Abstract
Although cure rates for acute lymphoblastic leukemia (ALL) have increased, development of resistance to drugs and patient relapse are common. The environment in which the leukemia cells are present during the drug treatment is known to provide significant survival benefit. Here, we have modeled this process by culturing murine Bcr/Abl-positive acute lymphoblastic leukemia cells in the presence of stroma while treating them with a moderate dose of two unrelated drugs, the farnesyltransferase inhibitor lonafarnib and the tyrosine kinase inhibitor nilotinib. This results in an initial large reduction in cell viability of the culture and inhibition of cell proliferation. However, after a number of days, cell death ceases and the culture becomes drug-tolerant, enabling cell division to resume. Using gene expression profiling, we found that the development of drug resistance was accompanied by massive transcriptional upregulation of genes that are associated with general inflammatory responses such as the metalloproteinase MMP9. MMP9 protein levels and enzymatic activity were also increased in ALL cells that had become nilotinib-tolerant. Activation of p38, Akt and Erk correlated with the development of environment-mediated drug resistance (EMDR), and inhibitors of Akt and Erk in combination with nilotinib reduced the ability of the cells to develop resistance. However, inhibition of p38 promoted increased resistance to nilotinib. We conclude that development of EMDR by ALL cells involves changes in numerous intracellular pathways. Development of tolerance to drugs such as nilotinib may therefore be circumvented by simultaneous treatment with other drugs having divergent targets.
Introduction
A major challenge facing patients with acute lymphoblastic leukemia (ALL) is the development of resistance to drug therapy. ALL can be divided into different subcategories. Philadelphia-chromosome (Ph) positive ALL belongs to a poor-prognosis subcategory and is caused by the aberrant fusion of the BCR and ABL genes.Citation1,Citation2 Even specific drugs, such as nilotinib, imatinib and dasatinib that target the Bcr/Abl protein, in general only produce a transient response.Citation3,Citation4
Therapeutic drugs initially are able to effectively reduce the numbers of peripheral blood leukemic cells, but relapse for Ph-positive ALL while on treatment is frequent.Citation5-Citation7 A primary well-known mechanism of drug resistance in this subclass of ALL is the emergence of a clone that has acquired point mutations in the Abl ATP binding pocket, which renders the specific drugs relatively ineffective.Citation8-Citation10
This type of drug resistance typically emerges after weeks or months of treatment and has been named acquired drug resistance because an intrinsic property of the ALL cells has been modified. Meads et al.Citation11 argued that a phase preceding the acquired drug resistance can be distinguished, if cancer cells are supported by the microenvironment in which they reside while being treated with drugs. The type of drug resistance that evolves in this phase is called environment-mediated drug resistance (EMDR) and is mediated both by cell-cell contact and by growth factors and other products in trans. EMDR is likely to be a major source of relapse.
In patients, leukemic lymphoblasts exposed to therapeutic drugs generally are located in the proximity of other cells and extracellular matrix. We have previously developed a transgenic mouse model for the type of ALL caused by the Bcr/Abl oncoproteinCitation12 and are able to culture ALL cells in vitro if stromal support is provided. This co-culture system can also be used to model the emergence of EMDR. By using a moderate dose of drug, we were able, over the course of 2–3 weeks, to generate ALL cells that were tolerant to imatinib, lonafarnib, nilotinib and a CKIIα kinase inhibitor in the presence of stroma, whereas similar doses of drug are able to kill the cells when no stroma is present.Citation13-Citation16 In the current study, we report on the changes that occur in such cultures as the ALL cells develop EMDR.
Results
Emergence of EMDR in pro-B lymphoblastic leukemia cells is accompanied by drug-specific as well as common changes in the expression of multiple genes
The BCR/ABL oncogene encodes a constitutively active tyrosine kinase which activates a variety of downstream signaling molecules, thereby facilitating survival and proliferation of the leukemia cells. We treated the lymphoblastic leukemia cell lines B2 and 8093 that were established from individual BCR/ABL P190 transgenic mice with two drugs, nilotinib and lonafarnib, in vitro in the presence of stroma. If a moderate dose of drug is used for treatment, not all of the leukemia cells are eradicated, and EMDR reproducibly emerges after 8–14 d of continued drug treatment, after which the cells are able to proliferate in that concentration of the drug (lonafarnib, Fig. S1 and ref. Citation17). The drug nilotinib forms a complex in the ATP-binding pocket of the Abl moiety of Bcr/Abl and inhibits its tyrosine kinase activity.Citation18 Lonafarnib is an anti-cancer drug that inactivates farnesyltransferase, an enzyme responsible for the prenylation of proteins such as Ras.Citation19
To examine if EMDR is associated with changes in gene expression, we treated both ALL cell lines in our in vitro model in triplicate with nilotinib or with lonafarnib and isolated RNA before treatment (beginning), during acquisition of drug resistance (midpoint; m, d3/4) and at the final drug-resistant phase (end; e, d20/30). RNA was subjected to microarray analysis as previously described.Citation20 A comparison of the expression profiles of nilotinib-resistant 8093 cells with the original non-drug-resistant population showed that around 3000 genes were differentially (> 2x up or down) regulated, whereas in the second ALL cell line, B2, only around 480 genes showed altered expression. Lonafarnib-resistance was accompanied by smaller changes in expression, with around 250 genes in 8093 and 175 in B2 being affected (). While the ALL cell line B2 was from a transgenic mouse on an outbred genetic background, 8093 was from an animal at f6 on C57Bl/6J. Thus, overall, the genetically homogenous cells showed more changes than cells from a mixed genetic background, and treatment with the Bcr/Abl-specific drug nilotinib caused more differences than with the farnesyltransferase inhibitor.
Figure 1. EMDR to two drugs with a different mechanism of action is accompanied by differential expression of common genes. Schematic illustration summarizing the results of gene expression profiling on two different pro-B lymphoblastic leukemia cell lines (8093 and B2) from BCR/ABL transgenic mice, that developed EMDR during treatment with nilotinib or lonafarnib in the presence of stroma. For each condition, the most up- and downregulated genes (lower part and upper part of the heatmap, respectively) are shown. Red and green represent the individual up-/downregulation values, respectively.
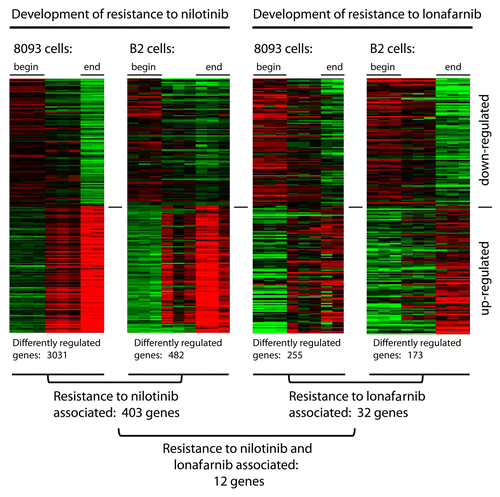
When we extracted those genes that were in common between the two cell lines, there were 403 genes associated with nilotinib-resistance development in common between 8093 and B2. Resistance development to lonafarnib was restricted to 32 common genes for 8093 and B2 (). We also examined whether there were any genes commonly regulated between nilotinib- and lonafarnib-treated cells. Surprisingly, although these drugs have a very different mechanism of action, we identified 12 genes in common, which all were increased in EMDR-generated lymphoblasts (). Expression of some genes was maintained at a high level at the end point, when the cells were fully viable and actively proliferating again. These results show that ALL cells, as measured by gene array, exhibit multiple and heterogeneous responses to drug treatment, as well as activation of common pathways during the development of EMDR.
Table 1. Genes showing increased expression during the acquisition of EMDR to both nilotinib or lonafarnib.
EMDR is associated with differential regulation of genes typically associated with inflammation
To analyze EMDR-related changes in gene expression in more detail, we used Ingenuity Pathway Analysis software. This initial analysis demonstrated that a remarkable number of the genes in the nilotinib-treated 8093 and B2 cells, of which expression was significantly altered, belonged to categories typically associated with inflammationCitation44 (see Fig. S3). As illustrated in , this included products involved in the metabolism of leukotrienes and prostaglandins; in platelet and mast cell function; cytokines, chemokines and their receptors; Toll-like and IgE Fc-receptors and signaling; complement; proteases, peptidases and tryptases; lysozome/phagosome-associated products; and other products involved in the activation of macrophages as well as products involved in negative regulation of inflammation.Citation44 Of note, also 6 of the 12 genes associated with EMDR to both nilotinib and lonafornib () are related to inflammation.
Figure 2. Large-scale upregulation of inflammatory-associated gene products in 8093 and B2 BCR/ABL lymphoblastic leukemia cells that developed EMDR to nilotinib. Functional grouping of genes related to inflammation, which show significant changes in expression during the development of EMDR. Shown are the average up-/downregulation values of each time point during nilotinib treatment of the two ALL cell lines 8093 and B2: b = begin of treatment (day 0); m = midpoint (maximal reduction of cell viability as a consequence of drug treatment); e = endpoint (full recovery of drug treated ALL cells that developed EMDR). Red and green represent the individual up-/downregulation values, respectively. Absolute log-transformed, corrected microarray values were used of biological triplicates ± SEM.
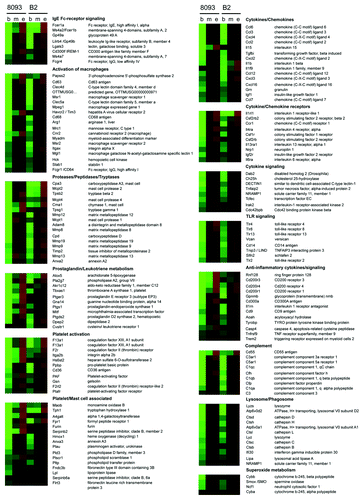
In many cases, complete signaling pathways including their stimuli and receptors were transcriptionally upregulated during EMDR. For example, most of the factors involved in prostaglandin/leukotriene/thromboxane synthesis, which are critical mediators of acute and chronic inflammation,Citation44,Citation45 were increased in expression during EMDR (, Fig. S4). These included phospholipase A2 (pla2g7), which initially converts diacylglycerol and phospholipids to arachidonic acid; the lipooxigenase alox5, which is involved in the synthesis of leukotrienes from arachidonic acid; cyclooxygenase 1 (cox1/ptgs1), which converts arachidonic acid into prostaglandin H2; prostaglandin D synthetase 2 (ptgds2), which converts prostaglandin H2 into prostaglandin D2; and thromboxane synthase 1 (tbxas1), platelet-activating factor (paf) and pro-platelet basic protein (ppbp), which are important for the generation of thromboxane from prostaglandin H2. In addition, several related receptors were upregulated during EMDR (ptger3, f2r, cysltr1 and ptafr).
Also, products related to signaling via CD36, a critical mediator of sterile inflammation,Citation46 were upregulated during EMDR. Binding of CD36 to its ligands oxLDL and amyloid-β allows TLR4/6 heterodimerization and stimulates sterile inflammation by induction of IL-1β production and the generation of reactive oxygen species. Interestingly, besides cd36, also a mammalian homolog of amyloid-β, the amyloid-β like precursor protein 2 (aplp2), tlr4, il-1β and several components of the reactive oxygen species-generating NADPH oxidase complex including p91phox, p47phox and p22phox were upregulated during EMDR ().
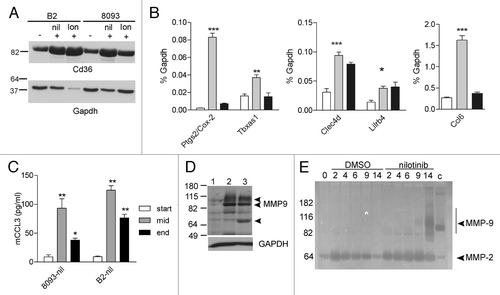
Some of the genes identified by gene array were chosen for further validation using western blotting, ELISA and quantitative RT-PCR. As shown in , western blot analysis confirmed that the increased expression of cd36 measured by the array corresponded with increased protein expression during nilotinib and lonafarnib-induced EMDR. Using quantitative RT-PCR and ELISA, validation of tbax1, ptgs2 (cox-2), clec4d, lilrb4, ccl6 (mRNA) and Ccl3 (ELISA), all known mediators in inflammation, further supported the microarray results ().
Figure 3. Increased expression of inflammation-associated genes during EMDR. (A) western blot analysis of samples from B2 on day 14 DMSO (-), on day 14 nilotinib (+) and on day 19 lonafarnib (+); from 8093 on day 14 DMSO (-), on day 9 nilotinib (+) and on day 14 lonafarnib (+). (B) Real-time RT/PCR on 8093 cells treated with 16 nM nilotinib in the presence of stroma as indicated. White bar, DMSO-treated cells; gray bar, t = 3 d of nilotinib treatment; black bar, t = 9 d. Results are expressed as % of that of gapdh gene expression. Bars represent mean ± SEM of triplicate samples. Samples independent of those used for the microarray. (C) Murine ccl3 levels were measured during the course of EMDR in the supernatant of the indicated cells treated with nilotinib using an ELISA. Samples independent of those used in other experiments. *p < 0.05; **p < 0.01; ***p < 0.001 when compared with start. (D) western blot analysis of Mmp9 protein. Lane 1, untreated 8093 cells; lane 2, 8093 cells at day 11 resistant to nilotinib; lane 3, 8093 cells at day 18 resistant to lonafarnib. Mmp9 proteins include 92 kDa, 82 N-terminal cleaved and 65 kDa C-terminal cleaved forms (indicated with arrowheads). Bottom panel, GAPDH loading control of the same samples. (E) Zymography on samples taken on the indicated days of 8093 cells treated with DMSO or 0.3 μM nilotinib. The arrow points to Mmp9 activity. Positive control for Mmp9 activity (c) mouse spleen lysates. The position of molecular weight standards is indicated to the left. A negative of the original zymogram is shown. The image B, D, and E show a representative result of experiments repeated at least once.
Increased activity of Mmp9
One interesting EMDR-associated gene identified by our analysis, which is related to both inflammation and leukemia development, is Mmp9. This metalloproteinase is well-known for its role in chronic and acute inflammatory disease and the inflammatory component in cancers.Citation47,Citation48 Moreover, Poyer et al.Citation49 and Pegahi et al.Citation50 reported that childhood ALL samples make and secrete Mmp2/Mmp9. Schneider et al.Citation51 further showed that the in vitro secretion of Mmp9 is a prognostic marker for childhood ALL, with high secretion of Mmp9 associated with a lower survival rate.Citation51 While neither B2 nor 8093 showed significant mmp9 expression at t = 0 without drug treatment, there was an increase in the levels of mmp9 in both samples when the cells had been treated for 3 d with nilotinib, when the viability of the culture had decreased to 5–10% of that of the culture at t = 0. The expression of other mmps including mmp12, mmp13 and mmp19 was also increased after treatment with nilotinib and with lonafarnib (not shown). We verified that the increased mmp9 expression correlated with increased Mmp9 protein ().
Because Mmps can release stroma-associated cytokines from the matrix, we considered the possibility that increased Mmp9 activity could result in increased pro-survival signals to the leukemic cells. Mmp9 is synthesized as an inactive pro-polypeptide. To investigate if the induced Mmp9 had enzymatic activity, we performed zymography for gelatinase activity. Because drug treatment could not be performed in the absence of serum and serum contains a substantial amount of Mmp activity, we assayed Mmp9 levels in the lymphoblastic leukemia cells and not in the tissue culture supernatant. As shown in , 8093 cells treated with DMSO over the course of 14 d showed no evidence of the production of active Mmp9. In contrast, cells treated with nilotinib had a clear induction of Mmp9 activity.
BCR/ABL ALL cells show elevated expression of genes related to inflammation during treatment with nilotinib in vivo
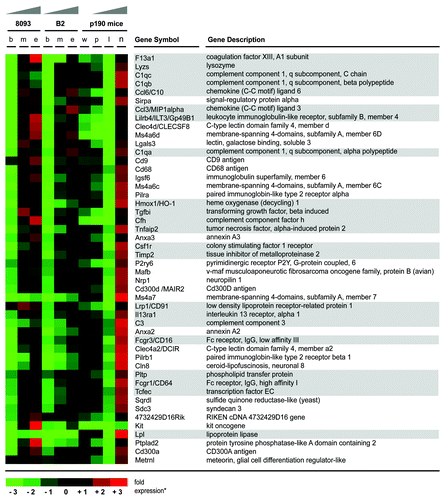
In a previous analysis, we performed gene expression profiling of pro-B cells from BCR/ABL P190 transgenic mice before onset of leukemia, during leukemia progression and after 8 d of treatment with nilotinib to monitor the distinct stages of leukemia development in vivo.Citation20 Interestingly, reanalysis of expression array data from these flow-sorted AA4.1+, CD19+ bone marrow cells directly isolated from normal wild type (wt) and BCR/ABL transgenic mice showed a concordant outcome with that of the EMDR in cultured cells (; Fig. S2). For example, short-term resistance to nilotinib was associated with increased expression of chemokines (ccl3 and ccl6) cytokine receptors (il13ra1 and csf1r), components of the complement system (c1qa, c1qc, c1qb, c1qh, cfh and c3), Fc-receptors (fcgr1 and fcgr3) and other genes related to inflammation (f13a1, ilt3, clec4d, tgfbi, tnfaip2, hmox1, timp2, tfec, pltp and lpl).
Figure 4. ALL cells in the bone marrow of BCR/ABL transgenic mice show elevated expression of genes related to inflammation during treatment with nilotinib. Gene array results of transcriptionally upregulated genes of transgenic BCR/ABL mice and, as a comparison, from the two murine BCR/ABL-positive leukemia cell lines, 8093 and B2, are shown. Up- and downregulation of gene expression during the development of EMDR to nilotinib is visualized by a heatmap (expression values are log transformed and up-/downregulation ratios are visualized by red and green color intensity, respectively). Gene array results represent the average level of gene expression from three individual experiments for the different stages during development of EMDR (ex vivo: b, untreated leukemia cells; m, mid point; e, development of nilotinib resistance/end point; in vivo: w, AA4.1+, CD19+ pro-B cells from wild type mice; p, pre-leukemic AA4.1+ CD19+ pro-B cells from BCR/ABL transgenic mice; l, AA4.1+ CD19+ pro-B cells lymphoblastic leukemia cells from transgenic mice; n, AA4.1+ CD19+ pro-B cells lymphoblastic leukemia cells from BCR/ABL mice treated for 8 d with nilotinib). Genes related to inflammation are accentuated by gray boxes.
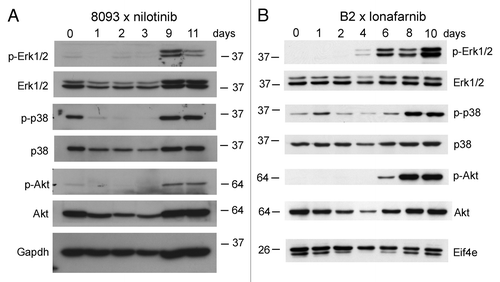
EMDR is accompanied by activation of Akt, Erk1/2 and p38MAPK pathways
The increased expression of genes during EMDR could be caused by massive activation of signal transduction pathways regulating stress and inflammation. The activation of the serine/threonine kinases Akt, Erk1/2 and p38 has been linked to oncogenic signalingCitation52 as well as to the regulation of inflammation. We therefore examined the activation of these kinases during the development of EMDR using western blotting. Interestingly, in the presence of stromal support, there was little activation of the Erk1/2 or of the Akt pathway in the ALL cells under steady-state conditions at t = 0 (). However, phosphorylation of Erk1/2 and Akt was highly induced at the point when the cells had become able to grow in the presence of nilotinib () or lonafarnib (). The MAPK p38 was activated before the cells had been exposed to drugs, but activation increased above the initial level during the development of EMDR. Thus, EMDR is specifically associated with the activation of the serine/threonine kinases Akt, Erk1/2 and p38.
Figure 5. Emergence of EMDR in lymphoblastic leukemia cells during treatment with nilotinib or lonafarnib is accompanied by activation of major signal transduction pathways. Western blot analysis of samples from 8093 cells treated with 20 nM nilotinib (A) or B2 treated with 0.25 μM lonafarnib (B) isolated during a time course of developing EMDR. Samples independent from those of the microarray; single experiment. The location of molecular weight markers (kDa) is indicated in the different panels. Numbers above the lanes show the day of sampling.
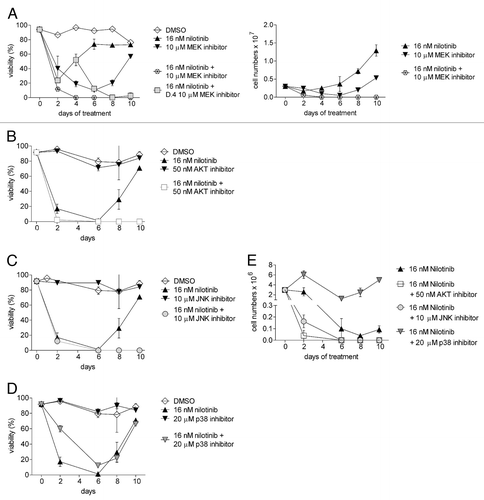
Inhibition of the Erk, JNK or Akt pathways inhibits the development of tolerance to nilotinib
To further examine the relationship between EMDR and the activation of these signal transduction pathways, we investigated the effect of their inhibition on the process of EMDR to nilotinib in 8093 ALL cells. Pilot experiments were performed to determine a suitable dose of inhibitor that, when used as monotreatment, did not eradicate the culture. Next, using that dose, its effect on EMDR in the presence of nilotinib was evaluated.
As shown in , treatment with 10 μM of the MEK inhibitor U0126 allowed ALL cells to develop drug tolerance within 10 d, as measured by regain of viability in the culture and resumption of cell proliferation. However, although cells treated with nilotinib alone similarly developed nilotinib-resistance, the addition of U0126 together with nilotinib, or after 4 d of monotreatment with nilotinib, killed the cells and prevented the emergence of nilotinib-resistance. A similar effect was obtained with an Akt inhibitor: when combined with nilotinib, viability dropped to 0 and no cell division was measured. Alone, the Akt inhibitor suppressed the proliferation of the ALL cells but had a small overall effect at the concentration used on the viability of the cells that remained (). We also tested inhibitors of the stress-activated pathways including p38 and JNK. illustrates that the effect of the JNK inhibitor was similar to that of the MEK and Akt inhibitors. The p38 inhibitor alone only had a small effect. However, in contrast to the other inhibitors, the inactivation of the p38 pathway reduced the effectiveness of nilotinib, and increased viability of nilotinib-treated cells ().
Figure 6. Inhibition of Erk, Akt and JNK pathways inhibits drug tolerance to nilotinib. 8093 ALL cells were treated with control DMSO, 16 nM nilotinib, or inhibitors of different pathways alone or combined with nilotinib. Inhibitors used include (A) 10 μM of the MEK inhibitor U0126, (B) 50 nM of the Akt inhibitor Triciribine (C) 10 μM of the JNK inhibitor SP600125 and (D) 20 μM of the p38 inhibitor CAY10571 (an SB203580 analog). The graphs shown in B-E were generated in a single experiment; the graphs share the nilotinib-treated and DMSO-treated samples. The graph in A represents a separate experiment. Left panels, viability (% viable cell counts/total cell counts). (E) Total viable cell counts from B-D. Results shown are representative of three independently performed experiments.
Discussion
More than one century ago, in 1863, Rudolf Virchow proposed, for the first time, a connection between inflammation and cancer. Within the past decade, numerous links have been reported between cancer and inflammatory processes in the microenvironment. For example, some types of cancers are thought to be initiated by chronic inflammation of the surrounding tissue and anti-inflammatory drugs are known to decrease the risk of developing some cancers (reviewed in refs. Citation53-Citation55). Interestingly, a widely used mouse model for the induction of plasmacytomas that resemble Burkitt lymphoma or diffuse large cell B cell lymphoma in man is based on the constitutive overexpression of the pro-inflammatory cytokine interleukin 6.Citation56,Citation57 Also, it is well-known, that some types of carcinomas attract and receive support from innate immune cells.Citation58
Although we here report an association between inflammation and leukemia, our study differs from those mentioned above in that the hallmarks of inflammation are found in the leukemia cells themselves. This outcome was unexpected. Some type of inflammatory response under such conditions could be anticipated from the microenvironment (stromal cells): when cancer cells are exposed to a therapeutically effective drug, numerous malignant cells will be killed, and this could result in a reaction from the microenvironment as if an aseptic “wound” is present, because of the dead and dying cells, and cell debris. However, we also performed gene expression profiling on the irradiated fibroblasts in the presence of nilotinib-treated 8093 cells and the fibroblasts did not show an inflammatory or any other major response on a transcriptional level to the presence of nilotinib-treated 8093 cells (GEO profiles GSE33329). Indeed, in our current study, we found that the leukemia cells themselves reacted to drug treatment in the presence of stroma by expressing inflammatory genes not typically associated with cells of this lineage. This effect was not restricted to the initial phase of acute “wounding” but for some genes persisted for up to 3–4 weeks after initiation of the drug treatment.
Numerous microarray analyses on RNA from ALL samples have been reported, many of which sought to discriminate different subcategories of ALL based on gene signatures. There are fewer studies that investigated drug resistance, and those that examined this issue mainly used samples of drug-resistant patients, not samples of patients that were being treated by drugs. However, two reports including that of Cheok et al.Citation59 and Rhein et al.Citation60 treated ALL patients for 1 or 8 d and compared the expression profiles of the treated ALL cells to those of the same patient at diagnosis. The study of Rhein et al.Citation60 used an approach that was conceptually somewhat similar to ours. They performed microarray analysis on relatively pure populations of ALL cells from the peripheral blood of the same patients at diagnosis and after 8 d of treatment with methotrexate. The CD11b (integrin αμ) and the IFNγR1 were two genes of which the expression was commonly increased among their samples. CD11b is a typical integrin expressed on innate immune cells. Interestingly, this integrin is a marker for minimal residual disease in childhood ALL.Citation61 CD11b expression was also increased in both nilotinib-resistant B2 and 8093 cells (not shown). Of the set of 82 commonly modified gene products in the samples of Rhein et al., there were 20 genes of which expression was increased at day 8, and 7 of these were also upregulated in our study in 8093 cells treated with nilotinib. Interestingly, this included lysozyme and IL8. A murine paralog of IL8 is cxcl2/MIP-2, which was highly increased in expression in 8093 cells resistant to nilotinib and in AA4.1+, CD19+ leukemic cells treated in vivo with nilotinib (not shown). Thus, in these human patient samples, there was increased expression of at least three inflammation-associated genes over the 8 d of treatment.
Such inflammatory response, as well as the response to the stress of drug treatment, could correlate with changes in signal transduction pathways in cancer cells. For example, Akt was reported to be activated by the proinflammatory environment in breast cancer.Citation62 We here detected activation of the Erk, p38 and Akt pathways in the mouse ALL cells that developed resistance to nilotinib and lonafarnib. Interestingly, the Erk, Akt and JNK pathways all contributed to the survival of nilotinib-stressed ALL cells, since inhibition of those pathways decreased the ability of the ALL cells to grow out in the presence of nilotinib. In contrast, our results show that the role of p38 in protection of ALL cells is complex, which is consistent with the context-dependent role of this pathway in other cell types. For example, although p38 activation is seen in different cancers, inactivation of p38 by gene targeting in mice results in increased tumorigenesis.Citation63 In contrast, inhibition of p38 activation in chronic lymphocytic leukemia and in ALL cells grown on stroma decreased survival and proliferation, respectively.Citation64,Citation65 Interestingly, the effect of p38 pathway inhibition on nilotinib-treated Bcr/Abl-positive leukemia measured here during EMDR is consistent with other studies in Bcr/Abl-positive leukemia cells. Although our study is the first to report this in EMDR, the therapeutic effect of imatinib, dasatinib, and IFNγ on Bcr/Abl-positive cells was also reported to be diminished in the presence of p38 inhibitors.Citation66-Citation68
Although our studies show a correlation between increased expression of genes associated with inflammation and EMDR, we have not demonstrated that this promotes EMDR, or, conversely, that EMDR causes the inflammatory response. Experiments using general non-steroidal anti-inflammatory drugs show that they can decrease EMDR, but the targets of such drugs are not precisely defined, and, moreover, we found increased expression of some of the genes after combined exposure with nilotinib (manuscript in preparation). Overall, we conclude that EMDR of Bcr/Abl-expressing lymphoblastic leukemia cells is accompanied by multiple changes in pathway activation and in transcription. Importantly, we also conclude that multiple combinations of drugs are able to overcome the ability of the ALL cells to reset their sensitivity to drugs such as nilotinib in the presence of stromal support, suggesting that the most effective strategies for eradication of ALL cells in the bone marrow should include the simultaneous exposure to multiple drugs.
Materials and Methods
Cells and drug treatment
Growth of B2 and 8093 mouse Bcr/Abl P190 transgenic pro-/pre-B acute lymphoblastic leukemia cells has been described before.Citation13,Citation16 Murine ALL cells were cultured on a mitotically inactivated irradiated mouse embryonic fibroblast (MEF) feeder layer. Cells were also plated on irradiated OP9 feeder layers in αMEM including 20% FBS, 1% L-glutamine and 1% penicillin/streptomycin as described.Citation69 Viability of cells was measured by Trypan blue exclusion. Viability is expressed as the percentage of viable cells of the total cell number. All measurements were done in triplicate wells. Values are expressed as mean ± SEM. Drug concentrations are indicated in the individual experiments. We used Triciribine as Akt inhibitor, SP600125 as JNK inhibitor, and CAY10571 as p38 inhibitor (Cayman Chemicals cat no 10010237, 10010466 and 10010400, respectively). The MEK1/2 inhibitor U0126 was from Cell Signaling (cat no 9903). Nilotinib and lonafarnib were obtained from Novartis and Schering-Plough, respectively.
Microarrays
All samples from individual time points were biological triplicates, except end points of lonafarnib and nilotinib treated 8093 cells (duplicates). B2 cells were treated with 0.25 μM lonafarnib and harvested on day 0, 3 and 30; B2 cells treated with 0.5 μM nilotinib were collected at day 0, 3 and 21; 8093 were treated with 1.0 μM lonafarnib and collected on day 0, 4 and 26; 8093 cells treated with 0.02 μM (20 nM) nilotinib were harvested on day 0, 3 and 20. In these cultures, similar to normal precursor B-lineage cells grown on stroma, there is a continuous trafficking of lymphoblasts from the medium to the top of the MEF layer, underneath it and back into the culture medium. Only cells loosely attached to the stroma or in the culture medium were collected. RNA was extracted using the Trizol reagent as per the manufacturer’s (Invitrogen Corporation) instructions. RNA was re-purified with phenol-chloroform extraction and ethanol precipitation. Microarray hybridization was performed by the Genome Core facility at the Research Institute of Children Hospital of Los Angeles. Briefly, RNA quality was first assessed using an Agilent Bioanalyzer (Agilent Technologies) and the 28S/18S ratios of all of the samples were between 1.3 and 2. RNA was converted to cDNA with Superscript Choice for cDNA Synthesis (Invitrogen) and subsequently converted to biotinylated cRNA with an Enzo High Yield RNA Transcript labeling kit (Enzo Diagnostics). After hybridization to the murine Mouse Gene 1.0 ST arrays (28,000 transcripts, Affymetrix), the gene chips were automatically washed and stained with streptavidin-phycoerythrin using a fluidics system. The chips were scanned with a Hewlett-Packard GeneArray Scanner (Hewlett-Packard). Results were analyzed using Partek and Ingenuity Systems (version 7.1) software programs. Only genes that show an up-/downregulation of > 2 times between the start and end point were used for further analysis. For final visualization of microarray data, average microarray values from individual time points were calculated and log transformed. Up-/downregulation values represent the ratio of the individual time point divided by the average of all time points from one condition. Ratios were then transformed to heatmaps using the Cluster software version 2.11 downloaded from http://rana.lbl.gov/EisenSoftware.htm.
Zymography
Cells were collected and lysed in 25 mM TRIS-HCl pH7.5, 100 mM NaCl, 1% (v/v) NP-40 for 15 min at 4°C. After centrifugation, supernatants were stored at -80°C. Twenty μg protein was run on 7% SDS-PAA gels with 0.1% gelatin, as described.Citation70
Antibodies and ccl3 measurements
Ccl3/Mip-1α levels in cell supernatant were measured using a mouse Ccl3/Mip-1α Quantikine ELISA kit (R&D Systems, Minneapolis, MN). CD36 antibodies were from Novus Biologicals, Inc. (Littleton, CO); Gapdh from Chemicon International/Millipore (Billercia, MA); pErk1/2; Erk1/2; p-p38; p38; p-Akt; Akt; Eif4e from Santa Cruz Biotechnology (Santa Cruz, CA).
Real-time RT/PCR
To quantify expression levels of selected genes, RNA was extracted from a nilotinib drug resistance-induction experiment independent from the ones performed for the microarrays. Cells were resuspended in RNAprotect Cell Reagent (Qiagen) prior to RNA extraction using an RNeasy Plus Mini Kit (Qiagen). An additional on-column treatment with DNase (Qiagen) was incorporated. RNA was reverse-transcribed into cDNA with a High Capacity 1st Strand Synthesis Kit (Applied Biosystems). Real-time RT/PCR was done as described in.Citation71 Murine primer sets used for amplification were as follows: gapdhU(5′-ACCCAGAAGACTGTGGATGG-3′) and gapdhD (5′-CACATTGGGGGTAGGAACAC-3′) yielding a 171 bp product; clec4dU (5′-GGTACTTGGACCTGCTGTCCTGT-3′) and clec4dD (5′-ATGGCGTCTTGTCCACCCACT-3′) yielding a 250 bp product; lilrb4U (5′-AGACCCAGACCCCACATTCCATGA-3′) and lilrb4D (5′-GTGTGCCCTGGTTACATTGTCTTTCA-3′) yielding a 276 bp product; ccl6U (5′-TCCTTGTGGCTGTCCTTGGGT-3′) and ccl6D (5′-ATGATGCCCGGCTTGATGCAC-3′) yielding a 202 bp product; cox2/ptgs2U (5′-TGCTGCCCGACACCTTCAACA-3′) and cox2/ptgs2D (5′-AACCCGGCCAGCAATCTGTCT-3′) yielding a 134 bp product; tbxasU (5′-TTACGGGACCAGCAAGCAGCA-3′) and tbxasD (5′-TCAAAGCCTTCCACGCCCACA-3′) yielding a 101 bp product. Results were normalized to gapdh.
| Abbreviations: | ||
| ABL | = | Abelson |
| ALL | = | acute lymphoblastic leukemia |
| BCR | = | breakpoint cluster region |
| EMDR | = | environment-mediated drug resistance |
| MEF | = | mouse embryonic fibroblast |
| Ph | = | Philadelphia chromosome |
Additional material
Download Zip (3.2 MB)Disclosure of Potential Conflict of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Donna Foster for excellent care of the mice and the USC Affymetrix MicroArray Core Facility at Childrens Hospital Los Angeles for expert assistance. Pavinder Kaur is acknowledged for participation in an early phase of this study. We thank Fei Fei and Min Lim for their contributions to the overall study. This work was supported by PHS grants CA090321 (to NH), by funding from a CHLA RCDA, a Jean Perkins Scholar and a StopCancer award (Y-mK); and by William Lawrence and Blanche Hughes Foundation (Y-mK, NH, JG).
Notes
† Current affiliation: Laboratory of Molecular Immunology; The Rockefeller University; New York, NY USA
‡ Current affiliation: Division of Hematology and Hematopoietic Cell Transplantation; City of Hope National Medical Center; Duarte, CA USA
§ Current affiliation: Laura Bassi Centre of Expertise; Theraped/Forschungsprogramm für Rezeptorbiochemie und Tumorstoffwechsel; Universitätsklinik für Kinder- und Jugendheilkunde; Paracelsus Medizinische Privatuniversität, Vienna, Austria
References
- Heisterkamp N, Groffen J. BCR/ABL gene structure and BCR function. In: Carella A, Daley G, Eaves C, Goldman J, Hehlman R, eds. Chronic myeloid leukemia: Biology and treatment, 2001:3-17.
- Ren R. Mechanisms of BCR-ABL in the pathogenesis of chronic myelogenous leukaemia. Nat Rev Cancer 2005; 5:172 - 83; http://dx.doi.org/10.1038/nrc1567; PMID: 15719031
- Ottmann OG, Pfeifer H. First-line treatment of Philadelphia chromosome-positive acute lymphoblastic leukaemia in adults. Curr Opin Oncol 2009; 21:Suppl 1 S43 - 6; http://dx.doi.org/10.1097/01.cco.0000357476.43164.6b; PMID: 19561414
- Schultz KR, Bowman WP, Aledo A, Slayton WB, Sather H, Devidas M, et al. Improved early event-free survival with imatinib in Philadelphia chromosome-positive acute lymphoblastic leukemia: a children’s oncology group study. J Clin Oncol 2009; 27:5175 - 81; http://dx.doi.org/10.1200/JCO.2008.21.2514; PMID: 19805687
- Gaynon PS. Childhood acute lymphoblastic leukaemia and relapse. Br J Haematol 2005; 131:579 - 87; http://dx.doi.org/10.1111/j.1365-2141.2005.05773.x; PMID: 16351633
- Pui CH, Evans WE. Treatment of acute lymphoblastic leukemia. N Engl J Med 2006; 354:166 - 78; http://dx.doi.org/10.1056/NEJMra052603; PMID: 16407512
- Den Boer ML, van Slegtenhorst M, De Menezes RX, Cheok MH, Buijs-Gladdines JG, Peters ST, et al. A subtype of childhood acute lymphoblastic leukaemia with poor treatment outcome: a genome-wide classification study. Lancet Oncol 2009; 10:125 - 34; http://dx.doi.org/10.1016/S1470-2045(08)70339-5; PMID: 19138562
- Kantarjian H, Giles F, Wunderle L, Bhalla K, O’Brien S, Wassmann B, et al. Nilotinib in imatinib-resistant CML and Philadelphia chromosome-positive ALL. N Engl J Med 2006; 354:2542 - 51; http://dx.doi.org/10.1056/NEJMoa055104; PMID: 16775235
- Jones D, Thomas D, Yin CC, O’Brien S, Cortes JE, Jabbour E, et al. Kinase domain point mutations in Philadelphia chromosome-positive acute lymphoblastic leukemia emerge after therapy with BCR-ABL kinase inhibitors. Cancer 2008; 113:985 - 94; http://dx.doi.org/10.1002/cncr.23666; PMID: 18615627
- Klemm L, Duy C, Iacobucci I, Kuchen S, von Levetzow G, Feldhahn N, et al. The B cell mutator AID promotes B lymphoid blast crisis and drug resistance in chronic myeloid leukemia. Cancer Cell 2009; 16:232 - 45; http://dx.doi.org/10.1016/j.ccr.2009.07.030; PMID: 19732723
- Meads MB, Gatenby RA, Dalton WS. Environment-mediated drug resistance: a major contributor to minimal residual disease. Nat Rev Cancer 2009; 9:665 - 74; http://dx.doi.org/10.1038/nrc2714; PMID: 19693095
- Heisterkamp N, Jenster G, ten Hoeve J, Zovich D, Pattengale PK, Groffen J. Acute leukaemia in bcr/abl transgenic mice. Nature 1990; 344:251 - 3; http://dx.doi.org/10.1038/344251a0; PMID: 2179728
- Kaur P, Feldhahn N, Zhang B, Trageser D, Müschen M, Pertz V, et al. Nilotinib treatment in mouse models of P190 Bcr/Abl lymphoblastic leukemia. Mol Cancer 2007; 6:67; http://dx.doi.org/10.1186/1476-4598-6-67; PMID: 17958915
- Mishra S, Pertz V, Zhang B, Kaur P, Shimada H, Groffen J, et al. Treatment of P190 Bcr/Abl lymphoblastic leukemia cells with inhibitors of the serine/threonine kinase CK2. Leukemia 2007; •••:178 - 80
- Mishra S, Zhang B, Cunnick JM, Heisterkamp N, Groffen J. Resistance to imatinib of bcr/abl p190 lymphoblastic leukemia cells. Cancer Res 2006; 66:5387 - 93; http://dx.doi.org/10.1158/0008-5472.CAN-05-3058; PMID: 16707466
- Zhang B, Groffen J, Heisterkamp N. Increased resistance to a farnesyltransferase inhibitor by N-cadherin expression in Bcr/Abl-P190 lymphoblastic leukemia cells. . Leukemia 2007; 21:1189 - 97; http://dx.doi.org/10.1038/sj.leu.2404667
- Zhang B, Groffen J, Heisterkamp N. Resistance to farnesyltransferase inhibitors in Bcr/Abl-positive lymphoblastic leukemia by increased expression of a novel ABC transporter homolog ATP11a. Blood 2005; 106:1355 - 61; http://dx.doi.org/10.1182/blood-2004-09-3655; PMID: 15860663
- O’Hare T, Walters DK, Deininger MW, Druker BJ. AMN107: tightening the grip of imatinib. Cancer Cell 2005; 7:117 - 9; http://dx.doi.org/10.1016/j.ccr.2005.01.020; PMID: 15710324
- Morgillo F, Lee HY. Lonafarnib in cancer therapy. Expert Opin Investig Drugs 2006; 15:709 - 19; http://dx.doi.org/10.1517/13543784.15.6.709; PMID: 16732721
- Trageser D, Iacobucci I, Nahar R, Duy C, von Levetzow G, Klemm L, et al. Pre-B cell receptor-mediated cell cycle arrest in Philadelphia chromosome-positive acute lymphoblastic leukemia requires IKAROS function. J Exp Med 2009; 206:1739 - 53; http://dx.doi.org/10.1084/jem.20090004; PMID: 19620627
- Febbraio M, Hajjar DP, Silverstein RL. CD36: a class B scavenger receptor involved in angiogenesis, atherosclerosis, inflammation, and lipid metabolism. J Clin Invest 2001; 108:785 - 91; PMID: 11560944
- Lee KH, Ono M, Inui M, Yuasa T, Takai T. Stimulatory function of gp49A, a murine Ig-like receptor, in rat basophilic leukemia cells. J Immunol 2000; 165:4970 - 7; PMID: 11046024
- Chang CC, Liu Z, Vlad G, Qin H, Qiao X, Mancini DM, et al. Ig-like transcript 3 regulates expression of proinflammatory cytokines and migration of activated T cells. J Immunol 2009; 182:5208 - 16; http://dx.doi.org/10.4049/jimmunol.0804048; PMID: 19380766
- Gu X, Laouar A, Wan J, Daheshia M, Lieberman J, Yokoyama WM, et al. The gp49B1 inhibitory receptor regulates the IFN-gamma responses of T cells and NK cells. J Immunol 2003; 170:4095 - 101; PMID: 12682239
- Katz HR. Inhibition of pathologic inflammation by leukocyte Ig-like receptor B4 and related inhibitory receptors. Immunol Rev 2007; 217:222 - 30; http://dx.doi.org/10.1111/j.1600-065X.2007.00522.x; PMID: 17498062
- Pahl MV, Vaziri ND, Yuan J, Adler SG. Upregulation of monocyte/macrophage HGFIN (Gpnmb/Osteoactivin) expression in end-stage renal disease. Clin J Am Soc Nephrol 2010; 5:56 - 61; http://dx.doi.org/10.2215/CJN.03390509; PMID: 19833906
- Ripoll VM, Irvine KM, Ravasi T, Sweet MJ, Hume DA. Gpnmb is induced in macrophages by IFN-gamma and lipopolysaccharide and acts as a feedback regulator of proinflammatory responses. J Immunol 2007; 178:6557 - 66; PMID: 17475886
- Kuan CT, Wakiya K, Dowell JM, Herndon JE 2nd, Reardon DA, Graner MW, et al. Glycoprotein nonmetastatic melanoma protein B, a potential molecular therapeutic target in patients with glioblastoma multiforme. Clin Cancer Res 2006; 12:1970 - 82; http://dx.doi.org/10.1158/1078-0432.CCR-05-2797; PMID: 16609006
- Arce I, Martínez-Muñoz L, Roda-Navarro P, Fernández-Ruiz E. The human C-type lectin CLECSF8 is a novel monocyte/macrophage endocytic receptor. Eur J Immunol 2004; 34:210 - 20; http://dx.doi.org/10.1002/eji.200324230; PMID: 14971047
- Graham LM, Brown GD. The Dectin-2 family of C-type lectins in immunity and homeostasis. Cytokine 2009; 48:148 - 55; http://dx.doi.org/10.1016/j.cyto.2009.07.010; PMID: 19665392
- Porcheray F, Viaud S, Rimaniol AC, Léone C, Samah B, Dereuddre-Bosquet N, et al. Macrophage activation switching: an asset for the resolution of inflammation. Clin Exp Immunol 2005; 142:481 - 9; PMID: 16297160
- Zucchetto A, Benedetti D, Tripodo C, Bomben R, Dal Bo M, Marconi D, et al. CD38/CD31, the CCL3 and CCL4 chemokines, and CD49d/vascular cell adhesion molecule-1 are interchained by sequential events sustaining chronic lymphocytic leukemia cell survival. Cancer Res 2009; 69:4001 - 9; http://dx.doi.org/10.1158/0008-5472.CAN-08-4173; PMID: 19383907
- Terpos E, Politou M, Viniou N, Rahemtulla A. Significance of macrophage inflammatory protein-1 alpha (MIP-1alpha) in multiple myeloma. Leuk Lymphoma 2005; 46:1699 - 707; http://dx.doi.org/10.1080/10428190500175049; PMID: 16263571
- Coelho AL, Schaller MA, Benjamim CF, Orlofsky AZ, Hogaboam CM, Kunkel SL. The chemokine CCL6 promotes innate immunity via immune cell activation and recruitment. J Immunol 2007; 179:5474 - 82; PMID: 17911634
- Dolgachev V, Thomas M, Berlin A, Lukacs NW. Stem cell factor-mediated activation pathways promote murine eosinophil CCL6 production and survival. J Leukoc Biol 2007; 81:1111 - 9; http://dx.doi.org/10.1189/jlb.0906595; PMID: 17234680
- Lossos IS, Czerwinski DK, Alizadeh AA, Wechser MA, Tibshirani R, Botstein D, et al. Prediction of survival in diffuse large-B-cell lymphoma based on the expression of six genes. N Engl J Med 2004; 350:1828 - 37; http://dx.doi.org/10.1056/NEJMoa032520; PMID: 15115829
- Dean RA, Cox JH, Bellac CL, Doucet A, Starr AE, Overall CM. Macrophage-specific metalloelastase (MMP-12) truncates and inactivates ELR+ CXC chemokines and generates CCL2, -7, -8, and -13 antagonists: potential role of the macrophage in terminating polymorphonuclear leukocyte influx. Blood 2008; 112:3455 - 64; http://dx.doi.org/10.1182/blood-2007-12-129080; PMID: 18660381
- Kassim SY, Fu X, Liles WC, Shapiro SD, Parks WC, Heinecke JW. NADPH oxidase restrains the matrix metalloproteinase activity of macrophages. J Biol Chem 2005; 280:30201 - 5; http://dx.doi.org/10.1074/jbc.M503292200; PMID: 15983040
- Meng QR, Gideon KM, Harbo SJ, Renne RA, Lee MK, Brys AM, et al. Gene expression profiling in lung tissues from mice exposed to cigarette smoke, lipopolysaccharide, or smoke plus lipopolysaccharide by inhalation. Inhal Toxicol 2006; 18:555 - 68; http://dx.doi.org/10.1080/08958370600686226; PMID: 16717027
- Hofmann HS, Hansen G, Richter G, Taege C, Simm A, Silber RE, et al. Matrix metalloproteinase-12 expression correlates with local recurrence and metastatic disease in non-small cell lung cancer patients. Clin Cancer Res 2005; 11:1086 - 92; PMID: 15709175
- Sarkar S, Nuttall RK, Liu S, Edwards DR, Yong VW. Tenascin-C stimulates glioma cell invasion through matrix metalloproteinase-12. Cancer Res 2006; 66:11771 - 80; http://dx.doi.org/10.1158/0008-5472.CAN-05-0470; PMID: 17178873
- Sato K, Shikano S, Xia G, Takao J, Chung JS, Cruz PD Jr., et al. Selective expression of vacuolar H+-ATPase subunit d2 by particular subsets of dendritic cells among leukocytes. Mol Immunol 2006; 43:1443 - 53; http://dx.doi.org/10.1016/j.molimm.2005.07.035; PMID: 16144709
- Karlstetter M, Walczak Y, Weigelt K, Ebert S, Van den Brulle J, Schwer H, et al. The novel activated microglia/macrophage WAP domain protein, AMWAP, acts as a counter-regulator of proinflammatory response. J Immunol 2010; 185:3379 - 90; http://dx.doi.org/10.4049/jimmunol.0903300; PMID: 20709948
- Paul WE. Fundamental immunology. Philadelphia: Lippincott Williams & Wilkins, 2003.
- Rajakariar R, Hilliard M, Lawrence T, Trivedi S, Colville-Nash P, Bellingan G, et al. Hematopoietic prostaglandin D2 synthase controls the onset and resolution of acute inflammation through PGD2 and 15-deoxyDelta12 14 PGJ2. Proc Natl Acad Sci U S A 2007; 104:20979 - 84; http://dx.doi.org/10.1073/pnas.0707394104; PMID: 18077391
- Stewart CR, Stuart LM, Wilkinson K, van Gils JM, Deng J, Halle A, et al. CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nat Immunol 2010; 11:155 - 61; http://dx.doi.org/10.1038/ni.1836; PMID: 20037584
- Goetzl EJ, Banda MJ, Leppert D. Matrix metalloproteinases in immunity. J Immunol 1996; 156:1 - 4; PMID: 8598448
- Hu J, Van den Steen PE, Sang QX, Opdenakker G. Matrix metalloproteinase inhibitors as therapy for inflammatory and vascular diseases. Nat Rev Drug Discov 2007; 6:480 - 98; http://dx.doi.org/10.1038/nrd2308; PMID: 17541420
- Poyer F, Coquerel B, Pegahi R, Cazin L, Norris V, Vannier JP, et al. Secretion of MMP-2 and MMP-9 induced by VEGF autocrine loop correlates with clinical features in childhood acute lymphoblastic leukemia. Leuk Res 2009; 33:407 - 17; http://dx.doi.org/10.1016/j.leukres.2008.08.019; PMID: 18829111
- Pegahi R, Poyer F, Legrand E, Cazin L, Vannier JP, Lamacz M. Spontaneous and cytokine-evoked production of matrix metalloproteinases by bone marrow and peripheral blood pre-B cells in childhood acute lymphoblastic leukaemia. Eur Cytokine Netw 2005; 16:223 - 32; PMID: 16266864
- Schneider P, Costa O, Legrand E, Bigot D, Lecleire S, Grassi V, et al. In vitro secretion of matrix metalloprotease 9 is a prognostic marker in childhood acute lymphoblastic leukemia. Leuk Res 2010; 34:24 - 31; http://dx.doi.org/10.1016/j.leukres.2009.07.039; PMID: 19748669
- Sharma SV, Gajowniczek P, Way IP, Lee DY, Jiang J, Yuza Y, et al. A common signaling cascade may underlie “addiction” to the Src, BCR-ABL, and EGF receptor oncogenes. Cancer Cell 2006; 10:425 - 35; http://dx.doi.org/10.1016/j.ccr.2006.09.014; PMID: 17097564
- Coussens LM, Werb Z. Inflammation and cancer. Nature 2002; 420:860 - 7; http://dx.doi.org/10.1038/nature01322; PMID: 12490959
- Robak P, Smolewski P, Robak T. The role of non-steroidal anti-inflammatory drugs in the risk of development and treatment of hematologic malignancies. Leuk Lymphoma 2008; 49:1452 - 62; http://dx.doi.org/10.1080/10428190802108854; PMID: 18608871
- Porta C, Larghi P, Rimoldi M, Totaro MG, Allavena P, Mantovani A, et al. Cellular and molecular pathways linking inflammation and cancer. Immunobiology 2009; 214:761 - 77; http://dx.doi.org/10.1016/j.imbio.2009.06.014; PMID: 19616341
- Kovalchuk AL, Kim JS, Park SS, Coleman AE, Ward JM, Morse HC 3rd, et al. IL-6 transgenic mouse model for extraosseous plasmacytoma. Proc Natl Acad Sci U S A 2002; 99:1509 - 14; http://dx.doi.org/10.1073/pnas.022643999; PMID: 11805288
- Ramiro AR, Jankovic M, Callen E, Difilippantonio S, Chen HT, McBride KM, et al. Role of genomic instability and p53 in AID-induced c-myc-Igh translocations. Nature 2006; 440:105 - 9; http://dx.doi.org/10.1038/nature04495; PMID: 16400328
- Nelson D, Ganss R. Tumor growth or regression: powered by inflammation. J Leukoc Biol 2006; 80:685 - 90; http://dx.doi.org/10.1189/jlb.1105646; PMID: 16864602
- Cheok MH, Yang W, Pui CH, Downing JR, Cheng C, Naeve CW, et al. Treatment-specific changes in gene expression discriminate in vivo drug response in human leukemia cells. Nat Genet 2003; 34:85 - 90; http://dx.doi.org/10.1038/ng1151; PMID: 12704389
- Rhein P, Scheid S, Ratei R, Hagemeier C, Seeger K, Kirschner-Schwabe R, et al. Gene expression shift towards normal B cells, decreased proliferative capacity and distinct surface receptors characterize leukemic blasts persisting during induction therapy in childhood acute lymphoblastic leukemia. . Leukemia: official journal of the Leukemia Society of America. Leukemia Research Fund, UK 2007; 21:897 - 905
- Rhein P, Mitlohner R, Basso G, Gaipa G, Dworzak MN, Kirschner-Schwabe R, et al. CD11b is a therapy resistance- and minimal residual disease-specific marker in precursor B-cell acute lymphoblastic leukemia. Blood 2010; 115:3763 - 71; http://dx.doi.org/10.1182/blood-2009-10-247585; PMID: 20228269
- Prueitt RL, Boersma BJ, Howe TM, Goodman JE, Thomas DD, Ying L, et al. Inflammation and IGF-I activate the Akt pathway in breast cancer. International journal of cancer Journal international du cancer 2007; 120:796-805.
- Yong HY, Koh MS, Moon A. The p38 MAPK inhibitors for the treatment of inflammatory diseases and cancer. Expert Opin Investig Drugs 2009; 18:1893 - 905; http://dx.doi.org/10.1517/13543780903321490; PMID: 19852565
- Ringshausen I, Dechow T, Schneller F, Weick K, Oelsner M, Peschel C, et al. Constitutive activation of the MAPkinase p38 is critical for MMP-9 production and survival of B-CLL cells on bone marrow stromal cells. . Leukemia: official journal of the Leukemia Society of America. Leukemia Research Fund, UK 2004; 18:1964 - 70; http://dx.doi.org/10.1038/sj.leu.2403544
- Gaundar SS, Bradstock KF, Bendall LJ. p38MAPK inhibitors attenuate cytokine production by bone marrow stromal cells and reduce stroma-mediated proliferation of acute lymphoblastic leukemia cells. Cell Cycle 2009; 8:2975 - 83; http://dx.doi.org/10.4161/cc.8.18.9545; PMID: 19713744
- Mayer IA, Verma A, Grumbach IM, Uddin S, Lekmine F, Ravandi F, et al. The p38 MAPK pathway mediates the growth inhibitory effects of interferon-alpha in BCR-ABL-expressing cells. J Biol Chem 2001; 276:28570 - 7; http://dx.doi.org/10.1074/jbc.M011685200; PMID: 11353767
- Parmar S, Katsoulidis E, Verma A, Li Y, Sassano A, Lal L, et al. Role of the p38 mitogen-activated protein kinase pathway in the generation of the effects of imatinib mesylate (STI571) in BCR-ABL-expressing cells. J Biol Chem 2004; 279:25345 - 52; http://dx.doi.org/10.1074/jbc.M400590200; PMID: 15056660
- Dumka D, Puri P, Carayol N, Lumby C, Balachandran H, Schuster K, et al. Activation of the p38 Map kinase pathway is essential for the antileukemic effects of dasatinib. Leuk Lymphoma 2009; 50:2017 - 29; http://dx.doi.org/10.3109/10428190903147637; PMID: 19672773
- Fei F, Stoddart S, Muschen M, Kim YM, Groffen J, Heisterkamp N. Development of resistance to dasatinib in Bcr/Abl-positive acute lymphoblastic leukemia. . Leukemia: official journal of the Leukemia Society of America. Leukemia Research Fund, UK 2010; 24:813 - 20; http://dx.doi.org/10.1038/leu.2009.302
- Hawkes SP, Li H, Taniguchi GT. Zymography and reverse zymography for detecting MMPs, and TIMPs. Methods Mol Biol 2001; 151:399 - 410; PMID: 11217315
- Behan JW, Yun JP, Proektor MP, Ehsanipour EA, Arutyunyan A, Moses AS, et al. Adipocytes impair leukemia treatment in mice. Cancer Res 2009; 69:7867 - 74; http://dx.doi.org/10.1158/0008-5472.CAN-09-0800; PMID: 19773440