Abstract
Tumor cells expressing TLRs is generally recognized to mediate tumor inflammation. However, whether and how tumor TLR signaling pathways negatively regulate tumor inflammation remains unclear. In this report, we find that TLR4 signaling of H22 hepatocarcinoma tumor cells is transduced through MyD88 pathway to actin cytoskeletons, leading to the release of microparticles (MPs), the cellular membrane-derived vesicles. As a result, tumor macrophages take up MPs and acquire MP-contained microRNA let-7b, which attenuates tumor inflammation by targeting proinflammatory cytokine IL-6. Thus, tumor TLR signaling, contrary to the original promoting effect, may play an opposite role in downregulating tumor inflammation through MP pathways.
Introduction
Despite its role in promoting the initiation, promotion, and progression of tumorigenesis,Citation1,Citation2 tumor inflammation must be kept in check lest the catastrophic damage from uncontrolled inflammation. Obviously, ways, means and mechanisms are needed to keep the persistent low-grade inflammation within the tumor microenvironment. However, the exact mechanisms underlying the regulation of tumor inflammation are still incompletely understood. Toll-like receptors (TLRs), the fundamental signaling pathway that triggers inflammation, have been demonstrated to be generally expressed in tumor cells with functional activities.Citation3-Citation5 In this respect, it is clear that the existence of its numerous ligands in the tumor microenvironment,Citation6,Citation7 including various fragments of degraded extracellular matrix (ECM) components, extracellular heat shock proteins (HSPs), high mobility group box 1 (HMGB1), and extracellular nuclear materials, points at TLR signaling as the most likely mediator of tumor inflammation. However, whether and how tumor cell TLR signaling negatively regulates tumor inflammation has hitherto remained unclear.
Besides tumor cells, innate immune cells, such as macrophages and dendritic cells, also act as important cell types regulating tumor inflammation.Citation8,Citation9 However, it remains unclear if and how tumor cells synergize with such inflammatory cells to orchestrate tumor inflammation. Recent studies have highlighted small non-coding RNAs, called microRNAs that play a critical role in regulating inflammation by binding to mRNA 3′-UTR of the target proinflammatory gene so as to inhibit their expression at translational levels.Citation10,Citation11 Interestingly, microRNAs are not restricted to their parent cells but can also be transferred to other single cells for functional expression through multiple transferring pathways, including microparticle mediated pathways.Citation12-Citation14
Studies have shown that during apoptosis or even when simply activated, eukaryotic cells shed vesicles of components of the plasma membranes encapsulating cytoplasmic elements into the extracellular space.Citation15-Citation17 These vesicles, known as microparticles, vary from 100 to 1000 nm in size.Citation18 Consistent with the primary cellular components, MPs also contain enzymes, proteins, RNAs and even DNAs, and are capable of transferring messenger molecules including microRNAs from one cell to another,Citation19-Citation21 functioning as vectors for delivering molecular messages among cells. Given the propensity for tumor cells to ubiquitously express inflammation-regulating microRNAs,Citation22-Citation25 in this study, we hypothesized that tumor cell-derived MPs transferred inflammation-regulating microRNAs to macrophages, thus tuning tumor inflammation. We showed that TLR4 signaling triggered the release of MPs by hepatocarcinoma tumor cells, which transferred microRNA let-7b to macrophages and resulted in the downregulation of proinflammatory cytokine IL-6.
Results and Discussion
H22 hepatocarcinoma tumor cell TLR4 signaling induces generation of MPs through MyD88-dependent pathway
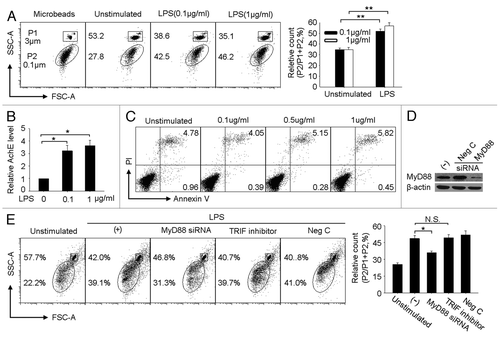
Among TLR family members, TLR4 is typically highly expressed in tumor cells.Citation3 Here, we wondered whether the activation of tumor cell TLR4 signaling induces the production of tumor MPs. To determine this possibility, a TLR4-expressing murine hepatocarcinoma tumor cell line H22 was tested here,Citation4 using microbeads of 0.1 and 3 μm as gating standards in order to count the number of MPs (). After 48 h culturing in the presence or absence of different concentrations of LPS, the supernatants were harvested and analyzed by flow cytometry for any release of MPs. The result showed that upon 0.1 or 1 μg/ml LPS stimulation, the release of MPs was significantly increased, compared with control groups (). To further convince this result, we used acetylcholine esterase ELISA kit, an alternative approach used for the quantitation of cellular vesicles,Citation26 to evaluate the quantity of MPs. Consistently, the levels of acetylcholine esterase was higher in the MPs from LPS stimulated group than those from PBS group (). To exclude the possibility that the released MPs above were derived from LPS-induced cell death, we additionally analyzed the apoptosis of LPS-stimulated H22 tumor cells by flow cytometry. The result showed that 0.1 or 1 μg/ml LPS did not induced H22 cell apoptosis after 48 h (). Therefore, TLR4 signaling does in fact stimulate the release of MPs in H22 tumor cells. But because both MyD88-dependent and TIR-domain-containing adaptor-inducing IFNγ (TRIF)-dependent pathways may transduce LPS signal,Citation27 it was necessary to further clarify and pin point the exact role of TLR4 downstream signaling to mediate the release of MPs. To this end, the expression of MyD88 in H22 tumor cells was knocked down by siRNA technology (). The knockdown of MyD88 expression resulted in a significant decrease of MP production (). This result was corroborated when TRIF inhibitors, used to block TLR4-TRIF signaling, were found not to affect the production of MPs (), suggesting, in turn, that the MyD88 pathway acts as the only adaptor molecule to transduce TLR4 signaling for MP release.
Figure 1. LPS induces H22 tumor cells releasing MPs through MyD88 dependent pathway. (A) LPS stimulated tumor cells releasing MPs. H22 tumor cells were cultured with 0.1 or 1 μg/ml LPS for 48 h. The MPs in the supernatants were analyzed by flow cytometry. In this assay, 3 μm and 0.1 μm microbeads were used as standard to gate MPs. The representative was shown in the left panel and the relative number of MPs released by H22 tumor cells was shown in the right panel. The detailed was described in the Materials and Methods. (B) MPs were quantitated by acetylcholine esterase assay. H22 tumor cells were cultured in the presence of different concentration of LPS. 48 h later, MPs were isolated from the supernatants to determine AchE levels by ELISA kit. (C) H22 tumor cells were cultured in the presence of different concentration of LPS. 48 h later, tumor cells were collected for apoptosis assay by flow cytometry. (D) The knockdown of MyD88 by MyD88 siRNA in H22 tumor cells was confirmed by western blot. (E) Blocking MyD88 rather than TRIF signaling pathway decreased the release of MPs by LPS. MyD88 siRNA-transfected or TRIF inhibitor-treated H22 tumor cells were stimulated with LPS (0.1 μg/ml) for 48 h. The released MPs in the supernatants were determined by flow cytometry. *, p < 0.05; **, p < 0.01.
TLR4 signaling activates actomyosin cytoskeleton for MP generation
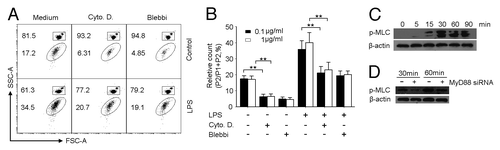
Next, the attention was directed at how TLR4 signaling resulted in the release of MPs by H22 tumor cells. Given the important role played by cytoskeletons in cellular exocytosis,Citation28,Citation29 we here questioned whether TLR4 signaling relied upon a cytoskeletal signaling in order to induce MPs generation. To test this, we treated H22 tumor cells in the presence of LPS with cytochalasin D to disrupt F-actin polymerization and blebbistatin to inhibit myosin II activity, considering the critical role of F-actin assembly and myosin II activation in cytoskeleton remodeling. As expected, the addition of either cytoskeleton inhibitor substantially decreased the production of MPs by LPS (). In line with these data, we found that upon TLR4 activation, phosphorylated myosin light chain (pMLC), a marker for the active form of myosin II,Citation30 was materially induced (). However, the blockade of TLR4 signaling by MyD88 siRNA inhibited the expression of pMLC (). Taken together, these data suggested that tumor cell TLR4 signaling mediates the generation of MPs via F-actin/myosin II pathway. But does this actually lead to the regulation of inflammation?
Figure 2. TLR4 signaling activates actomyosin cytoskeleton for MP generation in H22 tumor cells. (A and B) Cytoskeleton inhibitors hindered MP generation by LPS in H22 tumor cells. H22 cells were incubated with LPS (0.1 or 1 μg/ml) in the presence of cytochalasin D (Cyto D) or blebbistatin (Blebbi) for 48 h. The released MPs were determined by flow cytometry. The result represented three independent experiments. (C) The phosphorylation of myosin II light chain (MLC) in H22 tumor cells. H22 cells were exposed to LPS (0.1 µg/ml) for 0, 5, 15, 30, 60 and 90 min and subjected to western blot with anti-pMLC antibody. (D) Phosphorylation of MLC was MyD88 dependent. MyD88 siRNA-transfected H22 cells were stimulated with 0.1 µg /ml LPS for 30 or 60 min. The phosphorylation of MLC (pMLC) was determined by western blot.
Tumor cell TLR4 signaling-induced MPs inhibit proinflammatory IL-6 production of LPS-stimulated macrophages
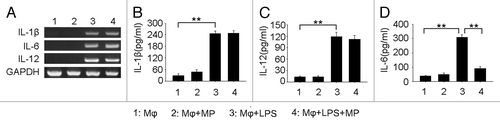
To address question above, we concentrated on tumor-infiltrating immune cells, especially macrophages, which are both (1) central players in the development of tumor inflammation and (2) capable of taking up MPs. Using LPS to stimulate macrophages, with or without TLR4-triggered, H22-released MPs, showed that the addition of MPs did not resulted in the alteration of proinflammatory cytokines IL-1β and IL-12 at the mRNA and protein levels (–). Interestingly, however, the expression of IL-6 was significantly suppressed at the protein levels rather than mRNA levels (). These data suggested that H22 tumor cell TLR4 signaling-induced MPs might affect tumor inflammation by inhibiting IL-6 production of macrophages.
Figure 3. Tumor cell TLR4 signaling-induced MPs inhibit proinflammatory IL-6 production of LPS-stimulated macrophages. (A) Tumor cell TLR4 signaling-induced MPs had no effect on proinflammatory cytokines at mRNA levels. RAW264.7 macrophages were treated with H22 tumor cell-derived MPs, 100ng/ml LPS, or both for 12 h. The mRNA expression of proinflammatory cytokines IL-1β, IL-6, IL-12 was detected by RT-PCR. (B-D) H22 tumor cell MPs inhibited IL-6 protein production of LPS stimulated macrophages. H22 tumor cell-derived MPs were co-cultured with RAW264.7 macrophages in the presence of LPS (1 μg/ml) for 48 h. The supernatants were harvested to detect IL-1β, IL-6 and IL-12 by ELISA. Experiments were repeated three times and results were expressed as mean ± SD.
Tumor cell TLR4 signaling-induced MPs includes abundant microRNAs
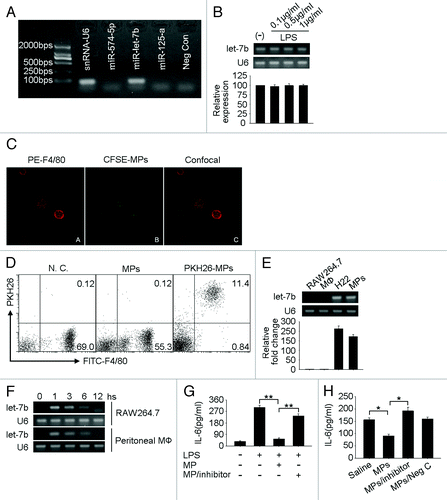
Because microRNAs regulate gene expression at the translational levels and normally not at the mRNA levels, the above IL-6 regulatory pattern promoted us to question whether TLR4 signaling-induced tumor cell MPs contained tumor cell microRNAs, leading to the regulation of IL-6 expression. To test this, we first isolated total RNAs from TLR4 signaling-induced MPs for miRNA microarray analysis. Based on the array result, miR-574–5p, let-7b, miR-125a, miR-24, miR-709, miR-191, miR-690, miR-1224 were shown to be abundant in MPs. Then, we used RT-PCR method to verify the array data, and the first three microRNAs with the highest expressional levels, namely, miR-574–5p, let-7b and miR-125a were checked. In contrast to miR-574–5p and miR-125a, which were not detected in the experiment settings, let-7b was shown a marked expression (). Given that reports had clearly demonstrated that IL-6 is the target of let-7b,Citation25,Citation31 it was not far-fetched to suggest that TLR4 signaling-triggered, tumor cell-released MPs must somehow transfer let-7b to macrophages, leading to the downregulation of IL-6 expression. In addition, we here also confirmed that LPS stimulation did not affect the expression of let-7b by H22 tumor cells (), suggesting TLR4 signaling only affects the formation of MPs and but not let-7b expression in H22 tumor cells. Therefore, although LPS stimulation certainly affected the formation of MPs, it also, significantly, transferred let-7b to macrophages allowing the conclusion that this may be the mechanism for the downregulation of IL-6.
Figure 4. Tumor cell-derived MPs downregulate IL-6 expression by transferring let-7b to macrophages. (A) let-7b was highly expressed in H22 tumor cell-released MPs. Five × 107 H22 tumor cells were stimulated with LPS (1 μg/ml) for 48 h and MPs in the supernatant were harvested for the RNA isolation. RT-PCR was conducted to detect the expression of miRNAs miR-574–5p, let-7b and miR-125a. U6 snRNA was used as control. (B) LPS stimulation did not affect the expression of let-7b in H22 tumor cells. H22 cells were treated with various concentrations of LPS (0.1, 0.5 and 1 μg/ml). Twelve hours later, total RNA were isolated to detect let-7b expression by RT-PCR (upper) and real time RT-PCR (bottom). (C) MPs were taken up by macrophages in vitro. CFSE-labeled H22 tumor cells were stimulated with LPS (0.1 μg/ml). The released MPs were collected and incubated with macrophages for 6 h. After stained with PE-conjugated anti-F4/80 antibody, cells were observed under confocal microscopy. (D) MPs were taken up by macrophages in vivo. MPs released by LPS-stimulated H22 tumor cells were stained with PKH26 and injected into peritoneal cavity of H22 tumor cell ascites bearing BALB/c mice. Six hours later, peritoneal cells were harvested and incubated in cell culture dish for 2 h to get rid of non-adherent cells. Adherent cells were collected and stained with FITC-conjugated F4/80 antibody for flow cytometric analysis. (E) The expression of let-7b in macrophages was analyzed by RT-PCR (upper) and real time RT-PCR (bottom). (F) MPs mediated the transfer of Let-7b to macrophages. H22 tumor cell-derived MPs were incubated with RAW264.7 or peritoneal macrophages for different times. The expression of let-7b in cells was analyzed by RT-PCR. (G and H) The inactivation of let-7b relieved the suppression on IL-6. H22 tumor cells were transfected with let-7b inhibitor. Twenty-four hours later, cells were treated with 0.1 µg/ml LPS to generate MPs. The MPs were then incubated with RAW264.7 cells in the presence of LPS for 36 h. IL-6 was determined in the supernatants by ELISA (G). Or the MPs were injected into tumor site. Thirty-six hours later, tumor tissues were minced and the supernatants were subjected to ELISA for the determination of IL-6 (H).
Tumor cell-released MPs downregulate IL-6 expression by transferring let-7b to macrophages both in vitro and in vivo
To further elucidate the above conclusion, we labeled peritoneal macrophages with PE-conjugated anti-F4/80 antibody (red) and H22 tumor cells with CSFE (green). We found that H22 derived MPs were taken up by macrophages (). To validate these in vitro data in vivo, TLR4 signaling-induced MPs were labeled with membrane red fluorescent dye PKH26 and injected into H22 tumors. Six hours later, tumor infiltrating leukocytes were isolated for flow cytometric analysis. As expected, F4/80+ macrophages were also shown PKH26 positive (), suggesting that macrophages are capable of taking up tumor cell-derived MPs in tumor microenvironment. This was corroborated by the different time points when incubated macrophages and TLR4 signaling-induced MPs and determined let-7b in macrophages. Macrophages selves did not express let-7b (). However macrophages, after taking up MPs, were shown to express let-7b (). Interestingly, this “transfection like” phenomenon was transient and only observed within the first 6 h by RT-PCR (), supporting the exogenous source of let-7b.
Then, we clarified that the exogenous let-7b was functional and mediated the downregulation of IL-6 in macrophages. For this purpose, we constructed let-7b inactive MPs by LPS stimulating H22 tumor cells that had been transfected with let-7b inhibitor or control oligonucleotide transfection. Interestingly, let-7b inactive MPs could not decrease the protein levels of IL-6, whereas control MPs still remained the inhibitory effect on IL-6 expression in LPS-stimulated macrophages (), suggesting that TLR4 signaling-induced MPs indeed transfer functional let-7b to macrophages for IL-6 downregulation. Besides in macrophages, the total IL-6 levels in tumor tissues were also measured, in consideration of the fact that the MPs also contained other microRNAs. As shown in , the gross IL-6 levels were decreased after the injection of control MPs, and the injection of let-7b inactive did not influence IL-6 levels in tumor tissue.
While most evidence points to inflammation as a promoter of carcinogenesis, there is contrary evidence to suggest an antitumor role for inflammation.Citation32,Citation33 Moreover, undue inflammation may also jeopardize tumor development. Therefore, tumors need a precise and perhaps narrow window whereby they can exploit, remodel or re-tune local inflammation so as to favor their insatiable uncontrollable growth. This report illustrates one of such mechanisms in that tumor cells release MPs that exquisitely lower tumor inflammation via transferring tumor microRNAs to nearby macrophages. The biosynthesis of MPs is a ubiquitous cellular event governed by patho/physiological conditions and upon extrinsic or intrinsic stimulations.Citation20,Citation21 Regardless of the existence of numerous signal molecules in tumor microenvironment, the definite signal(s) to trigger MP production by tumor cells still remains elusive. In this study, Tumor cell TLR4 signaling is identified as one, even if not the primary type responsible for MP production. This may have important implications, considering the existence of abundant ligands for TLR4 in tumor microenvironment. Perhaps more significantly, though TLR signaling has been demonstrated to be the fundamental pathway mediating inflammation, the findings here suggest that tumor cells also employ TLR, especially in the form of TLR4, to decrease inflammation. In conclusion, we propose that a relative homeostasis for inflammation is kept in tumor microenvironment via MPs’ loop, namely, the generation of MPs as the consequence of tumor inflammatory signaling, in turn, weakens tumor inflammation by targeting tumor inflammatory cells.
Materials and Methods
Animals and cell lines
BALB/c mice aged 6 to 8 weeks old were purchased from the Center of Medical Experimental Animals of Hubei Province (Wuhan, China) for studies approved by the Animal Care and Use Committee of Tongji Medical College. Murine tumor cell line H22 (hepatocarcinoma), RAW264.7 (macrophage) were purchased from China Center for Type Culture Collection (CCTCC, Wuhan, China) and cultured according to their guidelines.
Generation and isolation of MPs
MPs were prepared as described before.Citation34 Briefly, H22 tumor cells were cultured in the presence or absence of 0.1 or 1 μg/ml LPS for 24 h. The supernatants were harvested and centrifuged for 10 min at 600 g to get rid of cells and then for 2 min at 14,000 g to remove debris. At last the supernatant was further centrifuged for 60 min at 14,000 g to pellet MPs. The pellets were washed three times and re-suspended in in 300 μl pre-filtered (0.1 μm) PBS for the experiments.
Count of MPs
Isolated MPs were suspended in 250 μl PBS with 1 μl LB30 and LB1 Polystyrene Latex Beads (Sigma-Aldrich). After mixing, the number of MPs was counted by a flow cytometer in accordance with their respective bead sizes. The forward and side scatters were set at logarithmic gain. Three micrometer beads were gated as P1and 0.1μm beads were gated as P2. The number of MP was calculated as P2/P1 + P2 × total events.
Acetylcholine esterase assay
107 H22 tumor cells were cultured in the presence of different concentration of LPS for 48 h. MPs in the supernatants were isolated and lysed with 1% NP-40 containing protease inhibitor cocktail (Sigma-Aldrich). After centrifugation, the levels of AchE in the supernatants were determined by AchE ELISA kit (Hyperheal Biotech) according to the manufacturer’s instructions.
Apoptosis assay
H22 tumor cells were cultured with or without 0.1 or 1μg/ml LPS. After 48 h, cells were collected and stained with FITC-conjugated Annexin V and propidium iodide (PI) for flow cytometric analysis.
microRNA microarray
MPs were prepared as described above. Total RNA was isolated from MPs by using Trizol agent (Invitrogen). The microRNA microarray was performed according to our previous study.Citation35 Briefly, RNA samples were labeled with fluorescent dyes cy3 and hybridized with microRNA complementary probes printed on glass slide (Shanghai Bio Corporation). After hybridization, the slides were scanned with PerkinElmer ScanArray Scanner (PerkinElmer), and the images were analyzed with GenePix software. Each sample was assayed twice in triplicate per array.
Labeling of MPs
Isolated MPs were labeled with a red-fluorescent cell linker (PKH26, Sigma) according to the manufacturer’s protocol.
Western blot
Cell lysates and prestained molecular weight markers were separated by SDS-PAGE followed by transfer onto nitrocellulose membranes. The membranes were blocked in TBST (Tris-buffered saline with 0.5% of Triton X-100) containing 5% nonfat milk, and probed with antibodies. After incubation with the secondary antibody conjugated with horseradish peroxidase, membranes were extensively washed, and the immunoreactivity was visualized by enhanced chemiluminescence according to the manufacturer’s protocol (ECL kit, Santa Cruz). Antibody β-actin (SC-47778) was purchased from Santa Cruz, MyD88 (T3223) from Epitomics, and phosphorylated myosin light chain 2 from Cell Signaling (3674)
ELISA (ELISA)
The amounts of IL-1β, IL-6 and IL-12 in the supernatants were determined by ELISA kits (R&D Systems). In some cases, tumor tissues were homogenized in PBS (0.5 ml) containing 100 μm PMSF (Sigma), 1% (vol/vol) aprotinin (Sigma), 2 μg/ml leupeptin (Sigma), and 1 μg/ml pepstatin (Sigma). After centrifugation, the supernatants were assessed.
Analysis of microRNAs by RT-PCR and quantitative RT-PCR
Total microRNAs were isolated from MPs or H22 tumor cells using microRNA isolation kit (Ambion). Reverse transcription primers for miR-574–5p, let-7b, miR-125a were designed, respectively, with RNA mfold version 2.3 server (supplementary information), and its specificity was identified according to our previous study.Citation34 One hundred nanograms of enriched microRNA was used for the cDNA synthesis. A 67-bp cDNA product was amplified by PCR with primers: miR-574–5p, 5′-TCTGAGTGTGTGTGTGTG-3′ (sense); let-7b 5′-CTCTGAGGTAGTAGGTTG-3′ (sense); miR-125a, 5′-TAGTCCCTGAGACCCTTT-3′ (sense); common antisense primer, 5′-GACTGTT CCTCTCTTCCTC-3′.
For real-time PCR, the above primers and the Taqman probe [6-FAM]TTGCGACTAC ACACACACACACA[BHQ1a-6FAM] were mixed with TaqMan® Universal PCR Master Mix (Applied Biosystems). The reaction mixtures were incubated at 95°C for 10 min, followed by 40 cycles of 95°C for 15 sec and 60°C for 1 min in Stratagene QRT-PCR instrument.
Transfection assay
MyD88 siRNA, miR-let-7b inhibitor and the corresponding scrambled control oligonucleotides were purchased from RiboBio Corporation. In brief, for transient transfection, 200 pmol of synthesized oligonucleotide was mixed with 100 ml of Nucleofector solution (Amaxa, Gaithersburg, MD, USA), and transfected into 5 × 106 H22 tumor cells by electroporation using Nucleofector instrument. After transfection, cells were allowed to recover by incubating for 4 h at 37°C, and then used for the following assays.
Statistics
Results were expressed as mean values plus or minus SD and interpreted by repeated-measure ANOVA. Differences were considered to be statistically significant when the p value was less than 0.05.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
This work was supported by the National Basic Research Program of China (2012CB932500), Funds for International Cooperation and Exchange of the National Natural Science Foundation of China (30911120482), the Program for New Century Excellent Talents in University (NCET-08-0219), the National Natural Science Foundation of China (31000640, 31171387), the Fundamental Research Funds for the Central Universities (HUST-2010JC024, HUST-2011TS027).
References
- Coussens LM, Werb Z. Inflammation and cancer. Nature 2002; 420:860 - 7; http://dx.doi.org/10.1038/nature01322; PMID: 12490959
- Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell 2010; 140:883 - 99; http://dx.doi.org/10.1016/j.cell.2010.01.025; PMID: 20303878
- Huang B, Zhao J, Li H, He KL, Chen Y, Chen SH, et al. Toll-like receptors on tumor cells facilitate evasion of immune surveillance. Cancer Res 2005; 65:5009 - 14; http://dx.doi.org/10.1158/0008-5472.CAN-05-0784; PMID: 15958541
- Huang B, Zhao J, Shen S, Li H, He KL, Shen GX, et al. Listeria monocytogenes promotes tumor growth via tumor cell toll-like receptor 2 signaling. Cancer Res 2007; 67:4346 - 52; http://dx.doi.org/10.1158/0008-5472.CAN-06-4067; PMID: 17483348
- Kelly MG, Alvero AB, Chen R, Silasi DA, Abrahams VM, Chan S, et al. TLR-4 signaling promotes tumor growth and paclitaxel chemoresistance in ovarian cancer. Cancer Res 2006; 66:3859 - 68; http://dx.doi.org/10.1158/0008-5472.CAN-05-3948; PMID: 16585214
- Sims GP, Rowe DC, Rietdijk ST, Herbst R, Coyle AJ. HMGB1 and RAGE in inflammation and cancer. Annu Rev Immunol 2010; 28:367 - 88; http://dx.doi.org/10.1146/annurev.immunol.021908.132603; PMID: 20192808
- Huang B, Zhao J, Unkeless JC, Feng ZH, Xiong H. TLR signaling by tumor and immune cells: a double-edged sword. Oncogene 2008; 27:218 - 24; http://dx.doi.org/10.1038/sj.onc.1210904; PMID: 18176603
- DeNardo DG, Johansson M, Coussens LM. Immune cells as mediators of solid tumor metastasis. Cancer Metastasis Rev 2008; 27:11 - 8; http://dx.doi.org/10.1007/s10555-007-9100-0; PMID: 18066650
- Solinas G, Germano G, Mantovani A, Allavena P. Tumor-associated macrophages (TAM) as major players of the cancer-related inflammation. J Leukoc Biol 2009; 86:1065 - 73; http://dx.doi.org/10.1189/jlb.0609385; PMID: 19741157
- Davidson-Moncada J, Papavasiliou FN, Tam W. MicroRNAs of the immune system: roles in inflammation and cancer. Ann N Y Acad Sci 2010; 1183:183 - 94; http://dx.doi.org/10.1111/j.1749-6632.2009.05121.x; PMID: 20146715
- Sonkoly E, Pivarcsi A. microRNAs in inflammation. Int Rev Immunol 2009; 28:535 - 61; http://dx.doi.org/10.3109/08830180903208303; PMID: 19954362
- Valadi H, Ekström K, Bossios A, Sjöstrand M, Lee JJ, Lötvall JO. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol 2007; 9:654 - 9; http://dx.doi.org/10.1038/ncb1596; PMID: 17486113
- Collino F, Deregibus MC, Bruno S, Sterpone L, Aghemo G, Viltono L, et al. Microvesicles derived from adult human bone marrow and tissue specific mesenchymal stem cells shuttle selected pattern of miRNAs. PLoS One 2010; 5:e11803; http://dx.doi.org/10.1371/journal.pone.0011803; PMID: 20668554
- Yuan A, Farber EL, Rapoport AL, Tejada D, Deniskin R, Akhmedov NB, et al. Transfer of microRNAs by embryonic stem cell microvesicles. PLoS One 2009; 4:e4722; http://dx.doi.org/10.1371/journal.pone.0004722; PMID: 19266099
- VanWijk MJ, VanBavel E, Sturk A, Nieuwland R. Microparticles in cardiovascular diseases. Cardiovasc Res 2003; 59:277 - 87; http://dx.doi.org/10.1016/S0008-6363(03)00367-5; PMID: 12909311
- Morel O, Toti F, Hugel B, Freyssinet JM. Cellular microparticles: a disseminated storage pool of bioactive vascular effectors. Curr Opin Hematol 2004; 11:156 - 64; http://dx.doi.org/10.1097/01.moh.0000131441.10020.87; PMID: 15257014
- Hugel B, Martínez MC, Kunzelmann C, Freyssinet JM. Membrane microparticles: two sides of the coin. Physiology (Bethesda) 2005; 20:22 - 7; http://dx.doi.org/10.1152/physiol.00029.2004; PMID: 15653836
- Ratajczak J, Wysoczynski M, Hayek F, Janowska-Wieczorek A, Ratajczak MZ. Membrane-derived microvesicles: important and underappreciated mediators of cell-to-cell communication. Leukemia 2006; 20:1487 - 95; http://dx.doi.org/10.1038/sj.leu.2404296; PMID: 16791265
- Mack M, Kleinschmidt A, Brühl H, Klier C, Nelson PJ, Cihak J, et al. Transfer of the chemokine receptor CCR5 between cells by membrane-derived microparticles: a mechanism for cellular human immunodeficiency virus 1 infection. Nat Med 2000; 6:769 - 75; http://dx.doi.org/10.1038/77498; PMID: 10888925
- Mause SF, Weber C. Microparticles: protagonists of a novel communication network for intercellular information exchange. Circ Res 2010; 107:1047 - 57; http://dx.doi.org/10.1161/CIRCRESAHA.110.226456; PMID: 21030722
- Théry C, Ostrowski M, Segura E. Membrane vesicles as conveyors of immune responses. Nat Rev Immunol 2009; 9:581 - 93; http://dx.doi.org/10.1038/nri2567; PMID: 19498381
- Volinia S, Calin GA, Liu CG, Ambs S, Cimmino A, Petrocca F, et al. A microRNA expression signature of human solid tumors defines cancer gene targets. Proc Natl Acad Sci U S A 2006; 103:2257 - 61; http://dx.doi.org/10.1073/pnas.0510565103; PMID: 16461460
- Iliopoulos D, Jaeger SA, Hirsch HA, Bulyk ML, Struhl K. STAT3 activation of miR-21 and miR-181b-1 via PTEN and CYLD are part of the epigenetic switch linking inflammation to cancer. Mol Cell 2010; 39:493 - 506; http://dx.doi.org/10.1016/j.molcel.2010.07.023; PMID: 20797623
- Löffler D, Brocke-Heidrich K, Pfeifer G, Stocsits C, Hackermüller J, Kretzschmar AK, et al. Interleukin-6 dependent survival of multiple myeloma cells involves the Stat3-mediated induction of microRNA-21 through a highly conserved enhancer. Blood 2007; 110:1330 - 3; http://dx.doi.org/10.1182/blood-2007-03-081133; PMID: 17496199
- Iliopoulos D, Hirsch HA, Struhl K. An epigenetic switch involving NF-kappaB, Lin28, Let-7 MicroRNA, and IL6 links inflammation to cell transformation. Cell 2009; 139:693 - 706; http://dx.doi.org/10.1016/j.cell.2009.10.014; PMID: 19878981
- Savina A, Furlán M, Vidal M, Colombo MI. Exosome release is regulated by a calcium-dependent mechanism in K562 cells. J Biol Chem 2003; 278:20083 - 90; http://dx.doi.org/10.1074/jbc.M301642200; PMID: 12639953
- Takeda K, Akira S. TLR signaling pathways. Semin Immunol 2004; 16:3 - 9; http://dx.doi.org/10.1016/j.smim.2003.10.003; PMID: 14751757
- Nightingale TD, White IJ, Doyle EL, Turmaine M, Harrison-Lavoie KJ, Webb KF, et al. Actomyosin II contractility expels von Willebrand factor from Weibel-Palade bodies during exocytosis. J Cell Biol 2011; 194:613 - 29; http://dx.doi.org/10.1083/jcb.201011119; PMID: 21844207
- Masedunskas A, Sramkova M, Parente L, Sales KU, Amornphimoltham P, Bugge TH, et al. Role for the actomyosin complex in regulated exocytosis revealed by intravital microscopy. Proc Natl Acad Sci U S A 2011; 108:13552 - 7; http://dx.doi.org/10.1073/pnas.1016778108; PMID: 21808006
- le Duc Q, Shi Q, Blonk I, Sonnenberg A, Wang N, Leckband D, et al. Vinculin potentiates E-cadherin mechanosensing and is recruited to actin-anchored sites within adherens junctions in a myosin II-dependent manner. J Cell Biol 2010; 189:1107 - 15; http://dx.doi.org/10.1083/jcb.201001149; PMID: 20584916
- Schulte LN, Eulalio A, Mollenkopf HJ, Reinhardt R, Vogel J. Analysis of the host microRNA response to Salmonella uncovers the control of major cytokines by the let-7 family. EMBO J 2011; 30:1977 - 89; http://dx.doi.org/10.1038/emboj.2011.94; PMID: 21468030
- Nelson D, Ganss R. Tumor growth or regression: powered by inflammation. J Leukoc Biol 2006; 80:685 - 90; http://dx.doi.org/10.1189/jlb.1105646; PMID: 16864602
- Aggarwal BB, Shishodia S, Sandur SK, Pandey MK, Sethi G. Inflammation and cancer: how hot is the link?. Biochem Pharmacol 2006; 72:1605 - 21; http://dx.doi.org/10.1016/j.bcp.2006.06.029; PMID: 16889756
- Tang K, Liu J, Yang Z, Zhang B, Zhang H, Huang C, et al. Microparticles mediate enzyme transfer from platelets to mast cells: a new pathway for lipoxin A4 biosynthesis. Biochem Biophys Res Commun 2010; 400:432 - 6; http://dx.doi.org/10.1016/j.bbrc.2010.08.095; PMID: 20801099
- Huang B, Zhao J, Lei Z, Shen S, Li D, Shen GX, et al. miR-142-3p restricts cAMP production in CD4+CD25- T cells and CD4+CD25+ TREG cells by targeting AC9 mRNA. EMBO Rep 2009; 10:180 - 5; http://dx.doi.org/10.1038/embor.2008.224; PMID: 19098714