Abstract
Therapeutic monoclonal antibodies (mAbs) currently dominate the biologics marketplace. Development of a new therapeutic mAb candidate is a complex, multistep process and early stages of development typically begin in an academic research environment. Recently, a number of facilities and initiatives have been launched to aid researchers along this difficult path and facilitate progression of the next mAb blockbuster. Complementing this, there has been a renewed interest from the pharmaceutical industry to reconnect with academia in order to boost dwindling pipelines and encourage innovation. In this review, we examine the steps required to take a therapeutic mAb from discovery through early stage preclinical development and toward becoming a feasible clinical candidate. Discussion of the technologies used for mAb discovery, production in mammalian cells and innovations in single-use bioprocessing is included. We also examine regulatory requirements for product quality and characterization that should be considered at the earliest stages of mAb development. We provide details on the facilities available to help researchers and small-biotech build value into early stage product development, and include examples from within our own facility of how technologies are utilized and an analysis of our client base.
Introduction
With declining sales of traditional small-molecule drugs, biologics including mAbs, are seen by many as critical for the future of the pharmaceutical industry. Sales of biologics exceeded $100 billion USD in 2010 Citation1 and 5 out of 10 of the world's top selling drugs are now antibody based molecules ().Citation2 Several large pharmaceutical companies have recently invested heavily through multibillion-dollar mergers and acquisitions to build product portfolios and pipelines in the biologics area, e.g., Pfizer's acquisition of Wyeth ($68 billion), Roche's takeover of Genentech ($46.8 billion) and Astra Zeneca's purchase of Medimmune ($15 billion) and Cambridge Antibody Technology (£580 million). Many of today's blockbuster therapeutic mAbs started life in an academic lab or the target antigen was discovered by basic research organizations. These early molecules took years to move from the bench to preclinical evaluation, clinical trials and final product approval. However, there is now significant pressure to evaluate lead compounds quickly and move more drugs to the market faster. For small biotechnology companies and university spinoff entities, obtaining stable and multiyear investment is a particular challenge. It is imperative to develop clinical mAb candidates quickly but also use techniques and methodologies that are compatible with future commercial development. Many companies are now only willing to invest heavily for products that have shown efficacy in Phase 2 trials or are likely to progress to Phase 3.
Creation of therapeutic mAb candidates is a time-consuming, multistep process that includes molecule discovery, optimization, expression, purification and characterization ().Citation3 The skills and knowledgebase to develop lead candidates or design a scalable bioprocess production and purification strategy are commonly outside the scope of a typical university laboratory or small company. This has created a need for contract manufacturing organizations (CMOs), and government sponsored facilities that can aid in early stage mAb development. There are currently a number of established high profile commercial antibody discovery and cell line development companies in operation, such as Morphosys, Boehringer Ingelheim, Selexis, Celltrion and Lonza (amongst others), who have experience and know-how to develop therapeutic mAbs. However, the caveats regarding these facilities are both the upfront cost, which can be prohibitive for a research organization, and the early incorporation of proprietary intellectual property (IP), which facilitates significant ongoing royalties as well as complex milestone driven payments that may extend for the life of the product. The IP surrounding mAb discovery and production is also complexCitation4 and these additional costs early in development can be a barrier to new mAb development.
To help bridge this gap and add value to projects in a cost-effective manner early in development, a range of government sponsored biologics development facilities have been established (). The Association of Academic Biologics Manufacturers maintains a web site (aabmonline.org) that also lists information about many of these facilities. These organizations may or may not operate at cGMP level, but contain the expertise and physical facilities to enable development of therapeutic mAbs and other biologics through production of high-quality material. The ability to produce early stage, preclinical and Phase 1 clinical study material in these facilities allows significant value to be built into a candidate, which in turn allows an enhanced value proposition when entering venture capital partnering or licensing negotiations. These organizations offer a wide variety of services, including mAb discovery, cell line development, bioprocess optimization and product characterization. Typically these organizations use small-scale bioreactors (<100 L) and single-use technology to generate material. Access to these facilities is often dependent on the client and is positively geared toward aiding small companies, academic and government sponsored research groups. In our own facility we focus on production of material typically in the 1–10 g range, using a range of conventional and single-use bioreactors. A discussion of industrially relevant bioprocesses is provided in the following sections.
Recombinant mAb Discovery: Development Options for Researchers
The path for the development of mAbs as biologic medicines has been both long and arduous and it has been over 100 years since Paul Ehrlich's magic bullet concept was proposed.Citation5 However, mAbs are now proving to be spectacularly successful in the clinic for treatment of human disease indications, principally cancer and inflammatory diseases such as rheumatoid arthritis, Crohn disease and psoriasis.Citation6–Citation8 Since the inception of antigen specific mouse mAbs in 1975,Citation9 there has been a progressive drive to develop new technologies to render these molecules to be more human, so as to minimize the human anti-murine antibody response (HAMA),Citation10 culminating in the development of fully human antibodies.Citation11 A number of techniques for antibody discovery are now used and routinely performed in basic research laboratories, companies or within some of the facilities described here ().
In order to generate mAbs that more closely resemble human antibodies, developments in molecular cloning initially facilitated the creation of mouse-human chimeric antibodies,Citation11,Citation12 and subsequently humanized mAbs, via complementarity determining region (CDR) grafting.Citation13 CDR grafted antibodies are now the predominant antibody approved for human administration.Citation14 Although mouse sequences are essentially removed from CDR-grafted antibodies (excluding the CDR loops, which can be regarded as species independent), there is still potential for anti-idiotype immune response due to the CDR regions.
While humanization resulted in molecules that were nearly human, in terms of sequence, the biggest recent advance for the recombinant antibody field has been our ability to create human antibodies. These molecules are essentially indistinguishable from immunoglobulins made in vivo by the humoral immune system. The advent of human antibodies has been made possible through advances in genetic engineering in combination with sophisticated molecular biological techniques. The two principal technologies for the isolation of human antibodies are in vitro display methods, such as phage, yeast and ribosome display, and “humanized” mice, i.e., transgenic mice modified to contain genomic regions containing the human humoral immune systems.
Display technologies enable high-diversity libraries containing antibody variable region fragments to be screened against a target antigen. Phage display is perhaps the most common technique and involves the display of antibody fragments derived from immunoglobulin gene libraries on the surface of fd filamentous phage, followed by antigen-driven selection.Citation15,Citation16 The linking of phenotype (displayed antibody) with genotype (the gene encoding the antibody is harboured in the phagemid within the phage particle) allows the isolation of specific antibodies and subsequent rescue of the gene encoding the antibody. This technique has allowed the isolation of antibodies to a wide range of targets and epitopes that may not have been possible using traditional in vivo immunisation.Citation17 Fragments isolated by display technologies can easily be reformatted into whole immunoglobulin backbones in a high-throughput and sequence independent matter.Citation18 Even though phage display technology was established more than 20 years ago and despite the widespread use in early stage antibody discovery, to date only two approved antibody products have been created via phage display. These antibodies are adalimumab (Humira®, Abbott), approved in 2002, which is used in treatment of autoimmune disorders, and belimumab (BENLYSTA®, Human Genome Sciences and GlaxoSmithKline), approved in 2011 as the first new treatment for systemic lupus erythematosus in 56 years.
More recently, exciting developments have been made in the area of yeast display technologies. This includes technology developed by Adimab that involves modification of published yeast display methods.Citation19,Citation20 This methodology enables rapid screening of antigens against high-diversity libraries (exceeding 10Citation14) and the corresponding fully assembled mAbs to be produced directly by the yeast. This negates the need for reformatting of antibody fragments into full antibodies and produces a panel of mAbs for the target antigen. The speed and efficiency of this technology has attracted substantial commercial interest.
The second major advance has been the creation of genetically engineered mouse strains that contain various genomic constructs encoding human immunoglobin loci.Citation21,Citation22 The basic premise of using these strains involves immunisation of the mouse with the target antigen, which elicits a humoral immune response; however, the antibodies produced are of human sequence. Hybridomas can then be produced through somatic cell fusion, human antibody-secreting hybridoma cell lines are isolated and the antibodies are cloned and transferred to mammalian cell production systems. There are now several versions of this “humanized” mouse technology, including XenoMouse® developed by Abgenix (acquired by Amgen), the VelocImmune® mouse platform from Regeneron and UltimAb (HuMAb-Mouse® and KM-Mouse®) developed by Medarex (acquired by Bristol-Myers Squibb) and Kirin. The molecular tour de force that was behind the creation of these strains cannot be understated, and each strain has particular nuances due to the underlying molecular genetics utilized, the discussion of which is beyond the scope of this review but has been covered elsewhere in references Citation21 and Citation22. These technologies have been successfully used to generate several approved clinical candidates and many more are in clinical development.Citation14,Citation22,Citation23 Very recently, embryonic stem cell technology developed in the laboratory of Professor Allan Bradley, Emeritus Director of the Sanger Institute, Cambridge UK has been used to develop next generation, transgenic, humanized mice with optimized genetics, for highly efficient generation of human antibodies.Citation24 The Kymouse™ platform is designed to maintain maximum human immunoglobulin diversity. In addition, these strains have been precisely engineered to retain critical B-cell regulatory signals/sequences in order to ensure efficient maturations and clonal diversity. The start-up company has just received initial funding from the Wellcome Trust and the technology is available under license to both academic and commercial operations.
Depending on the method used for mAb discovery there may be a requirement for both affinity maturation and sequence engineering of the antibody to improve pharmacokinetic or physicochemical characteristics of the molecule. These methods can involve secondary in vitro display based screens, random or site-specific molecular mutagenesis. A detailed discussion of these methods and techniques are beyond the scope of this review, but are covered in several excellent reviews and should be considered during early stage mAb discovery screens.Citation25–Citation27
Early Stage Cell Line Development Expression Technology
Once a candidate mAb is identified, mammalian cell expression systems are most commonly used to produce the recombinant protein.Citation28 While alternate production systems such as bacteria, yeast and plants have been developed, mammalian systems still dominate the industry and produce molecules with the requisite biological activity.Citation29 It should, however, be noted that new technologies may alter this equation in the future; for example, the engineered yeast strains developed by GlycoFi (now Merck & Co.) offer the ability to produce customized, on-demand and homogeneous glycosylation patterns.Citation30
Production levels in mammalian cell-based expression systems have improved more than 20-fold over the past two decades, mainly due to advancements in bioprocess and growth media.Citation28 Fully chemically defined growth media is now readily available from several suppliers, eliminating the risks and inconsistencies associated with animal derived components such as serum. Currently, suspension adapted cells originating from the ovary of a Chinese hamster (CHO) are the unlikely workhorse of the biopharmaceutical industry, due to their ease of use and established regulatory safety profile.Citation3,Citation28 Newer, human-derived cell lines such as PER.C6® offer some advantages, but are yet to be used in an approved product.Citation31
Transient gene expression.
For production of recombinant mAbs early in development, transient expression systems are used because they allow rapid production of protein that can be used for bioactivity and characterization studies. In a transient process, plasmids encoding the gene of interest are transfected en-masse into a production host cell line, such as human embryonic kidney (HEK) or CHO cells. This typically employs a chemical transfection reagent such as polyethylenimine (PEI) or Lipofectamine™ and relatively small-scale culture volumes (<10 L). The transfected culture is cultivated for a pre-determined period of time (around 7–21 days), based on cell viability or product stability, before harvesting. Output levels may be improved through simple optimisations, like temperature shift or media supplementation.Citation32–Citation34
Transient expression is also useful when screening a number of product variants, requiring minimal preparation and enabling fast turn-over. However, the process is inherently self-limiting, due to plasmid dilution during cell division and historically expression levels are low (commonly less than 10 mg/L). Direct scale-up in volume can make up for inefficiency in output per se; however, cost of plasmid preparation and transfection reagents place economical constraints on the process. Furthermore, as volume increases, batch turn-over efficiency will decrease due to physical limitation of the processing equipment capabilities. HEK cells have been the platform cell line of choice for transient gene expression because they generally outperform CHO cells; however, several recent reports have demonstrated high-level production in CHO.Citation32,Citation35 For example, CHO-based episomal systems that facilitate extra-chromosomal plasmid replication and allow small starting cell numbers and efficient expansion and production have been developed.Citation32,Citation36 The ability to perform early stage transient production in CHO cells is preferred because this cell line is predominantly used during large-scale commercial production.
Stable cell line development.
While transient technology allows rapid production of multiple proteins in small quantities, the development of stable cell lines is required for production of larger amounts of material. For a process to be commercially viable, a cell line expressing in excess of 1 g/L should be developed; however, yields in excess of 10 g/L have been reported in reference Citation3. Commonly, stable expression platform technologies involve auxotrophic selection or epigenetic engineering.
Auxotrophic selection requires transfecting a cell line with a plasmid containing both the genes of interest and a “survival” gene, typically a metabolic enzyme. Transfectants are then selected under minimal or incomplete media, which omits key survival nutrient components, and commonly includes increasing amounts of specific enzyme inhibitors. The stepwise addition of these inhibitors further increases the survival pressure and facilitates stringent selection of a sub-population of cells, which may exhibit high expression of the survival gene and the protein of interest. Under this stringent selection, cells are known to physically expand the DNA around the site of integration thereby making many copies of the survival gene and by proxy, the co-located gene of interest. This mechanism is known as amplification and has been a key factor allowing high-level recombinant protein expression in mammalian cells.Citation28,Citation37
In industry, the two mostly commonly used auxotrophic selection techniques are the dihydrofolate reductase (DHFR) and glutamine synthetase (GS) systems.Citation28 When a plasmid encoding the DHFR enzyme is co-transfected with a gene of interest into DHFR-mutant CHO cells, e.g., DG44 or CHO DUKX-B11, high expressers can be selected under increasing concentration of the DHFR antagonist methotrexate, when transfectants are grown in hypoxanthine and thymidine deficient media. With the GS system on the other hand, the GS gene is co-transfected with protein of interest into wild-type cells and addition of the specific GS inhibitor methionine sulphoximine can be used to effectively select high-expressing cells in glutamine-free, but glutamate-supplemented, media.Citation38
Other technologies have also been developed to streamline creation of high-producing stable cell lines. These involve epigenetic engineering utilising epigenetic regulators, such as scaffold or matrix attachment regions (S/MARS) developed by Selexis or ubiquitous chromatin opening elements (UCOE™) available from Millipore.Citation39,Citation40 These are typically large DNA sequence motifs isolated from endogenous genes that are thought to increase accessibility of transcriptional machinery and decrease probability of gene silencing after integration of the transgene. Preferential site of integration also means epigenetic engineering has the potential to yield a more homogeneous population in terms of expression profile when compared with conventional methods that rely on random genome integration and amplification.
Once a cell line is developed, regulations stipulate that it must be cloned, i.e., derived from a single cell. This is typically achieved through limiting dilution and outgrowth in 96-well plates. Performing this step in serum-free conditions can be challenging for inexperienced researchers. Commercially-available cloning media is now available from Sigma/SAFC and the use of additives, such as recombinant albumin, may also be required. However, new technologies developed in this area allow an intelligent and high-throughput approach to single-cell cloning.Citation41 Within our own facility we utilize ClonePix™ FL (Genetix) based technology, which relies on fluorescent detection of target protein secreted from cells in a semi-solid media and fluorescent-activated cell sorting using surface based and intracellular markers.Citation42–Citation44
Despite refinement of selection methodology and technology advances for isolation of clones, creation of cell lines with requisite stability, product quality and expression characteristics remains a rate-limiting step in mAb production and there is a need for new techniques and methodologies that will facilitate faster, more predictable clone isolation. Recent advances in genome-scale modelling and custom gene editing may provide an avenue for creation of next-generation mammalian cell production systems.Citation31,Citation45 For example, recently a triple gene knockout CHO cell line was created using zinc finger nuclease technology, which enables efficient selection and high-level production of afucoslyated antibodies for enhanced ADCC.Citation46
While these stable cell line development systems are used routinely in industry, complexities in terms of IP, cell line ownership, licensing and royalty payments can limit options for early stage developers, where maximum financial and business flexibility is desirable. Life Technologies have recently launched an innovative product platform in this area. Known as the Gibco® Freedom™ Cell Line Development Kit, this provides all the reagents, cell lines and protocols to enable development of a high-yielding CHO clone. Perhaps of most interest, this kit comes in both a research and commercial use form and with a simplified pricing and license structure. However, even the protocols in this kit require a large amount of operator skill. Staff within facilities outlined in can provide advice and practical expertise during stable cell line production. The analysis of our own client/contract profile shown in indicates that a bridging facility such as ours mainly serves clients very early in development. In our experience, a large proportion of clients at this phase are under tight financial constraints. It is therefore important to provide a cost-effective, in-house cell line development service free from IP constraints during research phase activities with a focus on implementing a universal approach for cell line development, regardless the expression system chosen for the projects. Speed is also of the essence when performing early stage development. Our facility's aim is to rapidly produce a cell line with a sufficient expression level to deliver a target quantity of recombinant protein for preclinical studies up to Phase 1 clinical development. This approach allows progression of the project, while maintaining a balance between technological and financial constraints.
Production and Purification of Early Stage Material
Upstream technologies.
Perhaps the most significant recent advance affecting early stage development of recombinant biologics such as mAbs has been improvements in single-use bioprocessing equipment.Citation31 Traditional biologics production suites were built around steam in place (SIP) high-grade stainless steel or autoclavable glass stirred tank reactors (STR) and corresponding metal hold-tanks and piping. Integration of single-use technology plays a critical role in providing operational flexibility. Modular design of single-use products allows rapid capacity adjustment, customized process flows and parallel multi-unit operations. The elimination of sterilisation, cleaning and validation reduces operation complexity, minimizes chance of human error and, most importantly, reduces initial capital expenditure. The “plug-and-play” nature of single-use products makes cross-training between operators feasible, which reduces the full-time equivalents (FTEs) required for a given process. The advantages afforded over conventional stainless-steel processing equipment allow the production team to be highly adaptive and facilitates efficient resource management. Analysis of our own client profile concluded that a bridging facility needs to take a high-throughput approach for small-scale operations (process volume less than 10 L) while enabling pilot-scale (>10 L) demands at the same time.
For small-scale suspension mammalian cell culture, high-capacity shaking CO2 and humidity controlled incubators are most commonly used, such as those provided by Infors and Kunher. Culture volumes from only a few mLs to 1 L can be easily performed and range of innovative plastic-ware is now available, including 50 mL “Spin Bioreactor Tubes” from TPP and disposable high-capacity shake flasks from companies such as NUNC, Corning and Thompson Scientific. Square glass bottles have also proven to be excellent vessels for simple small-scale production.Citation47
During clone evaluation, a number of small-scale bioreactor systems have been developed, enabling multiple parallel runs with bioprocess control. This includes systems such as the Micro24 from PALL (formerly Micro Reactor Technologies) and ambr micro bioreactor from TAP Biosystems, which both allow runs of 24 mini-bioreactors in parallel using volumes less than 15 mL. Scale-down mini-reactors systems are also available from companies such as DASGIP, Sartorius AG and Applikon Biotechnology. These systems allow cost-effective yet powerful bioprocess optimization of mAb-expressing cell lines prior to scale-up. With sufficient know-how it is possible to conduct more complex design of experiment (DOE) studies to maximize efficiency.
When scale-up production is required (1–25 L), traditional STRs can be replaced with WAVE-type bioreactor systems, such as those provided by GE Healthcare and Sartorius AG. These consist of pressurized disposable bags that sit on a heated rocking platform. Use of a pre-sterilized and validated bag system has the advantages of minimizing downtime and cross-contamination between batches. These systems are widely used in both industry and academia and are the main production system used in our facility. WAVE-type systems, however, do have a volume limitation and a number of STRs that incorporate disposable bag technology have been developed. These are now available up to 2,000 L scale, e.g., Hyclone S.U.B. (Thermo), and are suitable for commercial production.Citation3,Citation31 Advances in non-invasive, optical-based sensor instrumentation have also permitted bag-based systems to incorporate the bioprocess control capabilities once only possible in traditional reactors. Industry acceptance of these technologies has grown rapidly and new pilot plant developments and CMO facilities are increasingly opting for single-use systems in place of fixed systems. DSM Biologics is currently building such a biologics plant of the future (BPOF) in Brisbane Australia that will operate up to 2,000 L scale using only single-use reactor systems.
Downstream technologies.
High-level purification of mAbs from culture supernatants represents a significant challenge.Citation3,Citation48 Within small- to medium-scale facilities, the downstream process can be initiated by single-use depth filters for clarification, delivering filtrates with pre-defined clarity specification with minimal product loss. These are easily scalable and replace expansive and laborious centrifuge-based processes. A typical mAb purification process in industry involves three chromatographic steps, including affinity capture (usually Protein A) followed by dual phase ion exchange incorporating both capture and flow through modes and final ultra- and viral-filtration.Citation3,Citation48 Chromatography resins remain expensive and technology advances have lagged the rest of the industry, aside from engineered versions of alkali-stabilized and protease-resistant Protein A, which allows more usage cycles and efficient cleaning. There has also been renewed interest in use of multimodal resins, such as hydroxyapatite, in an attempt to reduce the required number of steps from 3 to 2.Citation48 Ceramic hydroxyapatite (CHT) can perform mixed-mode purification, involving non-specific interactions between positively charged calcium ions and negatively charged phosphate ions on CHT with negatively charged carboxyl groups and positively charged amino groups of target proteins. CHT can be effective in removal of dimers and aggregates as well as endotoxin.Citation49
Developments in chromatography equipment systems now allow for DOE approaches to be easily adopted, using off the shelf systems such as the ÄKTA™ Avant and Unicorn 6 control software from GE Healthcare, greatly enhancing data-driven approaches to optimizing purification. Furthermore, fully sanitisable systems such as the ÄKTA™ Pilot, also from GE Healthcare, enables purification of high-quality material at small to medium scale while being cGMP compatible. These types of systems are essential items for biologics bridging facilities.
There are also new platforms that have been developed to provide integrated single-use solutions during downstream processing that are appropriate for a wide-range of production scales while incorporating industry best-practice. Two such systems are the Mobius® Single-Use Manufacturing platform from Millipore and FlexAct® system from Sartorius. These can be purchased as customizable “cart” based solutions that cover a number of downstream unit operations, including buffer preparation, cell harvest, virus inactivation, media preparation, virus removal, buffer exchange (i.e., tangential flow filtration). Incorporation of these solutions in bridging facilities is ideal as it allows incorporation of industrially-relevant and scalable bioprocess procedures.
While acceptance of single-use technology continues to grow in the industry, there remain hurdles to address, including concerns about potential leachables and extractables, materials loss, chemical and physical compatibilities and the high on-going cost of goods.Citation3 Availability and batch-to-batch consistency of plastics has also been an issue. Nevertheless, these technologies enable small facilities with relatively few FTEs and low capital expenditure to quickly and easily produce high-quality material for preclinical and early stage clinical testing.
Early Stage Consideration of Regulatory Requirements
Production of therapeutic mAbs for preclinical assessment requires a rigorous understanding of the product. Recent changes have meant that Phase 1 clinical material does not need to be produced under the same regulatory framework as for commercial manufacturing. In July 2008, the US Food and Drug Administration (FDA) released the Guidance for Industry CGMP for Phase 1 Investigational Drugs, which specifies that biologic investigational new drug applications are exempt from 21 CFR part 211. In Europe, the European Medicines Agency (EMA) also released documents providing information for early stage biologics development (Guideline on the Requirements for Quality Documentation Concerning Biological Investigational Medicinal Products in Clinical Trials, EMA/CHMP/BWP/534898/2008; Guidelines on Strategies to Identify and Mitigate Risks for First-in-Human Clinical Trials with Investigational Medicinal products EMEA/CHMP/2007). The full spectrum of regulatory requirements specifically for product safety should be considered during product development; further information can be found in several recent reviews in reference Citation50–Citation54.
To address the requirements for characterization of therapeutic mAb products, the EMA and FDA have produced the specific guidance documents “Production and Quality Control of Monoclonal Antibodies and Related Substances” and “Points to Consider in the Manufacture and Testing of Monoclonal Antibody Products for Human Use,” respectively. What is clear from these guidance documents and the experience of industry over the past 30 years is that extensive chemistry, manufacturing and control (CMC) information for therapeutic mAbs is required. It should also be noted that this type of information relating to the drug substance should be considered even very early in development. For early development of mAbs, this requires extensive physiochemical characterization of the molecule and an understanding of the importance of these properties. For instance, a mAb with outstanding kinetics and activity in cell-based assays may exhibit a high-degree of proteolytic breakdown products during purification or high-levels of aggregation. Issues such as these that contribute to heterogeneity of mAbs can quickly end drug development programs.Citation55 Factors to consider for characterization of therapeutic mAbs are included in . For a non-specialist laboratory, development of this type of testing regimen can be costly and time-consuming, but also add value for future commercial development. Given the complexity of these assays, outsourcing the project early can help overcome many of these hurdles. Interpretation of the product attribute requirements can be challenging because, for the majority of these parameters, there is no defined, acceptable level of contaminant or impurity. In most instances, the regulator guidance documents outline that acceptance criteria should be established and the rationale for the assays used should be described. The exception is for endotoxin, in which a specific value of 5EU per kg body weight per hour is stipulated in the Pharmacopoeia.Citation56 For host cell DNA, a recommended value of less than 100 pg of residual host DNA per dose of protein is suggested, and the analytical method used should exhibit a sensitivity of 10 pg.Citation57 The WHO Expert Committee on Biological Standardization revised this figure so that currently 10 ng of residual DNA per dose is acceptable.Citation58
The requirements for testing and validation of therapeutic mAbs are complex, with each regulatory jurisdiction varying substantially from the next. In manufacturing, industry has moved toward a quality by design (QbD) framework in which complex DOE analysis is followed in order to set acceptable parameters for each process.Citation59 While this approach is beyond the capabilities of the facilities described here, it underlies the importance of understanding process and product attributes. In summary, even during early stage non-GMP production it is critical to develop an awareness of these regulatory frameworks and ensure that the processes used will be translatable and the basic product quality attributes of the molecule being developed have been assessed. Many of the staff within the facilities listed in will be able to provide valuable assistance during this stage of development.
The AIBN Biologics Facility: An Operational Review
Our own facility is a government-funded (both Federal and State), university-affiliated laboratory, with a mission of translating early stage research into potential protein therapeutics toward clinical development. We have 20 staff with mixed backgrounds in molecular biology, antibody engineering, cell biology, process engineering and clinical development both from academia and industry. We provide a sponsored fee-for-service arrangement, tiered in a manner dependent upon the client. Our main goal is to assist institutional researchers or start-up biopharmaceutical companies to move from basic discoveries into clinical focus. The physical facility consists of 200 m2 of a high efficiency particulate air (HEPA)-filtered cleanroom environment, covering tissue culture laboratories, general preparation/wet areas, cold-rooms and dedicated upstream and downstream suites. We have a strong focus on single-use systems and have bioreactor capacity up to 100 L. We have also maintained a strong focus on mAbs and mAb-related molecules as the target proteins, as well as the use of mammalian cells for production.
Three years after establishment of the facility, we have worked with >20 companies encompassing >150 projects, processed >1,000 L of culture supernatant and produced >200 grams of antibodies. Profiles of past and current client projects were categorized and analysed based on their stage of clinical development, nature of services requested and size of production needed (). This provided a reference for predicting future demand of the services, required resource allocation, capital expenditure and nature of internal R&D activities. In retrospective analysis, contracts were categorized into Proof-of-Concept, Research and Development (R&D) and Preclinical. “Proof of Concept” involved antigen production and purification, antibody discovery and characterisation and target protein production. Contracts in the “R&D” category required services of cell line and process development, small batch production and further characterisation and assay development. The “Preclinical” category generally consisted of production scale-up, downstream bioprocess development, formulation and stability studies. shows that the majority (40% + 39%) of contracts were concentrated on what might be termed early preclinical stage. This reflects the demographics of the research activities in general, where a large number of projects were discovery in nature and only a small portion might advance further towards clinical development. , on the other hand, shows that while the preclinical category accounted for only one-fifth of the contracts, it represented 83.1% of the facility output; it also accounted for a large proportion of contract research income (data not shown). The average process volume (cultured cells in L) required for each category was 2.3 L (Proof-of-Concept), 5.9 L (R&D) and 58.7 L (Preclinical). The analysis also indicated that the facility needed to maintain a high-throughput approach with small projects (<10 L per production) and yet maintain process flexibility and capability to perform larger scale production runs when required. This was achieved in no small part due to adoption of single-use technologies. Our facility, and the other facilities in , clearly fill an important gap in the early development of the next generation of therapeutic antibodies and demonstrate the value of this type of bridging capability.
It is imperative that biologic bridging facilities not only achieve outcomes for clients, but that they deliver an effective mechanism to translate this work to industry partners for future commercialisation. The interface between academic groups and the pharmaceutical industry can be difficult to navigate. In our facility, we manage this interaction in two main ways. Firstly, we conduct a range of research activities that align with topics of interest to industry, such as rapid cell line development and advanced cell line engineering. Attending relevant conferences and publishing this work assists in the establishment of research partnerships and relationships with industry. In turn, this enables potential avenues of commercialisation of appropriate projects and products. Secondly, many universities have now established formal and well-regarded commercialization arms that have direct links to relevant industry partners. For example, the University of Queensland established Uniquest as a stand-alone entity that can translate commercial outcomes from research with valuable IP. An early formalisation of these arrangements is also key to avoid potential IP issues. There are numerous examples where university technology has been improperly commercialized, leading to lengthy litigation. In our facility, we have developed template agreements that clearly define the IP and encumbered technology, which enables effective relationships with both mAb developers and commercialisation partners. We also have an in-house legal team that is able to work with industry to develop these agreements. From our own experience, flexibility is required by both parties to establish long-term and productive relationships.
Conclusions and Perspectives
Therapeutic mAbs are a rapidly expanding sector of the pharmaceutical market place, with hundreds of clinical candidates currently in development.Citation8 There is also an ever-expanding number of antibody related molecules currently being investigated, including Fc fusion proteins, bispecific antibodies, antibody fragments, single chain antibodies and novel engineered protein scaffolds.Citation60,Citation61 In addition, both radio- and cytotoxic conjugates are re-emerging to expand efficacy and indications of existing therapeutic molecules.Citation62 mAbs are also being investigated for use in other important disease areas such as bacterial and viral infection.Citation63 Finally, some of the early mAb blockbusters are approaching patent expiry, signaling the way for a raft of next generation biosimilars and bio-superiors.Citation31,Citation64 The future for therapeutic mAbs looks bright and continued discovery and development of clinical candidates is a rapidly growing area.
It is also clear that there is a need for discovery of new, highly specific and differentiating biomarkers for disease, and this area also looks set to undergo a revolution. New technologies now enable whole genome expression data to be generated quickly and cheaply. This will allow programs such as the International Cancer Genome Consortium to build accurate whole genome data sets for a wide range of cancers.Citation65 In other diseases, whole genome sequencing will allow profiling of large patient cohorts to identify new and clinically tractable biomarkers. These advances may allow generation of large panels of therapeutic mAbs that could be tailored, to an individual depending on the expression profile of their disease state. The public availability of much of this data, as well as the low-cost of next-generation DNA sequencing technologies, also offers an excellent opportunity for members of the academic and small-biotech communities to make important discoveries and translate this into the next mAb blockbuster.
Academic groups and research clinicians still remain at the forefront of biomarker discovery and these labs are a hotbed of early stage therapeutic mAb development. Because of this, large companies are attempting to reconnect with academia through co-sponsored facilities based on university campuses. In November 2010 Pfizer announced development of the Center for Therapeutic Innovation (CTI) based at The University of California, San Francisco aimed at fast-tracking clinical translation of biologic candidates and incorporating an agreement to provide $85 million in research support and milestone payments over the next five years. In June 2011 Pfizer announced a second CTI to be based in Boston, committing $100 million, working with a number of groups including Beth Israel Deaconess, Boston University, Children's Hospital, Harvard University, Tufts Medical Center, Tufts University, the University of Massachusetts Medical School and Partners HealthCare (parent company of Massachusetts General and Brigham and Women's hospitals). Pfizer will now have first rights to develop biologics from the most promising research to come from this huge pool of researchers. In addition, Pfizer are currently looking at other sites to expand this program. MedImmune (AstraZenca) have recently partnered with Inserm Transfert, the private subsidiary of the French National Institute of Health and Medical Research (Inserm), funding up to ten projects over 3 years. These large-scale collaborations may be a natural progression of an earlier alternative approach during the 2000s in which several pharmaceutical firms recruited prominent senior academics to run research within the companies, e.g., Pfizer's targeting of Stanford Academic Corey Goodman, Genentech's hire of Stanford's Richard Scheller and Merck's recruitment of MIT Professor Peter Kim. These latest moves of funding academic research programs signal what many consider to be a general failure of internal industry based R&D programs to generate the next blockbuster despite enormous direct investment. These developments are also an excellent opportunity for academia to further engage with industry to expedite translational research.
In this review, we discussed many of the facets that should be considered during early development of a therapeutic mAb and also highlighted the challenges faced. In addition, there are now a number of well-resourced and well-equipped facilities that are able to guide early stage development of these biologics to aid transition into the clinic. These bridging facilities are essential for development of the next generation of therapeutic mAbs and offer a mechanism to connect researchers with commercial partners while building value early in product development.
Abbreviations
| ADCC | = | antigen dependent cell-mediated cytotoxicity |
| CHO | = | Chinese hamster ovary |
| CDR | = | complementarity determining regions |
| CMOs | = | contract manufacturing organizations |
| DOE | = | design of experiment |
| DHFR | = | dihydrofolate reductase |
| CMC | = | chemistry, manufacturing and control |
| FACS | = | fluorescent activated cell sorting |
| FTE | = | full-time equivalent |
| GS | = | glutamate synthetase |
| HAMA | = | human anti-murine antibodies |
| IP | = | intellectual property |
| mAb | = | monoclonal antibody |
| QbD | = | quality by design |
| STR | = | stirred tank reactors |
| SIP | = | steam in place |
| TNF | = | tumour necrosis factor |
Figures and Tables
Figure 1 Early stage development of a therapeutic mAb spanning antigen identification through discovery, expression and purification. This preclinical stage represents an iterative process in which results from testing and evaluation feed back on the molecular elements of the mAb.
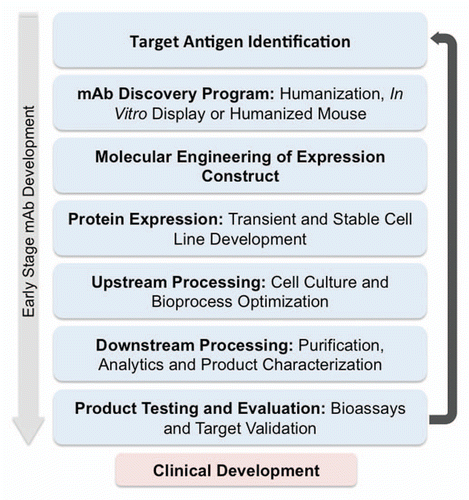
Figure 2 A client-based analysis of client projects conducted by the AIBN Biologics Facility over the past three years. (A) Contract distribution based on phase of development. (B) Total output percentage based on the litres of culture used per project category.
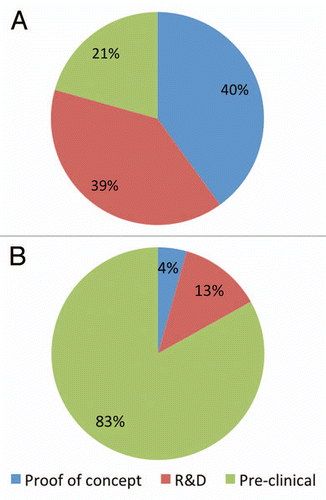
Table 1 Top selling drug products worldwide
Table 2 Academic and government funded facilities for early stage development of biologics
Table 3 Analytical characterization of mAbs
Acknowledgments
This work was supported through NCRIS funding to the AIBN Biologics Facility at the University of Queensland. Special thanks to Martina Jones for critical assessment of the manuscript.
References
- Intelligence LMB. Top 30 Biologics 2010. R&D Pipeline News 2011;
- Maggon K. Twenty Best Selling Drugs 2010 2011; Knol Publishing http://knol.google.com/k/krishan-maggon/top-ten-twenty-best-selling-drugs-2010/3fy5eowy8suq3/141
- Kelley B. Industrialization of mAb production technology: the bioprocessing industry at a crossroads. mAbs 2009; 1:443 - 452
- Petering J, McManamny P, Honeyman J. Antibody therapeutics—the evolving patent landscape biotechnology annual review 2011. N Biotechnol 2011; 28:538 - 544
- Strebhardt K, Ullrich A. Paul Ehrlich's magic bullet concept: 100 years of progress. Nat Rev Cancer 2008; 8:473 - 480
- Aggarwal S. What's fueling the biotech engine-2009–2010. Nat Biotechnol 2010; 28:1165 - 1171
- Walsh G. Biopharmaceutical benchmarks 2010. Nat Biotechnol 2010; 28:917 - 924
- Reichert JM. Metrics for antibody therapeutics development. mAbs 2010; 2:695 - 700
- Kohler G, Milstein C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 1975; 256:495 - 497
- Tjandra JJ, Ramadi L, Mckenzie IFC. Development of Human Antimurine Antibody (Hama) Response in Patients. Immunol Cell Biol 1990; 68:367 - 376
- Winter G, Milstein C. Man-made antibodies. Nature 1991; 349:293 - 299
- Morrison SL, Johnson MJ, Herzenberg LA, Oi VT. Chimeric human antibody molecules: mouse antigen-binding domains with human constant region domains. Proc Natl Acad Sci USA 1984; 81:6851 - 6855
- Jones ST, Bendig MM. Rapid PCR-cloning of full-length mouse immunoglobulin variable regions. Biotechnology 1991; 9:88 - 89
- Getts DR, Getts MT, McCarthy DP, Chastain EML, Miller SD. Have we overestimated the benefit of human(ized) antibodies?. mAbs 2010; 2:682 - 694
- McCafferty J, Griffiths AD, Winter G, Chiswell DJ. Phage antibodies: filamentous phage displaying antibody variable domains. Nature 1990; 348:552 - 554
- Marks JD, Hoogenboom HR, Bonnert TP, McCafferty J, Griffiths AD, Winter G. By-passing immunization. Human antibodies from V-gene libraries displayed on phage. J Mol Biol 1991; 222:581 - 597
- Bradbury AR, Sidhu S, Dubel S, McCafferty J. Beyond natural antibodies: the power of in vitro display technologies. Nat Biotechnol 2011; 29:245 - 254
- Jones ML, Seldon T, Smede M, Linville A, Chin DY, Barnard R, et al. A method for rapid, ligation-independent reformatting of recombinant monoclonal antibodies. J Immunol Methods 2010; 354:85 - 90
- Chao G, Lau WL, Hackel BJ, Sazinsky SL, Lippow SM, Wittrup KD. Isolating and engineering human antibodies using yeast surface display. Nat Protoc 2006; 1:755
- Feldhaus MJ, Siegel RW, Opresko LK, Coleman JR, Feldhaus JM, Yeung YA, et al. Flow-cytometric isolation of human antibodies from a nonimmune Saccharomyces cerevisiae surface display library. Nat Biotechnol 2003; 21:163 - 170
- Kellermann SA, Green LL. Antibody discovery: the use of transgenic mice to generate human monoclonal antibodies for therapeutics. Curr Opin Biotech 2002; 13:593 - 597
- Lonberg N. Fully human antibodies from transgenic mouse and phage display platforms. Curr Opin Immunol 2008; 20:450 - 459
- Jakobovits A, Amad RG, Yang X, RoskoS L, Schwab G. From XenoMouse technology to panitumumab, the first fully human antibody product from transgenic mice. Nat Biotechnol 2007; 25:1134 - 1143
- Bradley A. Kymouse Platform to generate highly selective, potent and well-tolerated human antibody-based biopharmaceuticals PEGS Summit 2011 Boston
- Igawa T, Tsunoda H, Kuramochi T, Sampei Z, Ishii S, Hattori K. Engineering the variable region of therapeutic IgG antibodies. mAbs 2011; 3:243 - 252
- Loo L, Robinson MK, Adams GP. Antibody engineering principles and applications. Cancer J 2008; 14:149 - 153
- Presta L. Antibody engineering for therapeutics. Curr Opin Struct Biol 2003; 13:519 - 525
- Wurm FM. Production of recombinant protein therapeutics in cultivated mammalian cells. Nat Biotechnol 2004; 22:1393 - 1398
- Rita Costa A, Elisa Rodrigues M, Henriques M, Azeredo J, Oliveira R. Guidelines to cell engineering for monoclonal antibody production. Eur J Pharm Biopharm 2010; 74:127 - 138
- Potgieter TI, Cukan M, Drummond JE, Houston-Cummings NR, Jiang Y, Li F, et al. Production of monoclonal antibodies by glycoengineered Pichia pastoris. J Biotechnol 2009; 139:318 - 325
- Hou JJC, Codamo J, Pilbrough W, Hughes B, Gray PP, Munro TP. New frontiers in cell line development: Challenges for biosimilars. J Chem Technol Biotechnol 2011; 86:895 - 904
- Codamo J, Munro TP, Hughes BS, Song M, Gray PP. Enhanced CHO cell-based transient gene expression with the Epi-CHO expression system. Mol Biotechnol 2011; 48:109 - 115
- Backliwal G, Hildinger M, Hasija V, Wurm FM. High-density transfection with HEK-293 cells allows doubling of transient titers and removes need for a priori DNA complex formation with PEI. Biotechnol Bioeng 2008; 99:721 - 727
- Wulhfard S, Tissot S, Bouchet S, Cevey J, De Jesus M, Hacker DL, et al. Mild hypothermia improves transient gene expression yields several fold in Chinese hamster ovary cells. Biotechnol Prog 2008; 24:458 - 465
- Rajendra Y, Kiseljak D, Baldi L, Hacker DL, Wurm FM. A simple high-yielding process for transient gene expression in CHO cells. J Biotechnol 2011; 153:22 - 26
- Codamo J, Hou JJC, Hughes BS, Gray PP, Munro TP. Efficient mAb production in CHO cells incorporating PEI-mediated transfection, mild hypothermia and the co-expression of XBP-1. J Chem Technol Biotechnol 2011; 86:923 - 934
- Schimke RT. Gene amplification in cultured animal cells. Cell 1984; 37:705 - 713
- de la Cruz Edmonds MC, Tellers M, Chan C, Salmon P, Robinson DK, Markusen J. Development of transfection and high-producer screening protocols for the CHOK1SV cell system. Mol Biotechnol 2006; 34:179 - 190
- Antoniou M, Harland L, Mustoe T, Williams S, Holdstock J, Yague E, et al. Transgenes encompassing dual-promoter CpG islands from the human TBP and HNRPA2B1 loci are resistant to heterochromatin-mediated silencing. Genomics 2003; 82:269 - 279
- Zahn-Zabal M, Kobr M, Girod PA, Imhof M, Chatellard P, de Jesus M, et al. Development of stable cell lines for production or regulated expression using matrix attachment regions. J Biotechnol 2001; 87:29 - 42
- Browne SM, Al-Rubeai M. Selection methods for high-producing mammalian cell lines. Trends Biotechnol 2007; 25:425 - 432
- Pilbrough W, Munro TP, Gray P. Intraclonal protein expression heterogeneity in recombinant CHO cells. PLoS One 2009; 4:8432
- Sleiman RJ, Gray PP, McCall MN, Codamo J, Sunstrom NA. Accelerated cell line development using two-color fluorescence activated cell sorting to select highly expressing antibody-producing clones. Biotechnol Bioeng 2008; 99:578 - 587
- Song M, Raphaelli K, Jones ML, Aliabadi-Zadeh K, Leung KM, Crowley D, et al. Clonal selection of high producing, stably transfected HEK293 cell lines utilizing modified, high-throughput FACS screening. J Chem Technol Biotechnol 2011; 86:935 - 941
- Dietmair S, Nielsen LK, Timmins NE. Engineering a mammalian super producer. J Chem Technol Biotechnol 2011; 86:905 - 914
- Liu PQ, Chan EM, Cost GJ, Zhang L, Wang J, Miller JC, et al. Generation of a triple-gene knockout mammalian cell line using engineered zinc-finger nucleases. Biotechnol Bioeng 2010; 106:97 - 105
- Muller N, Derouazi M, Van Tilborgh F, Wulhfard S, Hacker DL, Jordan M, et al. Scalable transient gene expression in Chinese hamster ovary cells in instrumented and non-instrumented cultivation systems. Biotechnol Lett 2007; 29:703 - 711
- Liu HF, Ma J, Winter C, Bayer R. Recovery and purification process development for monoclonal antibody production. mAbs 2010; 2:480 - 499
- Gagnon P. Monoclonal antibody purification with hydroxyapatite. N Biotechnol 2009; 25:287 - 293
- Chamberlain P. Preclinical strategies and safety issues in developing therapeutic monoclonal antibodies. N Biotechnol 2011; 28:481
- Snodin DJ, Ryle PR. Understanding and applying regulatory guidance on the nonclinical development of biotechnology-derived pharmaceuticals. BioDrugs 2006; 20:25 - 52
- Chapman K, Pullen N, Coney L, Dempster M, Andrews L, Bajramovic J, et al. Preclinical development of monoclonal antibodies: considerations for the use of non-human primates. mAbs 2009; 1:505 - 516
- Lynch CM, Grewal IS. Preclinical safety evaluation of monoclonal antibodies. Handb Exp Pharmacol 2008; 19 - 44
- Lynch CM, Hart BW, Grewal IS. Practical considerations for nonclinical safety evaluation of therapeutic monoclonal antibodies. mAbs 2009; 1:2 - 11
- Liu H, Gaza-Bulseco G, Faldu D, Chumsae C, Sun J. Heterogeneity of monoclonal antibodies. J Pharm Sci 2008; 97:2426 - 2447
- Petsch D, Anspach FB. Endotoxin removal from protein solutions. J Biotechnol 2000; 76:97 - 119
- Zoon K. FDA. Points to Consider in the Manufacture and Testing of Monoclonal Antibody Products for Human Use 1997;
- WHO. WHO Expert Committee on Biological Standardization. Highlights of the 46th meeting, October 1996. Wkly Epidemiol Rec 1997; 141 - 145
- Rathore AS. Roadmap for implementation of quality by design (QbD) for biotechnology products. Trends Biotechnol 2009; 27:546 - 553
- Holliger P, Hudson PJ. Engineered antibody fragments and the rise of single domains. Nat Biotechnol 2005; 23:1126 - 1136
- Skerra A, Gebauer M. Engineered protein scaffolds as next-generation antibody therapeutics. Curr Opin Chem Biol 2009; 13:245 - 255
- Beck A, Haeuw JF, Wurch T, Goetsch L, Bailly C, Corvaia N. The next generation of antibody-drug conjugates comes of age. Discov Med 2010; 10:329 - 339
- Nelson AL, Dhimolea E, Reichert JM. Development trends for human monoclonal antibody therapeutics. Nat Rev Drug Discov 2010; 9:767 - 774
- Roger SD. Biosimilars: current status and future directions. Expert Opin Biol Ther 2010; 10:1011 - 1018
- Cloonan N, Waddell N, Grimmond SM. The clinical potential and challenges of sequencing cancer genomes for personalized medical genomics. IDrugs 2010; 13:778 - 781
- Flatman S, Alam I, Gerard J, Mussa N. Process analytics for purification of monoclonal antibodies. J Chromatogr B Analyt Technol Biomed Life Sci 2007; 848:79 - 87