Abstract
Necrotic cells are known to activate the innate immune system and trigger inflammation by releasing damage associated molecular patterns (DAMPs). However, how necrotic cells influence the induction of antigen-specific CD8+ T cell-mediated adaptive immune responses under sterile conditions, in the absence of pathogen associated molecular patterns (PAMPs), remains poorly understood. Here, we examined antigen-specific CD8+ T-cell responses to primary sterile necrotic tumor cells both in vitro and in vivo. We found that primary necrotic cells alone fail to generate CD8+ T cell-dependent immune responses toward cell-associated antigens. We show that necrotic cells trigger CD8+ T-cell immunity only in the presence of PAMPs or analogs, such as p(dI-dC) and/or unmethylated CpG DNA. The electroporation of tumor cells with these PAMPs prior to necrosis induction triggered antigen-specific CD8+ T-cell responses through a TLR9/MyD88-dependent pathway. In addition, we found that necrotic cells contain factors that can block the cross-priming of CD8+ T cells even under non-sterile conditions and can serve as a possible mechanism of immunosuppression. These results suggest that antigen-specific CD8+ T-cell responses to primary necrotic tumor cells can be induced in the presence of PAMPs and thus have a substantial impact on the development of antitumor vaccination strategies.
Introduction
Tumor cell death can control antitumor immune responses. Understanding how dying/dead cells activate or silence the immune system helps in the development of efficient antitumor vaccination strategies and in the manipulation of unwanted immune responses in the course of transplantation, infection and autoimmunity.Citation1
Necrosis has been defined as an immunogenic form of cell death associated with the rupture of the cell membrane and release of intracellular contents into the microenvironment. It has been suggested that intracellular contents released from necrotic cells contain specific molecules that serve as endogenous danger signals (i.e., damage-associated molecular patterns or DAMPs) and alarm the immune system to respond.Citation2-Citation4 Several studies have demonstrated the existence of DAMPs and have elucidated their mode of action. In particular, heat shock proteins,Citation5 HMGB1,Citation6 uric acid,Citation7 genomic DNA,Citation8 mRNA,Citation9 nucleoside analogs,Citation10 ATP,Citation11 F-actinCitation12 and hyaluronanCitation13 have been characterized as DAMPs that lead to the activation of immune responses.
Under physiological conditions cells undergo a particular routine of programmed cell death known as apoptosis.Citation14,Citation15 Apoptosis is considered to be a universal mechanism by means of which an organism can clear old and damaged cells while keeping the immune system quiescent. The reason for this quiescence is proposed to be a sequestration of DAMPs during apoptosis, facilitating the induction of tolerance.Citation16 Over time, it has become clear that the definition of apoptosis as immunologically silent and necrosis as immunogenic does not properly reflect the actual situation.
Studies of the last decade have shown that apoptotic cells also can serve as an antigen source for the cross-priming of, rather than for the induction of cross-tolerance in, CD8+ T cells.Citation17,Citation18 It was found that apoptotic cells trigger Toll-IL1 receptor signaling-independent adaptive immune responses.Citation19 Additional studies showed that the immunogenicity of apoptosis is mediated by the activation of caspasesCitation20 and depends on the activity of the NLRP3 inflammasome.Citation21 Translocation of calreticulin on the surface of apoptotic cells was shown to act as an ‘eat me’ signal, eventually leading to CD8+ T cell activation.Citation22 All these data indicate that apoptotic cell death can induce potent immune responses.
Autophagy, a molecular pathways of cell self-degradation,Citation23 has recently been shown to be required for the cross-presentation of cell-associated antigensCitation24 as well as for the generation of antitumor immune responses during chemotherapy-induced tumor cell death.Citation25 In this scenario, activation of the immune system was shown to be dependent on the release of ATP by dying cells, which in turn is regulated by autophagy.Citation25
It is also known that the uptake of necrotic cells might occur through a phosphatidylserine-dependent mechanism that does not ultimately lead to production of pro-inflammatory cytokines. 26.27 In several studies, we and others have shown that necrosis fails to protect from tumor development in prophylactic antitumor vaccination experiments.Citation19,Citation28,Citation29 Therefore, the exact mechanisms of interaction between the immune system and necrotic cells as well as the consequences of such an interaction for antigen-specific CD8+ T cell-mediated immune responses remain unknown.
Here, we have studied the effect of primary necrosis in the absence of pathogen-associated molecular patterns (PAMPs), namely “sterile necrosis,” on the cross-priming of CD8+ T cells. We show that sterile necrosis of tumor cells as induced by 3 freeze-thawing cycles (F/T) abrogated their ability to prime CD8+ T cells both in vitro and in vivo. In addition, we show that if cells undergo necrosis under “non-sterile” conditions, they can activate antigen-specific CD8+ T cells in vivo. Introduction of double stranded unmethylated DNA prior to the induction of necrosis reversed the non-immunogenic state of sterile necrotic cells in vivo. Finally, we show that necrotic cells contain factors that can block the CD8+ T-cell priming capacity of non-sterile necrotic cells in vivo.
Our results suggest that antigen-specific CD8+ T cell responses to primary necrotic tumor cells can be induced in the presence of PAMPs and thus have a substantial impact on development of successful antitumor treatment strategies.
Results
Primary sterile necrosis does not activate CD8+ T cells in vitro
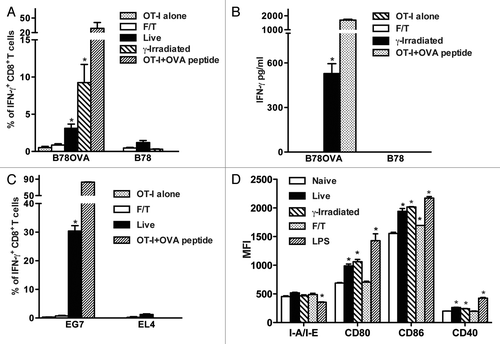
We evaluated if sterile necrotic cells induce the cross-priming of antigen-specific CD8+ T cells in vitro. Freeze-thawed B78H1OVA (B78OVA expressing the model antigen ovalbumin) or wild type B78H1 (B78) cells (5 × 104) were cultured with OT-I splenocytes (5 × 105) for 48 h and interferon γ (IFNγ) expression by CD8+ T cells was determined. As shown in , necrotic cells did not induce IFNγ expression in CD8+ T cells in contrast to live or γ-irradiated cells. This result was confirmed by ELISA (). Next, we asked if similar results can be observed with different cell lines. We cultured EG7 (OVA-expressing EL4 cells) or B16OVA cells with OT-I splenocytes and found that this did not lead to the expression of IFNγ by antigen-specific CD8+ T cells ( and data not shown), while live cells used as a positive control induced OVA-specific CD8+ T cell responses. Moreover, necrotic B78 cells did not upregulate co-stimulatory molecules on CD11c+ splenic dendritic cells (DCs) except for a slight upregulation of CD86. In contrast, DCs exposed to live or γ-irradiated B78 cells or lipopolysaccharide (LPS) upregulated CD80, CD86 and CD40 and downregulated I-A/I-E ().
Figure 1. Primary sterile necrotic tumor cells fail to stimulate OVA-specific CD8+ T cells in vitro. (A and B) 5 × 104 F/T, live or γ-irradiated B78OVA or B78 cells were co-cultured in vitro with 5 × 105 OT-I splenocytes for 48 h and interferon γ (IFNγ)-secreting CD8+ T cells (A) or IFNγ concentration (B) were analyzed using IFNγ intracellular staining and IFNγ ELISA respectively. Data are pooled results of 3 experiments. (C) As in (A), OT-I splenocytes were co-cultured for 48 h with F/T or live EG7 cells and % of IFNγ producing cells was determined. EL4 cells were used as an OVA-negative control cell line. Data are representative of 3 independent experiments. (D) 5 × 104 F/T, live or γ-irradiated B78 cells were co-cultured with unsorted splenocytes. After 24 h, CD11c+ cells were stained and analyzed for I-A/I-E, CD80, CD86 and CD40 expression. As a positive control, DCs were stimulated with 100 ng/mL lipopolysaccharide (LPS). Data are representative of 2 independent experiments. *p < 0.05 as compared with OT-I cells alone (Student’s t test).
Sterile necrotic cells do not lead to antigen-specific CD8+ T cell-mediated immunity against cell-associated antigens in vivo
Next, we investigated whether sterile necrotic cells can induce antigen-specific CD8+ T cell responses in vivo. For this OVA-specific cell lysis () and intracellular IFNγ secretion () by CD8+ T cells were evaluated in mice after injection of live, γ-irradiated or necrotic B78OVA cells. Mice vaccinated with necrotic cells did not show antigen-specific lysis in the spleen and draining lymph nodes (DLNs) (). Accordingly, CD8+ T lymphocytes from these mice failed to produce IFNγ after peptide restimulation in vitro (). In contrast, CD8+ T lymphocytes from mice vaccinated with live or γ-irradiated tumor cells had high antigen-specific lytic activity and produced IFNγ after peptide restimulation. It is important to mention that live B78OVA cells were as potent for immune response generation as γ-irradiated cells ().When EG7 cells were used, we detected significantly lower antigen-specific lysis in mice vaccinated with necrotic tumor cells as compared with animals receiving live EG7 cells ().
Figure 2. Primary sterile necrosis does not induce antigen-specific CD8+ T cell mediated immune responses in vivo. (A) Mice were vaccinated with 2 × 106 F/T, γ-irradiated or live B78OVA cells and antigen-specific lytic activity was determined using in vivo CTL assay after seven days in draining lymph nodes (DLNs) and spleens. Figure A shows an experiment representative of at least 3 independent experiments with 3 mice per group. (B) Lymphocytes from the DLNs and spleens of naïve, F/T, γ-irradiated or live tumor cell vaccinated mice were isolated 7 d after vaccination. Cells were restimulated in vitro with 0.1 μg/mL OVA peptide for 4 h and the frequency of interferon γ (IFNγ)-secreting CD8+ T cells was analyzed. Data are from one experiment representative of 2 independent experiments. (C). Mice were vaccinated with 5 × 106 F/T or live EG7 cells and antigen-specific lysis was measured as in (A). Pooled data of 2 independent experiments with 3 mice per group are shown. (D) C57BL/6 mice were vaccinated with 2 × 106 allogeneic H2Kd-positive live or F/T CT26OVA cells and the lytic activity of H2Kb-restricted, OVA-specific CD8+ T cells was evaluated. Data are representative of at least 3 independent experiments with 3 mice per group. *p < 0.05 as compared with F/T (necrotic) group (Student’s t test).
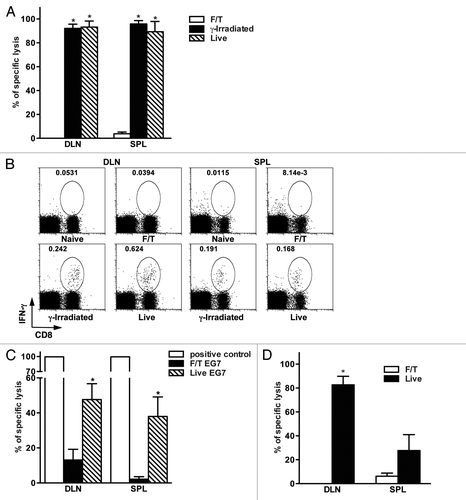
We also tested if allogeneic sterile necrotic tumor cells can induce antigen-specific CD8+ T-cell responses. For this aim, OVA-specific immune responses were evaluated in C57BL/6 mice (H2Kb) vaccinated with live or necrotic CT26OVA cells, which express H2Kd on their surface. Necrotic CT26OVA cells were unable to induce OVA-specific CD8+ T cell activation even after prime-boost vaccination. On the contrary, live CT26OVA cells triggered robust OVA-specific cytotoxic T lymphocyte (CTL) responses ().
To evaluate different types of necrosis inducers, B78OVA cells were exposed to distilled water, triggering necrosis via osmotic shock. Mice vaccinated with cells exposed to osmotic shock failed to develop antigen-specific responses, similar to mice vaccinated with F/T necrotic cells (data not shown).
Antigens are present in sterile necrotic cells but are not a substrate for efficient cross-priming of naïve CD8+ T cells
Our results show that primary sterile necrotic cells fail to induce CD8+ T-cell activation in vitro as well as in vivo. Therefore, we decided to explore the possible mechanisms responsible for such an effect, and asked whether necrotic tumor cells could be converted into immunogenic vaccines. First, we analyzed the presence of our model antigen (OVA) in necrotic B78OVA tumor cells. For this, the presence of OVA was assessed in both the soluble (supernatant) and particulate (pellet) fractions of necrotic cells cultured at 37°C at different time points. We found a band of an approximately 43 KDa corresponding to OVA in both fractions, even after 72 h of culture ().
Figure 3. Antigens are present in sterile necrotic cells but cannot be a substrate for cross-priming of naïve CD8+ T cells. (A) F/T cells were cultured at 37°C and the soluble and particulate fractions were prepared at indicated time points. After subsequent denaturation, electrophoresis and immunoblotting, the model antigen (OVA protein) was detected in both fractions. Samples were normalized for the cell number used in necrosis induction. (B) Mice were primed on day 0 and boosted on days 1, 2, 3 with 5 × 106 F/T cells. Seven days after the last vaccination, on day 10, antigen-specific lysis was tested in the spleen and draining lymph nodes (DLNs). As a positive control in this experiment and in all subsequent experiments, mice were vaccinated with 2 × 106 γ-irradiated B78OVA cells. Pooled data of 2 experiments with 3 mice per group are shown. *p < 0.05 as compared with F/T group (Student’s t test).
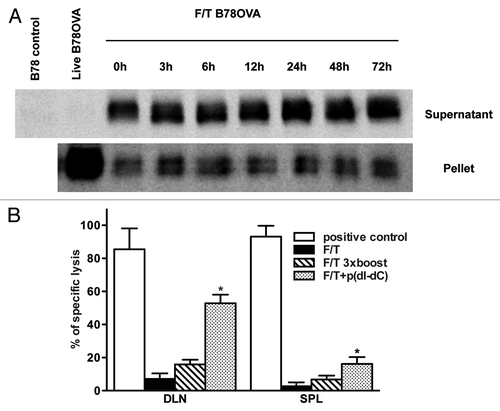
In a second experiment, mice were primed and boosted 3 times with 5x106 necrotic B78OVA cells during 4 consecutive days. This protocol increased the intensity and duration of antigen exposure. However, even this vaccination protocol did not generate antigen-specific CD8+ T cell-dependent immune responses ().
Next, we asked whether the non-immunogenicity of necrotic cells can be reversed by unmethylated double-stranded synthetic DNA, as a model of PAMPs. We electroporated p(dI-dC) into cells prior to necrosis induction and injected them into mice. We observed significantly higher antigen-specific lysis of OVA-pulsed targets in DLNs (but not in the spleen). This was in contrast to control mice, which received previously unmodified necrotic cells (). These results indicate that antigen degradation/elimination alone is not responsible, as we had hypothesized initially, for the failure of necrotic tumor cells to prime CD8+ T cells in vivo. Antigens are still present in necrotic cells, yet other signals are needed in order to induce antigen-specific immune responses.
Non-immunogenicity of sterile necrotic cell death is reversed using double-stranded unmethylated DNA through activation of TLR9/MyD88-dependent pathway
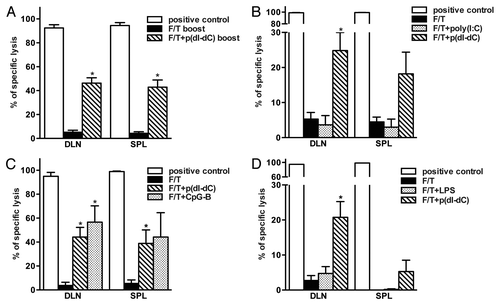
As shown in , p(dI-dC) can reverse the loss of immunogenicity as induced by necrotic cell death. To prove this further and analyze the underlying mechanisms, we repeated the experiment with different TLR ligands. In order to exclude any possible influence of rapid antigen clearance, we performed prime boost experiments, and all subsequent studies were performed according to this protocol. As indicated in , mice primed and boosted with p(dI-dC)-electroporated necrotic tumor cells showed enhanced immune responses as compared with control mice. Cells from mice vaccinated with poly(I:C)-electroporated necrotic tumor cells () failed to lyse antigen-pulsed targets whereas cells from mice vaccinated with CpG-B-electroporated necrotic cells exhibited enhanced lysis (). This suggests that CpG-B-vaccinated mice develop efficient CD8+ T cell-dependent immune responses. In a separate experiment, mice were vaccinated with necrotic cells mixed with p(dI-dC) or LPS so that each mouse received 2x106 cells with 50 μg of model DNA or 100 μg LPS. Seven days after boost, cells from mice vaccinated with necrotic cells mixed with p(dI-dC) but not with LPS exerted CTL activity in vivo ().
Figure 4. Failure of sterile necrosis to induce cross-priming of CD8+ T cells in vivo can be reversed using model DNA p(dI-dC) or CpG-B. (A–C) Mice were primed on day 0 and boosted on day 3 with 1 × 106 F/T B78OVA cells electroporated with p(dI-dC) (A), poly(I:C) (B) or CpG-B (C). In vivo CTL analysis was performed on day 10. (D) Mice were primed on day 0 and boosted on day 3 with 2 × 106 F/T cells mixed with p(dI-dC) or LPS. In vivo CTL was performed as in (A) on day 10. Pooled data of 3 (A), 5 (B), or 2 experiments (C and D) with 3 mice per group are shown. *p < 0.05 as compared with F/T group (Student’s t test).
To elucidate the mechanism of action of model DNA p(dI-dC), prime-boost experiments with electroporated necrotic cells were performed in Myd88−/−, Tlr2−/−, Tlr4−/−, Tlr2−/−Tlr4−/− and Tlr9−/− mice (). These experiments revealed that TLR9 and MyD88 signaling are responsible for the restoration of the immunogenicity of necrotic cells since no antigen-specific lysis was observed in Tlr9−/− and Myd88−/− mice after vaccination. The particulate (pellet), but not the soluble (supernatant), fraction of non-sterile, primary necrotic tumor cells was capable to induce antigen-specific CD8+ T cell responses in vivo ().
Figure 5. Reversal of immunogenicity is TLR9-dependent and relies on MyD88 signaling. (A-C) Wild type, Myd88−/− (A), Tlr2−/−, Tlr4−/−(B), Tlr2−/−Tlr4−/− or Tlr9−/− (C) C57BL/6 mice were primed on day 0 and boosted on day 3 with 1 × 106 p(dI-dC)-electroporated F/T B78OVA cells. Antigen-specific immune responses were analyzed by in vivo CTL assay on day 10. (D) 1 × 106 whole cells, soluble or particulate fractions from p(dI-dC)-electroporated F/T cells were injected into mice on days 0 and 3 and killing activity of CD8+ T cells was evaluated on day 10 as shown in (A). Pooled data from 2 independent experiments are shown with minimum of 3 mice per group. *p < 0.05 as compared with F/T group (Student’s t test). E. B78OVA cells were electroporated with CpG-B or with PBS as in and were subjected to F/T to induce necrosis. Mice were vaccinated with 1 × 106 F/T cells on days 0, 3 and were challenged on the opposite flank with 1 × 104 live B16OVA cells on day 7. As a control, naïve mice or mice vaccinated with γ-irradiated B78OVA cells were used and tumor-free survival was monitored. Data are pooled from 2 independent experiments. *p < 0.05 Log-Rank (Mantel-Cox) test.
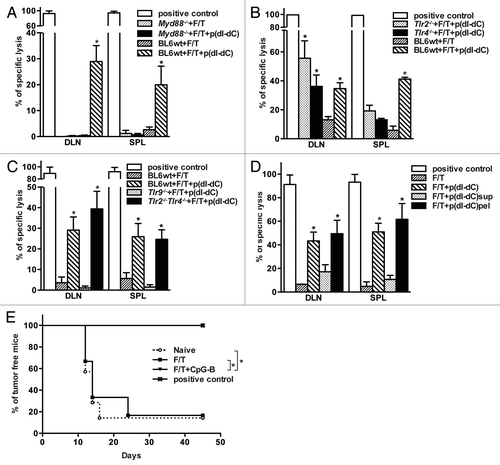
Finally, we vaccinated mice with CpG-B-electroporated (non-sterile) or sterile necrotic B78OVA cells and challenged them with live B16OVA cells. Mice vaccinated with non-sterile necrotic cells were protected from tumor development. Positive control groups, which were vaccinated with γ-irradiated B78OVA cells, also remained tumor-free after challenge with live B16OVA cells. In contrast, all mice vaccinated with primary sterile necrotic cells developed tumors ().
Necrotic cells contain factors that block cross-priming of antigen-specific CD8+ T cells against cell-associated antigens
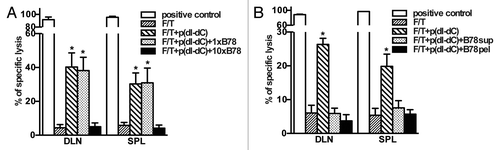
Our results show that unmethylated double-stranded DNA in necrotic cells is required for the elicitation of immune responses. Next, we asked whether necrotic cells contain factors that abolish p(dI-dC)-dependent CD8+ T cell activation. To address this question, 2 × 106 p(dI-dC)-electroporated necrotic B78OVA cells were mixed with 2 × 106 or 2 × 107 B78 whole necrotic cells and injected into mice. In separate experiments, the soluble or particulate fractions of B78 necrotic cells were used for co-injection instead of whole cells. As shown in , mice showed reduced antigen-specific lysis after vaccination with 2 × 107 B78 necrotic cells suggesting that necrotic cells release factors which can dampen the immunogenicity of our PAMP model. These factors were present both in both the soluble and particulate fractions of necrotic cells (), since co-injection of either of them abolished CD8+ T cell-mediated lysis in vivo.
Figure 6. Necrotic cells release factors, which block cross-priming of antigen-specific CD8+ T cells in vivo. (A and B) Necrotic B78OVA cells (F/T) electroporated with p(dI-dC) were mixed with whole B78 necrotic cells (A), or with soluble/particulate fractions from B78 necrotic cells (B) and injected into mice. In vivo CTL was performed 7 d after boost. Pooled data of 3 (A) or 2 experiments (B) with 3 mice per group are shown. *p < 0.05 as compared with 3 × F/T group (Student’s t test).
Discussion
The influence of cell death on the immune system has been a subject of intense investigation. Necrosis is considered to be an immunogenic event and endogenous DAMPs released from dead/dying cells are proposed to trigger adaptive immune responses. Since the initial presentation of the danger theory, various DAMPs have been discovered and their effect on the antigen-specific CD8+ T-cell activation has been studied.
However, in most of the studies performed so far, the immunogenicity of dead cells and of DAMPs has been assayed in vitro, using TCR transgenic animals with non-physiologically high frequency of antigen-specific T cells.Citation4 In some studies, in vitro generated monocyte-derived DC (MoDCs) were used.Citation30 Alternatively, the necrotic cells used were contaminated with mycoplasma,Citation31 or recombinant DAMPs were derived from bacterial sources with no control over the LPS content. Therefore, it is very difficult to draw conclusions from such experiments.Citation32-Citation34 DAMPs have also been tested in vaccination protocols with soluble or bead-bound antigens.Citation2,Citation35 In addition, several studies have been performed based on in vitro or in vivo methods of tumor cell death induction (such as UV irradiation,Citation36 vascular targeting,Citation29,Citation37 radiofrequency or cryoablation).Citation38,Citation39 Such approaches fail to induce ‘pure’ homogeneous primary necrotic cell death and therefore are not appropriate for examining the mechanisms of immunogenicity in detail.
Recent studies clearly indicate that the immunogenicity of dying/dead cells does not correlate with the type of cell death. In several reports,Citation19,Citation20,Citation22,Citation24 it has been shown that cells undergoing types of death other than necrosis can induce subsequent CD8+ T cell-mediated immunity whereas cells succumbing to freeze-thawing cycles fail to do so.
Here, we have specifically analyzed the immunological outcome of sterile primary necrotic cell death in vitro and in vivo. In particular, we wanted to evaluate the influence of necrotic cells on the cross-priming of naïve CD8+ T cells, since triggering of long-lasting antigen-specific cellular immune responses is of great importance for efficient antitumor vaccination strategies. We found that freeze-thawed necrotic cells fail to activate CD8+ T cells in vitro. This effect was not dependent on the cell line used. It is important to mention that even H2Kb-expressing necrotic cells (EG7, B16OVA), which might present antigen directly to CD8+ T cells as live cells, were unable to induce IFNγ production by OT-I splenocytes. To our surprise, CD11c+ splenic DCs exposed to necrotic cells in vitro did not upregulate co-stimulatory molecules (except for a slight upregulation of CD86), which is a typical response of DCs to PAMPs. This could be due to the induction of qualitatively different responses after exposure to sterile necrotic cells. We also observed that untreated (live) or γ-irradiated cells activate DCs similar to LPS. It is important to mention that untreated or γ-irradiated cells consists of a mixture of hypothetically ‘naive’, ‘stressed’, ‘apoptotic’ or ‘necrotic’ cells, which are probably responsible for similar effects on DC activation, whereas freeze-thawed cells consist of 100% trypan blue-positive, necrotic cells.
Mice injected with primary necrotic tumor cells failed to develop antigen-specific CD8+ T-cell activation. This was also seen when a different method, osmotic shock, was used to induce necrosis. Interestingly, even allogeneic necrotic tumor cells failed to trigger OVA-specific CD8+ T-cell responses. These cells are very immunogenic and induce efficient immune responses when injected as such in mice. Our data suggests that primary sterile necrotic cells are not suitable substrates for cross-priming despite the release of ‘hypothetical’ DAMPs.
A number of questions remain unanswered about the in vivo mechanisms of cross-priming, including the substrate specificity (e.g., whole proteins, DRIPs, protein fragments or even peptides)Citation40,Citation41 as well as the antigen concentration,Citation42,Citation43 form (soluble or particulate)Citation44 and persistence.Citation45 Therefore, we asked whether a limitation of antigen levels after necrosis induction might be the reason why T cells were not activated. Prime-boost vaccination of mice using high number (5 × 106) of necrotic cells for 4 d failed to trigger an immune response. At the same time, two injections of 1 × 106 necrotic B78OVA cells electroporated with model DNA induced an immune response. This indicates that a limited availability of the antigen is unlikely to be the sole explanation of the observed phenomena and that the introduction of PAMPs into necrotic cells, converting them into quasi-non-sterile inducers, is necessary for optimal responses. In line with this, our model antigen (ovalbumin) was detected in necrotic cells even after 72 h. However, non-sterile necrotic cells elicited an immune response upon repeated administration only.
The restoration of immunogenicity by model p(dI-dC) or CpG-B DNA that we observed is in live with a recent study.Citation46 In this report, de Brito et al. show that precursors of CD8α+ DCs in vitro require stimulation by unmethylated CpG-B DNA for cross-presentation of necrotic cell-associated antigens.46After electroporation of p(dI-dC), the particulate, but not the supernatant, fraction was immunogenic and as efficient as whole necrotic-cell vaccination. This was the case in spite of the fact that most of the antigen was released into the soluble fraction. A possible explanation for this might relate to the specific form of the antigen, as supported by recent reports.Citation42,Citation44 These studies have shown that particulate antigens can be much more efficiently cross-presented than their soluble counterparts.Citation44 However, in our system, addition of PAMPs was needed even for the particulate form of the antigen to cross-prime CD8+ T cells.
Our data show that - despite the presence of antigens in primary sterile necrotic cells - PAMPs are required for the induction of successful antigen-specific CD8+ T-cell responses. Further studies are needed to address the exact mechanisms of how PAMPs, and in this particular case p(dI-dC) and CpG-B, promote T-cell activation.
Finally, necrotic cells precluded the cross-priming of CD8+ T cells under non-sterile conditions when model DNA was used as a PAMP. This effect was seen with both the soluble and particulate fractions of necrotic tumors. These results suggest that the cross-priming of CD8+ T cells depends not only on the presence of antigens and PAMPs, but also on intracellular factors released during necrosis. Our data suggest that some of these factors negatively regulate CD8+ T cell activation and define whether adaptive immune responses are induced or not - a hypothesis that is a subject of further investigation.
In summary, the results described here not only improve our understanding of the biology of immune responses to dying tumor cells, but are relevant for the design of more efficient cell-based cancer vaccines. This knowledge is essential for the development of novel combination therapy protocols, in which immune-based therapies are combined with conventional strategies for the induction of cell death.
Materials and Methods
Reagents, cell lines and media
Double-stranded alternating deoxypolynucleotide p(dI-dC) was obtained from Amersham, LPS and poly(I:C) from Sigma. CpG ODN-1668 (CpG-B) was synthesized by Operon Biotechnologies.
EG7 cells (OVA transfected clone of EL4 cells) were obtained from ATCC. B78H1wt (B78) is MHC-I negative amelanotic clone derived from mouse B16 melanoma cells.Citation47 B78OVA and CT26OVA cells, which express OVA in the cytoplasm, were generated as previously described.Citation29,Citation48 B16OVA melanoma cells were kindly provided by Dr. Schueler (DKFZ). All cell lines were routinely tested for mycoplasma contamination.
Mice
6–10 weeks old female C57BL/6 mice were obtained from Charles River laboratories. OVA TCR-transgenic OT-I miceCitation49,Citation50 were obtained by Dr. Sauer (MHH). Myd88−/−, Tlr2−/−, Tlr4−/−, Tlr9−/−, Tlr2−/−4−/− mice have previously been described.Citation51-Citation55 Mice were housed under specific pathogen free conditions. All animal experiments were performed according to institutional guidelines and approved by local Animal Care and Use Committee.
Antibodies, fluorescent dyes and flow cytometry analysis
Anti-mouse CD8-APC conjugates was from purchased from eBioscience, anti-mouse I-A/I-E, CD80, CD40, CD86, CD11c and IFNγ antibodies, Rat IgG2a isotype control antibodies as well 7-AAD were obtained from PharMingen. CFSE was from Molecular probes (Karlsruhe). Flow cytometry was performed using FACScan (BD Biosciences) and data were analyzed with Cell Quest (BD Biosciences) and FlowJo (TriStar, Inc.) software.
Induction of cell death
Tumor cells were exposed to three freeze-thawing cycles (F/T, for necrosis induction) in liquid nitrogen or as a control left untreated or exposed to 75Gy γ-irradiation. In one set of experiments, tumor cells (1 × 107/mL) were exposed to distilled water for 40 min at 37°C in order to develop necrosis. Necrosis induction was confirmed by trypan blue staining.
Analysis of antigen-specific immune responses in vitro
Five × 104 necrotic (F/T), γ-irradiated or untreated OVA-expressing cells were co-cultured in vitro with 5 × 105 OT-I splenocytes for 48 h and IFNγ secretion was determined using either intracellular cytokine staining (PharMingen), or IFNγ ELISA (R&D Systems) according to manufacturer’s instructions.
Cell electroporation
B78OVA cells were electroporated with 40 μg unmethylated DNA or dsRNA in 0.4 cm gap cuvette using Bio-Rad gene-pulser (Bio-Rad laboratories). After electroporation, cells were exposed to three freeze-thawing cycles and used for vaccination. In separate experiments, electroporated-necrotic cells were centrifuged at 16,000 g for 20 min and soluble and particulate fractions were separated. Particulate fractions were resuspended in PBS and volume was adjusted to the initial volume of whole cells. Particulate as well as soluble fractions were then used in vaccination experiments.
Vaccination protocols
Mice were vaccinated with the indicated live, γ-irradiated or necrotic cells subcutaneously in the left flank. Antigen-specific lysis was analyzed by in vivo cytotoxic lymphocyte assay (in vivo CTL) or by intracellular cytokine analysis in draining lymph nodes (DLN) and spleen from vaccinated mice. Alternatively, vaccinated mice were challenged with 1 × 104 live B16OVA cells and monitored for tumor-free survival.
In vivo CTL assay
In vivo CTL assay was performed as described before.Citation56 Briefly, single cell suspensions of the splenocytes from donor mice were isolated and used as a target. Target splenocytes were pulsed with OVA257–264 (SIINFEKL from Biosyntan) or left unpulsed. The cells were then labeled with different concentrations of CFSE and were injected into mice intravenously (1–2x107 per each mouse total). After 18 h, DLNs from the vaccination site and spleens were isolated and CTL activity of host CD8+ T cells was analyzed using flow cytometry. Antigen-specific lysis was calculated by measuring CFSEhigh OVA pulsed cell peak reduction as compared with CFSElow OVA un-pulsed cells. Results were expressed as % OVA-specific lysis. Mice vaccinated with γ-irradiated B78OVA cells were used as positive controls.
Immunoblotting analysis
Necrotic B78OVA cells were cultured at 37°C and supernatant or pellet was collected after 0, 3, 6, 12, 24, 48 and 72 h using centrifugation at 16,000 g for 20 min. Equal amounts of necrotic cell supernatant and pellet fractions were separated by SDS gel electrophoresis. Ovalbumin was detected using a rabbit anti-OVA antibody (Chemicon) followed by HRP conjugated goat anti-rabbit antibody (Dako). Membranes were developed using ECL detection reagent from Amersham and analyzed using Gel Doc 2000 (Bio-Rad laboratories). As a negative control B78 cell lysates were used.
Statistical analysis
All data are expressed as means ± SEM. Significance of differences between groups was analyzed using two-tailed, unpaired Student’s t-test. Confidence interval was set up to 95% and differences were considered to be significant when the p value was < 0.05. Tumor free survival was analyzed using Log-Rank (Mantel-Cox) test.
| Abbreviations: | ||
| DAMPs | = | damage associated molecular patterns |
| DLN | = | draining lymph node |
| F/T | = | 3 cycles of freeze-thawing |
| PAMPs | = | pathogen associated molecular patterns |
| p(dI-dC) | = | double-stranded alternating copolymer poly(dI-dC)⋅(dI-dC) |
| SPL | = | spleen |
Disclosure of Potential Conflicts of Interest
Authors declare they have no potential conflicts of interest.
Acknowledgments
This research was supported (in part) by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research and by the Networking Fund of the Helmholtz Association within the Helmholtz Alliance on Immunotherapy of Cancer to MPM.
References
- Ma Y, Conforti R, Aymeric L, Locher C, Kepp O, Kroemer G, et al. How to improve the immunogenicity of chemotherapy and radiotherapy. Cancer Metastasis Rev 2011; 30:71 - 82; http://dx.doi.org/10.1007/s10555-011-9283-2; PMID: 21298323
- Gallucci S, Lolkema M, Matzinger P. Natural adjuvants: endogenous activators of dendritic cells. Nat Med 1999; 5:1249 - 55; http://dx.doi.org/10.1038/15200; PMID: 10545990
- Matzinger P. Tolerance, danger, and the extended family. Annu Rev Immunol 1994; 12:991 - 1045; http://dx.doi.org/10.1146/annurev.iy.12.040194.005015; PMID: 8011301
- Sauter B, Albert ML, Francisco L, Larsson M, Somersan S, Bhardwaj N. Consequences of cell death: exposure to necrotic tumor cells, but not primary tissue cells or apoptotic cells, induces the maturation of immunostimulatory dendritic cells. J Exp Med 2000; 191:423 - 34; http://dx.doi.org/10.1084/jem.191.3.423; PMID: 10662788
- Basu S, Binder RJ, Suto R, Anderson KM, Srivastava PK. Necrotic but not apoptotic cell death releases heat shock proteins, which deliver a partial maturation signal to dendritic cells and activate the NF-kappa B pathway. Int Immunol 2000; 12:1539 - 46; http://dx.doi.org/10.1093/intimm/12.11.1539; PMID: 11058573
- Scaffidi P, Misteli T, Bianchi ME. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 2002; 418:191 - 5; http://dx.doi.org/10.1038/nature00858; PMID: 12110890
- Shi Y, Evans JE, Rock KL. Molecular identification of a danger signal that alerts the immune system to dying cells. Nature 2003; 425:516 - 21; http://dx.doi.org/10.1038/nature01991; PMID: 14520412
- Ishii KJ, Suzuki K, Coban C, Takeshita F, Itoh Y, Matoba H, et al. Genomic DNA released by dying cells induces the maturation of APCs. J Immunol 2001; 167:2602 - 7; PMID: 11509601
- Karikó K, Ni H, Capodici J, Lamphier M, Weissman D. mRNA is an endogenous ligand for Toll-like receptor 3. J Biol Chem 2004; 279:12542 - 50; http://dx.doi.org/10.1074/jbc.M310175200; PMID: 14729660
- Lee J, Chuang TH, Redecke V, She L, Pitha PM, Carson DA, et al. Molecular basis for the immunostimulatory activity of guanine nucleoside analogs: activation of Toll-like receptor 7. Proc Natl Acad Sci U S A 2003; 100:6646 - 51; http://dx.doi.org/10.1073/pnas.0631696100; PMID: 12738885
- Wilkin F, Duhant X, Bruyns C, Suarez-Huerta N, Boeynaems JM, Robaye B. The P2Y11 receptor mediates the ATP-induced maturation of human monocyte-derived dendritic cells. J Immunol 2001; 166:7172 - 7; PMID: 11390464
- Ahrens S, Zelenay S, Sancho D, Hanč P, Kjær S, Feest C, et al. F-actin is an evolutionarily conserved damage-associated molecular pattern recognized by DNGR-1, a receptor for dead cells. Immunity 2012; 36:635 - 45; http://dx.doi.org/10.1016/j.immuni.2012.03.008; PMID: 22483800
- Jiang D, Liang J, Fan J, Yu S, Chen S, Luo Y, et al. Regulation of lung injury and repair by Toll-like receptors and hyaluronan. Nat Med 2005; 11:1173 - 9; http://dx.doi.org/10.1038/nm1315; PMID: 16244651
- Woo M, Hakem R, Mak TW. Executionary pathway for apoptosis: lessons from mutant mice. Cell Res 2000; 10:267 - 78; http://dx.doi.org/10.1038/sj.cr.7290054; PMID: 11191349
- Joza N, Kroemer G, Penninger JM. Genetic analysis of the mammalian cell death machinery. Trends Genet 2002; 18:142 - 9; http://dx.doi.org/10.1016/S0168-9525(01)02618-X; PMID: 11858838
- Liu K, Iyoda T, Saternus M, Kimura Y, Inaba K, Steinman RM. Immune tolerance after delivery of dying cells to dendritic cells in situ. J Exp Med 2002; 196:1091 - 7; http://dx.doi.org/10.1084/jem.20021215; PMID: 12391020
- Albert ML, Sauter B, Bhardwaj N. Dendritic cells acquire antigen from apoptotic cells and induce class I-restricted CTLs. Nature 1998; 392:86 - 9; http://dx.doi.org/10.1038/32183; PMID: 9510252
- Nowak AK, Lake RA, Marzo AL, Scott B, Heath WR, Collins EJ, et al. Induction of tumor cell apoptosis in vivo increases tumor antigen cross-presentation, cross-priming rather than cross-tolerizing host tumor-specific CD8 T cells. J Immunol 2003; 170:4905 - 13; PMID: 12734333
- Janssen E, Tabeta K, Barnes MJ, Rutschmann S, McBride S, Bahjat KS, et al. Efficient T cell activation via a Toll-Interleukin 1 Receptor-independent pathway. Immunity 2006; 24:787 - 99; http://dx.doi.org/10.1016/j.immuni.2006.03.024; PMID: 16782034
- Casares N, Pequignot MO, Tesniere A, Ghiringhelli F, Roux S, Chaput N, et al. Caspase-dependent immunogenicity of doxorubicin-induced tumor cell death. J Exp Med 2005; 202:1691 - 701; http://dx.doi.org/10.1084/jem.20050915; PMID: 16365148
- Ghiringhelli F, Apetoh L, Tesniere A, Aymeric L, Ma Y, Ortiz C, et al. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1beta-dependent adaptive immunity against tumors. Nat Med 2009; 15:1170 - 8; http://dx.doi.org/10.1038/nm.2028; PMID: 19767732
- Obeid M, Tesniere A, Ghiringhelli F, Fimia GM, Apetoh L, Perfettini JL, et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat Med 2007; 13:54 - 61; http://dx.doi.org/10.1038/nm1523; PMID: 17187072
- Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell 2011; 147:728 - 41; http://dx.doi.org/10.1016/j.cell.2011.10.026; PMID: 22078875
- Uhl M, Kepp O, Jusforgues-Saklani H, Vicencio JM, Kroemer G, Albert ML. Autophagy within the antigen donor cell facilitates efficient antigen cross-priming of virus-specific CD8+ T cells. Cell Death Differ 2009; 16:991 - 1005; http://dx.doi.org/10.1038/cdd.2009.8; PMID: 19229247
- Michaud M, Martins I, Sukkurwala AQ, Adjemian S, Ma Y, Pellegatti P, et al. Autophagy-dependent anticancer immune responses induced by chemotherapeutic agents in mice. Science 2011; 334:1573 - 7; http://dx.doi.org/10.1126/science.1208347; PMID: 22174255
- Hirt UA, Leist M. Rapid, noninflammatory and PS-dependent phagocytic clearance of necrotic cells. Cell Death Differ 2003; 10:1156 - 64; http://dx.doi.org/10.1038/sj.cdd.4401286; PMID: 14502239
- Brouckaert G, Kalai M, Krysko DV, Saelens X, Vercammen D, Ndlovu MN, et al. Phagocytosis of necrotic cells by macrophages is phosphatidylserine dependent and does not induce inflammatory cytokine production. Mol Biol Cell 2004; 15:1089 - 100; http://dx.doi.org/10.1091/mbc.E03-09-0668; PMID: 14668480
- Scheffer SR, Nave H, Korangy F, Schlote K, Pabst R, Jaffee EM, et al. Apoptotic, but not necrotic, tumor cell vaccines induce a potent immune response in vivo. Int J Cancer 2003; 103:205 - 11; http://dx.doi.org/10.1002/ijc.10777; PMID: 12455034
- Gamrekelashvili J, Krüger C, von Wasielewski R, Hoffmann M, Huster KM, Busch DH, et al. Necrotic tumor cell death in vivo impairs tumor-specific immune responses. J Immunol 2007; 178:1573 - 80; PMID: 17237406
- Somersan S, Larsson M, Fonteneau JF, Basu S, Srivastava P, Bhardwaj N. Primary tumor tissue lysates are enriched in heat shock proteins and induce the maturation of human dendritic cells. J Immunol 2001; 167:4844 - 52; PMID: 11673488
- Salio M, Cerundolo V, Lanzavecchia A. Dendritic cell maturation is induced by mycoplasma infection but not by necrotic cells. Eur J Immunol 2000; 30:705 - 8; http://dx.doi.org/10.1002/1521-4141(200002)30:2<705::AID-IMMU705>3.0.CO;2-P; PMID: 10671230
- Singh-Jasuja H, Scherer HU, Hilf N, Arnold-Schild D, Rammensee HG, Toes RE, et al. The heat shock protein gp96 induces maturation of dendritic cells and down-regulation of its receptor. Eur J Immunol 2000; 30:2211 - 5; PMID: 10940912
- Vabulas RM, Braedel S, Hilf N, Singh-Jasuja H, Herter S, Ahmad-Nejad P, et al. The endoplasmic reticulum-resident heat shock protein Gp96 activates dendritic cells via the Toll-like receptor 2/4 pathway. J Biol Chem 2002; 277:20847 - 53; http://dx.doi.org/10.1074/jbc.M200425200; PMID: 11912201
- Vabulas RM, Ahmad-Nejad P, Ghose S, Kirschning CJ, Issels RD, Wagner H. HSP70 as endogenous stimulus of the Toll/interleukin-1 receptor signal pathway. J Biol Chem 2002; 277:15107 - 12; http://dx.doi.org/10.1074/jbc.M111204200; PMID: 11842086
- Shi Y, Zheng W, Rock KL. Cell injury releases endogenous adjuvants that stimulate cytotoxic T cell responses. Proc Natl Acad Sci U S A 2000; 97:14590 - 5; http://dx.doi.org/10.1073/pnas.260497597; PMID: 11106387
- Sancho D, Joffre OP, Keller AM, Rogers NC, Martínez D, Hernanz-Falcón P, et al. Identification of a dendritic cell receptor that couples sensing of necrosis to immunity. Nature 2009; 458:899 - 903; http://dx.doi.org/10.1038/nature07750; PMID: 19219027
- Blakey DC, Westwood FR, Walker M, Hughes GD, Davis PD, Ashton SE, et al. Antitumor activity of the novel vascular targeting agent ZD6126 in a panel of tumor models. Clin Cancer Res 2002; 8:1974 - 83; PMID: 12060643
- den Brok MH, Sutmuller RP, Nierkens S, Bennink EJ, Frielink C, Toonen LW, et al. Efficient loading of dendritic cells following cryo and radiofrequency ablation in combination with immune modulation induces anti-tumour immunity. Br J Cancer 2006; 95:896 - 905; http://dx.doi.org/10.1038/sj.bjc.6603341; PMID: 16953240
- den Brok MH, Sutmuller RP, Nierkens S, Bennink EJ, Toonen LW, Figdor CG, et al. Synergy between in situ cryoablation and TLR9 stimulation results in a highly effective in vivo dendritic cell vaccine. Cancer Res 2006; 66:7285 - 92; http://dx.doi.org/10.1158/0008-5472.CAN-06-0206; PMID: 16849578
- Norbury CC, Basta S, Donohue KB, Tscharke DC, Princiotta MF, Berglund P, et al. CD8+ T cell cross-priming via transfer of proteasome substrates. Science 2004; 304:1318 - 21; http://dx.doi.org/10.1126/science.1096378; PMID: 15166379
- Lev A, Takeda K, Zanker D, Maynard JC, Dimberu P, Waffarn E, et al. The exception that reinforces the rule: crosspriming by cytosolic peptides that escape degradation. Immunity 2008; 28:787 - 98; http://dx.doi.org/10.1016/j.immuni.2008.04.015; PMID: 18549799
- Kurts C, Miller JF, Subramaniam RM, Carbone FR, Heath WR. Major histocompatibility complex class I-restricted cross-presentation is biased towards high dose antigens and those released during cellular destruction. J Exp Med 1998; 188:409 - 14; http://dx.doi.org/10.1084/jem.188.2.409; PMID: 9670054
- Heath WR, Carbone FR. Cross-presentation, dendritic cells, tolerance and immunity. Annu Rev Immunol 2001; 19:47 - 64; http://dx.doi.org/10.1146/annurev.immunol.19.1.47; PMID: 11244030
- Li M, Davey GM, Sutherland RM, Kurts C, Lew AM, Hirst C, et al. Cell-associated ovalbumin is cross-presented much more efficiently than soluble ovalbumin in vivo. J Immunol 2001; 166:6099 - 103; PMID: 11342628
- Yewdell JW. The seven dirty little secrets of major histocompatibility complex class I antigen processing. Immunol Rev 2005; 207:8 - 18; http://dx.doi.org/10.1111/j.0105-2896.2005.00309.x; PMID: 16181323
- de Brito C, Tomkowiak M, Ghittoni R, Caux C, Leverrier Y, Marvel J. CpG promotes cross-presentation of dead cell-associated antigens by pre-CD8α+ dendritic cells [corrected]. [corrected] J Immunol 2011; 186:1503 - 11; http://dx.doi.org/10.4049/jimmunol.1001022; PMID: 21187449
- Silagi S. Control of pigment production in mouse melanoma cells in vitro. Evocation and maintenance. J Cell Biol 1969; 43:263 - 74; http://dx.doi.org/10.1083/jcb.43.2.263; PMID: 4981070
- Klein C, Bueler H, Mulligan RC. Comparative analysis of genetically modified dendritic cells and tumor cells as therapeutic cancer vaccines. J Exp Med 2000; 191:1699 - 708; http://dx.doi.org/10.1084/jem.191.10.1699; PMID: 10811863
- Clarke SR, Barnden M, Kurts C, Carbone FR, Miller JF, Heath WR. Characterization of the ovalbumin-specific TCR transgenic line OT-I: MHC elements for positive and negative selection. Immunol Cell Biol 2000; 78:110 - 7; http://dx.doi.org/10.1046/j.1440-1711.2000.00889.x; PMID: 10762410
- Hogquist KA, Jameson SC, Heath WR, Howard JL, Bevan MJ, Carbone FR. T cell receptor antagonist peptides induce positive selection. Cell 1994; 76:17 - 27; http://dx.doi.org/10.1016/0092-8674(94)90169-4; PMID: 8287475
- Hemmi H, Takeuchi O, Kawai T, Kaisho T, Sato S, Sanjo H, et al. A Toll-like receptor recognizes bacterial DNA. Nature 2000; 408:740 - 5; http://dx.doi.org/10.1038/35047123; PMID: 11130078
- Hoshino K, Takeuchi O, Kawai T, Sanjo H, Ogawa T, Takeda Y, et al. Cutting edge: Toll-like receptor 4 (TLR4)-deficient mice are hyporesponsive to lipopolysaccharide: evidence for TLR4 as the Lps gene product. J Immunol 1999; 162:3749 - 52; PMID: 10201887
- Takeuchi O, Hoshino K, Kawai T, Sanjo H, Takada H, Ogawa T, et al. Differential roles of TLR2 and TLR4 in recognition of gram-negative and gram-positive bacterial cell wall components. Immunity 1999; 11:443 - 51; http://dx.doi.org/10.1016/S1074-7613(00)80119-3; PMID: 10549626
- Adachi O, Kawai T, Takeda K, Matsumoto M, Tsutsui H, Sakagami M, et al. Targeted disruption of the MyD88 gene results in loss of IL-1- and IL-18-mediated function. Immunity 1998; 9:143 - 50; http://dx.doi.org/10.1016/S1074-7613(00)80596-8; PMID: 9697844
- Spiller S, Dreher S, Meng G, Grabiec A, Thomas W, Hartung T, et al. Cellular recognition of trimyristoylated peptide or enterobacterial lipopolysaccharide via both TLR2 and TLR4. J Biol Chem 2007; 282:13190 - 8; http://dx.doi.org/10.1074/jbc.M610340200; PMID: 17353199
- Oehen S, Brduscha-Riem K. Differentiation of naive CTL to effector and memory CTL: correlation of effector function with phenotype and cell division. J Immunol 1998; 161:5338 - 46; PMID: 9820507