Abstract
Mice with an impaired Type I interferon (IFN) signaling (IFNAR1- and IFNβ-deficient mice) display an increased susceptibility toward v-ABL-induced B-cell leukemia/lymphoma. The enhanced leukemogenesis in the absence of an intact Type I IFN signaling is caused by alterations within the tumor environment. Deletion of Ifnar1 in tumor cells (as obtained in Ifnar1f/f CD19-Cre mice) failed to impact on disease latency or type. In line with this observation, the initial transformation and proliferative capacity of tumor cells were unaltered irrespective of whether the cells expressed IFNAR1 or not. v-ABL-induced leukemogenesis is mainly subjected to natural killer (NK) cell-mediated tumor surveillance. Thus, we concentrated on NK cell functions in IFNAR1 deficient animals. Ifnar1-/- NK cells displayed maturation defects as well as an impaired cytolytic activity. When we deleted Ifnar1 selectively in mature NK cells (by crossing Ncr1-iCre mice to Ifnar1f/f animals), maturation was not altered. However, NK cells derived from Ifnar1f/f Ncr1-iCre mice showed a significant cytolytic defect in vitro against the hematopoietic cell lines YAC-1 and RMA-S, but not against the melanoma cell line B16F10. Interestingly, this defect was not related to an in vivo phenotype as v-ABL-induced leukemogenesis was unaltered in Ifnar1f/f Ncr1-iCre compared with Ifnar1f/f control mice. Moreover, the ability of Ifnar1f/f Ncr1-iCre NK cells to kill B16F10 melanoma cells was unaltered, both in vitro and in vivo. Our data reveal that despite the necessity for Type I IFN in NK cell maturation the expression of IFNAR1 on mature murine NK cells is not required for efficient tumor surveillance.
Keywords: :
Introduction
Type I IFNs were identified and intensively studied as cytokines induced during viral infections. Type I IFN exerts biological functions even in the absence of acute infection, suggesting an important role in various physiological processes. Moreover, Type I IFN is constitutively expressed at a low level to ensure the maintenance of cellular homeostasis.Citation1 In hematopoietic organs, this weak IFN signaling affects the development of specific cell lineages and regulates the cell cycle of hematopoietic stem cells.Citation2-Citation5 However, how IFN signaling contributes to immunosurveillance of distinct types of tumors is only partially understood, despite the clinical use of IFN for cancer therapy.Citation6,Citation7
The concept that the immune system not only protects the host against tumor development but also shapes tumor immunogenicity is the rationale for the cancer immunoediting hypothesis.Citation8,Citation9 Type I IFN has been reported to be essential for cancer immunoediting. Mice lacking IFNα receptor 1 (Ifnar1−/−) display a significantly higher susceptibility to develop primary methylcholanthrene-induced sarcomas than wild type mice. Upon transplantation of IFNAR1-deficient tumors into wild type mice, the responsiveness of recipients to Type I IFN leads to tumor rejection. In contrast, upon transplantation into Ifnar1−/− mice IFNAR1-deficient tumors grow.Citation10 Further experiments using bone marrow chimeras led to the conclusion that the hematopoietic cells of the host need to be sensitive to Type I IFN for tumor rejection and that endogenously produced or therapeutically administered Type I IFN mediates its antitumor effects mainly through acting on host hematopoietic cells.Citation9,Citation11
Several mechanisms have been postulated to explain how Type I IFN stimulates the immune system. On the one hand, Type I IFN has been implicated in innate immunity, potentiating natural killer (NK) cell proliferation and activity. On the other hand, Type I IFN appears to influence adaptive immune responses by increasing the expression of MHC class I (and II) molecules, enhancing antigen presentation, and promoting the proliferation, expansion and survival of CD8+ T cells.Citation9
NK cell development in the adult mouse is best characterized in the bone marrow, although some reports suggest sites where alternative developmental routes take place.Citation12,Citation13 Developmental or differentiation stages have been described based on sequential acquisition of NK cell specific markers and functional competence. The earliest NK cell progenitor (NKP) is defined as a bone marrow cell expressing CD122, the shared β-chain of the interleukin (IL)-2 and IL-15 receptor, and lacking any lineage specific markers. The next stage of differentiation is an immature NK (iNK) cell characterized by the expression of NK1.1, but not DX5, followed by inhibitory and activating Ly49 receptors. As iNK cells differentiate further they display NK1.1, DX5 and Ly49 receptors, and are then called mature NK (mNK) cells. Ncr1 (also known as NKp46) is expressed during the iNK cell stage and its expression persists constitutively. mNK cells leave the bone marrow and reside in the blood as well as in the spleen, liver, lung and various other organs, where they continue to mature.Citation12
Swann and colleagues demonstrated that Type I IFN is an early and critical regulator of NK cell antitumor activity, for example in the methylcholanthrene-induced sarcoma model or against the NK cell-sensitive RMA-S cancer cell line. Moreover, Type I IFN is critical for the efficacy of cytokine-based immunotherapy of lung metastases provoked by 3LL or B16F10 tumor cells.Citation14 However, the role of Type I IFN in NK cell antitumor responses, including that against B cell leukemia, still remains largely elusive.
In this study, we addressed the importance of Type I IFN signaling on the NK cell-mediated immunosurveillance of v-Abl oncogene-driven leukemia.Citation15 This tumor model is of great clinical relevance as it represents leukemogenesis in patients in whom the ABL gene is activated by the t(9;22) chromosomal translocation (Philadelphia chromosome). The resulting fusion protein, BCR/ABL, is a constitutively active tyrosine kinase and drives both chronic and acute lymphoid leukemia.Citation16,Citation17
Our study reveals an enhanced susceptibility of mice lacking functional Type I IFN signaling for v-Abl-induced leukemia/lymphoma. Increased tumor incidence can be linked to defects in NK cell-mediated tumor surveillance. Using a novel Ncr1-iCre mouse model, we further demonstrate an unexpected role for Type I IFN in NK cell development.
Results
Increased frequency ofv-Abl-induced B cell leukemia/lymphoma in Ifnar1−/− and Ifnb−/− mice
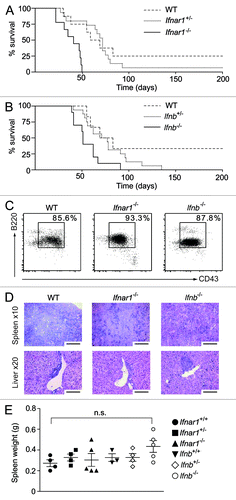
We have previously reported that Tyk2−/− mice are more susceptible to v-Abl-induced leukemia/lymphoma than control mice.Citation15 As the Janus kinase TYK2 is critical in the signaling pathway elicited by Type I IFN, we speculated that impairments in Type I IFN signaling might relate to the higher incidence and shortened latency of v-Abl-induced tumors in Tyk2−/− mice. To test this, we challenged newborn wild type, Ifnar1+/−, Ifnar1−/−, Ifnb+/− and Ifnb−/− mice with a replication-deficient, ecotropic form of the Abelson murine leukemia virus (A-MuLV) via intraperitoneal injection. B lymphoid leukemia/lymphoma formation was significantly accelerated in mice with impaired Type I IFN signaling as compared with control animals (). Notably, Ifnar1−/− mice display a more severe phenotype than Ifnb−/− mice. This is most likely due to the biological activity of other Type I IFNs, including IFNα. Transformed B cells were found to be B220+/CD43+/CD19+ by flow cytometry, and this did not differ in any of the genotypes tested (). Similarly, upon disease onset, the phenotype of the disease was homogenous, and, in affected mice, symptoms such as splenomegaly, enlarged liver and lymph nodes and increased white blood cell (WBC) numbers as well as the extent of leukemic cell infiltration into hematopoietic organs were comparable across all experimental groups (; Figs. S1A and B and data not shown).
Figure 1. In vivo analysis of oncogenic A-MuLV challenge. Newborn wild type (WT), Ifnar1+/−, Ifnar1−/−, Ifnb+/− and Ifnb−/− mice were injected intraperitoneally with a replication incompetent mouse pathogenic Abelson virus (A-MuLV) and monitored for disease development. (A, B) Kaplan-Meier plots of (A) WT, Ifnar1+/− and Ifnar1−/− littermates (n = 8, n = 15 and n = 9, respectively) and (B) WT, Ifnb+/− and Ifnb−/− littermates (n = 6, n = 16 and n = 10, respectively) infected with A-MuLV. Analyses of median survival demonstrate that leukemia proceeds significantly faster in Ifnar1−/− and Ifnb−/− mice than in WT control animals (A: Ifnar1−/− 41 d; wild type 72 d; p < 0.001; B: Ifnb −/− 51 d; WT 76 d; p < 0.05). (C) Representative FACS plots of lymph nodes infiltrated with leukemic cells. Percentages indicate the portion of CD19+B220+CD43+ cells, gated on living cells in the lymph nodes. (D) H&E-stained histologic sections show comparably dense tumor cell infiltrations in spleens and livers of WT, Ifnar1−/− and Ifnb−/− mice. Scale bars represent 100 μm in x10 and 50 μm in x20 magnification. (E) Spleen weights after sacrificing the diseased mice. Results are reported as means ± SEM.
Ifnar1−/− and Ifnb−/− bone marrow cells are readily transformed by the v-Abl oncogene
Genotype-dependent differences in disease latency may be explained by the fact that mice have increased B cell numbers or are more readily transformed upon the oncogenic challenge. Thus, we tested whether the responsiveness to and/or the production of Type I IFN is essential for cell transformation by A-MuLV in vitro. Colony formation assays were performed and failed to reveal any major differences in the transformation capacity. As illustrated in infection of Ifnar1−/−, Ifnb−/− and respective control bone marrow cells with A-MuLV resulted in comparable numbers of colonies. In addition, in vitro transformation of Ifnar1−/− and Ifnb−/− bone marrow cells resulted in equally efficient outgrowth of stable cell lines (wild type: 9/10; 90%; Ifnar1−/−: 8/9; 88%; Ifnb−/−: 9/11; 82%). Immunophenotyping of these cell lines revealed homogenous populations of B220+/CD19+/CD43+ and Gr1−/CD11b− pro-B lymphocytes (data not shown).
Figure 2. Characterization of B lymphoid transformation, proliferation and cell cycle-profiles in the absence of Type I IFN signaling. A-MuLV–induced colony formation of (A) WT, Ifnar1+/−, Ifnar1−/− and (B) WT, Ifnb+/−, Ifnb−/− bone marrow cells in methylcellulose. Summarized data obtained from A-MuLV–induced colony formation assays of (C) WT, Ifnar1+/−, Ifnar1−/− and (D) WT, Ifnb+/−, Ifnb−/− bone marrow cells showed non-significant (n.s.) differences. Data represent means ± SEM of four samples (one representative experiment out of three is shown). (E) [3H]Thymidine incorporation of bone marrow–derived WT (26.23 cpm ± 1.09; n = 18), Ifnar1−/− (27.93 cpm ± 0.86; n = 18) and Ifnb−/− (28.96 cpm ± 0.69; n = 18) A-MuLV–transformed cell lines. Data revealed no differences and summarize results of three independent experiments (means ± SEM). (F) Representative cell cycle profiles of A-MuLV transformed WT, Ifnar1−/− and Ifnb−/− bone marrow cell lines analyzed by FACS (sub-G1: WT: 6.5% ± 0.4; Ifnar1−/−: 4.5% ± 1.3; Ifnb−/−: 5.4% ± 0.6; G0/G1: WT: 42.4% ± 2.5; Ifnar1−/−: 31.4% ± 2.1; Ifnb−/−: 38.1% ± 3.9; S-phase: WT: 34.0% ± 1.8; Ifnar1−/−: 41.4% ± 2.7; Ifnb−/−: 36.3% ± 0.8; G2/M: WT: 17.1% ± 0.8; Ifnar1−/−: 23.0% ± 0.7; Ifnb−/−: 20.2% ± 3.7). Data revealed no differences and represent means ± SEM (G) CFSE dye dilution assay. Tumor cells were labeled with CFSE and cultured for up to 48 h without cytokines in standard culture medium. After 4, 8, 24 and 48 h, cells were harvested and the incremental loss of CFSE intensity corresponding to cell divisions was analyzed by flow cytometry.
![Figure 2. Characterization of B lymphoid transformation, proliferation and cell cycle-profiles in the absence of Type I IFN signaling. A-MuLV–induced colony formation of (A) WT, Ifnar1+/−, Ifnar1−/− and (B) WT, Ifnb+/−, Ifnb−/− bone marrow cells in methylcellulose. Summarized data obtained from A-MuLV–induced colony formation assays of (C) WT, Ifnar1+/−, Ifnar1−/− and (D) WT, Ifnb+/−, Ifnb−/− bone marrow cells showed non-significant (n.s.) differences. Data represent means ± SEM of four samples (one representative experiment out of three is shown). (E) [3H]Thymidine incorporation of bone marrow–derived WT (26.23 cpm ± 1.09; n = 18), Ifnar1−/− (27.93 cpm ± 0.86; n = 18) and Ifnb−/− (28.96 cpm ± 0.69; n = 18) A-MuLV–transformed cell lines. Data revealed no differences and summarize results of three independent experiments (means ± SEM). (F) Representative cell cycle profiles of A-MuLV transformed WT, Ifnar1−/− and Ifnb−/− bone marrow cell lines analyzed by FACS (sub-G1: WT: 6.5% ± 0.4; Ifnar1−/−: 4.5% ± 1.3; Ifnb−/−: 5.4% ± 0.6; G0/G1: WT: 42.4% ± 2.5; Ifnar1−/−: 31.4% ± 2.1; Ifnb−/−: 38.1% ± 3.9; S-phase: WT: 34.0% ± 1.8; Ifnar1−/−: 41.4% ± 2.7; Ifnb−/−: 36.3% ± 0.8; G2/M: WT: 17.1% ± 0.8; Ifnar1−/−: 23.0% ± 0.7; Ifnb−/−: 20.2% ± 3.7). Data revealed no differences and represent means ± SEM (G) CFSE dye dilution assay. Tumor cells were labeled with CFSE and cultured for up to 48 h without cytokines in standard culture medium. After 4, 8, 24 and 48 h, cells were harvested and the incremental loss of CFSE intensity corresponding to cell divisions was analyzed by flow cytometry.](/cms/asset/0cefc3ba-fd90-4c23-aad9-c81d46479558/koni_a_10921284_f0002.gif)
In vivo A-MuLV transforms B lymphoid precursor cells.Citation18,Citation19 As Ifnb−/− mice were reported to bear alterations within the B lymphoid lineageCitation20 we investigated B cell development in Ifnar1−/− and Ifnb−/− mice. According to Hardy and colleagues, specific developmental stages of B cells can be distinguished by differential cell surface expression of maturation markers.Citation21-Citation23 The developmental stages range from pre-pro-B cells (B220+/CD43hi/CD19−/BP-1−), early pro-B cells (B220+/CD43hi/CD19+/BP-1−), late pro-B cells (B220+/CD43hi/CD19+/BP-1+), pre-B cells (B220+/CD43lo/IgM−/IgD−), immature B cells (B220+/CD43lo/IgM+/IgD−) to mature B cells (B220+/CD43lo/IgM+/IgD+). In our hands, bone marrow of Ifnar1−/− mice displayed significantly decreased numbers of pre-pro-B cells, whereas in Ifnb−/− mice the numbers of early pro-B cells were strongly reduced (Figs. S2A–D). We failed to detect any significant differences in the numbers of late pro-B, pre-B, immature and mature B cells in the spleen and lymph nodes in wild type vs. Ifnar1−/− or Ifnb−/− mice. The size and weight of hematopoietic organs were also comparable (data not shown). In summary, these observations suggest an important role for Type I IFN signaling at the earliest stages of B cell development (common lymphoid progenitor up to early pro-B cell transition), but does not explain the reduced disease latency of Ifnar1−/− or Ifnb−/− mice upon A-MuLV infection.
To investigate whether Type I IFN influences the proliferative potential of transformed cells, [3H]thymidine incorporation assays were performed. No differences in [3H]thymidine uptake were detectable between wild type, Ifnar1−/− and Ifnb−/− derived cell lines (). These experiments were complemented by a CFSE dye dilution assay that confirmed the comparable proliferative potential of the transformed cell lines (). In line with this, doubling times were superimposable: wild type: 22 h ± 0.06; Ifnar1−/−: 21.9 h ± 0.05; Ifnb−/−: 22.26 h ± 0.2. Propidium iodide (PI) staining was done to analyze not only cell cycle distribution but also to determine the amount of apoptotic cells (which in this assay is reflected by the sub-G1 population). No significant differences were observed in these parameters (). Taken together, these in vitro experiments revealed neither an essential role for Type I IFN in A-MuLV-induced cellular transformation nor an alteration in proliferative capacity and/or apoptotic behavior.
Loss of Type I IFN response does not alter tumor cell intrinsic properties
We next asked whether impaired Type I IFN signaling in the tumor cells themselves affects disease development. To abrogate Type I IFN signaling in the tumor cell compartment we crossed conditional Ifnar1 knockout (Ifnar1f/f) mice to CD19-Cre transgenic animals. Newborn Ifnar1f/f CD19-Cre and littermate controls were infected with A-MuLV to promote leukemia. The survival curves of Ifnar1f/f CD19-Cre mice and control animals were superimposable (). In line with this observation, the phenotype of diseased mice was comparable, as reflected by tumor cell infiltration into hematopoietic organs (). Ifnar1 deletion was confirmed by flow cytometry and PCR (Figs. S3A and B). Taken together, these data are in line with the concept that Type I IFN signaling is of minor importance in the tumor cell compartment itself.
Figure 3. In vivo analysis of oncogenic A-MuLV challenge in Ifnar1f/f CD19-Cre animals. (A) Kaplan-Meier plot of newborn Ifnar1f/f CD19-Cre and Ifnar1f/f littermate mice infected via i.p. with A-MuLV (Ifnar1f/f CD19-Cre: n = 14, Ifnar1f/f: n = 13). (B) H&E-stained histologic sections of spleens and livers derived from diseased mice. Scale bars represent 100 μm in x10 and 50 μm in 20x magnification. (C) Percentages of tumor cells infiltrating bone marrow (BM), spleen (SP) and lymph nodes (LN) of Ifnar1f/f and Ifnar1f/f CD19-Cre mice.
In addition to a negligible role of Type I IFN signaling in tumor cells, the autocrine production of Type I IFN by tumor cells was also shown not to be related to a tumor cell-intrinsic effect. In vitro A-MuLV-transformed cell lines were generated from IFNβ reporter mice, where the Ifnb gene is replaced by the luciferase gene on one allele.Citation24 These cells were transferred subcutaneously into nu/nu recipient mice. After injection of a bioluminescent substrate, mice were monitored in a dedicated imager. Even though the functionality of the system was confirmed by a virus-based test, no sign of luciferase activity, i.e., no IFNβ production, could be detected (Fig. S3C).
NK cell maturation is impaired in Ifnar1−/− mice
We have previously shown that NK cells are the main mediators of immunosurveillance against A-MuLV-induced B lymphoid leukemia,Citation15,Citation25,Citation26 and we have linked the increased tumor susceptibility of Tyk2−/− mice to an impaired NK cell function.Citation15
To elucidate the role of Type I IFN signaling in NK cells in the Abelson tumor model, we crossed Ifnar1f/f mice to Ncr1-iCre transgenic animals.Citation27 The efficient deletion of Ifnar1 in the Ifnar1f/f Ncr1-iCre mice was verified by PCR (Fig. S5A) and flow cytometry (Figs. S5B and C). We then characterized the influence of the deletion on the NK cell compartment. No significant differences in NK cell development () in the bone marrow were detected in Ifnar1f/f Ncr1-iCre and Ifnar1f/f as well as in Ifnar1−/− mice (). IFNAR1 expression in bone marrow was assessed by flow cytometry. As expected and in line with the expression pattern of NKp46/NCRI, the deletion of Ifnar1 was not detectable in NKPs or iNK cells but was evident in mNK cells of the bone marrow, with the highest deletion efficiency in the most mature NK cell stage ().
Figure 4. Role of IFNAR1 in NK cell development. (A) Scheme of NCRI expression during NK cell development. (B) Frequency of BM NK precursor (NKP), immature (iNK) and mature NK (mNK) cells obtained from Ifnar1−/−, Ifnar1f/f Ncr1-iCre and WT mice. Bar graphs show the percentage of NKPs (DX5- NK1.1-, gated on Lin-CD122+), iNKs (DX5-NK1.1+, gated on Lin-CD122+) and mNKs (DX5+NK1.1+, gated on Lin-CD122+) in BM. (C) IFNAR1 expression on NKP, iNK and mNK subpopulations obtained from Ifnar1−/−, Ifnar1f/f Ncr1-iCre and Ifnar1f/f mice. MFI = mean fluorescence intensity. (D) Maturation status of splenic Ifnar1f/f, Ifnar1f/f Ncr1-iCre and Ifnar1−/− NK cells by flow cytometric analysis of CD27/CD11b expression. Gated on the entity of splenic NK1.1+ CD3- NK cells (set to 100%), bar graphs display the percentages of NK cells in four stages corresponding to progressive maturation: first CD27-CD11b-, second CD27+CD11b-, third CD27+CD11b+ and fourth CD27-CD11b+. (E) Bar graphs show the percentage of KLRG1+ cells (left panel) and DNAM-1+ cells (middle panel) in the NK1.1+CD3- NK cell compartment of the spleen. Bar graph on the right displays NKG2D expression level (mean fluorescence intensity, MFI) on splenic NK cells from Ifnar1−/−, Ifnar1f/f Ncr1-iCre and Ifnar1f/f mice. Pooled results from at least two independent experiments are presented as means ± SEM (*p < 0.05, **p < 0.01, ***p < 0.001).
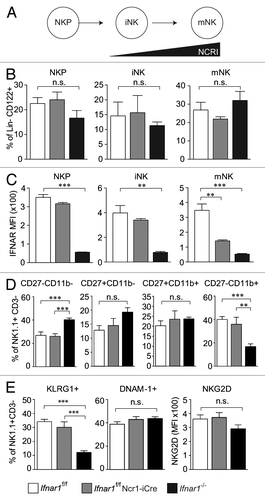
To further investigate the role of Type I IFN response in NK cells, NK cell maturation in the periphery was assessed in Ifnar1−/− as well as Ifnar1f/f Ncr1-iCre mice. Cytofluorometric analysis of freshly isolated splenic NK cells from Ifnar1f/f Ncr1-iCre and Ifnar1f/f control mice did not reveal any obvious differences in NK-cell numbers. However, in line with a previous publication,Citation14 splenic NK cell numbers were reduced in Ifnar1−/− mice (Fig. S5D). In addition, we revealed a pronounced defect in final maturation of Ifnar1−/− NK cells. Complete knockout of Ifnar1 resulted indeed in significantly reduced numbers of mature NK cells (CD27-CD11b+), whereas the NK cell-specific Ifnar1 deletion had no impact on NK cell maturationCitation28,Citation29 (). NK cells were further analyzed for expression of the NK cell surface markers KLRG1, DNAM-1, NKG2D, CD122 (IL-2Rβ), CD16/CD32, Ly49C/I and Ly49D. No differences in the expression of the tested markers were detected ( and data not shown) with one exception: Ifnar1−/− mice harbored significantly fewer KLRG1+ NK cells (), which correlates with the lower numbers of mature NK cells. Again, no differences between Ifnar1f/f Ncr1-iCre and Ifnar1f/f mice were observed. Our data demonstrate that Type I IFN is essential for NK cell maturation but does not require the presence of the Type I IFN receptor on the NK cells themselves.
Ifnar1−/− and Ifnb−/− NK cells show an impaired tumor target cell lysis
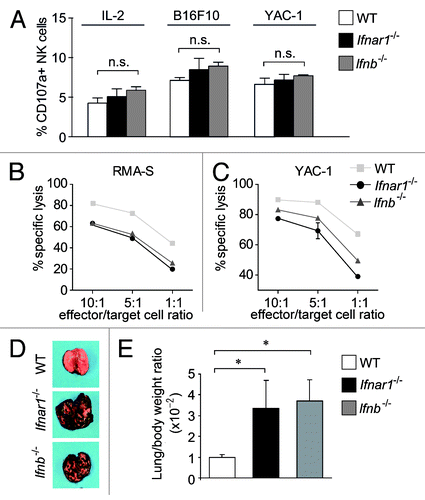
To assess NK cell function in the absence of Type I IFN signaling, we purified splenic NK cells from Ifnar1−/−, Ifnb−/− and control mice. In order to kill their targets, NK cells first need to aggregate with them and degranulate. When we assessed degranulation by flow cytometric analysis of CD107a on the NK cell surface, we did not observe any differences regardless of the NK cell genotype or target cells used (). In addition, we failed to detect alterations in the expression of cytolytic mediators such as perforin, granzyme A and B as well as IFNγ in NK cells derived from Ifnar1−/−, Ifnb−/−, Ifnar1f/f Ncr1-iCre and control mice (Fig. S6). Differences only became evident when we looked at target cell lysis itself. As documented in cytotoxicity assays, Ifnar1−/− and Ifnb−/− NK cells were significantly less capable of killing their targets as compared with wild type NK cells (). To verify this functional defect in vivo, we made use of a non-hematopoietic tumor model by injecting B16F10 melanoma cells into Ifnar1−/−, Ifnb−/− and wild type mice (). B16F10 cells are known to be under the immunologic control of NK cell-mediated clearance.Citation30,Citation31 Two weeks after the injection of B16F10 cells, the tumor cell infiltrates in the lungs of recipient mice were compared. At this time point, Ifnar1−/− and Ifnb−/− mice were severely affected, and showed significant infiltration of tumor cells in the lungs, which was further reflected by an increase in organ weight. These observations indicate that NK cells of Ifnar1−/− and Ifnb−/− mice are significantly impaired in their effector function.
Figure 5. Characterization of Ifnar1−/− and Ifnb−/− NK cells. (A) A FACS-based degranulation assay measuring surface expression of the late endosomal marker CD107a was performed. Bar graph displays percentage of NK cells expressing CD107a under basal conditions (left), or after co-incubation with B16F10 and YAC-1 target cells (middle and right panel, respectively). (B, C) The in vitro cytotoxicity of WT, Ifnar1−/− and Ifnb−/− NK cells was measured in a flow cytometric-based assay using RMA-S and YAC-1 cells as targets. The specific lysis at different effector:target-cell ratios (10:1, 5:1, 1:1) differed significantly between the analyzed NK cells. Individual points represent means from triplicate wells ± SEM. One representative experiment out of three is depicted. (*p < 0.05). (D) Representative isolated lungs of WT, Ifnar1−/− and Ifnb−/− mice 14 d after intravenous injection of B16F10 melanoma cells. (E) The lung/body weight ratio was significantly higher in Ifnar1−/− and Ifnb−/− compared with WT mice after challenge with B16F10 tumor cells (p < 0.05).
Loss of IFNAR1 in NK cells leaves leukemia latency unaffected
To test the impact of Type I IFN signaling in the NK cell compartment during leukemia development Ifnar1f/f Ncr1-iCre as well as Ifnar1f/f control mice were infected with A-MuLV. In contrast to our expectations, Ifnar1f/f Ncr1-iCre mice succumbed to leukemia/lymphoma with a latency that was comparable to that of their littermate controls (). At best we detected a tendency for Ifnar1f/f mice to die slightly later. Again, the phenotype of the disease was comparable in mice with a different genotype and so was the extent of tumor-cell infiltration into hematopoietic organs (Figs. S4A–D).
Figure 6. In vivo analysis of Ifnar1f/f Ncr1-iCre animals. (A) Kaplan-Meier plot of Ifnar1f/f Ncr1-iCre and Ifnar1f/f littermate mice infected via i.p. injection of A-MuLV. Log-rank test did not reveal a statistically significant difference in disease latency (p = 0.279; Ifnar1f/f Ncr1-iCre: n = 8, Ifnar1f/f: n = 9). (B, C) The in vitro cytotoxicity of WT, Ifnar1−/− and Ifnar1f/f Ncr1-iCre NK cells was measured in a flow cytometric-based assay using RMA-S and YAC-1 cells as targets. The specific lysis at different effector:target-cell ratios (10:1, 5:1, 1:1) differed significantly between the analyzed NK cells. Individual points represent means from triplicate wells ± SEM. One representative experiment out of three is depicted. (*p < 0.05). (D) Lung/body weight ratio and (E) number of tumor nodules on lungs after B16F10 melanoma cell injection into Ifnar1f/f Ncr1-iCre, Ifnar1f/f and Ifnar1−/− mice. (F) The in vitro cytotoxicity of Ifnar1f/f Ncr1-iCre, Ifnar1f/f and Ifnar1−/− NK cells was measured in a flow cytometric-based assay using B16F10 cells as targets. Individual points represent means from two preparations per group and triplicate wells ± SEM (*p < 0.05, **p < 0.01, ***p < 0.001).
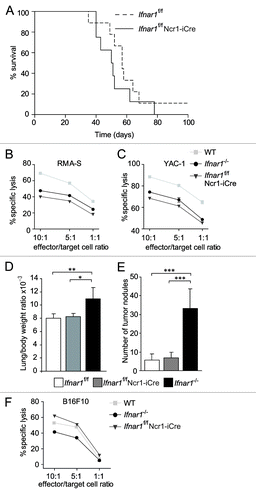
To test whether the NK cell functions are unaltered in Ifnar1f/f Ncr1-iCre mice, we prepared splenic NK cells from these animals. Interestingly, in vitro expanded splenic NK cells derived from Ifnar1f/f Ncr1-iCre mice displayed defects in their capacity to kill YAC-1 and RMA-S cells that are comparable to Ifnar1−/− NK cells (). These findings point to a direct contribution of Type I IFN to NK cell cytotoxicity, yet cannot explain the unaltered disease latency.
When we tested another NK cell-controlled tumor model, i.e., the B16F10 melanoma model, the NK cell-specific deletion of Ifnar1 failed to impact on the development of tumor nodules in the lungs. This was in contrast with the phenotype observed in complete Ifnar1 knockout animals, which displayed a significantly increased tumor-cell infiltration into the lung (; ). In addition, killing assays with B16F10 cells revealed a reduced cytotoxic activity of Ifnar1−/− NK cells, but not of NK cells derived from Ifnar1f/f Ncr1-iCre mice, reflecting the in vivo observations ().
Thus, the outcome of v-Abl-induced leukemia/lymphoma is different in Ifnar1f/f Ncr1-iCre vs. Ifnar1−/− mice. This suggests that the sole loss of Type I IFN responsiveness in NK cells that express Ncr1 does not phenocopy the complete loss of Ifnar1 signaling in the Abelson tumor model.
Discussion
In this work, we found that the NK cell-dependent surveillance of v-Abl-induced leukemia/lymphoma is significantly compromised in mice deficient for Type I IFN signaling but that tumor cell-intrinsic Type I IFN signaling is of minor importance.
We took advantage of conditional Ifnar1 knockout mice to dissect the role of Type I IFN in the tumor cells themselves and in the NK cell compartment. Previous in vitro work revealed that Type I IFN prevents cellular transformation through mechanisms involving enhanced cellular expression of the tumor suppressor p53.Citation32 This appears of no relevance in the Abelson leukemia model. We failed to detect a significant increase in cell transformation in the absence of constitutive Type I IFN signaling in vitro (). Similarly, the absence of Ifnar1 in CD19+ leukemic cells did not accelerate disease progression suggesting only a minor impact – if any - of Type I IFN on the tumor cells themselves. Prior to the era of the BCR/ABL tyrosine kinase inhibitor imatinib (Gleevec®), IFNα has been used for decades to treat patients suffering from BCR/ABL-driven chronic myeloid leukemia. Today, the therapeutic approach has changed and IFNα is considered as second line treatment, to be used when cells become resistant to BCR/ABL inhibition. The rationale for IFNα treatment was partially based on the fact that IFNα exerts a direct growth inhibitory and pro-apoptotic effect on the tumor cells. In line with other reports,Citation9,Citation11 our results do not corroborate this hypothesis but rather point toward a dominant role of Type I IFN at the level of the immune system. This is strongly supported by the fact that disease progression was not altered in mice lacking IFNAR1 in the tumor cells.
As we have demonstrated in the past that NK cells play a key role in shaping leukemogenesis induced by the Abelson oncogene, we next took a closer look at the NK cell compartment. Initial work by Jablonska and colleagues indicate that Ifnb−/− mice are prone to develop B16F10 melanoma, a model predominantly controlled by NK cell-mediated surveillance.Citation33 We confirmed and extended these data using IFNAR1-deficient animals and we propose that the increased susceptibility of Ifnar1−/− mice to v-Abl-induced leukemogenesis is related to the NK cell compartment. Interestingly, only NK cells derived from Ifnar1−/− mice but not from Ifnar1f/f Ncr1-iCre mice showed a maturation defect. This may be explained by the fact that the Ncr1 gene (and thus the Cre recombinase) is only expressed in the late phases of NK cell development, when the cells mature. One may speculate that the time point at which NK cells require an input from Type I IFN has already passed. In line with our findings (Fig. S5D), the absence of either IFNAR1 or IFNAR2 has been reported to reduce NK cell numbers in the spleen.Citation14 Our data therefore clearly show that while systemic Type I IFN is required for NK cell development, the expression of IFNAR1 during late maturation stages on NKp46+ NK cells themselves is not.
Type I IFN is—at least in vitro—capable to enhance the cytotoxic functions of NK cells.Citation14 If Ifnar1 is only deleted in the NK cell compartment, however, the effects are less clear. Whereas YAC-1 and RMA-S cell killing by NK cells requires the presence of IFNAR1 on their surface, this was not the case for the killing of B16F10 cells. B16F10 melanoma cells express ligands to DNAM-1 and NCRs, and lack MHC class I,Citation34,Citation35 whereas YAC-1 cells have been shown to display considerable amounts of NKG2D ligands,Citation36 as do v-ABL transformed cells.Citation26 One may speculate that different ligands on the surface of target cells may contribute to the observed differences in NK cell-mediated cytotoxicity. Although the molecular mechanism remains obscure, signaling crosstalk downstream of IFNAR1 and NK cell receptors may be envisioned. The defect is clearly not related to the mobilization of the cytolytic granules toward the target cell, nor to a reduced expression of the cytolytic mediators such as perforin and granzymes.
Interestingly, this in vitro defect did not translate into a major disadvantage in vivo. When exposed to the Abelson retrovirus, Ifnar1f/f Ncr1-iCre animals managed to resist the oncogenic challenge significantly better than Ifnar1−/− mice. This suggests that the strong cytotoxic defects imposed by the loss of IFNAR1 are compensated in vivo. The results obtained with IL-2-expanded NK cells are clearly different from the complex situation in vivo, where many cytokines and modulating factors are present. We reason that the in vitro observations unmask a contribution of Type I IFN to cytotoxicity that is compensated in vivo. In addition, we did rule out that an impaired deletion of Ifnar1 accounts for these differences. One possible explanation is that in Ifnar1−/− mice other cell types that do require Type I IFN are critically involved in tumor surveillance. Conventional dendritic cells (DCs), macrophages as well as stromal cells like fibroblasts all may be involved in tumor surveillance in addition to NK cells. Type I IFN responses in these cell types are obviously not affected in Ifnar1f/f Ncr1-iCre mice, which may account for the unaltered disease latency as compared with wild type controls.
The interaction between activated DCs and NK cells has been described to increase the efficiency of NK cell antitumor effector functions both in vitro and in vivo, in independent experimental models.Citation37,Citation38 On one hand, IL-2 has been reported to be a key regulator of DC-dependent NK cell activation upon bacterial challenge.Citation39 On the other hand, NK cell priming was described to depend on functional Type I IFN responses by DCs and the subsequent production and trans-presentation of IL-15 by DCs to resting NK cells.Citation40 In the B16F10 melanoma model, neutrophils have also been implicated in tumor surveillance.Citation33 Further clarification is required on which complex networks of immune cells and cytokines are responsible to guarantee full-fledged tumor surveillance. Our data suggest that IFN regulates NK cell-mediated tumor surveillance with a high plasticity, as the lack of Type I IFN responsiveness in NK cells—despite having a major impact in vitro—appears to be compensated for in vivo.
Materials and Methods
Mice and mouse experiments
Ifnar1−/−Citation41 and Ifnb−/−Citation42 mice, as well as mice carrying loxP-flanked Ifnar1Citation43,Citation44 crossed with transgenic mice expressing Cre recombinase under the control of either Ncr1Citation31 or Cd19 promoters,Citation45 were on C57BL/6 background and maintained under specific pathogen-free conditions at Biomodels Austria, University of Veterinary Medicine Vienna, Austria. Experiments were performed using gender- and age-matched mice. All animal experiments were done in accordance with protocols approved by the Animal Welfare Committee of the Medical University of Vienna and the Austrian Federal Ministry for Science and Research.
Different disease models were used: 1) newborn mice were infected with 100 μL of replication-incompetent ecotropic retrovirus encoding for v-Abl by intraperitoneal injection (i.p.) as described by Sexl et al.Citation46 Mice were checked daily for disease onset. Diseased mice were sacrificed. Peripheral blood, lymphoid and hematopoietic organs were analyzed for infiltrating leukemic cells by FACS and by histopathology. 2) For transplantation experiments 105 B16F10 melanoma cells (in 100 μl PBS) were injected via tail vein (i.v.) into wild type, Ifnar1−/−, Ifnb−/− or Ifnar1f/f and Ifnar1f/f Ncr1-iCre recipient mice. The mice were sacrificed 14 d post injection and analyzed for lung weights and macroscopically visible tumor nodules on lung surface.
In vitro transformation assay and generation of v-ABL transformed cell lines
Single-cell suspensions of murine bone marrow of tibiae and femora were infected as described previously.Citation46 Thereafter, cells were either maintained in complete RPMI-1640 (PAA Laboratories) medium or plated in cytokine-free methylcellulose (MethoCultTM, StemCell Technologies Inc.) as described previously.Citation47 Cloning efficiency was evaluated by counting colonies using a Leica Fluovert microscope (magnification x4). Images were taken using a Zeiss Axio Imager.Z1 microscope. The assays were performed three times in duplicates. The ability of transformed cells to form cell lines was tested by transferring an aliquot of the infected cells (106) to growth factor–free medium. The culture was observed for the outgrowth of stable cell lines. Mock-infected cells were used as controls. Outgrown cell lines were analyzed by flow cytometry for the expression of B lineage (CD19, CD43; BD PharMingen) and myeloid lineage markers (Mac1, Gr1; BD PharMingen).
Virus preparation and cell culture
Maintenance of A010 A-MuLV producer cells and collection of the viral supernatant was performed as described previously.Citation48 Transformed bone marrow cell lines were maintained in complete RPMI-1640 as described (RPMI-1640 supplemented with 10% heat-inactivated FCS, 100 U/ml penicillin-streptomycin, 2mmol/L L-glutamine and 5 micromol/L beta mercaptoethanol).
B16F10 cells were maintained in DMEM supplemented with 10% heat-inactivated FCS, 100 U/ml penicillin-streptomycin, 2 mmol/L L-glutamine and 5 µmol/L β-mercaptoethanol. YAC-1 and RMA-S cells were maintained in RPMI-1640 supplemented with 10% heat-inactivated FCS, 100 U/ml penicillin-streptomycin, 2 mmol/L L-glutamine and 5 µmol/L β-mercaptoethanol.
NK cell purification, expansion and function
Murine NK cells were isolated, purified and expanded as previously described.Citation26 Briefly, single-cell suspensions were prepared from at least 2 spleens per genotype. For NK cell purification, erythrocyte-depleted single-cell suspensions were incubated with anti-DX5-coated MACS beads (Miltenyi Biotec) and purified by LS columns on a MACS separator (Miltenyi Biotec). NK cells were expanded for 6–10 d in medium (RPMI 1640 containing 10% FCS, β-mercaptoethanol, L-glutamine, penicillin/streptomycin) supplemented with rhIL-2 (henceforth IL-2) (Proleukin, Novartis, 5000 U/ml). The purity of NK cells was assessed by flow cytometry and was routinely > 90% - 95%. To monitor NK cell cytotoxicity a FACS-based assay in analogy to a previous publicationCitation49 was employed. Briefly, YAC-1, RMA-S or B16F10 tumor cells were stained with carboxyfluorescein diacetate succinimidyl ester (CFSE, 1.66µM) according to the manufacturer’s instructions (Molecular Probes, CellTrace™ CFSE Cell Proliferation Kit). Five x 104 CFSE-stained tumor cells/96 round-bottom well were incubated with NK cells in effector/target ratios of 10:1, 5:1 and 1:1 in duplicates. After 4 h all samples were subjected to 7-aminoactinomycin D (7-AAD, 0.1 µg, eBioscience) for 5 min and analyzed in a FACSCanto II (BD Biosciences). Percentage of specific NK cell lysis was calculated as follows: % specific lysis = (% 7-AAD+ CFSE+ cells after coincubation with NK cells) — (% 7-AAD+ CFSE+ cells without addition of NK cells).
Antibodies and flow cytometric analysis
For flow cytometry, single-cell suspensions were prepared from various tissues. Purified rat-anti-mouse CD16/CD32 (2.4G2) (BD PharMingen) was added to avoid nonspecific binding of monoclonal antibodies to FcγR. Samples were analyzed using BD Bioscience FACSCanto II and BD FACSDiva software. Following antibodies were used: (a) B cell immuno-phenotyping: CD19 (1D3), CD43 (S7), B220 (RA3–6B2), BP1 (6C3), IgM (II/41), IgD (II-26c), CD11b (M1/70), Gr1 (RB6–8C5) (all BD PharMingen). (b) NK cell analysis: NK1.1 (PK136), CD3 (500A2), CD16/CD32 (2.4G2), CD122 (TM-β1), Ly49C/I (5E6), Ly49D (4E5), CD107a (1D4B), biotin mouse lineage panel (145–2C11, RA3–6B2, RB5–8C5, TER-119) (all from BD Biosciences); CD11b (M1/70), CD27 (LG.7F9), CD49b (DX-5), KLRG1 (2F1), DNAM-1 (10E5), NKG2D (CX5), NKp46 (29A1.4) (eBioscience); IFNAR1 (MAR1-SA3) (Biolegend).
To obtain cell cycle profiles, cells were harvested in 0.5 ml hypotonic lysis solution (50 μg/mL propidium iodide in 0.1% sodium citrate, 0.1% triton X-100, 100 μg/mL RNase) and analyzed by FACS.
For dye dilution assays 107 cells were incubated with CFSE (5 µM in PBS). The reaction was stopped with 5% FCS. Cells were maintained in complete RPMI-1640 (PAA). Proliferation of cell lines was examined for 3 d analyzing an aliquot each day by FACS.
Degranulation was measured according to Betts et al.Citation50 In brief, 105 in vitro expanded NK cells were treated with different stimuli (medium alone, B16F10 or YAC-1) in the presence of 0.5 µg anti-mouse CD107a-FITC (BD Biosciences) and 0.6 µL GolgiStop (BD Biosciences) per 200 µL NK cell medium. After 4 h incubation all samples were stained for NK1.1 and CD3 and analyzed by flow cytometry.
[3H]Thymidine incorporation assay
v-ABL transformed cells were plated at a density of 5 x 104 cells in 96 round-bottom wells and incubated overnight. Then [3H]thymidine [1 μCurie/mL] was added for another 8 h of incubation. Cells were analyzed by harvesting them onto glass fiber filter mats using a Skatron 96-well harvester (Skatron Instruments AS). Filter mats were soaked in 2.5 mL Rotiszint® eco plus scintillation fluid (Roth) and filter-bound [3H]thymidine was quantified by three minute-counts in a scintillation counter (1900 CA Tri-Carb® Liquid Scintillation Analyzer; Packard Cranberra Company).
Histology
Representative samples of spleens, tumors, livers and lungs were fixed with 3.7% formaldehyde for 24 h, washed in PBS and embedded in paraffin. Five µm-sections were stained with Hematoxylin and Eosin using standard protocols. Images (magnification x100) were taken using a Zeiss Axio Imager.Z1 microscope. Slides were scanned using TissueFAXSTM software (TissueGnostics GmbH, Vienna, Austria; www.tissuegnostics.com).
Statistical procedures
Where appropriate, unpaired Student’s t-test or One-way ANOVA with Tukey post test were performed using GraphPad Prism version 5.00 for Windows (GraphPad Software, San Diego California USA, www.graphpad.com) Differences in Kaplan-Meier plots were analyzed for statistical significance using the log rank test of survival. p values lower than 0.05 were considered statistically significant (*p < 0.05, **p < 0.01, ***p < 0.001).
| Abbreviations: | ||
| 7-AAD | = | 7-aminoactinomycin D |
| A-MuLV | = | Abelson murine leukemia virus |
| BM | = | bone marrow |
| CFSE | = | carboxyfluorescein diacetate succinimidyl ester |
| DC | = | dendritic cell |
| iCre | = | codon-improved Cre |
| IFN | = | interferon |
| IFNAR1 | = | interferon α and β receptor 1 |
| IL | = | interleukin |
| iNK | = | immature natural killer |
| LN | = | lymph node |
| MACS | = | magnetic-activated cell sorting |
| MFI | = | mean fluorescence intensity |
| MHC | = | major histocompatibility complex |
| mNK | = | mature natural killer |
| NK | = | natural killer |
| NKP | = | natural killer cell precursor |
| SP | = | spleen |
| TYK2 | = | Tyrosine kinase 2 |
| v-ABL | = | Abelson murine leukemia viral oncogene homolog 1 |
| WBC | = | white blood cells |
Additional material
Download Zip (4.7 MB)Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We are grateful to Thomas Decker for insightful discussions and Shinya Sakaguchi for critically reading the manuscript. We thank Winfried Pickl for technical help, as well as Michaela Schlederer, Jaqueline Horvath, Sabine Fajmann, Isabelle Töplitz for technical assistance with immunohistochemistry and Christina Kapusta for genotyping of the mice. Financial support for this project was provided by a Marie Curie International Incoming Fellowship to TM, DOC-fFORTE fellowships to EMP and OS, the GEN-AU ‘Austromouse’ of the BM.W_Fa to M.M., the SFB F2810 and F2808 of the Austrian Science Foundation (FWF) to V.S. and M.M., the GEN-AU ‘Placebo’ of the BM.W_Fa to V.S., the Vienna Science and Technology Fund (WWTF) LS-07–037 to V.S. and P19534-B13 of the FWF to D.S.
N.N., T.M., N.M., O.S., E.E., E.Z.B., E.M.P., D.G., W.W., H.P.K., V.S and D.S. designed and performed research; N.N., T.M., N.M., O.S., E.E., E.M.P., D.G., E.Z.B., S.L., V.S. and D.S. analyzed data; UK, S.L., S.W., M.M., B.S. and V.S. provided vital new reagents and analytic tools; T.M., E.M.P., V.S. and D.S. wrote the paper.
Supplemental Material
Supplemental material may be downloaded here:
www.landesbioscience.com/journals/oncoimmunology/article/21284/
Notes
† These authors contributed equally to this work.
References
- Gough DJ, Messina NL, Clarke CJ, Johnstone RW, Levy DE. Constitutive Type I interferon modulates homeostatic balance through tonic signaling. Immunity 2012; 36:166 - 74; http://dx.doi.org/10.1016/j.immuni.2012.01.011; PMID: 22365663
- Hida S, Ogasawara K, Sato K, Abe M, Takayanagi H, Yokochi T, et al. CD8(+) T cell-mediated skin disease in mice lacking IRF-2, the transcriptional attenuator of interferon-alpha/beta signaling. Immunity 2000; 13:643 - 55; http://dx.doi.org/10.1016/S1074-7613(00)00064-9; PMID: 11114377
- Honda K, Mizutani T, Taniguchi T. Negative regulation of IFN-alpha/beta signaling by IFN regulatory factor 2 for homeostatic development of dendritic cells. Proc Natl Acad Sci U S A 2004; 101:2416 - 21; http://dx.doi.org/10.1073/pnas.0307336101; PMID: 14983024
- Mizutani T, Tsuji K, Ebihara Y, Taki S, Ohba Y, Taniguchi T, et al. Homeostatic erythropoiesis by the transcription factor IRF2 through attenuation of Type I interferon signaling. Exp Hematol 2008; 36:255 - 64; http://dx.doi.org/10.1016/j.exphem.2007.11.004; PMID: 18207304
- Sato T, Onai N, Yoshihara H, Arai F, Suda T, Ohteki T. Interferon regulatory factor-2 protects quiescent hematopoietic stem cells from Type I interferon-dependent exhaustion. Nat Med 2009; 15:696 - 700; http://dx.doi.org/10.1038/nm.1973; PMID: 19483695
- Ferrantini M, Capone I, Belardelli F. Interferon-alpha and cancer: mechanisms of action and new perspectives of clinical use. Biochimie 2007; 89:884 - 93; http://dx.doi.org/10.1016/j.biochi.2007.04.006; PMID: 17532550
- Rizza P, Moretti F, Belardelli F. Recent advances on the immunomodulatory effects of IFN-alpha: implications for cancer immunotherapy and autoimmunity. Autoimmunity 2010; 43:204 - 9; http://dx.doi.org/10.3109/08916930903510880; PMID: 20187707
- Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science 2011; 331:1565 - 70; http://dx.doi.org/10.1126/science.1203486; PMID: 21436444
- Smyth MJ. Type I interferon and cancer immunoediting. Nat Immunol 2005; 6:646 - 8; http://dx.doi.org/10.1038/ni0705-646; PMID: 15970935
- Dunn GP, Bruce AT, Sheehan KC, Shankaran V, Uppaluri R, Bui JD, et al. A critical function for Type I interferons in cancer immunoediting. Nat Immunol 2005; 6:722 - 9; http://dx.doi.org/10.1038/ni1213; PMID: 15951814
- Dunn GP, Koebel CM, Schreiber RD. Interferons, immunity and cancer immunoediting. Nat Rev Immunol 2006; 6:836 - 48; http://dx.doi.org/10.1038/nri1961; PMID: 17063185
- Sun JC, Lanier LL. NK cell development, homeostasis and function: parallels with CD8⁺ T cells. Nat Rev Immunol 2011; 11:645 - 57; http://dx.doi.org/10.1038/nri3044; PMID: 21869816
- Huntington ND, Vosshenrich CA, Di Santo JP. Developmental pathways that generate natural-killer-cell diversity in mice and humans. Nat Rev Immunol 2007; 7:703 - 14; http://dx.doi.org/10.1038/nri2154; PMID: 17717540
- Swann JB, Hayakawa Y, Zerafa N, Sheehan KC, Scott B, Schreiber RD, et al. Type I IFN contributes to NK cell homeostasis, activation, and antitumor function. J Immunol 2007; 178:7540 - 9; PMID: 17548588
- Stoiber D, Kovacic B, Schuster C, Schellack C, Karaghiosoff M, Kreibich R, et al. TYK2 is a key regulator of the surveillance of B lymphoid tumors. J Clin Invest 2004; 114:1650 - 8; PMID: 15578097
- Faderl S, Talpaz M, Estrov Z, O’Brien S, Kurzrock R, Kantarjian HM. The biology of chronic myeloid leukemia. N Engl J Med 1999; 341:164 - 72; http://dx.doi.org/10.1056/NEJM199907153410306; PMID: 10403855
- Specchia G, Mininni D, Guerrasio A, Palumbo G, Pastore D, Liso V. Ph positive acute lymphoblastic leukemia in adults: molecular and clinical studies. Leuk Lymphoma 1995; 18:Suppl 1 37 - 42; http://dx.doi.org/10.3109/10428199509075301; PMID: 7496353
- Witte ON. Functions of the abl oncogene. Cancer Surv 1986; 5:183 - 97; PMID: 3465440
- Rosenberg N, Witte ON. The viral and cellular forms of the Abelson (abl) oncogene. Adv Virus Res 1988; 35:39 - 81; http://dx.doi.org/10.1016/S0065-3527(08)60708-3; PMID: 2852893
- Deonarain R, Verma A, Porter AC, Gewert DR, Platanias LC, Fish EN. Critical roles for IFN-beta in lymphoid development, myelopoiesis, and tumor development: links to tumor necrosis factor alpha. Proc Natl Acad Sci U S A 2003; 100:13453 - 8; http://dx.doi.org/10.1073/pnas.2230460100; PMID: 14597717
- Hardy RR. B-cell commitment: deciding on the players. Curr Opin Immunol 2003; 15:158 - 65; http://dx.doi.org/10.1016/S0952-7915(03)00012-8; PMID: 12633665
- Hardy RR, Hayakawa K. B cell development pathways. Annu Rev Immunol 2001; 19:595 - 621; http://dx.doi.org/10.1146/annurev.immunol.19.1.595; PMID: 11244048
- Hardy RR, Li YS, Allman D, Asano M, Gui M, Hayakawa K. B-cell commitment, development and selection. Immunol Rev 2000; 175:23 - 32; http://dx.doi.org/10.1111/j.1600-065X.2000.imr017517.x; PMID: 10933588
- Lienenklaus S, Cornitescu M, Zietara N, Łyszkiewicz M, Gekara N, Jabłónska J, et al. Novel reporter mouse reveals constitutive and inflammatory expression of IFN-beta in vivo. J Immunol 2009; 183:3229 - 36; http://dx.doi.org/10.4049/jimmunol.0804277; PMID: 19667093
- Kovacic B, Stoiber D, Moriggl R, Weisz E, Ott RG, Kreibich R, et al. STAT1 acts as a tumor promoter for leukemia development. Cancer Cell 2006; 10:77 - 87; http://dx.doi.org/10.1016/j.ccr.2006.05.025; PMID: 16843267
- Zebedin E, Simma O, Schuster C, Putz EM, Fajmann S, Warsch W, et al. Leukemic challenge unmasks a requirement for PI3Kdelta in NK cell-mediated tumor surveillance. Blood 2008; 112:4655 - 64; http://dx.doi.org/10.1182/blood-2008-02-139105; PMID: 18684865
- Eckelhart E, Warsch W, Zebedin E, Simma O, Stoiber D, Kolbe T, et al. A novel Ncr1-Cre mouse reveals the essential role of STAT5 for NK-cell survival and development. Blood 2011; 117:1565 - 73; http://dx.doi.org/10.1182/blood-2010-06-291633; PMID: 21127177
- Hayakawa Y, Smyth MJ. CD27 dissects mature NK cells into two subsets with distinct responsiveness and migratory capacity. J Immunol 2006; 176:1517 - 24; PMID: 16424180
- Chiossone L, Chaix J, Fuseri N, Roth C, Vivier E, Walzer T. Maturation of mouse NK cells is a 4-stage developmental program. Blood 2009; 113:5488 - 96; http://dx.doi.org/10.1182/blood-2008-10-187179; PMID: 19234143
- Kodama T, Takeda K, Shimozato O, Hayakawa Y, Atsuta M, Kobayashi K, et al. Perforin-dependent NK cell cytotoxicity is sufficient for anti-metastatic effect of IL-12. Eur J Immunol 1999; 29:1390 - 6; http://dx.doi.org/10.1002/(SICI)1521-4141(199904)29:04<1390::AID-IMMU1390>3.0.CO;2-C; PMID: 10229107
- Eckelhart E, Warsch W, Zebedin E, Simma O, Stoiber D, Kolbe T, et al. A novel Ncr1-Cre mouse reveals the essential role of STAT5 for NK-cell survival and development. Blood 2011; 117:1565 - 73; http://dx.doi.org/10.1182/blood-2010-06-291633; PMID: 21127177
- Takaoka A, Hayakawa S, Yanai H, Stoiber D, Negishi H, Kikuchi H, et al. Integration of interferon-alpha/beta signalling to p53 responses in tumour suppression and antiviral defence. Nature 2003; 424:516 - 23; http://dx.doi.org/10.1038/nature01850; PMID: 12872134
- Jablonska J, Leschner S, Westphal K, Lienenklaus S, Weiss S. Neutrophils responsive to endogenous IFN-beta regulate tumor angiogenesis and growth in a mouse tumor model. J Clin Invest 2010; 120:1151 - 64; http://dx.doi.org/10.1172/JCI37223; PMID: 20237412
- Lakshmikanth T, Burke S, Ali TH, Kimpfler S, Ursini F, Ruggeri L, et al. NCRs and DNAM-1 mediate NK cell recognition and lysis of human and mouse melanoma cell lines in vitro and in vivo. J Clin Invest 2009; 119:1251 - 63; http://dx.doi.org/10.1172/JCI36022; PMID: 19349689
- Gilfillan S, Chan CJ, Cella M, Haynes NM, Rapaport AS, Boles KS, et al. DNAM-1 promotes activation of cytotoxic lymphocytes by nonprofessional antigen-presenting cells and tumors. J Exp Med 2008; 205:2965 - 73; http://dx.doi.org/10.1084/jem.20081752; PMID: 19029380
- Diefenbach A, Jamieson AM, Liu SD, Shastri N, Raulet DH. Ligands for the murine NKG2D receptor: expression by tumor cells and activation of NK cells and macrophages. Nat Immunol 2000; 1:119 - 26; http://dx.doi.org/10.1038/77793; PMID: 11248803
- Fernandez NC, Lozier A, Flament C, Ricciardi-Castagnoli P, Bellet D, Suter M, et al. Dendritic cells directly trigger NK cell functions: cross-talk relevant in innate anti-tumor immune responses in vivo. Nat Med 1999; 5:405 - 11; http://dx.doi.org/10.1038/7403; PMID: 10202929
- van den Broeke LT, Daschbach E, Thomas EK, Andringa G, Berzofsky JA. Dendritic cell-induced activation of adaptive and innate antitumor immunity. J Immunol 2003; 171:5842 - 52; PMID: 14634094
- Granucci F, Zanoni I, Pavelka N, Van Dommelen SL, Andoniou CE, Belardelli F, et al. A contribution of mouse dendritic cell-derived IL-2 for NK cell activation. J Exp Med 2004; 200:287 - 95; http://dx.doi.org/10.1084/jem.20040370; PMID: 15289500
- Lucas M, Schachterle W, Oberle K, Aichele P, Diefenbach A. Dendritic cells prime natural killer cells by trans-presenting interleukin 15. Immunity 2007; 26:503 - 17; http://dx.doi.org/10.1016/j.immuni.2007.03.006; PMID: 17398124
- Müller U, Steinhoff U, Reis LF, Hemmi S, Pavlovic J, Zinkernagel RM, et al. Functional role of Type I and Type II interferons in antiviral defense. Science 1994; 264:1918 - 21; http://dx.doi.org/10.1126/science.8009221; PMID: 8009221
- Erlandsson L, Blumenthal R, Eloranta ML, Engel H, Alm G, Weiss S, et al. Interferon-beta is required for interferon-alpha production in mouse fibroblasts. Current biology: CB 1998; 8:223-6.
- Prinz M, Schmidt H, Mildner A, Knobeloch KP, Hanisch UK, Raasch J, et al. Distinct and nonredundant in vivo functions of IFNAR on myeloid cells limit autoimmunity in the central nervous system. Immunity 2008; 28:675 - 86; http://dx.doi.org/10.1016/j.immuni.2008.03.011; PMID: 18424188
- Kamphuis E, Junt T, Waibler Z, Forster R, Kalinke U. Type I interferons directly regulate lymphocyte recirculation and cause transient blood lymphopenia. Blood 2006; 108:3253 - 61; http://dx.doi.org/10.1182/blood-2006-06-027599; PMID: 16868248
- Rickert RC, Rajewsky K, Roes J. Impairment of T-cell-dependent B-cell responses and B-1 cell development in CD19-deficient mice. Nature 1995; 376:352 - 5; http://dx.doi.org/10.1038/376352a0; PMID: 7543183
- Sexl V, Piekorz R, Moriggl R, Rohrer J, Brown MP, Bunting KD, et al. Stat5a/b contribute to interleukin 7-induced B-cell precursor expansion, but abl- and bcr/abl-induced transformation are independent of stat5. Blood 2000; 96:2277 - 83; PMID: 10979977
- Hoelbl A, Kovacic B, Kerenyi MA, Simma O, Warsch W, Cui Y, et al. Clarifying the role of Stat5 in lymphoid development and Abelson-induced transformation. Blood 2006; 107:4898 - 906; http://dx.doi.org/10.1182/blood-2005-09-3596; PMID: 16493008
- Sexl V, Kovacic B, Piekorz R, Moriggl R, Stoiber D, Hoffmeyer A, et al. Jak1 deficiency leads to enhanced Abelson-induced B-cell tumor formation. Blood 2003; 101:4937 - 43; http://dx.doi.org/10.1182/blood-2001-11-0142; PMID: 12576323
- Schuster C, Berger A, Hoelzl MA, Putz EM, Frenzel A, Simma O, et al. The cooperating mutation or “second hit” determines the immunologic visibility toward MYC-induced murine lymphomas. Blood 2011; 118:4635 - 45; http://dx.doi.org/10.1182/blood-2010-10-313098; PMID: 21878673
- Betts MR, Brenchley JM, Price DA, De Rosa SC, Douek DC, Roederer M, et al. Sensitive and viable identification of antigen-specific CD8+ T cells by a flow cytometric assay for degranulation. J Immunol Methods 2003; 281:65 - 78; http://dx.doi.org/10.1016/S0022-1759(03)00265-5; PMID: 14580882