Abstract
The mammalian antibody repertoire comprises immunoglobulin (Ig) molecules of multiple isotypes and subclasses with varying functional properties. Among the four subclasses of the human IgG isotype, we found that IgG2 exhibits a particular resistance to human and bacterial proteases that readily cleave the IgG1 hinge region in vitro. Autoantibodies (IgGs) that recognize points of proteolytic cleavage in the IgG1 hinge are widespread in the healthy human population, suggesting that IgG1 fragmentation and the generation of cryptic antigens for host immune surveillance commonly occur in vivo. We previously reported that autoantibodies to cleaved IgG1s can restore Fc-mediated effector functions that are lost following proteolytic cleavage of the hinge. In contrast, it was not possible to demonstrate an analogous cohort of autoantibodies to IgG2 hinge epitope analogs, and there appeared to be no functional component in human serum with the ability to reconstitute Fc effector functions to a cell-bound IgG2 fragment. Thus, the results indicate that among the IgG subclasses, human IgG2 is uniquely resistant to a number of known pathological proteases and that autoimmune recognition to potential cleavage points in the IgG2 hinge appears to be absent in human circulation.
Introduction
Therapeutic monoclonal antibodies (mAbs) are one of the fastest growing categories of biotherapeutic agents and, at present, all approved therapeutic mAbs are of immunoglobulin (Ig)G.Citation1 The human IgG class of antibodies contains four individual isotypes or subclasses. The subclasses are numbered based on their prevalence in human serum, such that IgG1 is present at the highest amount (∼60%) followed by IgG2 (∼25%), IgG3 (∼10%) and IgG4 (∼5%).Citation2 All four subclasses share a similar structure characterized by two antigen-binding (Fab) arms linked to a single constant domain (Fc) by the hinge region. Apart from similarities in the overall structure of IgGs, each subclass possesses structural idiosyncrasies, as well as unique biophysical and functional properties. For instance, each subclass has a distinct hinge region and, in many cases, the hinge region imparts substantial differences in both the structure and function of individual subclasses. The hinge region can be subdivided into three regions comprising two parallel chains: the upper hinge adjacent to the Fab arms, the core hinge that contains disulfide bonds that link the two heavy chains and the lower hinge that extends into the Fc domain. The hinge region of IgG1 contains the core sequence of C226-P227-P228-C229 (EU numbering) Citation3 and a lower hinge/adjacent CH2 region that is capable of interacting with all three activating FcγRs (FcγRI, FcγRIIa and FcγRIIIa),Citation4 as well as the C1q component of complement.Citation5 Interactions with activating FcγRs can elicit the cell-killing mechanisms of antibody-dependent cellular cytotoxicity (ADCC) and antibody-dependent cellular phagocytosis (ADCP), whereas interactions with C1q and the resultant complement cascade can lead to complement-dependent cytotoxicity (CDC). Compared to IgG1, IgG2 contains a three amino acid truncation in the upper hinge, retains the same “CPPC” core hinge, displays a single amino acid deletion in the lower hinge/CH2 region and contains multiple amino acid differences in both the upper and lower hinge region. The hinge region of IgG2 contributes to structural isoforms mediated by differential disulfide linkages,Citation6,Citation7 as well as the ability to form covalent dimers with other IgG2s.Citation8 Whereas IgG2s are considered silent in terms of activating CDC and natural killer (NK) cell-mediated ADCC due to weak interactions with C1q and FcγRIIIa, respectively, IgG2 can productively engage FcγRIIa, particularly the FcγRIIa H131 polymorphism,Citation9 and drive myeloid-mediated cell killing, as was seen with the IgG2 anti-epidermal growth factor receptor (EGFR) panitumumab.Citation10 Currently, human antibodies of both the IgG1 and IgG2 subclasses have been approved as cancer therapeutics (e.g., anti-HER2, anti-CD20, anti-EGFR IgG1 mAbs; anti-EGFR IgG2 mAb).Citation1
Our group and others have demonstrated that the hinge region of human IgG1 is susceptible to proteolysis by a number of physiologically-relevant proteases associated with both pathologic micro-organisms (e.g., IdeS from Streptococcus pyogenes and glutamyl endopeptidase V8 (GluV8) from Staphylococcus aureus) and invasive cancers [e.g., the matrix metalloproteinases (MMPs)].Citation11 Proteolysis in the lower hinge occurs in a two-step process whereby one heavy chain is first cleaved, generating a single-cleaved intermediate,Citation12,Citation13 then cleavage of the second heavy chain results in complete dissociation of the Fc domain, generating a F(ab')2 fragment. Even a single cleavage in one heavy chain is sufficient to abrogate IgG1-mediated ADCC and CDC in vitro, as well as block mAb-mediated cell clearance in vivo.Citation13 Cleaved IgGs have been found in a number of pathological settings including cystic fibrosis,Citation14 breast cancerCitation13 and synovial fluid from patients with arthritis.Citation15
It has long been recognized that human IgG2 is distinguished from other isotypes by its relatively greater resistance in vitro to proteolysis by papainCitation16 and pepsin,Citation17 enzymes frequently used to generate Fab and F(ab')2 fragments, respectively. However, these two proteases are of questionable physiological relevance. The purpose of the present study was two-fold. First, we investigated the proteolytic sensitivity of human IgG1 and IgG2 to a number of physiologically-relevant proteases associated with pathogenic microorganisms and invasive cancers. We had previously demonstrated a correlation between cryptic epitopes exposed in IgG1 by endogenous or bacterial proteases with a widespread presence of serum human anti-(IgG1)-hinge (HAH) autoantibodies in healthy individuals.Citation11,Citation18,Citation19 The results suggested that proteolysis of IgG1 in vivo and autoantibody generation to the cleavage products were related processes. In the present study, we questioned whether an analogous susceptibility of IgG2 to pathologic proteases in vivo would result in a similar presence of IgG cleavage product-specific autoantibodies.
Results
Human IgG2 is resistant to proteolysis within the lower hinge/CH2 region by physiologically-relevant proteases.
Previous investigations have detailed a number of physiologically-relevant human and bacterial proteolytic enzymes that catalyze specific cleavages in the human IgG hinge domain, although the reported rates of proteolysis in solution varied considerably among different enzymes and IgG subclasses.Citation12,Citation13,Citation15,Citation19–Citation22 depicts the patterns of proteolysis of a human IgG1 mAb after 24 h using several human and bacterial proteases. Specific lower hinge cleavage was indicated by the appearance of an intermediate single-cleaved IgG (scIgG) (MMP-7, MMP-13 and GluV8 at 24 h) or by a primarily terminal F(ab')2 derivative (MMP-3, MMP-12 and IdeS from S. pyogenes). The precise positions of peptide bond scissions within the hinge region of IgG1 have been reported for several proteases.Citation12,Citation13,Citation15,Citation19,Citation20,Citation22
In contrast to IgG1, IgG2 was not cleaved under the same conditions with any of the human enzymes tested (). One bacterial protease, IdeS, was capable of cleaving human IgG2; however, it was previously noted that IgG2 was more resistant to proteolysis by IdeS than any other IgG subclass.Citation22 Under the conditions used, IgG2 appeared to be completely resistant to hydrolysis by the other bacterial protease, GluV8. Thus, these results reveal a particular resistance on the part of human IgG2 to human proteolytic enzymes that are capable of fragmenting human IgG1.
Comparison of human anti-hinge autoantibody recognition of peptide analogs of the human IgG1 and IgG2 lower hinge/CH2 regions.
The hinge region sequences of human IgG1 and IgG2 are depicted in . Although both IgG1 and IgG2 share the same core hinge sequence CPPC, there is considerable sequence divergence between the adjoining upper and lower hinge/CH2 regions. IgG2 has a 3 amino acid truncation in the upper hinge and a single amino acid truncation in the lower hinge/CH2 region. There are also sequence differences within the lower hinge/CH2 sequence where MMP-3 and MMP-12 cleave IgG1 between P232 and E233; other differences occur at points in IgG1 at which MMP-7 cleaves between L234 and L235, GluV8 between E233 and L234, and IdeS between G236 and G237.Citation11 These differences may underlie the proteolytic resistance of IgG2 relative to IgG1.
The inability to generate proteolytic derivatives of IgG2 with a panel of proteases posed a challenge for the preparation of immune targets for the capture of potential anti-IgG2-hinge autoantibodies. However, peptide analogs of the IgG1 hinge were previously deployed for the identification of autoantibodies that bind to IgG1 proteolytic fragments. Analogously, to compare autoantibody reactivity to the hinge/CH2 region of IgG1 and IgG2, we constructed a series of 14-mer peptides encompassing C-terminal positions ranging from alanine 231 to valine 240 (conforming to EU numbering in IgG1). Peptides were synthesized with an N-terminal Ac-lysine (N-ε-biotin) for adherence to streptavidin-coated 96-well plates.
Previous studies pointed to a substantial IgG3 component within the autoimmune anti-hinge autoantibodies in healthy individuals.Citation19,Citation23 represents the ELISA results of serum pooled from 20 healthy donors on the hinge peptide analogs of IgG1 and IgG2 and detected with an anti-Fc reagent that specifically recognizes IgG3. There was substantial autoimmune reactivity to IgG1 peptides terminating at amino acids corresponding to sites within the lower hinge where known susceptibility to certain proteases exists. However, no similar autoimmune recognition was found toward peptide analogs with C-terminal positions in the IgG2 lower hinge. A similar discordance between IgG1 and IgG2 was apparent when autoantibodies were detected with a probe that recognizes the Fc region of all IgG subclasses (). These results indicated minimal immune recognition of the IgG2 hinge peptide analogs as compared to IgG1 analogs.
Whole blood or serum HAH autoantibody restoration of Fc-dependent effector functions to F(ab')2 fragments.
A previously reported functional property of HAH autoantibodies was an ability to restore Fc effector functions to cell-bound F(ab')2 fragments.Citation19 Indeed, this was demonstrated in the case of affinity-purified anti-IgG1 specific HAH in ADCC,Citation19 complement assays,Citation19,Citation24 and phagocytosis.Citation25 For tests in the IgG1 system, fragments were generated with a number of proteases, including MMP-3, GluV8, IdeSCitation19 and pepsin.Citation25 The inability to generate IgG2 fragments with most of the physiologically-relevant proteases, as well as the apparent absence of comparable HAH to the IgG2 hinge peptides, precluded an analogous, purified system approach for IgG2.
As a test for the presence of functional anti-IgG2 hinge autoantibodies that may not have been evident in ELISAs, we were limited to F(ab')2 fragments generated with the IdeS protease. We had previously shown that affinity purified HAH autoantibodies could restore ADCC capacity to IgG1 F(ab')2 IdeS fragments and, as described above, IdeS was the only physiological enzyme found to be capable of cleaving IgG2 to any appreciable extent ( and C). Our previous studies were performed in the absence of competing IgG,Citation19 and it has been demonstrated that excess IgG inhibits ADCC capacity.Citation26 To test the ability of HAH autoantibodies to restore ADCC capacity to IgG1 or IgG2 F(ab')2IdeS fragments in the presence of excess IgG, a whole blood ADCC assay was developed in which the blood contributed both the immune effector cells and HAH autoantibodies (final concentration of 50% whole blood). The target cells in the whole blood ADCC assay were CD20-expressing WIL2-S cells. As shown in and B, the IgG1 anti-CD20 mAbs achieved maximal lysis at approximately 1 µg/ml with two independent whole blood donors. IgG2 anti-CD20 mAbs did not mediate levels of ADCC above the background levels seen with the IgG1 isotype control mAb. Both IgG1 and IgG2 anti-CD20 mAbs showed comparable binding to WIL2-S cells, indicating that the difference in ADCC by IgG2 anti-CD20 was not due to reduced antigen-binding (data not shown). Whole blood from human donor number two was capable of reconstituting ADCC activity to F(ab')2 IdeS of IgG1 anti-CD20 (), whereas whole blood from donor one showed minimal reconstitution of ADCC (). There was no detectable restoration of whole blood ADCC capacity by either donor when WIL2-S cells were opsonized with IgG2 anti-CD20 F(ab')2 IdeS fragments.
The disparity in the restoration of function in the whole blood ADCC assays prompted the question as to how many IgG3 HAH autoantibodies were present in the serum from each donor. In order to quantify anti-IgG1 IdeS cleavage site autoantibodies, we employed the previously affinity purified HAH against the IdeS cleavage site in IgG1 as a standard.Citation19 The results indicated that donor one had approximately 8 µg/ml and donor two had approximately 20 µg/ml HAH autoantibodies directed against the IdeS cleavage site in IgG1. We were unable to quantify HAH autoantibodies directed against the IdeS cleavage site in IgG2 since detection of such HAH autoantibodies were at the limits of detection, as was seen with serum pooled from human donors ().
To further probe for HAH autoantibodies with capability to restore Fc-dependent effector functions to IgG1 and IgG2 F(ab')2 IdeS fragments, we performed CDC assays using serum from human donors as the source for HAH autoantibodies. CD20-expressing WIL2-S cells were used as target cells for CDC. As shown in and D, intact IgG1 anti-CD20 mAbs achieved similar maximal cell lysis in the presence of serum from both donors. Intact IgG2 anti-CD20 did not demonstrate any detectable CDC capacity in the presence of serum from either donor. Serum from either donor was capable of restoring CDC capacity to IgG1 anti-CD20 F(ab')2 IdeS fragments. Serum from donor two exhibited a greater degree of restoration than serum from donor one, perhaps consistent with the greater amount of IgG3 HAH in the serum from donor two. No restoration of CDC activity was detected when IgG2 anti-CD20 F(ab')2 IdeS fragments were used.
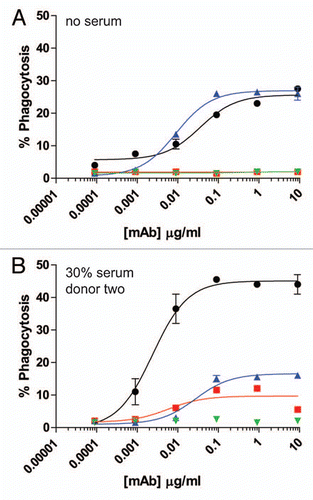
For further analysis of Fc-dependent effector functions, we tested the ability of excess serum from human donors to reconstitute ADCP. The target cells in these assays were the CD142-expressing human breast carcinoma cell line MDA-MB-231. The effector cells for the ADCP assay were purified human monocytes that were differentiated into macrophages by incubation with 20 ng/ml GM-CSF for six days, followed by overnight incubation with 100 ng/ml IFNγ. The resultant macrophages expressed both CD11b and CD14, as well as all three activating FcγRs and the inhibitory FcγRIIb (data not shown). Many previously reported flow cytometry-based ADCP assays labeled effector cells with lipophilic dyes such as PKH26 or PKH67, and detected macrophages with fluorescent-coupled antibodies to receptors present on the cell surface of macrophages, such as CD11b and CD14.Citation27–Citation29 For the present studies, preliminary experiments in which cells were labeled with the lipophilic dye PKH67 were problematic, particularly in the presence of serum, because high levels of background phagocytosis were detected even in the absence of opsonizing antibody (data not shown). Therefore, we utilized MDA-MB-231 cells that expressed green fluorescent protein (GFP) to identify target cells by flow cytometry. ADCP was evaluated by opsonizing GFP-expressing MDA-MB-231 cells with anti-CD142 antibodies in the presence of effector macrophages. Macrophages were detected with Alexa Fluor 647 conjugated anti-CD11b and anti-CD14 mAbs. Macrophages that had phagocytosed MDA-MB-231 cells were double positive for GFP and Alexa Fluor 647. As seen in , this methodology resulted in very low levels of background phagocytosis, even in the presence of human serum. shows representative fluorescent microscopy images of macrophages and GFP-expressing MDA-MB-231 cells in the absence of mAb (left part) and in the presence of 9 µg/ml IgG1 anti-CD142 (right part). The white arrows in the right part indicate macrophages that had phagocytosed MDA-MB-231 cells as evidenced by the GFP-signal (green) detected within the macrophage (red). In the absence of competing serum, intact IgG1 anti-CD142 achieved maximal phagocytosis at mAb concentrations approaching 1 µg/ml (). Intact IgG2 anti-CD142 was capable of achieving ADCP above background, but the maximal phagocytosis was well below the level seen with intact IgG1 anti-CD142 (). Inclusion of serum from each of the two human donors was capable of restoring ADCP to IgG1 anti-CD142 F(ab')2 IdeS fragments, whereas no restoration of function was detected on the IgG2 anti-CD142 F(ab')2 IdeS fragments ( and E).
It was recently demonstrated that human IgG2 mAbs directed against cancer targets could elicit cell destruction when myeloid lineage cells were employed as effector cells, particularly with IgG2 anti-EGFR panitumumab.Citation10 In our studies, the IgG2 anti-CD20 displayed no ADCC capacity in whole blood over background ( and B) and the macrophage-mediated ADCP induced by IgG2 anti-CD142 was minimally above background (). Because panitumumab had been previously reported as capable of eliciting cell-death with myeloid derived cells, we next compared human IgG1 anti-EGFR cetuximab with panitumumab in our macrophage-mediated ADCP assays. illustrates that, in the absence of serum, the level of ADCP induced by both cetuximab and panitumumab was comparable, and the F(ab')2 IdeS fragments of each parent mAb had no detectable ADCP capacity. ADCP was tested in the presence of serum from the human donor that previously displayed the most robust capacity to restore Fc-dependent cell killing to IgG1 F(ab')2 IdeS fragments. As shown in , human serum increased both the potency (decreased EC50) and efficacy (increased maximal lysis) of IgG1 anti-EGFR compared to the same mAb in the absence of serum. In contrast, serum reduced both the potency and efficacy of IgG2 anti-EGFR. The reason for the increase in ADCP seen with cetuximab in the presence of excess serum was unknown, but it was possible that some serum component other than HAH was contributing to this effect. Because of this observation, seen with multiple serum donors (data not shown), we could not rule out that the restoration of function detected on the IgG1 F(ab')2 IdeS fragments was limited to HAH autoantibodies, which do not bind to the intact IgG counterpart. For instance, some degree of restoration of function might be attributable to autoantibodies binding to regions in the F(ab')2 other than the cleavage point.
Taken together, these functional results demonstrated that serum containing HAH autoantibodies can restore the Fc-dependent effector functions to IgG1 F(ab')2 IdeS fragments, but not to IgG2 F(ab')2 IdeS fragments. Furthermore, the HAH autoantibody-mediated restoration of function was evident even in the presence of excess IgG.
Discussion
Together, IgG1 and IgG2 constitute more than 85% of total circulating IgG in healthy individuals. Many therapeutic mAbs are also of these two (human) subclasses.Citation1 It is of interest that these two subclasses exhibit disparate effector profiles. For example, IgG1s are well-recognized for their ability to mediate target cell killing by multiple mechanisms including ADCC, CDC and ADCP.Citation30,Citation31 In contrast, IgG2 is mostly unable to mediate ADCC and CDC presumably due to the differences in the lower hinge amino acid sequence compared to the one in IgG1 that is important for engaging Fcγ receptors on immune effector cells (e.g., natural killer cells) and complement C1q,Citation31 although IgG2s have often been linked to bacterial clearance by immune cells.Citation2,Citation32
IgG1 exhibits particular sensitivity to inactivating protease cleavages within or adjacent to the lower hinge sequences that are involved in Fcγ receptor interactions and C1q binding.Citation11 The scission of one or both lower hinge heavy chains also exposes cryptic/neo-epitopes that become targets for immune recognition.Citation19 Indeed, sera from healthy individuals contain autoantibodies that recognize these precise cleavage sites in IgG1.Citation19 This autoimmune phenomenon was linked to potential biological significance with the demonstration that the human anti-hinge (HAH) autoantibodies could restore cell killing functions (ADCC and CDC) to otherwise inactive IgG1 fragments. A likely explanation for the functional restoration appeared to be the formation of a cell-bound immune complex comprising an intact, competent Fc from the anti-hinge Ab. In fact, we and others noted that HAH IgG autoantibodies are disproportionately constituted by IgG3s,Citation19,Citation23 which is a minor isotype that is typified by strong effector functions.Citation33 Thus, it appears that endogenous pathologic proteases or similar bacterially-derived enzymes both inactivate host IgGs while at the same time provoking immune recognition to specific sites of protease cleavage. The current study attempted to extend the previous IgG1 findings to IgG2 to determine if analogous proteolytic and autoimmune inter-relationships also exist for this second most prevalent human isotype.
Comparative in vitro proteolytic digestions of IgG1 and IgG2 were performed using equivalent (catalytic) protease:IgG ratios and at physiological pH and ionic strength. With the candidate proteases, the results consistently indicated that IgG2 was more resistant to lower hinge proteolysis compared with IgG1. For most proteases, there was no detectable cleavage of IgG2. Sequence alignments of IgG1 and IgG2 () indicate that IgG2 possesses sequence variations at each of the amino acid cleavage positions mapped within IgG1. It is interesting to note that mutation of these positions in IgG1 (E233-L234-L235-G236) to the IgG2 counterparts (P233-V234-A235 with G236 deleted) has been shown to render an IgG1 silent with regard to FcγR binding and FcγR-mediated effector functions.Citation34,Citation35
One potential caveat to the in vitro proteolysis studies is that they were all performed in solution. It is possible that solution-phase reactions with purified proteases and antibodies may not accurately reflect the conditions in pathologic tissue environments in which the substrate antibodies may be clustered on cell surfaces and may be presented differently than in solution. Moreover, different proteases or their combinations might exist in pathological tissue environments than envisioned with purified systems. A different approach was felt needed to assess the likelihood of IgG2 proteolysis in vivo.
The tactic that was chosen for IgG2 paralleled the one previously employed for IgG1, i.e., use of autoantibody reactivity to reveal cryptic/neoantigens in the lower hinge. The peptide analog strategy for the IgG2 hinge proved to be a necessary adaptation since it was already shown difficult to generate proteolytic derivates of IgG2 with physiological proteases (). We focused on peptides with defined C-termini since these would also be presented on cell-bound single-cleaved IgG and F(ab')2 fragments, and it was felt that these would be most relevant by remaining associated to antigens within pathological environments. The studies using the IgG1 hinge peptide analogs had already provided evidence that many identified sites of protease cleavage were encompassed within a region recognized by anti-hinge autoantibodies. For IgG2, however, we found that there was little if any IgG immune recognition to the lower hinge ( and C). The binding results were obtained using the same serum samples that demonstrated abundant autoantibody binding to IgG1 peptides (and to proteolytic fragments of IgG1). Although autoantibody recognition/binding appeared minimal for IgG2 hinge epitopes, these results did not rule out the presence of undetected functional immune reactivity that could restore complement or ADCC-mediated cell killing to proteolytic IgG2 fragments. Functional studies of immune restoration were then undertaken.
Whole blood ADCC assays are becoming increasingly employed to assess Fc-dependent effector functions of mAbs due to several perceived benefits of whole blood ADCC over traditional ADCC assays that utilize purified PBMCs in the absence of excess IgGs.Citation10,Citation36 These benefits include the contribution of various effector cell populations, including polymorphonuclear cells (PMNs) that are not included in traditional PBMC preparations, as well as the presence of autologous excess IgGs that could compete for FcR occupancy.Citation26 We choose whole blood ADCCs for our studies because this assay format could provide both multi-lineage effector cells as well as HAH autoantibodies in the presence of excess IgG. Our results demonstrated that human IgG1 anti-CD20 mAbs could effectively eliminate target cells in the presence of excess IgGs, although higher concentrations of antibody were needed for maximal cell lysis compared to ADCC assays devoid of excess IgGs, as was reported previously in reference Citation26. The human IgG2 anti-CD20 mAbs did not mediate target cell lysis above background in these assays. One human donor was found to be capable of restoring whole blood ADCC activity to IgG1 anti-CD20 F(ab')2 IdeS fragments (), whereas another donor had minimal restoration of function (). This discrepancy could be due to the fact that the donor with minimal restoration of function had less IgG3 HAH autoantibodies compared to the donor that could restore lysis to IgG1 anti-CD20 F(ab')2 IdeS fragments. Perhaps the lower level of specific HAH autoantibodies found in donor number one was insufficient to overcome the suppressive effect of excess IgGs. However, neither donor was capable of restoring target cell lysis to IgG2 anti-CD20 F(ab')2 IdeS.fragments.
Human donor serum from both donors was capable of restoring CDC function only to the anti-CD20 IgG1 F(ab')2 IdeS fragments. Excess serum from donor two appeared to mediate a stronger restoration of function than serum from donor one, perhaps also in accordance with donor two's higher concentration of IgG3 HAH autoantibodies directed against the IdeS cleavage site in IgG1. We also observed an augmented CDC response on the anti-CD20 IgG1 F(ab')2 IdeS fragments relative to the CDC induced by the intact IgG1 anti-CD20 counterpart ( and D). This may be reminiscent of the CDC amplification effect of anti-hinge antibodies on F(ab')2 fragments previously described in reference Citation24.
ADCP afforded the opportunity to use an effector cell population that expressed multiple FcγRs, in particular, FcγRIIa, which has been previously shown to both productively engage human IgG2,Citation9 as well as elicit monocyte/macrophage cell-killing, specifically with anti-EGFR mAbs.Citation10 The IgG2 anti-CD142 mAb did not elicit a strong ADCP response compared with the IgG1 anti-CD142 mAb (). In contrast, in the absence of serum, the IgG2 anti-EGFR panitumumab elicited an ADCP response similar to IgG1 anti-EGFR cetuximab (). The presence of excess serum caused an unexplained increase in the ADCP capacity of cetuximab that was not observed with the anti-CD142 mAb. Cetuximab contains an oligosaccharide, galactose-α-1,3-galactose, in the Fab region, and a subset of patients have been identified that have IgE antibodies specific for the oligosaccharide.Citation37 Additionally, natural anti-galactose-α-1,3-galactose (anti-Gal) IgGs are also present in humans.Citation38 It would be of interest if the increase in ADCP capacity of cetuximab in the presence of donor serum was due to anti-Gal antibodies. Due to this increase in ADCP observed with intact IgG1, the anti-EGFR system was not suitable for evaluating the potential restoration of function by HAH autoantibodies. However, the results in the absence of excess serum could provide novel insights about the properties of human IgG2 antibodies directed against cell surface targets. The ability of the IgG2 anti-EGFR mAb to facilitate cell-death through myeloid lineage cells at a level similar to that seen with an IgG1 anti-EGFR mAb was consistent with a previous report in reference Citation10. The disparate ability of two different IgG2 anti-tumor mAbs to drive ADCP perhaps suggests that the ability of an IgG2 to effectively engage macrophage-mediated cell destruction should be evaluated on a case-by-case basis. Furthermore, if an IgG1 can elicit ADCP against a particular target, it may not be predictive that an IgG2 directed against the same target could elicit the same level of ADCP.
Recombinant mAbs possessing human Fc regions were the primary substrates for defining in vitro protease susceptibility in this study, and it cannot be ignored that several of these same antibodies are widely used for treating human disease. To the degree that IgG1 mAb therapies are effective via their binding to target cells and mediating immune clearance, it could be a matter of concern if proteolytic activity within pathological environments opposes IgG1 activity. Further, the roles of anti-IgG1 HAH autoantibodies remain to be fully defined with regard to their functions, such as immune indicators of chronic IgG1 susceptibility to proteases or as a compensatory mechanism to restore the activity of cleaved antibodies. In contrast, IgG2—via its evident resistance to proteases—may afford a more stable or sustained presence in hostile environments although at a cost of decreased cell-killing functions. In any case, the results of our investigations suggest a dynamic interplay between host immunity and IgG proteolysis in natural pathology as well the relevance of IgG proteolysis for the design of monoclonal antibody therapeutics that target protease-enriched environments.
In conclusion, our results demonstrate that human IgG2s are resistant to a number of physiologically-relevant proteases when incubated with purified enzymes in vitro. Previous studies have utilized peptide analogs corresponding to discrete C-terminal amino acids that would be exposed by cleavage in the lower hinge/CH2 region to identify autoimmune activity in human IgG1.Citation19 Peptide analogs of the IgG2 revealed minimal anti-IgG2 hinge activity, suggesting that IgG2s are also resistant to proteolysis in vivo. These findings could have important implications for the isotype selection of therapeutic antibodies whose intended targets are found within microenvironments enriched with proteases capable of cleaving human IgGs.
Materials and Methods
Antibodies.
The antibodies used for comparative proteolysis were a human anti-CD142 IgG1 (CDR-grafted, humanized containing the same variable region as the mAb previously described in ref. Citation39) and an anti-CD142 IgG2 containing the same variable region (CDR-grafted, humanized). The antibodies used for whole blood ADCC and CDC assays were an anti-human CD20 IgG1 (human/murine chimeric IgG1 (rituximab) from Genentech, clinical grade) and an anti-CD20 IgG2 (human/murine chimeric IgG2 with the same variable regions as the anti-CD20 IgG1 grafted onto an IgG2 backbone). The antibodies used in the ADCP assay were an anti-CD142 IgG1 (CDR-grafted, humanized), an anti-CD142 IgG2 containing the same variable region (CDR-grafted, humanized) and anti-human EGFR IgG1 (human/murine chimeric IgG1 (cetuximab) from ImClone Systems Inc., and Bristol-Myers Squibb Company, clinical grade) and an anti-human EGFR IgG2 (human IgG2 (panitumumab) from Amgen, clinical grade).
Human materials (whole blood, serum and leukopacks).
Human whole blood and serum for use in ADCC, CDC and ELISA assays were collected from healthy volunteers with necessary permissions and approvals. A third party Institutional Review Board approved the collection protocols, as well as the informed consent form. Leukopacks were obtained commercially from Biologics Specialty Corporation.
Proteases and IgG digestions.
Protease digestions of mAbs were performed as previously described in reference Citation13 and Citation19. Recombinant MMP-3 was expressed and purified at Centocor; MMP-7 (cat. BML-SE181), MMP-12 (cat. BML-SE138) and MMP-13 (cat. BML-SE246) were all obtained from Enzo Life Sciences. GluV8 (cat. E-006) was obtained from BioCentrum Ltd., and IdeS (cat. AO-FR1-999) was obtained from Genovis. The percent intact IgG was calculated based on the electropherogram profiles obtained by capillary electrophoresis (Agilent Technologies) by dividing the percent intact IgG1 remaining after 24 h in the presence of specific proteases by the percent intact IgG remaining after 24 h in the absence of protease. Data were analyzed and plotted using GraphPad Prism v5.
ELISA.
ELISAs for human Ig binding to peptide analogs of the IgG1 and IgG2 hinge were performed as previously described in reference Citation19. Hinge analog peptides were prepared as previously described in reference Citation19. Briefly, 96 well plates were coated with 10 µg/ml NeutraAvidin (Invitrogen, cat. A-2666) overnight at 4°C and washed and blocked as previously described in reference Citation15. Pooled human serum was diluted 1:50 in 3% BSA and incubated on the peptide analog-coated plates for 1 h at room temperature. Bound human antibodies were detected by either goat HRP-conjugated anti-human Fc (Jackson Immunoresearch, cat. 109-035-008, 1:20,000) or murine HRP-conjugated anti-human IgG3 (Invitrogen, cat. 053620, 1:1,000). The plates were developed using SIGMAFAST OPD (Sigma, cat. P9187) and stopped by addition of 3 M HCl. OD 490 was detected on a Spectra max Plus 384 (PerkinElmer). Data were analyzed and plotted using GraphPad Prism v5.
Whole blood ADCC assay.
The target cells for the whole blood ADCC assays were WIL2-S cells (American Type Culture Collection (ATCC), cat. CRL-8885). Target cells were pre-labeled with BATDA (PerkinElmer, cat. C136-100) for 30 min at 37°C as previously described in reference Citation19. Cells were washed twice and resuspended in Roswell Park Memorial Institute medium (RPMI), 10% heat-inactivated FBS, 0.1 mM nonessential amino acids and 1 mM sodium pyruvate (all from Invitrogen). A total of 50 µl of cells were added to the wells of 96-well U-bottom plates at a final concentration of 0.02 × 106 cells/well. An additional 50 µl of medium was added with and without mAbs and incubated for 20 min at room temperature. One hundred µl of whole blood was added to each well, and the plates were centrifuged at 150 g for 3 min and then incubated at 37°C for 3 h. At the end of the incubation, cells were again centrifuged at 150 g for 3 min. A total of 20 µl of supernatant was removed per well, and cell lysis was measured by addition of DELFIA Europium-based reagent (PerkinElmer, cat. C135-100). Fluorescence was measured using an Envision 2101 Multilabel Reader (PerkinElmer). Data were normalized to maximal cytotoxicity with 0.67% Triton X-100 (Sigma, cat. T8532) in the absence of whole blood and minimal control determined by spontaneous release of BATDA from targets cells in the absence of mAb. Data were fit to a sigmoidal dose-response model using GraphPad Prism v5.
CDC assay.
The target cells for the CDC assays were WIL2-S cells. A total of 50 µl of cells were added to the wells of 96-well U-bottom plates at a final concentration of 0.05 × 106 cells/well in RPMI, 10% heat-inactivated FBS, 0.1 mM nonessential amino acids and 1 mM sodium pyruvate. An additional 50 µl of medium was added with and without mAbs, then 100 µl of undiluted human serum was added as a source of HAH autoantibodies, and plates were incubated at room temperature for 2 h. After incubation, a total of 50 µl of a 10% rabbit complement (Invitrogen, cat. 31038-100) solution was added and plates were incubated at 37°C for 20 min. Twenty µl of Alamar Blue (Invitrogen, cat. DAL1100) was added, and plates were incubated overnight at 37°C and then analyzed on a SpectraMax M5 (Molecular Devices) at 570 nm. Data were normalized to maximal cytotoxicity with Triton X-100 (Sigma) and minimal control containing only cells, complement and plasma in the absence of mAb. Data were fit to a sigmoidal dose-response model using GraphPad Prism v5.
ADCP assay.
Human PBMCs were isolated from leukopacks using Ficoll gradient centrifugation. All ADCP assays in this study were performed with PBMCs isolated from the same donor. The FcγRIIa genotype of the donor was R131/H131, and the FcγRIIIa genotype of the donor was V158/V158, which was performed as a commercial service by GENEWIZ, Inc., (South Plainfield, NJ). CD14pos monocytes were purified from PBMCs by negative depletion using a CD14 isolation kit that did not deplete CD16pos monocytes (Stem Cell Technologies, cat. 19058). Monocytes were plated at 0.1 × 106 cells/cm2 in X-VIVO-10 medium (Lonza, cat. 04-380Q) containing 10% FBS and 20 ng/ml GM-CSF (R&D Systems, cat. 215-GM/CF) for 7 days. One hundred ng/ml of IFNγ (R&D Systems, cat. 285-IF/CF) was added for the final 24 h of differentiation. The target cells for the ADCP assay were MDA-MB-231 cells (ATCC, cat. HTB-26) that expressed GFP. A total of 50 µl of medium containing macrophages was added to the wells of 96-well plates for a final concentration of 0.1 × 106 cells/well, and 50 µl of MDA-MB-231 GFP-expressing cells were added for a final concentration of 0.025 × 106 cells/well, resulting in a ratio of 4 macrophages to 1 MDA-MB-231 cell. One hundred µl of medium containing 60% human serum with or without mAbs was added to the plates for a final serum concentration of 30%. Plates were then incubated at 37°C for 4 h. After incubation, cells were detached from the 96 well plates using Accutase (Sigma, cat. A6964). Macrophages were identified with anti-CD11b (BD Biosciences, cat.555385) and anti-CD14 (BD Biosciences, cat. 555395) antibodies coupled to Alexa Fluor 647 (Invitrogen, cat. A-20186) and then cells were acquired on a FACs Calibur (BD Biosciences). The data were analyzed using FloJo Software (Tree Star). Percent phagocytosis was determined by the following equation [(GFPpos, CD11bpos, CD14pos cells)/(GFPpos, CD11bpos, CD14pos cells plus GFP alonepos cells) × 100%]. Data were fit to a sigmoidal dose-response model using GraphPad Prism v5. In order to confirm uptake of tumor cells by the macrophages, fluorescent microscopy was performed. Briefly, 10,000 cells were removed from the ADCP set-up described above after incubation and detachment from the 96-well plate, but prior to addition of flow cytometry detecting mAbs. The 10,000 cells were plated onto the wells of a 96-well poly-D-lysine coated plate (Becton Dickinson, cat. 35-4640). Macrophages were detected with anti-CD11b (BD Biosciences, cat.555385) and anti-CD14 (BD Biosciences, cat. 555395) antibodies coupled to Alexa Fluor 568 (Invitrogen, cat. A-20184) and images were taken with a Nikon Eclipse TE2000-U epifluorescent microscope.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Financial Support
Centocor R&D Inc., provided funding for all research. All authors are employees of Centocor R&D Inc.
Abbreviations
| ADCC | = | antibody-dependent cellular cytotoxicity |
| ADCP | = | antibody-dependent cellular phagocytosis |
| CDC | = | complement-dependent cytotoxicity |
| ELISA | = | enzyme-linked immunosorbent assay |
| HAH | = | human anti-hinge |
Figures and Tables
Figure 1 Human IgG2 is resistant to cleavage by a number of physiologically-relevant proteases. (A) Purified human IgG1 was incubated with different proteases and analyzed by capillary electrophoresis under denaturing, non-reducing conditions. Specific enzymes are noted above individual lanes, and all digestions were carried out for 24 h at 37°C. The far right three lanes have purified human IgG1 standards, representing the following: Lane 9, single cleaved IgG1 (scIgG1); Lane 10, F(ab')2 fragment of IgG1; Lane 11, intact IgG1. The Fc monomer released under denaturing conditions is labeled as Fc(m). (B) Lanes 2–8 depict human IgG2 incubated with different proteases analyzed by capillary electrophoresis under denaturing, non-reducing conditions. The same standards used in (A) were run in Lanes 9–11. (C) Bar graph representation of the percent of intact IgG remaining after 24 h of proteolytic digestions. Open bars represent human IgG1 and shaded bars represent human IgG2. Bar heights correspond to the mean ± SD from four independent tests.
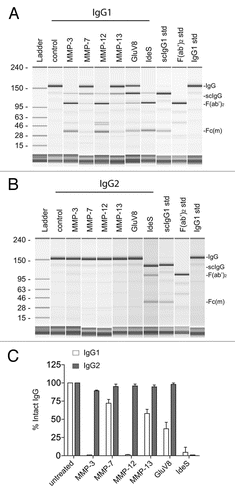
Figure 2 Human anti-hinge autoantibodies were detected against peptide analogs of the human IgG1 hinge region but not human IgG2. (A) The sequence of human IgG1 (top) and human IgG2 (bottom) ranging from amino acids 212–241 (EU numbering). (B) ELISA binding of IgG3 autoantibodies from serum pooled from 20 healthy donors to peptide analogs of the human IgG1 and IgG2 hinge/CH2 regions. Letters on the X-axis represent the free C-termini of 14-mer peptides. (C) ELISA binding of IgG autoantibodies from serum pooled from 20 healthy donors to peptide analogs of the human IgG1 and IgG2 hinge/CH2 regions.
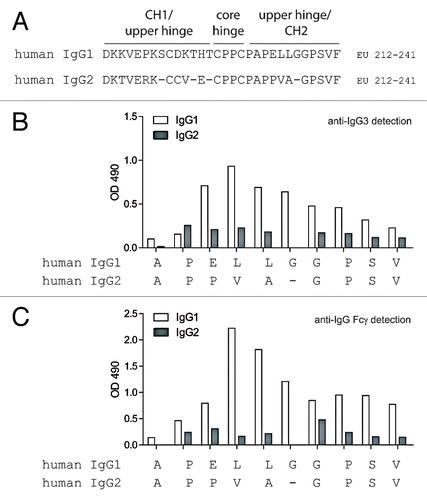
Figure 3 Whole blood ADCC and CDC using human IgG1 and IgG2 anti-CD20 intact mAbs and F(ab')2 fragments generated with IdeS. (A) Human donor one whole blood ADCC activity against WIL2-S cells using intact IgG1 anti-CD20 (black circles), IgG1 F(ab')2 of anti-CD20 (red squares), IgG2 anti-CD20 (blue up triangle), IgG2 F(ab')2 of anti-CD20 (green down triangle), and an IgG1 isotype control (black diamond) (n = 2). (B) Human donor two whole blood ADCC activity against WIL2-S cells (n = 2). (C) Cell-based CDC against WIL2-S cells containing 50% serum from human donor one (n = 2). (D) Cell-based CDC against WIL2-S cells containing 50% serum from human donor two (n = 2).
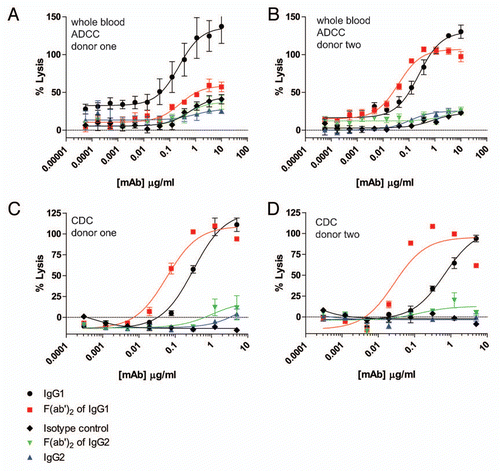
Figure 4 ADCP using human IgG1 and IgG2 anti-CD142 intact mAbs and F(ab')2 fragments generated with IdeS. (A) Representative flow cytometry dot plots depicting GFP-expressing MDA-MB-231 cells and human macrophages detected with anti-CD11b and anti-CD14 mAbs conjugated to Alexa Fluor 647. The left plot depicts cells incubated in the absence of anti-CD142 mAbs and the plot on the right depicts cells incubated with 9 µg/ml human IgG1 intact anti-CD142 mAb. (B) Representative fluorescent microscopy images of macrophages and GFP-expressing MDA-MB-231 cells in the absence of mAb (left panel) and incubated with 9 µg/ml IgG1 intact anti-CD142 (right part). Macrophages were detected with anti-CD11b and anti-CD14 antibodies conjugated to Alexa Fluor 568 (red), whereas the green indicates GFP-expressing MDA-MB-231 cells. The white arrows indicate macrophages that had phagocytosed MDA-MB-231 cells. (C) Human macrophage-mediated ADCP activity against GFP-expressing cells using intact IgG1 anti-CD142 (black circles), IgG1 F(ab')2 of anti-CD142 (red squares), IgG2 anti-CD142 (blue up triangle), and IgG2 F(ab')2 of anti-CD142 (green down triangle) in the absence of human serum. (D) ADCP activity against GFP-expressing cells in the presence of 30% human serum from donor one (n = 2). (E) ADCP activity against GFP-expressing cells in the presence of 30% human serum from donor two (n = 2).
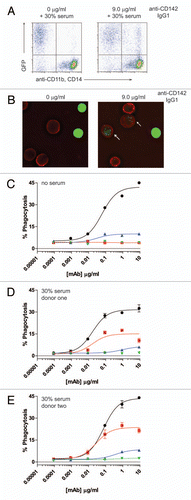
Figure 5 ADCP using human IgG1 and IgG2 anti-EGFR intact mAbs and F(ab')2 fragments generated with IdeS. (A) Human macrophage-mediated ADCP activity against GFP-expressing cells using intact IgG1 anti-EGFR (black circles), IgG1 F(ab')2 of anti-EGFR (red squares), IgG2 anti-EGFR (blue up triangle), and IgG2 F(ab')2 of anti-EGFR (green down triangle) in the absence of human serum (n = 2). (B) ADCP activity against GFP-expressing cells in the presence of 30% human serum from donor two (n = 2).
Acknowledgements
The authors would like to thank Bill Strohl and Omid Vafa for the contribution of reagents and discussions about IgG subclasses.
References
- Reichert JM. Metrics for antibody therapeutics development. mAbs 2010; 2:695 - 700
- Jefferis R. Antibody therapeutics: isotype and glycoform selection. Expert Opin Biol Ther 2007; 7:1401 - 1413
- Edelman GM, Cunningham BA, Gall WE, Gottlieb PD, Rutishauser U, Waxdal MJ. The covalent structure of an entire gammaG immunoglobulin molecule. Proc Natl Acad Sci USA 1969; 63:78 - 85
- Nimmerjahn F, Ravetch JV. Fcgamma receptors as regulators of immune responses. Nat Rev Immunol 2008; 8:34 - 47
- Bruggemann M, Williams GT, Bindon CI, Clark MR, Walker MR, Jefferis R, et al. Comparison of the effector functions of human immunoglobulins using a matched set of chimeric antibodies. J Exp Med 1987; 166:1351 - 1361
- Dillon TM, Ricci MS, Vezina C, Flynn GC, Liu YD, Rehder DS, et al. Structural and functional characterization of disulfide isoforms of the human IgG2 subclass. J Biol Chem 2008; 283:16206 - 16215
- Wypych J, Li M, Guo A, Zhang Z, Martinez T, Allen MJ, et al. Human IgG2 antibodies display disulfide-mediated structural isoforms. J Biol Chem 2008; 283:16194 - 16205
- Yoo EM, Wims LA, Chan LA, Morrison SL. Human IgG2 can form covalent dimers. J Immunol 2003; 170:3134 - 3138
- Bruhns P, Iannascoli B, England P, Mancardi DA, Fernandez N, Jorieux S, et al. Specificity and affinity of human Fcgamma receptors and their polymorphic variants for human IgG subclasses. Blood 2009; 113:3716 - 3725
- Schneider-Merck T, Lammerts van Bueren JJ, Berger S, Rossen K, van Berkel PH, Derer S, et al. Human IgG2 antibodies against epidermal growth factor receptor effectively trigger antibody-dependent cellular cytotoxicity but, in contrast to IgG1, only by cells of myeloid lineage. J Immunol 2009; 184:512 - 520
- Brezski RJ, Jordan RE. Cleavage of IgGs by proteases associated with invasive diseases: An evasion tactic against host immunity?. mAbs 2010; 2:212 - 220
- Gearing AJ, Thorpe SJ, Miller K, Mangan M, Varley PG, Dudgeon T, et al. Selective cleavage of human IgG by the matrix metalloproteinases, matrilysin and stromelysin. ImmunolLett 2002; 81:41 - 48
- Brezski RJ, Vafa O, Petrone D, Tam SH, Powers G, Ryan MH, et al. Tumor-associated and microbial proteases compromise host IgG effector functions by a single cleavage proximal to the hinge. Proc Natl Acad Sci USA 2009; 106:17864 - 17869
- Fick RB Jr, Baltimore RS, Squier SU, Reynolds HY. IgG proteolytic activity of Pseudomonas aeruginosa in cystic fibrosis. J Infect Dis 1985; 151:589 - 598
- Ryan MH, Petrone D, Nemeth JF, Barnathan E, Bjorck L, Jordan RE. Proteolysis of purified IgGs by human and bacterial enzymes in vitro and the detection of specific proteolytic fragments of endogenous IgG in rheumatoid synovial fluid. Mol Immunol 2008; 45:1837 - 1846
- Jefferis R, Weston PD, Stanworth DR, Clamp JR. Relationship between the papain sensitivity of human gammaG immunoglobulins and their heavy chain subclass. Nature 1968; 219:646 - 649
- Turner MW, Bennich HH, Natvig JB. Pepsin digestion of human G-myeloma proteins of different subclasses. I. The characteristic features of pepsin cleavage as a function of time. Clin Exp Immunol 1970; 7:603 - 625
- Brezski RJ, Knight DM, Jordan RE. The Origins, Specificity and Potential Biological Relevance of Human Anti-IgG Hinge Autoantibodies. ScientificWorldJournal 2011; 11:1153 - 1167
- Brezski RJ, Luongo JL, Petrone D, Ryan MH, Zhong D, Tam SH, et al. Human anti-IgG1 hinge autoantibodies reconstitute the effector functions of proteolytically inactivated IgGs. J Immunol 2008; 181:3183 - 3192
- Diemel RV, ter Hart HGJ, Derksen GJA, Koenderman AHL, Aalberse RC. Characterization of immunoglobulin G fragments in liquid intravenous immunoglobulin products. Transfusion 2005; 45:1601 - 1609
- Baici A, Knopfel M, Fehr K, Skvaril F, Boni A. Kinetics of the different susceptibilities of the four human immunoglobulin G subclasses to proteolysis by human lysosomal elastase. Scand J Immunol 1980; 12:41 - 50
- von Pawel-Rammingen U, Johansson BP, Bjorck L. IdeS, a novel streptococcal cysteine proteinase with unique specificity for immunoglobulin G. EMBO J 2002; 21:1607 - 1615
- Persselin JE, Stevens RH. Anti-Fab antibodies in humans. Predominance of minor immunoglobulin G subclasses in rheumatoid arthritis. Journal of Clinical Investigation 1985; 76:723 - 730
- Fumia S, Goede JS, Fischler M, Luginbuhl A, Frick S, Fodor P, et al. Human F(ab')(2)-containing immune complexes together with anti-hinge natural antibodies stimulate complement amplification in vitro and in vivo. Mol Immunol 2008; 45:2951 - 2961
- Yano S, Kaku S, Suzuki K, Terazaki C, Sakayori T, Kawasaki T, et al. Natural antibodies against the immunoglobulin F(ab')2 fragment cause the elimination of antigens recognized by the F(ab')2 from the circulation. Eur J Immunol 1995; 25:3128 - 3133
- Preithner S, Elm S, Lippold S, Locher M, Wolf A, da Silva AJ, et al. High concentrations of therapeutic IgG1 antibodies are needed to compensate for inhibition of antibody-dependent cellular cytotoxicity by excess endogenous immunoglobulin G. Mol Immunol 2006; 43:1183 - 1193
- McEarchern JA, Oflazoglu E, Francisco L, McDonagh CF, Gordon KA, Stone I, et al. Engineered anti-CD70 antibody with multiple effector functions exhibits in vitro and in vivo antitumor activities. Blood 2007; 109:1185 - 1192
- Richards JO, Karki S, Lazar GA, Chen H, Dang W, Desjarlais JR. Optimization of antibody binding to FcgammaRIIa enhances macrophage phagocytosis of tumor cells. Mol Cancer Ther 2008; 7:2517 - 2527
- Oflazoglu E, Stone IJ, Brown L, Gordon KA, van Rooijen N, Jonas M, et al. Macrophages and Fc-receptor interactions contribute to the antitumour activities of the anti-CD40 antibody SGN-40. Br J Cancer 2009; 100:113 - 117
- Presta LG. Molecular engineering and design of therapeutic antibodies. Curr Opin Immunol 2008; 20:460 - 470
- Strohl WR. Wood C. Isotype selection and Fc engineering: Design and construction of fit-for-purpose therapeutic antibodieas. Antibody Drug Discovery 2011; London Imperial College Press In Press
- Lai Z, Kimmel R, Petersen S, Thomas S, Pier G, Bezabeh B, et al. Multi-valent human monoclonal antibody preparation against Pseudomonas aeruginosa derived from transgenic mice containing human immunoglobulin loci is protective against fatal pseudomonas sepsis caused by multiple serotypes. Vaccine 2005; 23:3264 - 3271
- Clark MR. IgG effector mechanisms. Chem Immunol 1997; 65:88 - 110
- Armour KL, Clark MR, Hadley AG, Williamson LM. Recombinant human IgG molecules lacking Fcg receptor I binding and monocyte triggering activities. European Journal of Immunology 1999; 29:2613 - 2624
- Shields RL, Namenuk AK, Hong K, Meng YG, Rae J, Briggs J, et al. High resolution mapping of the binding site on human IgG1 for Fcgamma RI, Fc gamma RII, Fcgamma RIII and FcRn and design of IgG1 variants with improved binding to the Fcgamma R. J Biol Chem 2001; 276:6591 - 6604
- Peipp M, Lammerts van Bueren JJ, Schneider-Merck T, Bleeker WW, Dechant M, Beyer T, et al. Antibody fucosylation differentially impacts cytotoxicity mediated by NK and PMN effector cells. Blood 2008; 112:2390 - 2399
- Chun CH, Mirakhur B, Chan E, Le QT, Berlin J, Morse M, et al. Cetuximab-induced anaphylaxis and IgE specific for galactose-alpha-1,3-galactose. N Engl J Med 2008; 358:1109 - 1117
- Galili U. The alpha-gal epitope and the anti-Gal antibody in xenotransplantation and in cancer immunotherapy. Immunol Cell Biol 2005; 83:674 - 686
- Ngo CV, Picha K, McCabe F, Millar H, Tawadros R, Tam SH, et al. CNTO 859, a humanized anti-tissue factor monoclonal antibody, is a potent inhibitor of breast cancer metastasis and tumor growth in xenograft models. Int J Cancer 2007; 120:1261 - 1267