Abstract
The genome of colorectal carcinomas displaying pronounced microsatellite instability codes for an extraordinarily high number of mutated proteins that elicit tumor-specific cellular immune responses. We have recently demonstrated that leukemic cells are also vulnerable to T cells specific for tumor-associated antigens produced in the context of microsatellite instability. This finding extends our understanding of secondary and therapy-related leukemogenesis, linking it to the mutual interaction between immune control and escape.
An outstanding characteristic of tumors deficient in mismatch repair (MMR) and displaying pronounced microsatellite instability (MSI) is the elevated immunogenicity caused by their hypermutational state. Although MSI has been recognized long time ago in Lynch syndrome patients, it has only occasionally been examined in individuals bearing hematological malignancies and is usually considered as a rare event in the course of leukemogenesis.Citation1 This may be explained by 1) the elevated heterogeneity of this neoplasm, 2) the overall low number of cases analyzed, especially comparable cases of different disease subgroups, and 3) the markers used for the assessment of MSI. There is indeed no consensus panel for the determination of MSI in leukemia, with the actual value of mononucleotide repeats BAT-25 and BAT-26 being controversial.
In sharp contrast, many established leukemic cell lines appear to be MMR-deficient (MMR-D). Most of these cell lines have been established from patients with progressive (e.g., HSB-2 cells) or refractory (e.g., Nalm-6, DG-75, CTV-1, and CTV-2 cells) leukemia. One may speculate that these cells are intrinsically more prone to be propagated in culture than their MMR-proficient counterparts. It would certainly be interesting to analyze whether this is due to the elevated aggressiveness of the MSI+ cells, their pronounced mutational rate (allowing them to rapidly adapt to culture conditions), or possibly both.
An open question is the precise contribution of MSI to leukemogenesis. In some types of leukemia, MSI is a rather frequent finding. Up to 50% of therapy-related acute myeloid leukemia (AML) and myelodysplastic syndrome (MDS) cases exhibit an MSI+ phenotype (as compared with <5% of de novo AML/MDS cases).Citation2 This raises the question why MSI is a rare finding in primary leukemias and what favors the emergence of MSI during secondary leukemogenesis (which is frequent in breast carcinoma patients) and/or therapy-related AML/MDS? A loss of immunosurveillance resulting from transient or prolonged immunosuppression has been proposed as an optimal condition for the development of MSI+ leukemia.Citation3 This obviously occurs not only after stem cell transplantation (entailing an intense myelosuppressive conditioning regimen), but also following (at least some types of) chemo- and radiotherapy. In addition, several antineoplastic and immunosuppressive agents (e.g., alkylating substances like thiopurines) are known to stress the MMR system by producing DNA mismatches, which are recognized—but not removed—by MMR machinery.Citation2,Citation4 At least in vitro, this allows for the selection and clonal expansion of single MMR-D cells that are resistant to lethal DNA damage (e.g., that display methylation tolerance).Citation3 Of note, cells that become MMR-D are not malignant per se, but since they display a consistent increase in mutational rate, they can rapidly accumulate driver mutations in MSI target genes (). In fact, one may wonder why secondary or therapy-related leukemia is still a relatively rare disease.
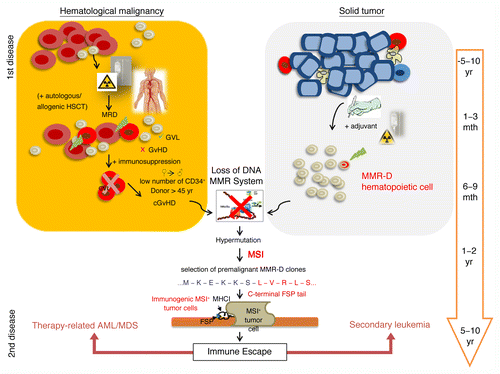
Figure 1. Potential development of mismatch repair-deficient secondary leukemia and therapy-related acute myeloid leukemia/myelodysplastic syndrome. Mismatch repair-deficient (MMR-D) hematopoietic cells are regularly primed and selected for by the therapeutic regimens employed against primary hematological and solid neoplasms. Patients affected by hematological disorders often receive stem cell transplantation in addition to chemo- and radiotherapy. In this setting, most residual leukemic cells (minimal residual disease, MRD) may be eliminated due to the graft-vs.-leukemia (GVL) effect. However, immunosuppression counteracts this process and, especially in cooperation with severe graft-vs.-host-disease (GvHD) reactions, drives the development of MMR-D leukemic clones. The treatment of hematological and solid cancer patients with alkylating agents may induce and select for MMR-D hematopoietic cells that display an intrinsically elevated mutational rate, preferentially affecting MSI target genes. Insertion and deletion mutations result in the production of truncated polypeptides, proteins with altered function as well as highly immunogenic frameshift peptide tails. Hence MSI+ cancer cells suffer from an increased immunological pressure and must acquire escape mechanisms. If successful, these cells will drive the emergence of clinically overt secondary leukemia or therapy-related MSI+ acute myeloid leukemia/myelodysplastic syndrome (AML/MDS).
For the most part, MSI-induced mutations are single base-pair insertions/deletions that cause frameshifts, hence driving the expression of truncated proteins with altered C-terminal sequences. We have previously established that such neo-peptide tails (frameshift peptides; FSPs) expressed by MSI+ colorectal carcinomas (CRCs) are highly immunogenic.Citation5-Citation7 More recently, we have hypothesized that MSI-induced FSPs may constitute antigens shared by different type of MSI+ cancer cells.Citation8 Thus, by taking advantage of established FSP-specific T-cell lines, we could efficiently target MSI+ leukemic cells that endogenously express the underlying FSP-related mutation.Citation8
Contrary to solid tumors (i.e., gastric carcinoma, CRC), in which malignant cells are embedded in epithelial and stromal cells, circulating MSI+ cancer cells cannot hide from the immune system. Rather, hematological MSI+ malignant cells are constantly subjected to an immunological control and this must be fatal for MSI+ cells displaying FSPs on their surface in the context of MHC molecules (). Supported by our previous results, we would assume that premalignant MMR-D/MSI+ cells are recognized and eliminated by the host immune system before the establishment of overt neoplasms. This may explain why MSI is rarely detectable in primary leukemia patients as well as the reduced number of distant metastasis found in patients suffering from MSI+ CRC.Citation9
To summarize and extrapolate the arguments discussed above, we would like to suggest a model for the development of MSI+ leukemias (). A state of immunosuppression would generally select for MMR-D precursor cells (either preexisting leukemic cells or normal lymphatic/hematopoietic cells) with an intrinsically high mutational rate. Normally, most MSI+ cells would be effectively removed by FSP-specific T cells. However, if MMR-D precursors manage to populate an immunoprivileged anatomical location, such as the hematopoietic stem cell niche, the emergence of an MSI+ leukemia would be favored. By analogy with MSI+ colorectal carcinogenesis, it can be supposed that the escape from immunosurveillance or the adaptation to permanent immunological pressure is central to MSI+ leukemogenesis. Despite the hypermutational state of MSI+ cells, this process is likely to take several years to decades in the presence of an intact immune system (). However, immunosuppression is expected to significantly accelerate the emergence of secondary and therapy-related MSI+ leukemia.
As a note, we have just initiated a comprehensive study to validate FSP candidates in clinical acute lymphoblastic leukemia and AML samples, with a focus on secondary and therapy-related cases. First, we aim at identifying novel MSI biomarkers and prospectively constitute to the develop of a consensus panel analogous to the Bethesda panel for MSI+ CRCs. Discovering precise genetic lesions that underlie MSI will undoubtedly contribute to a better understanding of MSI-driven leukemogenesis and facilitate the prognostic and predictive classification of primary as well as secondary and/or therapy-related leukemia patients. Moreover, the identification of highly immunogenic FSPs, be they specific for MSI+ leukemia or shared by different MSI+ neoplasm, will foster the development of MSI-specific vaccination strategies.
| Abbreviations: | ||
| AML | = | acute myeloid leukemia |
| CRC | = | colorectal carcinoma |
| FSP | = | frameshift peptide |
| MDS | = | myelodysplastic syndrome |
| MMR | = | mismatch repair |
| MMR-D | = | MMR-deficient |
| MSI | = | microsatellite instability |
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Sellick GS, Lubbe SJ, Matutes E, Catovsky D, Houlston RS. Microsatellite instability indicative of defects in the major mismatch repair genes is rare in patients with B-cell chronic lymphocytic leukemia: Evaluation with disease stage and family history. Leuk Lymphoma 2007; 48:1320 - 2; http://dx.doi.org/10.1080/10428190701361844; PMID: 17613760
- Casorelli I, Bossa C, Bignami M. DNA damage and repair in human cancer: molecular mechanisms and contribution to therapy-related leukemias. Int J Environ Res Public Health 2012; 9:2636 - 57; http://dx.doi.org/10.3390/ijerph9082636; PMID: 23066388
- Offman J, Opelz G, Doehler B, Cummins D, Halil O, Banner NR, et al. Defective DNA mismatch repair in acute myeloid leukemia/myelodysplastic syndrome after organ transplantation. Blood 2004; 104:822 - 8; http://dx.doi.org/10.1182/blood-2003-11-3938; PMID: 15090454
- Karran P. Thiopurines, DNA damage, DNA repair and therapy-related cancer. Br Med Bull 2006; 79-80:153 - 70; http://dx.doi.org/10.1093/bmb/ldl020; PMID: 17277075
- Garbe Y, Maletzki C, Linnebacher M. An MSI tumor specific frameshift mutation in a coding microsatellite of MSH3 encodes for HLA-A0201-restricted CD8+ cytotoxic T cell epitopes. PLoS One 2011; 6:e26517; http://dx.doi.org/10.1371/journal.pone.0026517; PMID: 22110587
- Schwitalle Y, Kloor M, Eiermann S, Linnebacher M, Kienle P, Knaebel HP, et al. Immune response against frameshift-induced neopeptides in HNPCC patients and healthy HNPCC mutation carriers. Gastroenterology 2008; 134:988 - 97; http://dx.doi.org/10.1053/j.gastro.2008.01.015; PMID: 18395080
- Schwitalle Y, Linnebacher M, Ripberger E, Gebert J, von Knebel Doeberitz M. Immunogenic peptides generated by frameshift mutations in DNA mismatch repair-deficient cancer cells. Cancer Immun 2004; 4:14; PMID: 15563124
- Maletzki C, Schmidt F, Dirks WG, Schmitt M, Linnebacher M. Frameshift-derived neoantigens constitute immunotherapeutic targets for patients with microsatellite-instable haematological malignancies: Frameshift peptides for treating MSI(+) blood cancers. Eur J Cancer 2013; In press http://dx.doi.org/10.1016/j.ejca.2013.02.035; PMID: 23561850
- Buckowitz A, Knaebel HP, Benner A, Bläker H, Gebert J, Kienle P, et al. Microsatellite instability in colorectal cancer is associated with local lymphocyte infiltration and low frequency of distant metastases. Br J Cancer 2005; 92:1746 - 53; http://dx.doi.org/10.1038/sj.bjc.6602534; PMID: 15856045