Abstract
The development of targeted therapies and immunotherapies has markedly advanced the treatment of metastasized melanoma. While treatment with selective BRAFV600E inhibitors (like vemurafenib or dabrafenib) leads to high response rates but short response duration, CTLA-4 blocking therapies induce sustained responses, but only in a limited number of patients. The combination of these diametric treatment approaches may further improve survival, but pre-clinical data concerning this approach is limited. We investigated, using Tyr::CreERT2PTENF−/−BRAFF-V600E/+ inducible melanoma mice, whether BRAFV600E inhibition can synergize with anti-CTLA-4 mAb treatment, focusing on the interaction between the BRAFV600E inhibitor PLX4720 and the immune system. While PLX4720 treatment strongly decreased tumor growth, it did not induce cell death in BRAFV600E/PTEN−/− melanomas. More strikingly, PLX4720 treatment led to a decreased frequency of tumor-resident T cells, NK-cells, MDSCs and macrophages, which could not be restored by the addition of anti-CTLA-4 mAb. As this effect was not observed upon treatment of BRAF wild-type B16F10 tumors, we conclude that the decreased frequency of immune cells correlates to BRAFV600E inhibition in tumor cells and is not due to an off-target effect of PLX4720 on immune cells. Furthermore, anti-CTLA-4 mAb treatment of inducible melanoma mice treated with PLX4720 did not result in enhanced tumor control, while anti-CTLA-4 mAb treatment did improve the effect of tumor-vaccination in B16F10-inoculated mice. Our data suggest that vemurafenib may negatively affect the immune activity within the tumor. Therefore, the potential effect of targeted therapy on the tumor-microenvironment should be taken into consideration in the design of clinical trials combining targeted and immunotherapy.
Introduction
The treatment of metastatic melanoma has progressed markedly in recent years due to the development of targeted therapies directed against (mutated) signaling proteins and immunotherapies such as monoclonal antibodies (mAb) specific for T-cell checkpoint molecules.Citation1
Blockade of CTLA-4 by monoclonal antibodies can stimulate an anti-tumor immune response in preclinical models.Citation2-Citation5 Two different anti-CTLA-4 antibodies have entered clinical trials, ipilimumab (Bristol-Myers Squibb) and tremelimumab (MedImmune). Ipilimumab was the first drug to lead to an improved overall survival in metastatic melanoma patients for 20 y.Citation6,Citation7 Although clinical responses (disease stabilization or regression) are often long-lasting, they can take several months to develop and only occur in a small proportion of treated patients.Citation8-Citation11 In detail, the phase III clinical trial data showed that ipilimumab induced tumor regression, as measured by RECIST criteria, in 11% of patients and disease stabilization in an additional 17.5%. The overall survival rate at 24 mo of follow up was 23.5% and long-term follow up from earlier phase 1 studies showed that responses were often sustained.Citation12 So far, no single predictive biomarker for a clinical response upon ipilimumab treatment has been identified. However, by comparing a small group of responders to non-responders it has recently been shown that melanomas having high baseline expression levels of immune-related genes, suggestive for immune cells infiltrating the tumor, are more likely to respond favorably to ipilimumab.Citation13
Vemurafenib and dabrafenib are small molecule inhibitors selective for the tumor-driving BRAFV600E mutation that is expressed in over 50% of the melanomas. The phase III clinical trial that evaluated vemurafenib showed that 48% of treated patients had a confirmed objective response and the median time to response was only 1.45 mo. However, these fast-developing responses are generally of short duration (progression free survival 5.3 mo), with almost all patients relapsing.Citation14,Citation15 As expected, presence of the BRAFV600E mutation is a prerequisite for a clinical response, but further mutation analyses showed that concurrent PTEN loss might reduce progression free survival.Citation16,Citation17
Based on the diametric properties of vemurafenib and ipilimumab with respect to response rate (resp. high and low), response duration (resp. short and long) and time to response onset (resp. short and long), it is thought that their combination will induce treatment synergy.Citation1,Citation18 In line with this concept, a number of studies support the idea that chemo or targeted therapies can stimulate anti-tumor immune responses by various mechanisms.Citation19-Citation24
First, Hong et al. observed that several chemotherapies can induce expression of T–cell-attracting chemokines, leading to improved tumor control due to the recruitment of tumor-reactive immune cells.Citation22 Second, studies by Zitvogel and Kroemer have suggested that cell death induced by chemotherapy can result in DC activation and subsequent cross-priming of tumor antigen-specific T cells.Citation20,Citation21,Citation23 In addition to the potential of targeted therapy to induce such immunogenic cell death, the treatment often leads to oncogene inactivation which has been shown, in murine tumor models, to result in an increased recruitment of immune cells, in particular CD4+ T cells, to the tumor site.Citation24 Furthermore, this recruitment showed to be essential to obtain sustained tumor regression upon driver oncogene inactivation. Finally, Coussens and colleagues demonstrated that the modulation of the tumor microenvironment toward a favorable immune signature (presence of CD8+ T cells in the absence of tissue-associated macrophages) improves the effect of chemotherapy.Citation19 Overall these data suggest that anti-tumor immune responses can contribute to the effect of targeted or chemotherapies.
Notably, a number of studies suggest that therapy induced tumor cell death has the potential to synergize with CTLA-4 blockade. Specifically, in mouse models of melanoma and prostate carcinoma, destruction of tumor tissue (e.g., by radiofrequency ablation, cryotherapy or radiotherapy) improved the anti-tumor activity of anti-CTLA-4 mAb treatment.Citation25,Citation26
Vemurafenib and its chemical analog PLX4720 have been shown to induce cell death in melanomas.Citation14,Citation27 Moreover, it has been demonstrated, in a small study population, that 15 d after starting BRAF inhibitor treatment the number of tumor-resident T cells was increased in a subgroup of patients.Citation28 Finally, in vitro studies demonstrated that PLX4720, in contrast to other targeted agents such as MEK or PI3 Kinase inhibitors, does not negatively affect T-cell function and may enhance surface expression of tumor associated antigens on melanoma cells.Citation29,Citation30
Collectively, the above data suggests that it will be attractive to explore a potential synergy between targeted therapies and immunotherapies. The advent of animal models harbouring mutations commonly found in human melanoma while being fully immuno-competent for the first time allows in vivo testing of combinations of targeted therapy such as MAP Kinase inhibition and immunotherapy, such as T-cell checkpoint blockade.Citation31,Citation32 Therefore, we aimed in the here presented work to investigate whether BRAFV600E inhibition can synergize with anti-CTLA-4 mAb treatment by focusing on the interaction between PLX4720 and the immune system. In detail, we used the C57BL6/J Tyr::CreERT2PTENF−/−BRAFF-V600E/+ inducible melanoma model to analyze the in vivo effect of selective BRAF inhibition on the presence of immune cells in the tumor and to determine whether the addition of anti-CTLA-4 mAb treatment improves tumor growth control.
Results
Selective BRAF inhibition in vivo results in strongly decreased tumor cell proliferation, but not induction of cell death, in BRAFV600E/PTEN−/− murine melanomas
We have recently described the C57BL6/J Tyr::CreERT2PTENF−/−BRAFF-V600E/+ melanoma model.Citation32 In this inducible model, all mice develop, within one month after tumor induction, a rapidly growing tumor with histology similar to human spindle cell melanoma. These melanomas are not only deficient for the expression of PTEN, but also harbour the BRAFV600E mutation, both genetic alterations commonly found in human melanoma.Citation33-Citation36 We initially showed that treatment of tumor-bearing Tyr::CreERT2PTENF−/−BRAFF-V600E/+ melanoma model mice with the BRAFV600E inhibitor PLX4720 led to a strong decrease in growth of melanomas, but did not induce tumor regression ().Citation32
Figure 1. Selective BRAF inhibition in vivo results in strongly decreased tumor cell proliferation, but not induction of cell death, in BRAFV600E/PTEN−/− murine melanomas. (A) Four to ten weeks old Tyr::CreERT2PTENF−/−BRAFF-V600E/+ mice were induced on the flank and 31 d later, when average tumor size was 10 mm2, tumor-bearing mice were placed on PLX4720 or mock treatment. Tumor outgrowth was followed over time and tumor size in mm2 is plotted against time from tumor induction for PLX4720 treated (blue line, n = 10) and control chow treated animals (red line, n = 14). (B) Tumors were taken from Tyr::CreERT2PTENF−/−BRAFF-V600E/+ mice at distinct time points after start of PLX4720 treatment (day 0, 2, 7 and 14). Proliferation of tumor cells was measured by Ki67 immunohistochemistry. Tumor cell death was assessed by immunohistochemistry for active caspase-3 and by a TUNEL assay. Positive staining is displayed in red. (C) The percentage of Ki67-expressing tumor cells was scored into four categories and this quantification was graphically displayed (category 0 = 0%, category 1 ≥ 0–4,9%, category 2 = 5–9,9% and category 3 = 10% or more).
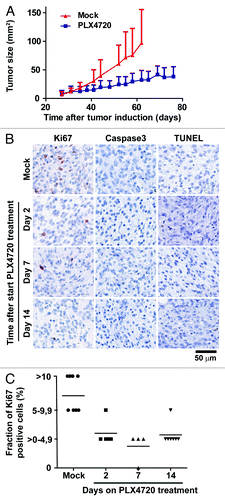
Subsequently, we investigated whether the strong decrease in tumor outgrowth upon PLX4720 treatment resulted from an increased tumor cell death or a proliferation arrest of the tumor cells by analyzing melanomas from tumor-bearing C57BL/6J Tyr::CreERT2PTENF−/−BRAFF-V600E/+ mice at distinct time points after start of PLX4720 treatment (day 0, 2, 7 and 14). Immunohistochemical analysis showed that the expression of the proliferation marker Ki67 was strongly decreased in tumors already after two days of PLX4720 treatment. This decreased proliferation of tumor cells was sustained throughout the period of analysis, as shown in representative stainings (). Per time point, multiple independent tumors were histologically analyzed and blinded quantification of Ki67 expression showed a sustained decrease of proliferating cells in the tumor from 5–15% at baseline to 0–5% at day 2, 7 and 14 of treatment (). This finding demonstrates the essential role of BRAFV600E in driving tumor cell proliferation in our model and is consistent with the strong decrease of tumor outgrowth in mice upon PLX4720 treatment.
The absence of tumor regression in melanoma-bearing mice suggested that extensive tumor cell death (necrosis or apoptosis) was not likely to be induced by PLX4720 treatment. Indeed, analysis of PLX4720 or mock treated tumors by immunohistochemistry for active caspase 3 and by a TUNEL assay did not demonstrate increased apoptosis (or necrosis) in treated melanomas ().
PLX4720 treatment leads to a decreased frequency of immune cells in BRAFV600E/PTEN−/− melanomas
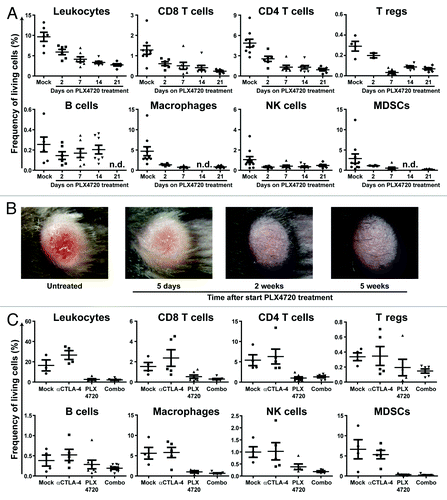
It has recently been shown that the presence of immune cells in the tumor microenvironment prior to anti-CTLA-4 mAb treatment is predictive for a clinical response.Citation13 To investigate the effect of targeting BRAFV600E on tumor-resident immune cells, we determined by flow cytometry the relative frequencies of various immune cell populations in size-matched tumors from mice that were mock-treated or treated with PLX4720 for 2, 7, 14 or 21 d.
Surprisingly, BRAFV600E inhibitor treatment led to a fast, substantial and sustained decrease of CD45+ leukocytes, from 9.7% (± 2.8) of all living cells in the tumor at baseline to 5.9% (± 1.6) and 2.7% (± 0.7) at respectively 2 and 21 d of treatment (resp. p = 0.0163 and p = 0.0001) (). In detail, the frequency of CD8+ and CD4+ T cells in the melanomas dropped during 21 d of treatment respectively from 1.3 (± 0.6) to 0.2% (± 0.1) and 4.9 (± 1.7) to 0.9% (± 0.5) (resp. p = 0.0017 and p = 0.0001). In general, a substantial part of the CD4+ T cells in the tumor consisted of regulatory T cells (Tregs) and in line with the other T-cell populations the frequency of this cell population decreased from 0.3 (± 0.1) to 0.07% (± 0.03) during treatment (p = 0.0006). The proportion of living cells in the tumor that were B220+CD19+ B-lymphocytes was only 0.25% (± 0.2) at baseline, but this frequency was not affected by the PLX4720 treatment. In addition, we observed a slight treatment-induced decrease in the frequency of NK-cells (1.0 (± 0.9) to 0.5% (± 0.2)), myeloid derived suppressor cells (MDSCs) [2.9 (± 3.6) to 0.2% (± 0.08)] and macrophages [4.7 (± 3.5) to 0.8% (± 0.3)]. In line with the observation that immune cell frequencies were reduced at the tumor site, tumors sustainably lost their erythematous and inflamed appearance upon PLX4720 treatment in the majority of cases as shown by pictures of a representative tumor at baseline and after 5, 14 and 35 d of PLX4720 treatment ().
Figure 2. PLX4720 treatment leads to a decreased frequency of immune cells in BRAFV600E/PTEN−/− melanomas and this cannot be restored by CTLA-4 blockade. (A) Tumor-bearing Tyr::CreERT2PTENF−/−BRAFF-V600E/+ mice were mock or PLX4720 treated for 2, 7, 14 or 21 d. Tumors were removed directly following euthanasia and single cell suspensions were analyzed by use of flow cytometry. Dead cells were removed from all analyses (except for intracellular stainings) by discarding propidium iodide positive cells. Leukocytes were defined as being CD45+ cells. CD4+, CD8+ and regulatory T cells were respectively defined as the CD4+CD8-, CD4-CD8+ or the CD4+CD25+FoxP3+ population. B cells were characterized by their expression of B220 and CD19 while NK-cells were distinguished by the expression of NK1.1 in the absence of CD4 or CD8 expression. Myeoloid derived suppressor cells (MDSCs) and macrophages were respectively defined as the CD11b+GR1+ or CD11b+F4/80+ cell population. The shown values represent the frequency of the assessed cell population as a percentage of all living cells in the single cell suspension of the tumor for individually analyzed mice. As the T reg population was distinguished by use of an intracellular stain the values in this plot are shown as a percentage of all cells in the tumor suspension. (B) A tumor from a Tyr::CreERT2PTENF−/−BRAFF-V600E/+ mouse was placed on PLX4720 treatment and tumor appearance was followed photographically over time. Depicted is the tumor phenotype at start, day 5, day 14 and day 35 of PLX4720 treatment (C) Tyr::CreERT2PTENF−/−BRAFF-V600E/+ mice received a mock-treatment, PLX4720-treatment, anti-CTLA-4 mAb treatment (twice weekly for 6 weeks) or the combination of PLX4720 and CTLA-4 blockade treatment. The frequency of immune cells as a percentage of living cells in the tumor was assessed by flow cytometry as described for panel A.
To investigate whether the decrease in frequency of tumor-resident T cells upon PLX4720 treatment was independent of the size of the tumor, we compared melanomas, which were on average 25 mm2, referred to as ‘small’ tumors, to larger tumors which were at least 60 mm2 in size when they were placed on PLX4720 treatment. In both cases, mice had been treated with PLX4720 or mock treated for at least 21 d. We found that for both small and large tumors PLX4720-treatment resulted in a substantial decrease in the frequency of tumor-resident T cells (Fig. S1).
Selective BRAF inhibitor-mediated decrease in frequency of tumor-resident T cells cannot be restored by CTLA-4 blockade
To study treatment synergy between BRAFV600E inhibition and CTLA-4 blockade, we investigated whether repetitive anti-CTLA-4 mAb injections could sustainably restore the decreased frequency of tumor-resident immune cells induced by PLX4720 treatment. We compared the frequency of immune cells, as the proportion of living cells in the tumor, in melanomas that were treated with PLX4720, anti-CTLA-4 mAb injections or a combination of these treatments (). Flow cytometric analyses showed that CTLA-4 blockade led to an increase in the frequency of CD45+ leukocytes compared with mock treated animals [from 16.4 (± 9.3) to 26.6% (± 8.7)]. In detail, tumor-resident T cells slightly increased from 1.5 (± 0.8) to 2.4% (± 1.8) for CD8+ T cells and 5.4 (± 2.7) to 6.3% (± 4.1) for CD4+ T cells, while the frequency of regulatory T cells remained unchanged [0.34 (± 0.1) to 0.35% (± 0.28)]. Furthermore, we observed that the addition of anti-CTLA-4 mAb treatment to PLX4720 treatment could not increase the reduced numbers of T cells in PLX4720 treated tumors [from 0.5 (± 0.4) to 0.3% (± 0.2) and 1.0 (± 0.8) to 1.3% (± 0.6) for respectively CD8+ and CD4+ T cells].
Reduced tumor immune cell frequencies upon selective BRAF inhibition correlates to the presence of the BRAFV600E mutation in tumor cells
The reduced frequencies of tumor-resident immune cells upon PLX4720 treatment could be a consequence of the inhibition of BRAFV600E in the melanoma cells or could result from an off-target effect of PLX4720 leading to loss of immune cells at the tumor site and possibly other organs. To investigate such a potential toxic effect of PLX4720 on T cells, we analyzed the frequencies of CD3+, CD4+ and CD8+ T cells in tumors, tumor draining lymph nodes (dLN), contralateral lymph nodes (cLN) and spleens from PLX4720 or mock-treated melanoma-bearing mice. While, once again, we found markedly reduced frequencies within the tumors upon PLX4720 treatment (data not shown), T-cell frequencies were not altered to such an extent in the lymphatic organs (). However, we did find a tendency toward decreased CD8+ T-cell proportions when exposing the mice to PLX4720. Overall, our findings are in line with in vitro data showing that PLX4720 does not hamper T-cell functioning.Citation29
Figure 3. Reduced tumor immune cell frequencies upon selective BRAF inhibition depends on the presence of the BRAFV600E mutation in tumor cells. (A) Tyr::CreERT2PTENF−/−BRAFF-V600E/+ mice received for at least 21 d a mock-treatment, PLX4720-treatment, anti-CTLA-4 mAb treatment (twice weekly for 6 weeks) or the combination of PLX4720 and CTLA-4 blockade treatment. Then the draining lymph node (dLN), contralateral lymph node (cLN) and spleen were removed directly following euthanasia and single cell suspensions were analyzed by use of flow cytometry as described for . (B) Nine week old male C57BL/6J mice were subcutaneously inoculated with 1x106 B16F10 cells in the shaven right flank. Four days after tumor inoculation mice were placed on PLX4720 or mock treatment and were subsequently sacrificed when tumors were at least 100 mm2 (500 mg) which was generally 10 to 21 d after tumor inoculation. The frequency of immune cells as a percentage of living cells in the tumor was assessed by flow cytometry as described for .
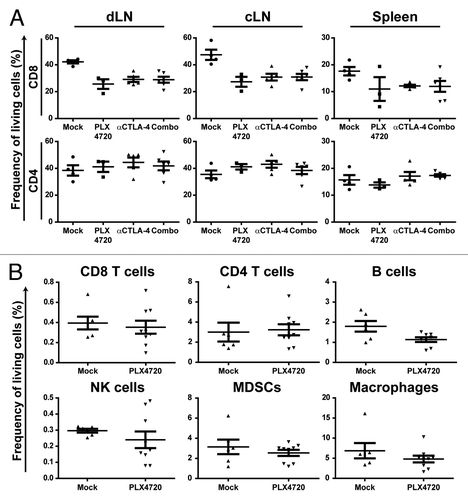
To evaluate whether the decreased frequency of tumor-resident immune cells upon PLX4720 treatment depends on the inhibition of BRAFV600E in the melanoma cells, we compared immune infiltrates in mock or PLX4720 treated BRAF wild-type tumors. As B16F10 melanoma cells express wild-type BRAF, these tumor cells cannot be affected by a blockade of BRAFV600E.Citation37 Importantly, the comparison of size-matched B16F10 tumors that were either PLX4720 or mock treated demonstrated no differences in the frequency of T cells, B cells, NK cells, MDSCs or macrophages (). These data imply that the effect of PLX4720 on tumor immune cell frequencies is not resulting from a direct toxic effect on immune cells and correlates to the presence of BRAFV600E in tumor cells.
Interestingly, we observed that B16F10 tumors that were treated with PLX4720 displayed a much higher growth rate than mock-treated tumors. In detail, 10 d after inoculation mock-treated tumors weighted 0.16 (± 0.07) grams while PLX4720 tumors weighted 0.30 (± 0.11) grams (p = 0.0032) (Fig. S2). This observation is in line with reported studies showing that BRAFV600E inhibition can lead to paradoxical MAP Kinase pathway activation, and subsequent proliferation, in BRAF wild-type tumor cells, suggesting that vemurafenib treatment may facilitate growth of BRAF wild-type tumors.Citation38,Citation39
Addition of anti-CTLA-4 mAb treatment to PLX4720 treatment does not further improve tumor growth control
In this study we observed that PLX4720 treatment of BRAFV600E/PTEN−/− melanomas did not lead to the induction of tumor cell death, but resulted in a decreased frequency of immune cells in the melanomas that could not be restored by repetitive anti-CTLA-4 mAb injections. These findings raised the question whether, despite the effect of PLX4720 treatment on tumor-resident immune cell frequencies, CTLA-4 blockade could still synergize with PLX4720 treatment in terms of tumor growth control. To address this question we compared the effect of CTLA-4 blockade combined with a tumor-vaccine on outgrowth of the B16F10 tumor to the effect of CTLA-4 blockade combined with PLX4720 on tumor outgrowth in the inducible melanoma mice.
To determine the effect of CTLA-4 blockade on B16F10 tumors, C57BL/6J mice were inoculated with 1x104 B16F10 cells in the flank. Then, at day 0, 3 and 6 mice were subcutaneously vaccinated with irradiated, GM-CSF expressing, B16F10 cells (Gvax-vaccination) and indicated cohorts also received intraperitoneal injections with anti-CTLA-4 mAb clone 9H10 (hamster-derived) or clone 9D9 (mouse-derived). Kaplan Meijer analyses of the B16F10 tumor-bearing mice demonstrated that Gvax-vaccination extended the survival duration of the C57BL/6J mice and that additional treatment with anti-CTLA-4 mAb clone 9D9 or 9H10 further improved their overall survival (p < 0.001) (). In accordance to previous data, these findings demonstrate that anti-CTLA-4 mAb treatment synergizes with the tumor-antigen rich Gvax-vaccination.Citation2
Figure 4. Addition of anti-CTLA-4 mAb treatment to PLX4720 treatment does not further improve tumor growth control, while it does when combined with Gvax-vaccination. (A) Nine week old female C57BL/6J mice were subcutaneously inoculated with 1x104 B16F10 cells in the shaven right flank at day 0. At day 0, 3 and 6 indicated mice received a subcutaneous injection in the contralateral flank with 150 Gray irradiated 1x106 GMCSF-expressing B16F10 cells (Gvax group, green line, n = 14) combined with intraperitoneal injections of 200 μg hamster-derived anti-CTLA-4 mAb clone 9H10 (blue line, n = 15) or 100 μg mouse-derived anti-CTLA-4 mAb clone 9D9 (purple line, n = 15). Tumor growth was followed over time by caliper measures and mice were sacrificed when tumors exceeded a 150 mm2 size or ulcerated. The survival of the different treatment groups was compared with that of mock treated mice (red line, n = 16) and depicted in a survival graph. (B) 4–10 week old Tyr::CreERT2PTENF−/−BRAFF-V600E/+ mice were induced on the flank and 31 d later, when average tumor size was 10 mm2, tumor-bearing mice were placed on mock treatment (red line, n = 11), PLX4720 treatment (dark blue line, n = 16), anti-CTLA-4 mAb clone 9H10 treatment (dark green line, n = 11) or PLX4720-treatment combined with either anti-CTLA-4 mAb clone 9H10 (light green line, n = 12) or clone 9D9 (light blue line, n = 8). Tumor growth was followed in two dimensions and graphed over time. (C) Survival of the mice described in was graphed over time.
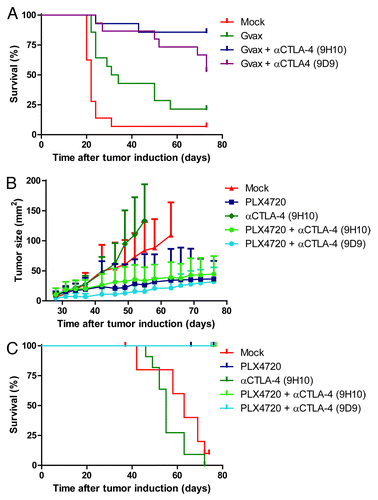
In parallel we assessed the effect of combined anti-CTLA-4 mAb and PLX4720 treatment in tumor-bearing C57BL/6J Tyr::CreERT2PTENF−/−BRAFF-V600E/+ mice. Mice were treated with PLX4720, injections of anti-CTLA-4 mAb clone 9H10 or clone 9D9 or the combination of PLX4720 with either of these antibodies. Subsequently, tumor outgrowth was followed over time and mice were sacrificed when the tumor ulcerated or exceeded a 150 mm2 size. When analyzing tumor growth in the melanoma model mice we observed that PLX4720 treatment led, as expected, to a strong decrease in tumor outgrowth which extended the survival of the mice (, dark blue line). However, addition of anti-CTLA-4 mAb treatment (either clone 9H10 or 9D9) could not further reduce tumor growth (bright green and bright blue lines). Therefore we conclude that in contrast to the setting in which CTLA-4 blockade was combined with Gvax-vaccination, the combination of PLX4720 and anti-CTLA-4 mAb treatment did not result in any detectable treatment synergy.
Discussion
Potential mechanisms by which PLX4720 treatment leads to reduced frequencies of immune cells in BRAFV600E/PTEN−/− melanomas
Following the three positive phase III trials for ipilimumab and vemurafenib, the clinical evaluation of the combination of these two drugs forms a logical next step.Citation6,Citation7,Citation15 The C57BL6/J Tyr::CreERT2PTENF−/−BRAFF-V600E/+ inducible melanoma model is the first transgenic mouse model in which this specific treatment combination can be tested in vivo. Using this mouse model, we observed that PLX4720 treatment led to a decreased frequency of tumor-resident T cells, NK cells, MDSCs and macrophages, which could not be restored by the addition of anti-CTLA-4 mAb treatment. Furthermore, anti-CTLA-4 mAb treatment did not further increase the anti-tumor effect of PLX4720 while CTLA-4 blockade did improve the effect of tumor-vaccination in B16F10 inoculated mice.
Although we observed a decreased frequency of tumor-resident immune cells in BRAFV600E/PTEN−/− melanomas, the frequency of the immune cell populations was comparable in mock and PLX4720 treated B16F10-tumors that are wild-type for BRAF. Therefore, we conclude that the decreased frequency of immune cells in tumors upon PLX4720 treatment did not result from an off-target effect of PLX4720 on immune cells and correlated with the inhibition of BRAFV600E in melanoma cells. BRAFV600E inhibition in tumor cells could potentially affect tumor-resident immune cell frequencies in two ways. First, blockade of BRAFV600E signaling might interfere with immune cell trafficking by directly reducing or altering the chemokine expression of the tumor cells. Hong et al. recently described an alteration of chemokine expression upon treatment with specific types of chemotherapy, but in that study the changed chemokine pattern resulted in the attraction of immune cells.Citation22 Second, PLX4720 treatment of BRAFV600E/PTEN−/− melanomas leads to a strong decrease of tumor cell proliferation in the absence of cell death induction, as was demonstrated in this study. This decreased proliferation of tumor cells is likely to result in decreased stromal changes and, supported by the lack of cell death, reduced expression of inflammatory molecules in the tumor microenvironment, which may thereby lead to lower immune cell frequencies in the tumor.
Absence of cell death induction by BRAFV600E inhibitor treatment
The absence of cell death induction upon BRAFV600E inhibitor treatment may not only play a role in the reduced frequency of tumor-resident immune cells, but, as a result, is likely to also contribute to the lack of treatment synergy when PLX4720 is combined with anti-CTLA-4 mAb treatment. It has been shown in different mouse models that CTLA-4 blockade is most effective in decreasing tumor outgrowth in settings in which an antigen rich environment is provided, for example by vaccination or the induction of tumor cell death.Citation2,Citation25,Citation26 As the blockade of BRAFV600E did not lead to tumor cell apoptosis or necrosis, such an antigen rich environment was not likely to be present in the BRAFV600E/PTEN−/− melanomas. This potentially contributed to the lack of the synergystic effect from anti-CTLA-4 mAb injections. In support of this notion, we did observe treatment synergy when combining CTLA-4 blockade with Gvax-vaccination in the B16F10 tumor model.
Possibly the additional PTEN-deficiency of the tumor cells plays an important role in inhibiting cell death induction upon PLX4720 treatment. In line with this idea, Paraiso et al. recently demonstrated that human BRAFV600E/PTEN-deficient melanoma cell lines showed limited cell death after PLX4720 treatment.Citation40 Moreover, Xing et al. recently published that concurrent mutational inactivation of PTEN is a mechanism for loss of BRAF dependence in melanomas harbouring the BRAFV600E mutation, indicating that this mutational profile will be less sensitive for BRAFV600E inhibitor treatment.Citation41
Human studies concerning BRAFV600E inhibitor treatment and tumor-resident immune cells
Although data concerning the effect of BRAFV600E inhibitor treatment on immune cell frequency in human melanoma is limited, Wilmott and Long et al. recently studied T-cell numbers in a small set of metastasized melanomas prior to BRAF inhibitor treatment, 3–15 d after start of treatment and in tumors which progressed on treatment.Citation28 In contrast to the reduced frequency of tumor-resident immune cells in the BRAFV600E/PTEN−/− murine melanomas, the study demonstrated increased T-cell frequencies in tumors after one week of treatment. These numbers dropped again to baseline-levels when tumors progressed. Unfortunately, the melanomas in this study were only profiled for their BRAF mutations and therefore it is unknown which proportion of these patients had a PTEN-deficient tumor. However, those melanomas which showed only a very low level of necrosis upon BRAF inhibitor treatment, resembling the growth control pattern of the murine BRAFV600E/PTEN−/− melanomas, had a far less pronounced increase of CD8+ and CD4+ T cells upon treatment. This suggests that, in accordance to our data, the induction of cell death plays an important role in the presence of immune cells in the tumor. It would be of interest to analyze, in larger patient cohorts, whether patients with BRAFV600E/PTEN-deficient melanomas indeed have decreased frequencies of T cells in their tumors upon selective BRAF inhibition as this could then be used a predictive biomarker for combination treatment.
Considering the rapid development of different treatments for metastasized melanoma, there has been great interest lately in the combination of targeted therapies with immune active compounds. For instance, the first Phase I/II study in which ipilimumab and vemurafenib are synchronously combined has recently started. A number of studies support the idea that chemotherapy or targeted therapy can stimulate anti-tumor immune responses by various mechanisms.Citation19-Citation24 However, our study demonstrates that BRAFV600E inhibitor treatment could be different and can lead to a reduced frequency of tumor-resident immune cells. Therefore, the use of vemurafenib could potentially hamper an anti-tumor immune response for some patients and in these cases the combination with anti-CTLA-4 mAb treatments may be ineffective. On a more general note, the data described here indicate that the potential effect of targeted therapy on the tumor microenvironment should be taken into consideration in the design of targeted and immunotherapy combination treatments.
Materials and Methods
Ethical statement
All described animal experiments were ethically approved by the Animal Experimentation Committee of the Netherlands Cancer Institute. Mice were treated in accordance with the Dutch law on animal experimentation.
Immunohistochemistry
The immunohistochemical staining of the formalin-fixed paraffin-embedded tissue samples was performed as described previously.Citation42 To stain for Ki67 and presence of active caspase-3 we used anti-Ki67 (Monosan, PSX1028) or anti-caspase-3 (Cell Signaling Technology, 9661L) as primary antibodies. Romulin AEC (Biocare Medical, RAEC810L) was used according to the manufacturers’ instructions to visualize positive staining, followed by a hematoxylin counterstain. The percentage of positive tumor cells was scored blindly using a light microscope. Microscopic images were acquired in the Zeiss Axiovision software by using the Zeiss Axiovert S 100 microscope and Zeiss color Axiocam.
Melanoma induction and growth analysis
Tumors were induced on the skin of the melanoma model mice as previously described.Citation31,Citation32 For three consecutive days, 2 μl of 5 mM 4-hydroxytamoxifen (4-HT) (Sigma-Aldrich, H6278) in pure DMSO (Sigma-Aldrich, 276855) was topically applied for 5 min to the shaven right flank of 4–10 week old mice. The tumor outgrowth was followed twice weekly by digital photographing of the tumor including a size reference. Tumor size was then analyzed in two dimensions using the ImageJ software (developed by National Institutes of Health, USA).
B16 tumor inoculation
B16F10 cells were cultured in IMDM (Invitrogen, 21980) medium supplemented with 10% heat-inactivated Fetal Bovine Serum (Sigma-Aldrich), 100 U/ml Penicillin and 100 μg/ml Streptomycin (Roche, 11074440001). After trypsinization, cells were washed twice in Hanks Balanced Salt Solution (HBSS) (Invitrogen, 14175). Then, nine week old male or female C57BL/6J mice were subcutaneously injected in the shaven right flank with the indicated amount (1x106 or 1x104) of B16F10 cells in 100 μl HBSS. Tumor growth was followed over time by caliper measurements and mice were sacrificed when tumors exceeded a 150 mm2 size or ulcerated.
Flow cytometric analysis of immune populations in tumors
Tumors, lymph nodes or spleens were removed from the animals directly after euthanasia. Subsequently, single cell suspensions from the collected tumor tissues were obtained by continuous slicing of the tumor followed by pressing the material through a 70 µm filter (BD Biosciences, 352350). The obtained single cell suspension was then stained with fluorochrome labeled antibodies (BD Biosciences). The following fluorochrome labeled antibodies were used to stain various cell populations: anti-CD3 clone 17A2, anti-CD4 clone GK1.5, anti-CD8 clone 53–6.7, anti-NK1.1 clone PK136, anti-CD11b clone M1/70, anti-B220 clone RA3–6B2, anti-CD19 clone 1D3, anti-Gr-1 clone RB6–8C5, anti-F4/80 clone BM8, anti-FoxP3 clone FJK-165, anti-CD25 clone PC61 and anti-CD45 clone 104. Intracellular staining of the FoxP3 molecule was performed according to the manufacturers’ protocol (eBioscience, 77–5775–40). Dead cells were excluded from the analysis by addition of propidium iodide (eBioscience, 00–6990–50) during the sample acquisition. Samples were acquired on a FACSCalibur (BD Biosciences) and analyzed using the FlowJo software (Tree Star Inc.).
PLX4720 treatment of tumor-bearing mice
For PLX4720 treatment mice were switched to either a chow diet containing 417 mg/kg PLX4720 (treatment arm) or the control chow containing no compound (control arm) which were both provided by Plexxikon. On average the food dosing is similar to a daily 50 mg/kg dosing by oral gavage.
Anti-CTLA-4 mAb treatment
Mice were injected intraperitoneal with the anti-CTLA-4 mAbs dissolved in HBSS. Dosing per injection was 200 μg for the hamster-derived anti-CTLA-4 mAb clone 9H10 (BioXcell, BE0131) and 100 μg for the mouse-derived anti-CTLA-4 mAb clone 9D9 (BioXcell, BE0164). When anti-CTLA-4 mAb treatment was combined with Gvax vaccination, mice received injections at day 0, 3 and 6 after B16F10 tumor inoculation. When treating Tyr::CreERT2PTENF−/−BRAFF-V600E/+ mice, mice received antibody injections twice a week for 6 weeks.
Gvax treatment
B16F10 cells genetically modified to express GM-CSF (kindly provided by T. van Hall, LUMC, the Netherlands) were cultured in IMDM medium supplemented with 10% heat-inactivated Fetal Bovine Serum (Sigma-Aldrich), 100 U/ml Penicillin and 100 μg/ml Streptomycin (Roche). At day 0, 3 and 6 after B16F10 tumor inoculation mice received a subcutaneous injection in the contralateral flank from the tumor containing 150 Gray irradiated 1x106 GMCSF-expressing B16F10 cells.
| Abbreviations: | ||
| mAb | = | mononclonal antibody |
| MDSCs | = | myeloid derived suppressor cells |
| dLN | = | tumor draining lymph node |
| cLN | = | contralateral lymph node |
Additional material
Download Zip (210.4 KB)Acknowledgments
This work was in part funded by the KWF Dutch Cancer Foundation (2008–3988), the Melanoma Research Alliance (197373) and by a collaborative grant from MedImmune, Cambridge.
We thank Dr Andrew Kaiser for his participation in scientific discussions. Furthermore, we are grateful to Dr Brian West (Plexxikon Inc.) for supplying us with the PLX4720 and control chow.
Disclosure of Potential Conflicts of Interest
The authors M.M. and R.S. are both employees of MedImmune, which recently acquired a license for the anti-CTLA-4 mAb tremelimumab. The other authors disclosed no other potential conflicts of interest.
Supplemental Material
Supplemental materials can be found at: http://www.landesbioscience.com/journals/oncoimmunology/article/20226/
References
- Blank CU, Hooijkaas AI, Haanen JB, Schumacher TN. Combination of targeted therapy and immunotherapy in melanoma. Cancer Immunol Immunother 2011; 60:1359 - 71; http://dx.doi.org/10.1007/s00262-011-1079-2; PMID: 21847631
- Curran MA, Montalvo W, Yagita H, Allison JP. PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc Natl Acad Sci USA 2010; 107:4275 - 80; http://dx.doi.org/10.1073/pnas.0915174107; PMID: 20160101
- Curran MA, Kim M, Montalvo W, Al-Shamkhani A, Allison JP. Combination CTLA-4 blockade and 4-1BB activation enhances tumor rejection by increasing T-cell infiltration, proliferation, and cytokine production. PLoS ONE 2011; 6:e19499; http://dx.doi.org/10.1371/journal.pone.0019499; PMID: 21559358
- Sutmuller RP, van Duivenvoorde LM, van Elsas A, Schumacher TN, Wildenberg ME, Allison JP, et al. Synergism of cytotoxic T lymphocyte-associated antigen 4 blockade and depletion of CD25(+) regulatory T cells in antitumor therapy reveals alternative pathways for suppression of autoreactive cytotoxic T lymphocyte responses. J Exp Med 2001; 194:823 - 32; http://dx.doi.org/10.1084/jem.194.6.823; PMID: 11560997
- van Elsas A, Sutmuller RP, Hurwitz AA, Ziskin J, Villasenor J, Medema JP, et al. Elucidating the autoimmune and antitumor effector mechanisms of a treatment based on cytotoxic T lymphocyte antigen-4 blockade in combination with a B16 melanoma vaccine: comparison of prophylaxis and therapy. J Exp Med 2001; 194:481 - 9; http://dx.doi.org/10.1084/jem.194.4.481; PMID: 11514604
- Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 2010; 363:711 - 23; http://dx.doi.org/10.1056/NEJMoa1003466; PMID: 20525992
- Robert C, Thomas L, Bondarenko I, O’Day S, M D JW, Garbe C, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med 2011; 364:2517 - 26; http://dx.doi.org/10.1056/NEJMoa1104621; PMID: 21639810
- Di Giacomo AM, Danielli R, Calabrò L, Bertocci E, Nannicini C, Giannarelli D, et al. Ipilimumab experience in heavily pretreated patients with melanoma in an expanded access program at the University Hospital of Siena (Italy). Cancer Immunol Immunother 2011; 60:467 - 77; http://dx.doi.org/10.1007/s00262-010-0958-2; PMID: 21170646
- O’Day SJ, Maio M, Chiarion-Sileni V, Gajewski TF, Pehamberger H, Bondarenko IN, et al. Efficacy and safety of ipilimumab monotherapy in patients with pretreated advanced melanoma: a multicenter single-arm phase II study. Ann Oncol 2010; 21:1712 - 7; http://dx.doi.org/10.1093/annonc/mdq013; PMID: 20147741
- Ribas A, Chmielowski B, Glaspy JA. Do we need a different set of response assessment criteria for tumor immunotherapy?. Clin Cancer Res 2009; 15:7116 - 8; http://dx.doi.org/10.1158/1078-0432.CCR-09-2376; PMID: 19934296
- Wolchok JD, Hoos A, O’Day S, Weber JS, Hamid O, Lebbé C, et al. Guidelines for the evaluation of immune therapy activity in solid tumors: immune-related response criteria. Clin Cancer Res 2009; 15:7412 - 20; http://dx.doi.org/10.1158/1078-0432.CCR-09-1624; PMID: 19934295
- Prieto PA, Yang JC, Sherry RM, Hughes MS, Kammula US, White DE, et al. CTLA-4 Blockade with Ipilimumab: Long-term Follow-up of 177 Patients with Metastatic Melanoma. Clin Cancer Res 2012; 18:2039 - 47; http://dx.doi.org/10.1158/1078-0432.CCR-11-1823; PMID: 22271879
- Ji RR, Chasalow SD, Wang L, Hamid O, Schmidt H, Cogswell J, et al. An immune-active tumor microenvironment favors clinical response to ipilimumab. Cancer Immunol Immunother 2011; http://dx.doi.org/10.1007/s00262-011-1172-6; PMID: 22146893
- Bollag G, Hirth P, Tsai J, Zhang J, Ibrahim PN, Cho H, et al. Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma. Nature 2010; 467:596 - 9; http://dx.doi.org/10.1038/nature09454; PMID: 20823850
- Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, et al, BRIM-3 Study Group. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med 2011; 364:2507 - 16; http://dx.doi.org/10.1056/NEJMoa1103782; PMID: 21639808
- Flaherty KT, Puzanov I, Kim KB, Ribas A, McArthur GA, Sosman JA, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med 2010; 363:809 - 19; http://dx.doi.org/10.1056/NEJMoa1002011; PMID: 20818844
- Nathanson KL. Tumor genetic analyses of patients with metastatic melanoma treated with the BRAF inhibitor GSK2118436 (GSK436). A.Martin, R. Letrero K. P. D'Andrea S. O'Day J. R. Infante G. S. Falchook M. Millward C. M. Curtis B. Ma R. C. Gagnon P. F. Lebowitz G. V. Long R. F. Kefford. Asco 2011. 2011. Ref Type: Abstract.
- Natarajan N, Telang S, Miller D, Chesney J. Novel immunotherapeutic agents and small molecule antagonists of signalling kinases for the treatment of metastatic melanoma. Drugs 2011; 71:1233 - 50; http://dx.doi.org/10.2165/11591380-000000000-00000; PMID: 21770473
- Denardo DG, Brennan DJ, Rexhepaj E, Ruffell B, Shiao SL, Madden SF, et al. Leukocyte Complexity Predicts Breast Cancer Survival and Functionally Regulates Response to Chemotherapy. Cancer Discov 2011; 1:54 - 67; http://dx.doi.org/10.1158/2159-8274.CD-10-0028; PMID: 22039576
- Green DR, Ferguson T, Zitvogel L, Kroemer G. Immunogenic and tolerogenic cell death. Nat Rev Immunol 2009; 9:353 - 63; http://dx.doi.org/10.1038/nri2545; PMID: 19365408
- Hannani D, Sistigu A, Kepp O, Galluzzi L, Kroemer G, Zitvogel L. Prerequisites for the antitumor vaccine-like effect of chemotherapy and radiotherapy. Cancer J 2011; 17:351 - 8; http://dx.doi.org/10.1097/PPO.0b013e3182325d4d; PMID: 21952286
- Hong M, Puaux AL, Huang C, Loumagne L, Tow C, Mackay C, et al. Chemotherapy induces intratumoral expression of chemokines in cutaneous melanoma, favoring T-cell infiltration and tumor control. Cancer Res 2011; 71:6997 - 7009; http://dx.doi.org/10.1158/0008-5472.CAN-11-1466; PMID: 21948969
- Kepp O, Galluzzi L, Martins I, Schlemmer F, Adjemian S, Michaud M, et al. Molecular determinants of immunogenic cell death elicited by anticancer chemotherapy. Cancer Metastasis Rev 2011; 30:61 - 9; http://dx.doi.org/10.1007/s10555-011-9273-4; PMID: 21249425
- Rakhra K, Bachireddy P, Zabuawala T, Zeiser R, Xu L, Kopelman A, et al. CD4(+) T cells contribute to the remodeling of the microenvironment required for sustained tumor regression upon oncogene inactivation. Cancer Cell 2010; 18:485 - 98; http://dx.doi.org/10.1016/j.ccr.2010.10.002; PMID: 21035406
- den Brok MH, Sutmuller RP, Nierkens S, Bennink EJ, Frielink C, Toonen LW, et al. Efficient loading of dendritic cells following cryo and radiofrequency ablation in combination with immune modulation induces anti-tumour immunity. Br J Cancer 2006; 95:896 - 905; http://dx.doi.org/10.1038/sj.bjc.6603341; PMID: 16953240
- Dewan MZ, Galloway AE, Kawashima N, Dewyngaert JK, Babb JS, Formenti SC, et al. Fractionated but not single-dose radiotherapy induces an immune-mediated abscopal effect when combined with anti-CTLA-4 antibody. Clin Cancer Res 2009; 15:5379 - 88; http://dx.doi.org/10.1158/1078-0432.CCR-09-0265; PMID: 19706802
- Lee JT, Li L, Brafford PA, van den Eijnden M, Halloran MB, Sproesser K, et al. PLX4032, a potent inhibitor of the B-Raf V600E oncogene, selectively inhibits V600E-positive melanomas. Pigment Cell Melanoma Res 2010; 23:820 - 7; http://dx.doi.org/10.1111/j.1755-148X.2010.00763.x; PMID: 20973932
- Wilmott JS, Long GV, Howle JR, Haydu LE, Sharma RN, Thompson JF, et al. Selective BRAF Inhibitors Induce Marked T-cell Infiltration into Human Metastatic Melanoma. Clin Cancer Res 2012; 18:1386 - 94; http://dx.doi.org/10.1158/1078-0432.CCR-11-2479; PMID: 22156613
- Boni A, Cogdill AP, Dang P, Udayakumar D, Njauw CN, Sloss CM, et al. Selective BRAFV600E inhibition enhances T-cell recognition of melanoma without affecting lymphocyte function. Cancer Res 2010; 70:5213 - 9; http://dx.doi.org/10.1158/0008-5472.CAN-10-0118; PMID: 20551059
- Hong DS, Vence LM, Falchook GS, Radvanyi LG, Liu C, Goodman VL, et al. BRAF(V600) INHIBITOR GSK2118436 TARGETED INHIBITION OF MUTANT BRAF IN CANCER PATIENTS DOES NOT IMPAIR OVERALL IMMUNE COMPETENCY. Clin Cancer Res 2012; http://dx.doi.org/10.1158/1078-0432.CCR-11-2515; PMID: 22355009
- Dankort D, Curley DP, Cartlidge RA, Nelson B, Karnezis AN, Damsky WE Jr., et al. Braf(V600E) cooperates with Pten loss to induce metastatic melanoma. Nat Genet 2009; 41:544 - 52; http://dx.doi.org/10.1038/ng.356; PMID: 19282848
- Hooijkaas AI, Gadiot J, Van der Valk M, Mooi WJ, Blank C. Targeting BRAFV600E in an inducible murine model of melanoma. 2012. Work in Revision.
- Bauer J, Büttner P, Murali R, Okamoto I, Kolaitis NA, Landi MT, et al. BRAF mutations in cutaneous melanoma are independently associated with age, anatomic site of the primary tumor, and the degree of solar elastosis at the primary tumor site. Pigment Cell Melanoma Res 2011; 24:345 - 51; http://dx.doi.org/10.1111/j.1755-148X.2011.00837.x; PMID: 21324100
- Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, et al. Mutations of the BRAF gene in human cancer. Nature 2002; 417:949 - 54; http://dx.doi.org/10.1038/nature00766; PMID: 12068308
- Ibrahim N, Haluska FG. Molecular pathogenesis of cutaneous melanocytic neoplasms. Annu Rev Pathol 2009; 4:551 - 79; http://dx.doi.org/10.1146/annurev.pathol.3.121806.151541; PMID: 19400696
- Smalley KS. Understanding melanoma signaling networks as the basis for molecular targeted therapy. J Invest Dermatol 2010; 130:28 - 37; http://dx.doi.org/10.1038/jid.2009.177; PMID: 19571822
- Castle JC, Kreiter S, Diekmann J, Löwer M, van de Roemer N, de Graaf J, et al. Exploiting the mutanome for tumor vaccination. Cancer Res 2012; 72:1081 - 91; http://dx.doi.org/10.1158/0008-5472.CAN-11-3722; PMID: 22237626
- Cichowski K, Jänne PA. Drug discovery: inhibitors that activate. Nature 2010; 464:358 - 9; http://dx.doi.org/10.1038/464358a; PMID: 20237552
- Poulikakos PI, Zhang C, Bollag G, Shokat KM, Rosen N. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature 2010; 464:427 - 30; http://dx.doi.org/10.1038/nature08902; PMID: 20179705
- Paraiso KH, Xiang Y, Rebecca VW, Abel EV, Chen YA, Munko AC, et al. PTEN loss confers BRAF inhibitor resistance to melanoma cells through the suppression of BIM expression. Cancer Res 2011; 71:2750 - 60; http://dx.doi.org/10.1158/0008-5472.CAN-10-2954; PMID: 21317224
- Xing F, Persaud Y, Pratilas CA, Taylor BS, Janakiraman M, She QB, et al. Concurrent loss of the PTEN and RB1 tumor suppressors attenuates RAF dependence in melanomas harboring (V600E)BRAF. Oncogene 2012; 31:446 - 57; http://dx.doi.org/10.1038/onc.2011.250; PMID: 21725359
- Gadiot J, Hooijkaas AI, Kaiser AD, van Tinteren H, van Boven H, Blank C. Overall survival and PD-L1 expression in metastasized malignant melanoma. Cancer 2011; 117:2192 - 201; http://dx.doi.org/10.1002/cncr.25747; PMID: 21523733