performed. As shown in , M60-A02 showed little inhibition, while C06 alone demonstrated substantial blockade of colony formation, indicating the ability of BxPC3 cells to form colonies in soft agar was mostly driven by the IGF-1R pathway. EI-04 and the M60-A02/C06 mAb combination also induced remarkable inhibition of anchorage-independent growth. No significant difference was observed in the activity of EI-04 and C06 tested at 100 nM. However, at the lower antibody concentration (0.3 nM), the M60-A02/C06 combination did not show any advantage over C06 alone, whereas EI-04 demonstrated significantly enhanced inhibitory activity compared to C06 alone or the M60-A02/C06 combination. The result is consistent with the improved potency of EI-04 in the IGF-1-driven cell growth inhibition study, again manifesting the avidity advantage of this tetravalent EI-BsAb.
Pharmacokinetics and superior anti-tumor growth efficacy in vivo.
The pharmacokinetics of EI-04 and M60-A02 were evaluated in nude mice for dosing regimen selection in efficacy studies. The serum concentrations of EI-04 and M60-A02 after a single dose of each at 5 and 20 mg/kg are shown in . M60-A02 and EI-04 both bind to cynomolgus and rodent EGFRs with similar activity to human EGFR (data not shown), and both show non-linear PK profiles in mice due to target-mediated disposition, as reported for other anti-EGFR mAbs in human and cynomolgus.Citation49,Citation50 Antibody serum concentration analysis using EGFR-specific and EGFR/IGF-1R bispecific binding assays gave similar results, indicating that the EI-04 bispecific antibody was intact in vivo throughout the course of the study. C06 does not bind to rodent IGF-1R and the half-life of C06 was previously reported to be ∼10 days in mice.Citation44 Therefore, M60-A02 and EI-04 were dosed twice weekly (BIW) to compensate for their faster clearance while C06 was dosed on a weekly schedule (QW) in the efficacy study.
BxPC3 and GEO tumor xenograft models were selected to evaluate the efficacy of EI-04. We and others had previously established anti-tumor activity with anti-EGFR and anti-IGF-1R single mAbs in both xenograft models indicating that these models are driven by both EGFR and IGF-1R signaling pathwaysCitation51 (Joseph I, et al. unpublished data). shows the anti-tumor efficacy of EI-04 compared to M60-A02, C06 alone or in combination in the BxPC3 model. At 20 mg/kg on a BIW schedule, EI-04 was statistically more efficacious than M60-A02 at 15 mg/kg (BIW) or C06 at 15 mg/kg (QW) alone. Interestingly, EI-04 also demonstrated statistical advantage over the M60-A02/C06 combination throughout the dosing period. EI-04 at all three dose levels (5, 10, 20 mg/kg) demonstrated a similar trend of superiority over mAbs alone (data not shown) or in combination () at their respective equal molar dosages in the BxPC3 model. It is important to note that the combinations at 7.5 mg/kg and 15 mg/kg exhibit statistically similar activity, suggesting the dosing of the combination has reached a plateau while the enhanced activity of EI-04 is truly synergistic; higher efficacy observed with EI-04 cannot be achieved with the mAb combination by increasing the mAb doses. In the GEO model, EI-04 at 20 mg/kg also demonstrated significantly enhanced efficacy compared to the single mAbs at 15 mg/kg, whereas comparable to the mAb combination (). Taken together, the data indicate that EI-04 can provide better therapeutic efficacy than anti-EGFR and anti-IGF-1R single antibodies alone or even in combination in selected tumors.
Discussion
Combination therapy is becoming standard practice in oncology due to the multi-factorial nature of cancer as a disease. Bispecific molecules are appealing as cancer therapeutics because they allow targeting of multiple antigens with fewer drugs, but there are other potential advantages over combinations of individual inhibitors. First, development of a single bispecific therapeutic is less complex than development of two drugs from both manufacturing and regulatory perspectives. Second, potential rational combination of bispecific molecules with other biologics offers the opportunity for a convenient and potentially cost effective method of targeting multiple antigens (>2) with biologics, whereas a combination of three biologics would likely be too costly. More importantly, the ability to co-engage or cross-link two targets with a single molecule may confer novel biological activities to the bispecific molecules. Until recently, clinical development of bispecific antibodies has largely been hindered because of issues concerning manufacturability, solubility and stability. We have found that stability-engineered bispecific antibodies can be produced with standard processes and at similar scales to standard IgG mAbs. The ability to manufacture stable, high quality IgG-like bispecific molecules with acceptable pharmaceutical properties will facilitate the clinical development of this promising class of therapeutic agents.
EGF-1R and IGF-1R are critical growth factor pathways targeted for the treatment of solid tumors. The crosstalk of the two receptors commonly co-expressed in human tumors often results in resistance to therapies targeting a single receptor, and combinations of individual inhibitors or dual-targeting bispecific reagents are being pursued for improved cancer therapy. Here we report that a stability-engineered EGFR × IGF-1R bispecific antibody (EI-04) with IgG-like biophysical properties was capable of concurrently blocking the two growth factor receptor pathways and demonstrated superior anti-tumor activity in vitro and in vivo compared to single mAbs. Distinct from other reported EGFR × IGF-1R bispecific molecules,Citation37,Citation41 this tetravalent bispecific antibody, perhaps due to increased binding avidity, also displayed greater activity than the combinations of single mAbs in several cases.
The avidity characteristic of the tetravalent EI-BsAb was demonstrated on BxPC3 tumor cells, where IGF-1R is expressed at a lower level than EGFR. The BsAb EI-04 exhibited higher binding potency than the anti-IGF-1R mAb C06 specifically to IGF-1R on BxPC3 cells. This effect could be due to potential cross-linking of the two receptors on the cell surface or simply increased local concentration of IGF-1R binding modules due to concurrent EGFR engagement by the bispecific. Importantly, we were able to demonstrate the functional significance of such high avidity binding by showing the superior potency of EI-04 at inhibiting IGF-1-simulated cell growth compared to C06 alone or in combination with anti-EGFR mAb M60-A02. To date, we have not yet been able to demonstrate reciprocity of this activity for EGFR binding in a tumor cell line expressing higher levels of IGF-1R than EGFR. We speculate that the ratios of the two receptors on cell surface of tumor cells, the relative affinities of the two binding arms for the respective receptors, or the geometry of the individual receptor/binding arm interactions may be key for the manifestation of avidity advantages in certain tumors and, importantly, perhaps define the therapeutic outcome.
It is noteworthy that EI-04 exhibited superior anti-tumor efficacy not only to the individual anti-EGFR and anti-IGF-1R mAbs but also to their combinations in the BxPC3 pancreatic tumor model. The superiority of EI-04 over the mAb combinations was observed at saturating dose levels, demonstrating the synergistic effect of dual targeting EGFR and IGF-1R by the single agent bispecific antibody, which could be attributable to potential co-engagement of two cell surface receptors with a single molecule, and/or improved tumor targeting conferred by the avidity advantage of the tetravalent BsAb. On the other hand, the anti-tumor efficacy of EI-04 and the mAb combination, though significantly better than that of single mAbs, was comparable in the GEO colon model, underlining the complexity in the target receptor biology in each tumor model.
Distinct from many IGF-1R inhibitory mAbs, which are known to induce rapid IGF-1R downregulation,Citation52–Citation55 the EI-04 BsAb using the anti-IGF-1R C06 scFv as the C-terminal scFv building block was found to be less effective at promoting IGF-1R downregulation than the parental mAb C06. It is likely that the C06 scFv as a C-terminal fusion and the C06 Fab engage IGF-1R in distinct ways, leading to differences in the degree of receptor cross-linking and consequently the rate of receptor internalization. Nonetheless, the EI-04 BsAb was able to efficiently block IGF-1R phosphorylation despite a lesser impact on receptor downregulation, indicating that IGF-1R inhibition by the EI-BsAb is mainly mediated by ligand blockade, and uncoupled from receptor downregulation.
It is also of note that in HN11 cells with constitutively active autocrine EGFR signaling, EI-04 inhibited EGFR autophosphorylation while the parental mAb M60-A02 appeared ineffective after 4 h treatment. It is unlikely that M60-A02 did not inhibit EGFR activation, as it substantially blocked downstream ERK phosphorylation under the treatment conditions. We speculate this may reflect a potential difference in the kinetics of inhibiting EGFR autophosphorylation by M60-A02 and EI-04 due to the likely cross-talk between EGFR and IGF-1R in HN11 cells. Additionally, a slight trend of superiority of EI-04 over the M60-A02/C06 mAb combination was observed when they were tested at 100 nM and 300 nM, but not at the lower antibody concentrations in the cell growth inhibition assay with HN11 cells. Future quantitative studies of the effects of EI-04 and the mAb controls at concentrations of ≥100 nM on EGFR and IGF-1R signaling would help to clarify the mechanisms for the observed differentiation between the BsAb and the mAb combinations, and to detect any possible non-specificity at high antibody concentrations.
While screening a panel of tumor cell lines from various tissue types for their sensitivity to EI-04, we found that a wide spectrum of tumor cell lines responded to EI-04 to a greater degree compared to single mAbs. Notably, the response of tumor cell lines to EI-04 was independent of their mutation status in K-Ras, a predictive biomarker for clinical resistance to EGFR mAb-based therapy in metastatic colorectal carcinoma.Citation28,Citation29 Our data suggests that EI-04 BsAb has potential utility in treating expanded patient populations compared with EGFR mAbs and may overcome tumor resistance to single EGFR mAb treatment.
Furthermore, we demonstrated that EI-04 could block cell cycle progression and induce cell cycle arrest in G1 phase more effectively than the individual antibodies. Thus, EI-04 has potential application, in combination with standard of care chemotherapy, in various tumor indications to sensitize tumor cells to chemo-induced apoptosis. In addition, we showed that EI-04 could simultaneously block both AKT and ERK signaling in tumor cells, paving the way for development of EI-04 in combination with emerging therapeutics targeting receptor downstream signaling molecules, e.g., Raf, MEK, PI3K and mTOR, for improved efficacy.
In summary, we have developed a tetravalent bispecific antibody targeting EGFR and IGF-1R for cancer therapy. The stability-engineered molecule has IgG-like biophysical properties acceptable for pharmaceutical development. The EI-04 BsAb is capable of concurrently and persistently blocking two growth factor receptor pathways and providing enhanced anti-tumor activity compared to single anti-EGFR and anti-IGF-1R mAbs alone or in combination due to its avidity features. Our results indicate that EI-04 as a single agent or in combination with other therapies may offer potential greater clinical benefit than the single mAbs, and thus warrant its clinical investigation.
Material and Methods
Reagents, antibodies and cell lines.
Recombinant human IGF-1R(1-903) was described previously in reference Citation47. Recombinant human EGFRvIII-his-FLAG protein was prepared at Biogen Idec. EGFRvIII is a tumor specific mutant variant of EGFR resulting from an in-frame deletion of exons 2–7. The gene encoding EGFRvIII-his-FLAG was created by PCR using a commercial EGFRvIII cDNA as template, incorporating the decahistidine and FLAG tag into the downstream primer. The EGFRvIII gene sequence included the native secretion signal and ended before the transmembrane domain with amino acids KIPS followed by the tag. This gene was cloned into a proprietary mammalian cell expression vector. Methods to generate CHO cells expressing the soluble EGFRvIII-his10-FLAG have been described previously in reference Citation56. EGFRvIII-his-FLAG was purified from CHO culture supernatant using Ni-NTA affinity, Q Sepharose anion exchange and Superdex 200 size exclusion chromatography steps. EGFR-Fc, HER2-Fc, HER3-Fc, HER4-Fc, EGF, IGF-1 and IGF-2 were purchased from R&D systems. Isotype control antibodies used in the study are all from Biogen Idec including C2B8, a human-mouse chimeric IgG1 antibody specific to human CD20 (rituximab), and 5C8, a human antibody against CD40L, with a IgG1 backbone or a chimeric aglycosylated IgG4.P/IgG1(CH3) constant domain with mutations (S228P, N297Q) to diminish binding to Fcγ receptors (agly.IgG4P/IgG1). Non-small cell lung carcinoma cell line NCI-H322M, breast adenocarcinoma line MCF-7 and colorectal cancer line GEO were obtained from National Cancer Institute (NCI). Pancreatic cancer cell line BxPC3, head and neck squamous cell carcinoma line HN11 and epidermoid carcinoma line A431 were obtained from American Type Culture (ATCC). All cells were routinely adapted and cultured in RPMI-1640 medium supplemented with 10% fetal bovine serum (FBS, Irvine Scientific) and 50 µg/ml Gentamicin (Invitrogen) for experiments.
Generation of inhibitory human anti-EGFR antibody M60-A02.
The heavy and light chain variable regions of anti-EGFR Fab RR456 were selected from a semi-synthetic human Dyax antibody phage libraryCitation57 by biopanning against recombinant human EGFRvIII, a tumor specific mutant variant of EGFR resulting from an in-frame deletion of exons 2–7. Clone M60-A02 is a variant of RR456 with increased binding affinity to EGFR, generated by heavy chain CDR1/CDR2 shuffling and the resulting library panned against increasingly dilute concentrations of EGFR.Citation58 The expression and purification of full-length IgG antibody of M60-A02 were as described previously in reference Citation47.
Construction and production of bispecific EGFR x IGF-1R antibody (EI-04).
To construct the EI-BsAb heavy chain, M60-A02 VH gene sequences were subcloned into a pre-digested expression plasmid containing the heavy chain signal peptide and a chimeric aglycosylated IgG4.P/IgG1 constant domain with the stability engineered anti-IGF-1R C06 scFvCitation44 appended to the carboxyl-terminus of the CH3 domain. Stable expression of an EGFR × IGF-1R BsAb (EI-04) in DHFR-deficient CHO DG44 cells was achieved by co-transfection with M60-A02 light chain plasmid and the aglycosylated M60-A02 heavy chain/C06 scFv fusion protein plasmid. Transfected cells were grown in culture media and enriched as a stable bulk culture pool using fluorescently labeled antibodies and reiterative fluorescent-activated cell sorting (FACS).Citation56 FACS was also used to generate individual cell lines. Cell pools or cell lines were scaled for antibody production and purification.
Purification and characterization of the EI-04 BsAb.
CHO supernatants containing the EI-04 BsAb were purified over columns containing MAbSelect resin (GE Healthcare) on an AKTA Explorer (GE Healthcare). The MAbSelect elute, containing <10% soluble aggregate, was further purified to remove aggregate using a TMAE anion-exchange column (GE Healthcare). Protein was concentrated using Amicon stirred cell or centrifuge-based concentrators (Millipore), and dialyzed into a final buffer. Purified EI-04 was diluted in reducing and non-reducing SDS-PAGE sample buffers, and then loaded onto a NuPAGE Bis-Tris 4–12% polyacrylamide gradient gel (Invitrogen). The gel was stained using SimplyBlue Safestain solution (Invitrogen) and de-stained in water. EI-04 BsAb was also subjected to tandem analytical size exclusion chromatography (SEC, Agilent Series 1100, Agilent Technologies) and static light scattering analysis (Wyatt Technology), with a Phenomenex BioSep-SEC-S3000 column (7.8 mm ID × 30 cm, 5 µm beads, Phenomenex) and SEC buffer (50 mM NaH2PO4, 50 mM Na2HPO4, 150 mM NaCl, 10 mM NaN3, pH 6.8). 102 µg BsAb (diluted to 1.0 mg/ml in PBS) was injected and run on the column at a flow rate of 0.5 ml/min over four replicate chromatography runs. Eluted protein was detected by UV absorbance at 280 nm. Endotoxin levels in samples were assessed using the Endosafe-PTS Portable Test System (Charles River Laboratories) chromogenic assay kit with a disposable Limulus Amebocyte Lysate (LAL) test cartridge. The endotoxin levels were consistently low, in accordance with the levels required for in vitro and in vivo studies.
Surface plasmon resonance (SPR) and biolayer interferometry binding assays.
SPR studies were performed on a Biacore 3000 instrument (GE Healthcare). EI-04 and its parental anti-EGFR antibody, M60-A02, were captured on a Biacore chip for kinetic studies of EGFR ectodomain binding to both molecules. Biotinylated goat anti-human IgG Fc (Jackson Immunoresearch Laboratories), diluted to 50 nM in Biacore buffer HBS-EP (10 mM Hepes, pH 7.4, 150 mM NaCl, 3 mM EDTA, 0.005% Surfactant P20, GE Healthcare Bio-Sciences AB), was immobilized on a streptavidin-coated Biacore Sensor Chip SA (GE Healthcare Bio-Sciences AB). EI-04 and M60-A02 in HBS-EP at 7.5 nM were captured on the anti-huIgG Fc chip. The binding of 0.37 to 30 nM of human EGFR-FLAG-His ectodomain, diluted in HBS-EP, was measured in association and dissociation phases at 30 µl/min. Data were processed by double referencing and analyzed with a Languir 1:1 interaction model using Biacore BiaEval software. Serial binding of EGFR and IGF-1R to EI-04 was measured by a sandwich Biacore assay. Biotinylated anti-His-tagged antibody, diluted in Biacore buffer HBS-EP was immobilized on a streptavidin-coated Biacore Sensor Chip SA. 30 nM IGF-1R-10His in HBS-EP was captured on the anti-His tag chip at 5 µl/min. 25 nM EI-04 BsAb in HBS-EP was flowed over the chip at 10 µl/min. In the next step, recombinant human EGFR-Fc at 1, 3 or 5 nM, diluted in HBS-EP, was flowed over the chip at 10 µl/min. Sensorgram data were processed by double referencing and analyzed using Biacore BiaEval software.
IGF-1R ectodomain binding was measured in a biolayer interferometry-based steady-state/equilibrium binding assay on an Octet Red instrument (ForteBio, Inc.), and 300 ng/ml of EI-04, C06 and M60-A02, all diluted in OB buffer (PBS, pH 7.4, 0.01% (w/v) NaN3, 1 mg/ml BSA, 0.02 % (v/v) Tween 20), were captured on anti-huIgG Fc Octet tips. Tips were washed in OB buffer and moved to wells containing different concentrations of recombinant, His-tagged IGF-1R ectodomain, from 100 to 0.017 nM in OB buffer. Binding of IGF-1R ectodomain to the tips was recorded as biolayer interferometry signals over a long association phase (250 min), and the binding signal at the end of the incubation (Rmax) was used as a measure of the fraction bound at steady state/equilibrium. Rmax was plotted versus the total concentration of IGF-1R, and the data were fit to a hyperbolic binding curve. As the IGF-1R ectodomain is a constitutive dimer, and the tip-bound BsAb and mAb molecules each contain two IGF-1R binding domains, the apparent affinity should reflect an avidity component for IGF-1R binding.
Differential scanning calorimetry.
EI-04 BsAb was analyzed by differential scanning calorimetry (DSC) on a VP-DSC capillary cell microcalorimeter (Microcal). The protein was analyzed at 1.0 mg/ml in 10 mM sodium citrate, 280 mM sucrose, pH 6.7. 4 × 400 µl samples of EI-04 were subjected to DSC analysis (10–100°C, 120°C/h, 10 min pre-scan per sample, 8 second filtering period, low feedback mode). Raw data were analyzed using Origin7 software (OriginLab Corporation) and fit to determine the thermostabilities of the different immunoglobulin domains within the bispecific antibody.
Competitive ligand-blocking assays.
The competitive IGF-1- and IGF-2-blocking ELISAs were performed as described previously in reference Citation47. A DELFIA assay was used to measure the ability of EI-04, M60-A02, cetuximab, panitumumab and unlabeled EGF to block Europium-EGF ligand binding to recombinant, purified EGFR ectodomain. Plates were coated with goat-anti human IgG and EGFR-Fc was added in 1% milk for receptor capture. Antibodies were titrated down 3-fold from 200 nM to 0.8 pM in the presence of 4 nM EGF-biotin. Streptavidin-Eu was used to detect EGFR-bound EGF-biotin. Plate fluorescence was read on the Wallac Victor fluorescence plate reader (Perkin Elmer) using the Europium protocol.
FACS-based binding studies.
For direct binding study, cells grown near confluence were lifted with cell dissociation buffer (Invitrogen), counted and plated in 96-well round bottom plates at 1 × 106 cells per well. EI-04, M60-A02 and C06 were tested at a starting concentration of 500 nM with serial half-log dilutions in FACS buffer (PBS/12% serum/0.05% sodium azide). Samples were incubated on ice for 45 min, then centrifuged at 1,500 rpm for 4 min at 4°C and washed 3 times with FACS buffer. The supernatant was aspirated and 100 µl of secondary antibody goat anti-human kappa-PE conjugate (Southern Biotech) at a 1:300 dilution was added to each corresponding well in FACS buffer. Samples were incubated for an additional 45 min on ice, and then washed as described above and resuspended in 100 µl FACS buffer containing propidium iodide (PI) (Molecular Probes) for dead cell exclusion. Samples were analyzed on the FACSCalibur flow cytometer using CellQuest software (Becton Dickinson). For competitive binding study, the parental mAbs anti-IGF-1R C06 and anti-EGFR M60-A02 were labeled with Alexa Fluor 647 (Invitrogen) according to the manufacturer's instructions. In the competitive binding assays, Alexa 647-labeled C06 or M60-A02 were held constant at 5 nM, mixed with unlabeled EI-04, C06 and M60-A02 serially diluted at 1:3 from a starting concentration of 1,000 nM and incubated with 0.5 × 106 of BxPC3 cells on ice for 45 min before FACS analysis.
Western blot analysis of IGF-1R and EGFR phosphorylation and downregulation.
NSCLC H322M cells were seeded into 12-well culture plates at 5 × 105 cells per well and grown in RPMI-1640 medium containing 10% FBS overnight. The next day, cells were serum-starved for 24 h and then treated with antibodies for 1 h at 37°C followed by stimulation with 200 ng/ml of IGF-1 or 100 ng/ml of EGF (R & D Systems) for 15–20 min. In a separate experiment, HNSCC-HN11 cells were seeded into 12-well culture plates at 5 × 105 cells per well and grown in RPMI-1640 medium containing 10% FBS overnight. The next day, cells were treated for 4 h with either 100 nM or 1 nM of EI-04, anti-EGFR and anti-IGF-1R mAbs in 10% FBS containing medium. Cellular proteins were extracted in a cell lysis buffer (Cell Signaling Technology), and protein concentrations in lysates were measured using the colorimetric BCA protein assay kit (Pierce Protein Research Products, Thermo-Fisher Scientific). Equal amounts of protein were separated on a NuPage 4–12% Tris-Bis gel (Invitrogen), and transferred to a Nitrocellulose membrane (0.45 µm pore). The blots were probed with primary antibodies for phospho-IGF-1R (Tyr1135/1136), phospho-EGFR (Tyr1173), total EGFR and total IGF-1R (Cell Signaling Technology) and then with a secondary antibody conjugate anti-rabbit-IgG-HRP (Jackson ImmunoResearch Laboratories) followed by development with Supersignal western Substrate Kit (Pierce Protein Research Products). Chemiluminescence images were captured using BioRad's VersaDoc 5000 imaging system.
AKT and ERK phosphorylation analysis by meso scale discovery (MSD).
Cellular proteins were extracted as described above. Phospho-AKT and total AKT levels in cell lysates were measured using a phospho-AKT (Ser 473)/total AKT duplex MSD kit (Meso Scale Discovery). Plates were loaded with 20 µg of total protein in duplicate, incubated overnight at 4°C with shaking and processed according to the manufacturer's protocol. Phospho-ERK and total ERK levels in cell lysates were measured using a phospho-ERK1/2 (Thr202/204, Tyr185/187)/total ERK1/2 duplex MSD kit (Meso Scale Discovery). Plates were loaded with 2.5 µg of total protein in duplicates, incubated overnight at 4°C with shaking and processed according to the manufacturer's protocol. Plates were read on the Sector Imager 2400 (Meso Scale Discovery). The percentage of phospho-protein over total protein was calculated using the formula: (2 × p-protein)/(p-protein + total protein) × 100.
Tumor cell growth inhibition assay.
Cells were seeded at 5,000–8,000 cells per well in 96-well plates and grown in RPMI-1640 medium containing 10% FBS overnight. The next day, cells were switched to serum-free medium supplemented with 100 ng/ml of IGF-1 or EGF (R & D Systems), or 10% FBS-containing medium. Then, various test antibodies including EI-04, C06, M60-A02, cetuximab, the combinations of C06 with M60-A02 or cetuximab, serially diluted starting from 300 nM, were added to the cell culture. After three days treatment, cell viability was determined with a Cell Titer Glo reagent (Promega). The percentage of growth inhibition in serum-free condition was calculated according to the formula [1 − (signal with Ab − signal in SFM)/(signal with IGF or EGF − signal in SFM)] × 100, while in the presence of serum it was calculated according to the formula [1 − (signal with Ab/signal withno Ab)] × 100.
Cell cycle analysis.
HN11 cells were seeded at 2 × 105 cells per well into six-well plates and cultured overnight in RPMI-1640 medium containing 10% FBS. Then cells were treated with a single dose of 100 nM of various antibodies for 48 h. Cells were fixed in pre-chilled (−20°C) 70% ethanol and stained with Propidium Iodide solution (20 µg/ml) for 30 min at room temperature before FACS analysis of DNA contents. The relative percentage of cells in sub-G1, G0/G1, S and G2/M phases was calculated from histograms using the FlowJo 7.7.2 software (Tree Star, Inc.).
Soft-agar colony formation assay.
A layer of 0.6% agar prepared with RPMI-1640 supplemented 10% fetal bovine serum was poured into 96-well culture plates and allowed to solidify. Then, BxPC3 cells mixed with 0.3% agar in medium containing 0.3 or 100 nM of various antibodies in the presence of both IGF-1 and EGF each at 100 ng/ml were plated at 4,000 cells per well on top of the 0.6% agar layer. The plates were incubated at 37°C in 5% CO2 incubator for 2 weeks. For detection of live colonies, alamarBlue reagent (Invitrogen) was added to cells and incubated for 8 h. Fluorescence signal indicative of colony growth was determined on a fluorescence plate reader (Flexstation 3, Molecular Devices) with excitation at 530 nm and emission at 590 nm.
Pharmacokinetics (PK) of EI-04 and M60-A02 in mice.
Mice were maintained in accordance with the Biogen Idec Institutional Animal Care and Use Committee guidelines for the humane treatment and care of laboratory animals. A single dose PK study was conducted with the BsAbs in female nude mice (Charles River Laboratories). The mice were dosed via tail intravenous injections of 5 and 20 mg/kg of M60-A02 or EI-04. At various time points post dosing, mice were sacrificed and blood was collected by cardiac puncture and separated for serum recovery. Serum samples were frozen at the time of collection and later tested by a bispecific ligand binding DELFIA assay for serum antibody titers. Briefly, plates were coated with His-tagged EGFR ectodomain and blocked. EGFR-coated and blocked plates were incubated with various dilutions of serum. EGFR-bound BsAb or mAb was detected after incubation with either biotinylated IGF-1R ectodomain + Europium labeled-streptavidin for EI-04 or Eu-anti-HuIgG Fc for M60-A02. Plates were incubated with enhancement solution for 15 min prior to measurement of the lanthanide fluorescence signal on a Wallac Victor instrument (Wallac, Perkin Elmer). Serum concentrations of the BsAb and mAb were determined from standard curves generated from known concentrations of EI-04 or M60-A02 on each plate. Pharmacokinetic properties were calculated using the noncompartmental module of WinNonLin (Pharsight Corp.).
Tumor xenograft study.
All animal studies were conducted under conditions and protocols approved by the Institutional Animal Care and Use Committee. The GEO xenografts were established in nude mice (8–10 week old, Charles River Laboratories) by injecting subcutaneously 5 × 106 cells in 50% matrigel in the flank region. CB-17 SCID mice (8–10 week old, Charles River Laboratories) were inoculated subcutaneously with 2 × 106 BxPC3 cells in 50% matrigel in the flank region to establish the BxPC3 tumors. Tumor-bearing mice were randomized into treatment groups (n = 9–10). The average tumor volume of the groups at the initiation of treatment was approximately ∼200 mm3. Mice were treated with control mAb 5C8 (IgG1) at 20 mg/kg on a twice a week (BIW) dosing schedule, M60-A02 at 3.75, 7.5 or 15 mg/kg BIW, C06 at 3.75, 7.5 or 15 mg/kg once weekly (QW), combination of M60-A02 and C06 each at 3.75, 7.5 or 15 mg/kg (BIW/QW), EI-04 at 5, 10 or 20 mg/kg BIW. The doses were selected on an equal molar basis for comparing BsAb and mAb with molecular weights of approximately 200 kDa and 150 kDa respectively. In general, tumor-bearing mice were treated with antibodies for a course of 6 weeks. Tumors were measured twice weekly and continued up to two weeks following termination of dosing. Statistical significance was established using the repeated measure one-way ANOVA-analysis.
Abbreviations
| BsAb | = | bispecific antibody |
| EGFR | = | epidermal growth factor receptor |
| IGF-1R | = | type I insulin-like growth factor receptor |
| EI-BsAb | = | EGFR × IGF-1R BsAb |
| scFv | = | single chain variable fragment |
| DELFIA | = | dissociation-enhanced lanthanide fluorescent immunoassay |
| SEC | = | size exclusion chromatography |
| DSC | = | differential scanning calorimetry |
| SPR | = | surface plasmon resonance |
Figures and Tables
Figure 1 Schematic diagram and biochemical analysis of EI-04. (A) Schematic diagram of EI-04, composed of an anti-EGFR Fab linked to an effectorless human Fc with a stability-engineered anti-IGF-1R scFv attached at the C-terminus. (B) SDS-PAGE analysis of EI-04 under non-reducing (lane 2) and reducing (lane 3) conditions. (C) Analytical size exclusion chromatogram of EI-04. Static light scattering measurements indicate that the material is monomeric with MW ∼200 kDa. (D) Differential scanning calorimetry (DSC) analysis of EI-04. Raw data are shown as a solid black line, while the deconvoluted peaks, each representing an EI-04 domain (Fab, CH2, CH3, scFv), are shown as dotted lines. The deduced domain assignments are indicated above each peak.
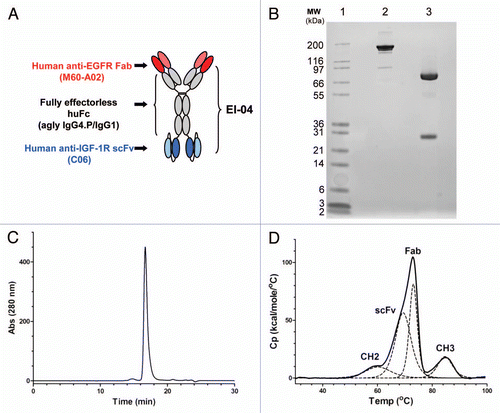
Figure 2 Binding activities of EI-04 BsAb and its parental anti-EGFR and anti-IGF-1R mAbs. (A and B) Biacore surface plasmon resonance sensorgrams of soluble EGFR ectodomain (0–30 nM) to chip-captured EI-04 (A) and anti-EGFR mAb M60-A02 (B). (C) Octet Red biolayer interferometry-based equilibrium analysis for soluble IGF-1R ectodomain binding to tip-bound EI-04 and anti-IGF-1R mAb C06. (D) Biacore surface plasmon resonance sensorgrams demonstrating serial binding of IGF-1R ectodomain and Fc-tagged EGFR ectodomain to EI-04.
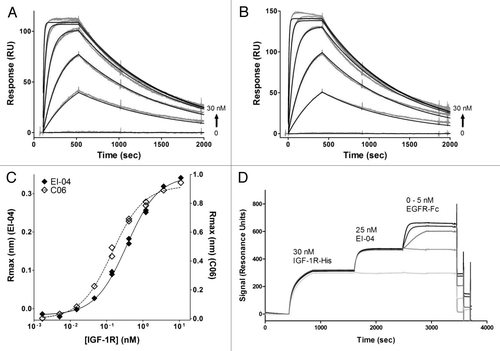
Figure 3 Dual inhibitory activities of EI-04 on EGFR and IGF-1R. (A) Blockade of europium-labeled EGF binding to EGFR-Fc by EI-04 in comparison to various anti-EGFR mAbs. (B) Blockade of a constant level of IGF-1 (25 nM, left part) or IGF-2 (60 nM, right part) binding to biotin-hIGF-1R-Fc by EI-04 in comparison to anti-IGF-1R mAb C06. (C) Inhibition of EGF-induced EGFR phosphorylation in H322M tumor cells by EI-04 in comparison to anti-EGFR mAb M60-A02. Serum-starved cells were treated with EI-04 or M60-A02, or control IgG (ctrl Ab) for 1 h followed by stimulation with 100 ng/ml of EGF for 15 min. Phospho-EGFR and total EGFR were analyzed by western blot. (D) Inhibition of IGF-1-induced IGF-1R phosphorylation in H322M tumor cells by EI-04 in comparison to anti-IGF-1R mAb C06. Serum-starved cells were treated with EI-04 or C06 or control IgG (ctrl Ab) for 1 h followed by stimulation with 200 ng/ml of IGF-1 for 20 min. Phospho-IGF-1R and total IGF-1R were analyzed by western blot.
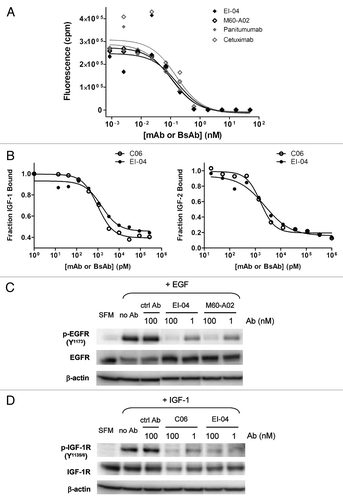
Figure 4 Concurrent blockade of EGFR and IGF-1R signaling pathways and enhanced inhibition of tumor cell growth and cell cycle progression by EI-04. (A) Simultaneous inhibition of phosphorylation of EGFR and IGF-1R in HN11 tumor cells by EI-04. Cells grown in culture medium supplemented with 10% FBS were treated with the indicated antibodies against EGFR and IGF-1R or a control IgG (ctrl Ab) for 4 h. Phospho-EGFR, phospho-IGF-1R, total EGFR, total IGF-1R and β-actin in cell lysates were analyzed by western blot. (B) Simultaneous blockade of phosphorylation of Akt and ERK in HN11 cells by EI-04. Cells were treated as described in (A). Phospho-Akt (left part) and phospho-ERK (right part) levels were quantified using MSD. Data are means ± SD (n = 2), representative results from two similar experiments. (C) Improved inhibition of serum-driven HN11 cell growth by EI-04 compared to single mAbs. Tumor cells grown in culture medium supplemented with 10% FBS were treated with serially diluted antibodies starting from 300 nM for three days prior to cell viability determination. Percent growth inhibition was calculated relative to no antibody treatment control. Data are means ± SD (n = 3), representative results from two similar experiments. Significance of difference between mAbs and EI-04: *p < 0.05, **p < 0.01, ***p < 0.001, by one-way ANOVA. (D) Enhanced blockade of serum-driven HN11 cell cycle progression by EI-04 compared to single mAbs. Tumor cells were grown in serum-free medium (SFM) or in culture medium supplemented with 10% FBS and treated with 100 nM of the indicated antibodies against EGFR and IGF-1R or a control IgG (ctrl Ab) for two days. Percentage of cells in each cell cycle phases was determined by FACS analysis.
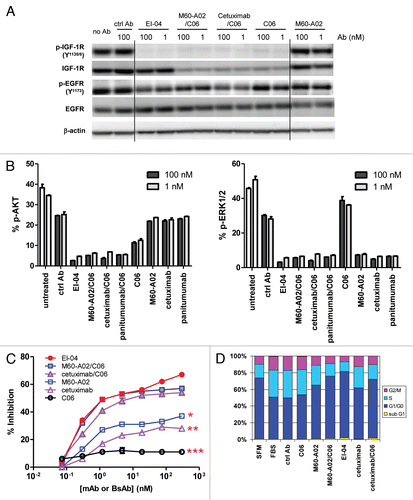
Figure 5 High avidity binding and synergistic inhibitory effect of EI-04 on BxPC3 tumor cells. (A) Flow cytometric measurement of the abilities of EI-04, M60-A02 and C06 to compete with Alexa 647-labeled C06 (left part) and Alexa 647-labeled M60-A02 (right part) for binding to BxPC3 tumor cells. Mean fluorescence intensity was measured when cells were stained with Alexa 647-C06 or Alexa 647-M60-A02 in the presence of various serially diluted competitor antibodies. Data are representative results from two similar experiments. (B) Inhibition of IGF-1-stimulated (left part) or EGF-stimulated (right part) BxPC3 tumor cell growth by EI-04, M60-A02, C06 or the M60-A02/C06 combination. Cells grown in serum-free medium supplemented with 100 ng/ml of IGF-1 or EGF were treated with serially diluted antibodies for three days prior to cell viability determination. Percent growth inhibition was calculated relative to no antibody treatment controls. Data are means ± SD (n = 3), representative results from two similar experiments. (C) Inhibition of BxPC3 cell colony formation by EI-04, M60-A02, C06 or the M60-A02/C06 combination. BxPC3 cells plated in soft agar were treated with a single dose of 0.3 or 100 nM of the indicated antibodies and allowed to grow in complete medium supplemented with 100 ng/ml of EGF and IGF-1 each for two weeks. Fluorescence signals correlating with live colonies are shown. Data are means ± SD (n = 4), representative results from two similar experiments.
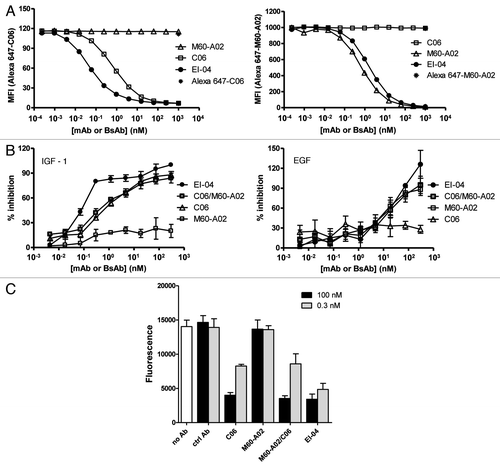
Figure 6 Comparable pharmacokinetic profiles of EI-04 and M60-A02 in nude mice. The serum concentrations of EI-04 and M60-A02 are shown for the indicated time points after a single tail vein injection of each antibody at 5 or 20 mg/kg. PK parameters from WinNonLin analysis are indicated in the bottom table.
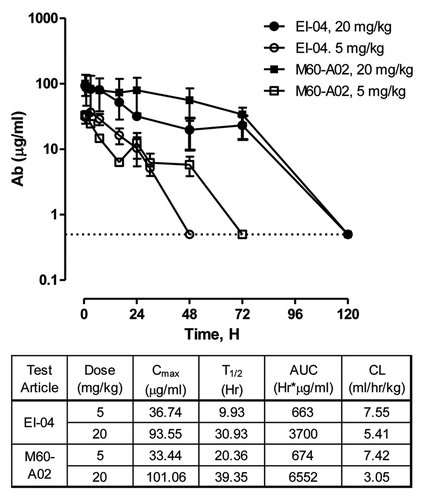
Figure 7 In vivo superior efficacy of EI-04 in two xenograft models. (A) Efficacy of EI-04 at 20 mg/kg in comparison to M60-A02 or C06 dosed at 15 mg/kg as single agents or in combination in the BxPC3 pancreatic cancer model. (B) Efficacy of EI-04 at 20 mg/kg, 10 mg/kg and 5 mg/kg in comparison to the M60-A02/C06 combination dosed at 15 + 15 mg/kg, 7.5 + 7.5 mg/kg and 3.75 + 3.75 mg/kg in the BxPC3 pancreatic cancer model. (C) Efficacy of EI-04 at 20 mg/kg in comparison to M60-A02 or C06 dosed at 15 mg/kg as single agents or in combination in the GEO colon cancer model. Tumor-bearing mice were given intraperitoneal administration of a control IgG (ctrl Ab, 5C8), M60-A02 and EI-04 twice a week (BIW) or C06 once a week (QW). The dosing schedules are indicated on the x-axis. The mean tumor volumes from each treatment group of approximately 10 mice were plotted as a function of time. The differences in the tumor growth rate between the mAb treatment groups and the EI-04 group dosed at the equal molar doses were indicated by *p < 0.05, **p < 0.01 or ***p < 0.001, if they are statistically significant as determined by one-way ANOVA.
Additional material
Download Zip (372 KB)References
- Normanno N, De Luca A, Bianco C, Strizzi L, Mancino M, Maiello MR, et al. Epidermal growth factor receptor (EGFR) signaling in cancer. Gene 2006; 366:2 - 16
- Baserga R, Peruzzi F, Reiss K. The IGF-1 receptor in cancer biology. Int J Cancer 2003; 107:873 - 877
- Pollak M. Insulin and insulin-like growth factor signalling in neoplasia. Nat Rev Cancer 2008; 8:915 - 928
- Hynes NE, MacDonald G. ErbB receptors and signaling pathways in cancer. Curr Opin Cell Biol 2009; 21:177 - 184
- Werner H, Bruchim I. The insulin-like growth factor-I receptor as an oncogene. Arch Physiol Biochem 2009; 115:58 - 71
- Vincenzi B, Zoccoli A, Pantano F, Venditti O, Galluzzo S. Cetuximab: from bench to bedside. Curr Cancer Drug Targets 2010; 10:80 - 95
- Gravalos C, Cassinello J, Garcia-Alfonso P, Jimeno A. Integration of panitumumab into the treatment of colorectal cancer. Crit Rev Oncol Hematol 2010; 74:16 - 26
- Iyer R, Bharthuar A. A review of erlotinib—an oral, selective epidermal growth factor receptor tyrosine kinase inhibitor. Expert Opin Pharmacother 2010; 11:311 - 320
- Campbell L, Blackhall F, Thatcher N. Gefitinib for the treatment of non-small-cell lung cancer. Expert Opin Pharmacother 2010; 11:1343 - 1357
- Gualberto A, Pollak M. Emerging role of insulin-like growth factor receptor inhibitors in oncology: early clinical trial results and future directions. Oncogene 2009; 28:3009 - 3021
- Rodon J, DeSantos V, Ferry RJ Jr, Kurzrock R. Early drug development of inhibitors of the insulin-like growth factor-I receptor pathway: lessons from the first clinical trials. Mol Cancer Ther 2008; 7:2575 - 2588
- Kindler HL, Richards DA, Stephenson J, Garbo LE, Rocha Lima CS, Safran H, et al. A placebo-controlled, randomized phase II study of conatumumab (C) or AMG 479 (A) or placebo (P) plus gemcitabine (G) in patients (pts) with metastatic pancreatic cancer (mPC). J Clin Oncol 2010; 28:4035
- Di Cosimo S, Bendell JC, Cervantes-Ruiperez A, Roda D, Prudkin L, Stein MN, et al. A phase I study of the oral mTOR inhibitor ridaforolimus (RIDA) in combination with the IGF-1R antibody dalotozumab (DALO) in patients (pts) with advanced solid tumors. J Clin Oncol 2010; 28:3008
- Adams TE, McKern NM, Ward CW. Signalling by the type 1 insulin-like growth factor receptor: interplay with the epidermal growth factor receptor. Growth Factors 2004; 22:89 - 95
- Riedemann J, Takiguchi M, Sohail M, Macaulay VM. The EGF receptor interacts with the type 1 IGF receptor and regulates its stability. Biochem Biophys Res Commun 2007; 355:707 - 714
- van der Veeken J, Oliveira S, Schiffelers RM, Storm G, van Bergen En Henegouwen PM, Roovers RC. Crosstalk between epidermal growth factor receptor- and insulin-like growth factor-1 receptor signaling: implications for cancer therapy. Curr Cancer Drug Targets 2009; 9:748 - 760
- Jones HE, Goddard L, Gee JM, Hiscox S, Rubini M, Barrow D, et al. Insulin-like growth factor-I receptor signalling and acquired resistance to gefitinib (ZD1839; Iressa) in human breast and prostate cancer cells. Endocr Relat Cancer 2004; 11:793 - 814
- Buck E, Eyzaguirre A, Rosenfeld-Franklin M, Thomson S, Mulvihill M, Barr S, et al. Feedback mechanisms promote cooperativity for small molecule inhibitors of epidermal and insulin-like growth factor receptors. Cancer Res 2008; 68:8322 - 8332
- Guix M, Faber AC, Wang SE, Olivares MG, Song Y, Qu S, et al. Acquired resistance to EGFR tyrosine kinase inhibitors in cancer cells is mediated by loss of IGF-binding proteins. J Clin Invest 2008; 118:2609 - 2619
- Morgillo F, Woo JK, Kim ES, Hong WK, Lee HY. Heterodimerization of insulin-like growth factor receptor/epidermal growth factor receptor and induction of survivin expression counteract the antitumor action of erlotinib. Cancer Res 2006; 66:10100 - 10111
- Desbois-Mouthon C, Baron A, Blivet-Van Eggelpoel MJ, Fartoux L, Venot C, Bladt F, et al. Insulin-like growth factor-1 receptor inhibition induces a resistance mechanism via the epidermal growth factor receptor/HER3/AKT signaling pathway: rational basis for cotargeting insulin-like growth factor-1 receptor and epidermal growth factor receptor in hepatocellular carcinoma. Clin Cancer Res 2009; 15:5445 - 5456
- Hendrickson AW, Haluska P. Resistance pathways relevant to insulin-like growth factor-1 receptor-targeted therapy. Curr Opin Invest Drugs 2009; 10:1032 - 1040
- Haluska P, Carboni JM, TenEyck C, Attar RM, Hou X, Yu C, et al. HER receptor signaling confers resistance to the insulin-like growth factor-I receptor inhibitor, BMS-536924. Mol Cancer Ther 2008; 7:2589 - 2598
- Kaulfuss S, Burfeind P, Gaedcke J, Scharf JG. Dual silencing of insulin-like growth factor-I receptor and epidermal growth factor receptor in colorectal cancer cells is associated with decreased proliferation and enhanced apoptosis. Mol Cancer Ther 2009; 8:821 - 833
- Ludovini V, Bellezza G, Pistola L, Bianconi F, Di Carlo L, Sidoni A, et al. High coexpression of both insulin-like growth factor receptor-1 (IGFR-1) and epidermal growth factor receptor (EGFR) is associated with shorter disease-free survival in resected non-small-cell lung cancer patients. Ann Oncol 2009; 20:842 - 849
- Takahari D, Yamada Y, Okita NT, Honda T, Hirashima Y, Matsubara J, et al. Relationships of insulin-like growth factor-1 receptor and epidermal growth factor receptor expression to clinical outcomes in patients with colorectal cancer. Oncology 2009; 76:42 - 48
- Ueda S, Hatsuse K, Tsuda H, Ogata S, Kawarabayashi N, Takigawa T, et al. Potential crosstalk between insulin-like growth factor receptor type 1 and epidermal growth factor receptor in progression and metastasis of pancreatic cancer. Mod Pathol 2006; 19:788 - 796
- Lievre A, Bachet JB, Boige V, Cayre A, Le Corre D, Buc E, et al. KRAS mutations as an independent prognostic factor in patients with advanced colorectal cancer treated with cetuximab. J Clin Oncol 2008; 26:374 - 379
- Freeman DJ, Juan T, Reiner M, Hecht JR, Meropol NJ, Berlin J, et al. Association of K-ras mutational status and clinical outcomes in patients with metastatic colorectal cancer receiving panitumumab alone. Clin Colorectal Cancer 2008; 7:184 - 190
- Heist RS, Christiani D. EGFR-targeted therapies in lung cancer: predictors of response and toxicity. Pharmacogenomics 2009; 10:59 - 68
- Chames P, Baty D. Bispecific antibodies for cancer therapy: the light at the end of the tunnel?. mAbs 2009; 1:539 - 547
- Hollander N. Bispecific antibodies for cancer therapy. Immunotherapy 2009; 1:211 - 222
- Thakur A, Lum LG. Cancer therapy with bispecific antibodies: Clinical experience. Curr Opin Mol Ther 2010; 12:340 - 349
- Heiss MM, Murawa P, Koralewski P, Kutarska E, Kolesnik OO, Ivanchenko VV, et al. The trifunctional antibody catumaxomab for the treatment of malignant ascites due to epithelial cancer: Results of a prospective randomized phase II/III trial. Int J Cancer 2010; 127:2209 - 2221
- Bargou R, Leo E, Zugmaier G, Klinger M, Goebeler M, Knop S, et al. Tumor regression in cancer patients by very low doses of a T cell-engaging antibody. Science 2008; 321:974 - 977
- Demarest SJ, Glaser SM. Antibody therapeutics, antibody engineering and the merits of protein stability. Curr Opin Drug Discov Devel 2008; 11:675 - 687
- Lu D, Zhang H, Koo H, Tonra J, Balderes P, Prewett M, et al. A fully human recombinant IgG-like bispecific antibody to both the epidermal growth factor receptor and the insulin-like growth factor receptor for enhanced antitumor activity. J Biol Chem 2005; 280:19665 - 19672
- Muller D, Karle A, Meissburger B, Hofig I, Stork R, Kontermann RE. Improved pharmacokinetics of recombinant bispecific antibody molecules by fusion to human serum albumin. J Biol Chem 2007; 282:12650 - 12660
- Ridgway JB, Presta LG, Carter P. ‘Knobs-into-holes’ engineering of antibody CH3 domains for heavy chain heterodimerization. Protein Eng 1996; 9:617 - 621
- Denlinger CS, Beeram M, Tolcher AW, Goldstein LJ, Slichenmyer WJ, Murray J, et al. A phase I/II and pharmacologic study of MM-111 in patients with advanced, refractory HER2-positive (HER2+) cancers. J Clin Oncol 2010; 28:169
- Emanuel SL, Engle LJ, Chao G, Zhu RR, Cao C, Lin Z, et al. A fibronectin scaffold approach to bispecific inhibitors of epidermal growth factor receptor and insulin-like growth factor-I receptor. mAbs 2011; 3:38 - 48
- Miller BR, Demarest SJ, Lugovskoy A, Huang F, Wu X, Snyder WB, et al. Stability engineering of scFvs for the development of bispecific and multivalent antibodies. Protein Eng Des Sel 2010; 23:549 - 557
- Michaelson JS, Demarest SJ, Miller B, Amatucci A, Snyder WB, Wu X, et al. Anti-tumor activity of stability-engineered IgG-like bispecific antibodies targeting TRAIL-R2 and LTbetaR. mAbs 2009; 1:128 - 141
- Dong J, Sereno A, Snyder WB, Miller BR, Tamraz S, Doern A, et al. Stable IgG-like bispecific antibodies directed towards the type I insulin-like growth factor receptor demonstrate enhanced ligand blockade and anti-tumor activity. J Biol Chem 2011; 286:4703 - 4717
- Sugawa N, Ekstrand AJ, James CD, Collins VP. Identical splicing of aberrant epidermal growth factor receptor transcripts from amplified rearranged genes in human glioblastomas. Proc Natl Acad Sci USA 1990; 87:8602 - 8606
- Wong AJ, Ruppert JM, Bigner SH, Grzeschik CH, Humphrey PA, Bigner DS, et al. Structural alterations of the epidermal growth factor receptor gene in human gliomas. Proc Natl Acad Sci USA 1992; 89:2965 - 2969
- Doern A, Cao X, Sereno A, Reyes CL, Altshuler A, Huang F, et al. Characterization of inhibitory anti-IGF-1R antibodies with different epitope specificity and ligand blocking properties: Implications for mechanism of action in vivo. J Biol Chem 2009; 284:10254 - 10267
- Yonesaka K, Zejnullahu K, Lindeman N, Homes AJ, Jackman DM, Zhao F, et al. Autocrine production of amphiregulin predicts sensitivity to both gefitinib and cetuximab in EGFR wild-type cancers. Clin Cancer Res 2008; 14:6963 - 6973
- Lammerts van Bueren JJ, Bleeker WK, Bogh HO, Houtkamp M, Schuurman J, van de Winkel JG, et al. Effect of target dynamics on pharmacokinetics of a novel therapeutic antibody against the epidermal growth factor receptor: implications for the mechanisms of action. Cancer Res 2006; 66:7630 - 7638
- Ma P, Yang BB, Wang YM, Peterson M, Narayanan A, Sutjandra L, et al. Population pharmacokinetic analysis of panitumumab in patients with advanced solid tumors. J Clin Pharmacol 2009; 49:1142 - 1156
- Tonra JR, Corcoran E, Deevi DS, Steiner P, Kearney J, Li H, et al. Prioritization of EGFR/IGF-IR/VEGFR2 combination targeted therapies utilizing cancer models. Anticancer Res 2009; 29:1999 - 2007
- Burtrum D, Zhu Z, Lu D, Anderson DM, Prewett M, Pereira DS, et al. A fully human monoclonal antibody to the insulin-like growth factor I receptor blocks ligand-dependent signaling and inhibits human tumor growth in vivo. Cancer Res 2003; 63:8912 - 8921
- Cohen BD, Baker DA, Soderstrom C, Tkalcevic G, Rossi AM, Miller PE, et al. Combination therapy enhances the inhibition of tumor growth with the fully human anti-type 1 insulin-like growth factor receptor monoclonal antibody CP-751,871. Clin Cancer Res 2005; 11:2063 - 2073
- Goetsch L, Gonzalez A, Leger O, Beck A, Pauwels PJ, Haeuw JF, et al. A recombinant humanized anti-insulin-like growth factor receptor type I antibody (h7C10) enhances the antitumor activity of vinorelbine and anti-epidermal growth factor receptor therapy against human cancer xenografts. Int J Cancer 2005; 113:316 - 328
- Maloney EK, McLaughlin JL, Dagdigian NE, Garrett LM, Connors KM, Zhou XM, et al. An anti-insulin-like growth factor I receptor antibody that is a potent inhibitor of cancer cell proliferation. Cancer Res 2003; 63:5073 - 5083
- Brezinsky SC, Chiang GG, Szilvasi A, Mohan S, Shapiro RI, MacLean A, et al. A simple method for enriching populations of transfected CHO cells for cells of higher specific productivity. J Immunol Methods 2003; 277:141 - 155
- Hoet RM, Cohen EH, Kent RB, Rookey K, Schoonbroodt S, Hogan S, et al. Generation of high-affinity human antibodies by combining donor-derived and synthetic complementarity-determining-region diversity. Nat Biotechnol 2005; 23:344 - 348
- Thie H, Voedisch B, Dubel S, Hust M, Schirrmann T. Affinity maturation by phage display. Methods Mol Biol 2009; 525:309 - 322