Abstract
Next generation sequencing enables identification of immunogenic tumor mutations targetable by individualized vaccines. In the B16F10 melanoma system as pre-clinical proof-of-concept model, we found a total of 563 non-synonymous expressed somatic mutations. Of the mutations we tested, one third were immunogenic. Immunization conferred in vivo tumor control, qualifying mutated epitopes as source for effective vaccines.
Cancer therapies attempt to exploit differences between tumor and normal cells. As cancer cells proliferate rapidly, many chemotherapies target rapidly dividing cells, causing toxicities for proliferating non-cancerous cells. As cancer is a disease caused by DNA aberrations,Citation1 mutations provide another difference between tumor and normal cells.
Tumors contain a large number of mutations, ranging from the 10s to the 100s,Citation2 that are unique to the tumor relative to the normal cells. Causative “driver” mutations shared by a subpopulation of patients can sometimes be targeted by small molecule inhibitors, such as the BRAF V600E mutation.Citation3 However, in the vast majority of cancer types there are no highly penetrant mutations. Rather, 95% of the mutations in a patient tumor appear to be unique to that tumor.Citation4 Thus, mutations may make ideal therapeutic targets, provided there was, for an individual patient, a platform to identify the mutations in the patient’s tumor and an effective way to efficiently target them.
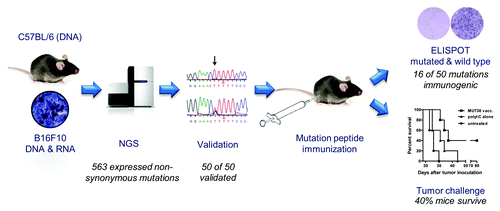
Therapeutic cancer vaccination, in which a patient’s immune system is taught to target cancer cells, represents a promising therapeutic modality. Existing cancer vaccines, including several in clinical trials, target antigens with tumor-specific expression. A key challenge is immune tolerance against self-proteins. Tumor specific mutation antigens, in contrast, are not subject to central tolerance mechanisms. Immune responses to them are prevalent in cancer patients.Citation5 Thus, the tumor mutanome may offer a large number of potential targets for personalized vaccine therapies. Our recent work,Citation6 summarized in , addressed following key questions: What method is suitable for identifying tumor mutations? Are they immunogenic? Does immunization with mutation-encoding antigens provide a survival benefit?
The “next-generation sequencing” (NGS) technology enabled us to profile cancer and normal cells to identify somatic mutations. Comparing mouse B16F10 melanoma cells to the parental reference C57BL/6 cells, we found 1,392 point mutations in transcripts. Of the 1,266 mutations in coding regions, 962 cause non-synonymous protein changes. Using NGS again, but for gene expression profiling (RNA-Seq), we found that 563 of the non-synonymous mutations occur in expressed genes. We developed a new algorithm to assign a false discovery rate (FDR) to each mutation. Of the 50 selected low-FDR (high confidence) mutations all passed validation.
A fraction of these mutations occur in established tumor suppressor genes, including Pten, Trp53 (p53) and Tp63; in genes associated with DNA repair, including Brca2, Atm, Ddb1 and Rad9b; and in genes associated with cancer pathways, including Rassf7 (RAS-253 associated protein), Ksr1 (kinase suppressor of ras 1), Mdm1 (TP53 binding nuclear protein) and Atm (PI3K pathway). Furthermore, a mutation exists in Trrap, a gene recently identified as a potential melanoma target.
We sought to determine the immunogenicity and specificity of each mutation. We designed long peptides 27 amino acids in length with either the mutated or wild-type amino acid positioned centrally, immunized naïve C57BL/6 mice, and assayed spleen cells using INFγ-ELISpot for mutation specific immunogenicity. Sixteen of the 50 mutant peptides (32%) were found to be immunogenic, with 11 of 16 eliciting immune responses directed preferentially against the mutated sequence relative to the wild-type sequence. Using our RNA vaccine platform, we confirmed endogenous processing of mutations into epitopes.Citation7 Observed T-cell frequencies were similar to the frequencies induced by the immunodominant Trp2180–88 epitope, demonstrating that many of the mutations are immunodominant epitopes of B16 melanoma. This is the first experimental data establishing the breadth of the immunogenicity of the tumor mutanome. Our findings match previous in silico predictions suggesting 30 to 50% of non-synonymous mutations are immunogenic.Citation8
We examined whether immunization with mutation-coding peptides would translate into a tumor survival benefit. By prophylactic vaccination complete tumor protection in 40% of the mice was achieved. In therapeutic models, we observed a remarkable growth inhibition induced by mutation-coding peptide immunization. These results demonstrate that vaccination against a single mutation encoding sequence is able to induce substantial anti-tumoral effects. To avoid tumor escape due to evolution under immunoediting pressure,Citation9 multiple mutations could be targeted. Noteworthy, we found no correlation between immunogenicity and potential oncological function, structural features or subcellular localization of the encoded protein. Thus, regardless of whether the mutation is a “driver” or “passenger” mutation, its utilization in a vaccine appears to provide an anti-tumor benefit.
In conclusion, in a pre-clinical model, we successfully demonstrate a blueprint process to identify somatic mutations, select mutations for an individual therapeutic vaccine, and immunize to provide an anti-tumor impact. Our data shows that the T cell druggable mutanome is substantial and can be exploited for patient benefit using individualized therapeutic cancer vaccines.
Figure 1. Discovery and characterization of the “T-cell druggable mutanome.”
References
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011; 144:646 - 74; http://dx.doi.org/10.1016/j.cell.2011.02.013; PMID: 21376230
- Ding L, Wendl MC, Koboldt DC, Mardis ER. Analysis of next-generation genomic data in cancer: accomplishments and challenges. Hum Mol Genet 2010; 19:R2 R188 - 96; http://dx.doi.org/10.1093/hmg/ddq391; PMID: 20843826
- Flaherty KT, Puzanov I, Kim KB, Ribas A, McArthur GA, Sosman JA, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med 2010; 363:809 - 19; http://dx.doi.org/10.1056/NEJMoa1002011; PMID: 20818844
- Stratton MR. Exploring the genomes of cancer cells: progress and promise. Science 2011; 331:1553 - 8; http://dx.doi.org/10.1126/science.1204040; PMID: 21436442
- Sensi M, Anichini A. Unique tumor antigens: evidence for immune control of genome integrity and immunogenic targets for T cell-mediated patient-specific immunotherapy. Clin Cancer Res 2006; 12:5023 - 32; http://dx.doi.org/10.1158/1078-0432.CCR-05-2682; PMID: 16951217
- Castle JC, Kreiter S, Diekmann J, Lower M, van de Roemer N, de Graf J, et al. Exploiting the mutanome for tumor vaccination. Cancer Res 2012; 72:5 1081 - 91; http://dx.doi.org/10.1158/0008-5472.CAN-11-3722; PMID: 22237626
- Kreiter S, Selmi A, Diken M, Sebastian M, Osterloh P, Schild H, et al. Increased antigen presentation efficiency by coupling antigens to MHC class I trafficking signals. J Immunol 2008; 180:309 - 18; PMID: 18097032
- Segal NH, Parsons DW, Peggs KS, Velculescu V, Kinzler KW, Vogelstein B, et al. Epitope landscape in breast and colorectal cancer. Cancer Res 2008; 68:889 - 92; http://dx.doi.org/10.1158/0008-5472.CAN-07-3095; PMID: 18245491
- Matsushita H, Vesely MD, Koboldt DC, Rickert CG, Uppaluri R, Magrini VJ, et al. Cancer exome analysis reveals a T-cell-dependent mechanism of cancer immunoediting. Nature 2012; Epub ahead of press.