Abstract
Optimal tumor eradication often results from the death of malignant cells, as induced by chemotherapeutic agents, coupled to the induction of antitumor immune responses. However, cancer cells frequently become resistant to the cytotoxic activity of chemotherapy. The aim of the present study was to evaluate whether zinc dichloride (ZnCl2), which was known to re-establish the chemosensitivity of cancer cells by reactivating p53, promotes immunogenic instances of cell death. We found that ZnCl2, in combination with chemotherapeutic agents such as cisplatin and adriamycin (ADR), favors the apoptotic demise of chemoresistant cells, while cisplatin and ADR alone fail to do so. The co-culture of immature dendritic cells (DCs) with cancer cells succumbing to the co-administration of chemotherapy and ZnCl2 led to DC activation, as indicated by the upregulation of the activation markers CD83 and CD86. In part, such process depended on cell death, as it was limited (but not abrogated) by the pan-caspase inhibitor Z-VAD-fmk. Moreover, DC activation relied on the ZnCl2-induced exposure of calreticulin (CRT) on the surface of cancer cells, correlating with the phosphorylation of eukaryotic translation initiation factor 2α (eIF2α), a marker of endoplasmic reticulum stress. The siRNA-mediated knockdown of CRT as well as the inhibition of CRT exposure with brefeldin A strongly impaired DC maturation, indicating CRT translocation as induced by that ZnCl2 is a key event in this setting. Altogether, these results suggest that ZnCl2, has the potential to enhance the therapeutic effects of antineoplastic agents not only by improving their cytotoxic activity but also by promoting CRT exposure.
Introduction
Despite consistent therapeutic progresses, several advanced solid tumors remain difficult to treat and are associated with dismal prognosis. Although chemotherapy yields high success rates in some oncological indications, it does not always succeed in tumor eradication, either because malignant cells have developed chemoresistance, or because not all chemotherapeutics stimulate anticancer immune responses.Citation1 In multiple instances, chemoresistance originates from the impairment of the oncosuppressor activity of p53. The complete lack of p53, the expression of mutant (mt) p53 variants as well as the deregulation of wild-type (wt) p53 are common in human cancers and are associated with increased resistance to chemo- and radiotherapy.Citation2 Significant efforts toward p53 reactivation are underway, because functional p53 is considered a key factor for the elicitation of efficient responses to chemotherapy and the apoptotic clearance of cancer cells.Citation3 In this regard, we have previously demonstrated that mt or misfolded p53 can be reactivated by the administration of zinc (in the form of zinc dichloride, ZnCl2), resulting in the reestablishment of the apoptotic response of mtp53-expressing cancer cells to chemotherapy.Citation4-Citation7
Ideally, besides promoting apoptosis, chemotherapy should be immunogenic, hence igniting an immune response against malignant cells.Citation1,Citation8 Antitumor immunity can be activated when the death of cancer cells is accompanied by a series of subtle changes in the composition of their surface and their microenvironment that allow components of the innate immune system, notably dendritic cells (DCs), to sense immunogenicity.Citation9 Among other features, immunogenic cell death manifest with the translocation of the endoplasmic reticulum (ER)-resident chaperone calreticulin (CRT) to the plasma membrane surface, followed by exposure or release of heat-shock proteins including HSP70 and HSP90.Citation10,Citation11 Those molecules either provide a direct signal for DC activation or act as vehicles for antigenic peptides, facilitating their engulfment by DCs and hence promoting T-cell activation. CRT is translocated on the cell surface following various types of ER stress, resulting in the emission of a pre-apoptotic immunogenic stimulus.Citation12 In particular, CRT exposure has been reported to follow the phosphorylation of eukaryotic translation initiation factor 2α (eIF2α) in the course of ER stress responses.Citation12 In this context, we have recently shown that the anticancer drugs bortezomib, an inhibitor of the proteasome, and Tyrphostin AG 490, targeting mitogen-activated protein kinase 9 (MAPK9, also known as JNK2) and signal transducer and activator of transcription 3 (STAT3) signaling, induce the immunogenic demise of primary effusion lymphoma (PEL) cells.Citation13 Although bortezomib-treated PEL cells died by apoptosis, the broad-spectrum caspase inhibitor Z-Val-Ala-DL-Asp-fluoromethylketone (Z-VAD-fmk) reduced the activation of co-cultured DCs to slight extents, suggesting that apoptosis itself was not the main event responsible for the immunogenicity of cell death. Rather, DC activation relied on the exposure of CRT, HSP70 and HSP90 of the surface of PEL cells.Citation13,Citation14
Driven by these considerations, we investigated whether ZnCl2, in combination with chemotherapeutic agents such as cisplatin and ADR might promote the immunogenic demise of cancer cells. We found that: (1) ZnCl2 re-establishes the sensitivity of chemoresistant cancer cells to antineoplastic agents, correlating with the reactivation of mtp53; (2) the co-administration of ZnCl2 and chemotherapy promoted an immunogenic instance of apoptosis, resulting in the activation of DCs; (3) cell death was important for the immunogenicity of chemoresistant cells succumbing to chemotherapy plus ZnCl2, although the pre-apoptotic exposure of CRT on the cell surface (as induced by ZnCl2) also played a major role. These data suggest that ZnCl2 cannot only exacerbate the cytotoxic effects of anticancer agents against p53-functionally deficient chemoresistant cells, but can also stimulate the emission of immunogenic signals.
Results
ZnCl2 exacerbates the cytotoxic effects of chemotherapy in chemoresistant p53-deficient cancer cells
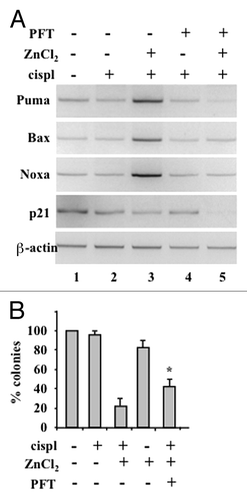
We have previously reported that ZnCl2 restores the chemosensitivity of several chemoresistant p53-functionally deficient cancer cells including breast carcinoma SKBR3 and glioblastoma U373MG cells, which carry mtp53, as well as colorectal carcinoma RKO cells subjected to the depletion of homeodomain-interacting protein kinase 2 (RKO-HIPK2i cells), which express a misfolding variant of p53, mainly reflecting the ability of ZnCl2 to mediate p53 reactivation.Citation4-Citation7 Here, we first evaluated whether ZnCl2 might reactivate endogenous p53 in glioblastoma ADF cells, as this cell line has been shown to carry an endogenous wt form of p53 devoid of transcriptional activity.Citation15,Citation16 To this aim, the transcription of p53 target genes was examined in ADF cells treated with cisplatin and ZnCl2, alone or in combination, by RT-PCR. Thus, while cisplatin alone failed to induce the expression of p53 target genes, the co-administration of cisplatin and ZnCl2 resulted in the transactivation of multiple pro-apoptotic p53 target genes, including BCL2-associated X protein (BAX), BCL2 binding component 3 (BBC3, best known as PUMA) and phorbol-12-myristate-13-acetate-induced protein 1 (PMAIP1, best known as NOXA), but not of cell cycle-regulatory genes such as cyclin-dependent kinase inhibitor 1A (CDKN1A, coding for p21Cip1) (, compare lane 3 with lane 2). p53 reactivation was confirmed with pifithryn-α (PFT),Citation17 a p53 inhibitor that was able to block the expression of p53 target genes in response to cisplatin and ZnCl2 (, compare lane 5 with lane 3). Long-term clonogenic assays coupled to densitometric analyses demonstrated that ZnCl2 in combination with cisplatin markedly reduces the clonogenic potential of ADF cells as compared with cisplatin alone (, compare lane 3 with lane 2). Of note, the decreased in the clonogenic potential of ADF cells treated with ZnCl2 plus cisplatin was partly (of about 2.1-fold) rescued by PFT (p = 0,034) (, compare lane 5 with lane 3). These data indicate that ZnCl2 may reactivate dysfunctional wtp53 thus restoring the chemosensitivity of ADF cells, in line with previous results obtained with U373 and RKO-HIPK2i cells.Citation4,Citation5 Although PFT efficiently inhibited p53 transactivation, it limited the cytotoxic effects of cisplatin plus ZnCl2 on ADF cells to a limited extent, suggesting that p53-independent mechanisms may contribute to cell death in this setting.
Figure 1. ZnCl2 reactivates p53 in chemoresistant glioblastoma cells. (A) Semi-quantitative RT-PCR analyses of p53 target genes in human glioblastoma ADF cells pre-treated with 100 μM ZnCl2 for 6 h before the addition of 4 μg/ml cisplatin (cisp) for 16 h. When appropriate, the p53 inhibitor pifithryn α (PFT) (30 μM) was added to ZnCl2 before the administration of chemotherapy. β-actin levels were monitored as an internal standard. (B) 20000 ADF cells were plated in 60-mm dished and - 24 h later - treated with 100 μM ZnCl2 for 16 h, followed by the addition of 4 μg/mL cisp for 2 h. Cells were then washed with PBS and placed in fresh medium. ZnCl2 was replaced in the culture medium every two days. Colonies were stained with crystal violet 14 d after seeding and quantified. Data are presented as means ± SD *p = 0.034, as compared with cells treated with cispl plus ZnCl2.
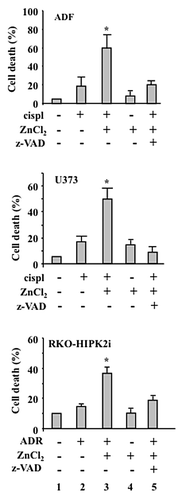
Next, we evaluated whether cell death as induced by ZnCl2 in combination with chemotherapy in ADF, U373 and RKO-HIPK2i cellsCitation4,Citation5 proceeds via apoptosis. To this aim, we performed terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assays, finding that ZnCl2 in combination with chemotherapy (in particular, cisplatin for ADF and U373 cells, or ADR for RKO-HIPK2i cells) induces significant extents of apoptotic DNA fragmentation in all these cell lines as compared with the administration of either chemotherapy or ZnCl2 alone (, compare lanes 3 with lanes 2). Moreover, the pre-treatment of ADF, U373 and RKO-HIPK2i cells with Z-VAD-fmk efficiently inhibited apoptosis as induced by chemotherapy plus ZnCl2 (, compare lanes 5 with lanes 3). Altogether, these data demonstrate that ADR or cisplatin employed as standalone interventions do not affect the viability of chemoresistant p53-functionally deficient cancer cells, while they efficiently induce apoptosis in the presence of ZnCl2.
Figure 2. ZnCl2 exacerbates the cytotoxic potential of chemotherapy in chemoresistant cancer cells. Percentage of TUNEL+ human glioblastoma ADF cells, human glioblastoma U373 cells and colorectal carcinoma RKO-HIPK2i cells treated with 100 μM ZnCl2 for 6 h before the addition of 4 μg/mL cisplatin (cispl) (in U373 and ADF cells) or 2 μM adriamycin (ADR) (in RKO-HIPK2i cells) for 24 h. When appropriate, the pan-caspase inhibitor Z-VAD-fmk (40 μM) was added to ZnCl2 before the administration of chemotherapy. Data are reported as means ± SD *p = 0.001, as compared with untreated cells.
Effect of cancer cells succumbing in the presence of ZnCl2 on DC maturation
As we found that ZnCl2 exacerbates the cytotoxic effects of chemotherapy against chemoresistant cancer cells bearing dysfunctional p53, we wondered whether cancer cells dying in the presence of ZnCl2 could activate the immune system. To address this question, we co-cultured immature dendritic cells (DCs) with cancer cells exposed to cisplatin or ADR, either alone or in combination with ZnCl2 and evaluated DC activation markers such as CD83 and CD86 by flow cytometry. We focused on DCs because their activation is stringently required for the elicitation of immune responses against apoptotic tumor cells.Citation18,Citation19 We found that cancer cells treated with chemotherapy or ZnCl2 alone do not activate DCs, as indicated by the reduced expression levels of CD83 and CD86 (, compare lanes 2 and 4 with lanes 1), in line with the limited ability of these interventions to trigger cell death (). On the contrary, tumor cells acquired the capacity to efficiently induce the maturation of DCs when they were treated with ZnCl2 plus cisplatin or ADR (, compare lanes 3 with lanes 2), paralleling the extents of cell death induction (). We then evaluated whether the activation of DCs in this context solely depend on cancer cell death. Interestingly, the pre-treatment of cancer cells with Z-VAD-fmk consistently inhibited cell death () yet impaired DC maturation to limited extents (, compare lanes 4 with lanes 3). Thus, it seems that that apoptosis is not the sole event responsible for the immunogenicity of cancer cells succumbing to chemotherapy in the presence of ZnCl2.
Figure 3. Chemoresistant cancer cells succumbing to chemotherapy in the presence of ZnCl2 promote dendritic cell (DC) maturation. (A) Human glioblastoma ADF cells, human glioblastoma U373 cells and colorectal carcinoma RKO-HIPK2i cells were treated with 100 μM ZnCl2 for 6 h, followed by the addition of 4 μg/mL cisplatin (cispl) (in U373 and ADF cells) or 2 μM adriamycin (ADR) (in RKO-HIPK2i cells) for 16 h. Thereafter, tumor cells were co-cultured with immature dendritic cells (DCs) for 24 h, followed by the cytofluorometric assessment of the DC maturation markers CD83 and CD86. Data from one representative experiment are reported as the mean percentage of CD83+ and CD86+ DCs ± SD *p = 0.001, as compared with untreated cells. (B) ADF cells were treated with 100 μM ZnCl2 alone or in combination with 40 μM Z-VAD-fmk for 6 h, followed by the administration of 4 μg/mL cisp for 16 h. Thereafter, cells were co-cultured with DCs and DC maturation markers assessed as in Panel A. Results from one representative experiment are shown as means ± SD (C) Upper panel: percentage of TUNEL+ wild-type (wt) RKO cells treated with 2 μM ADR for 24 h. When appropriate, 40 μM Z-VAD-fmk was added for 1 h before ADR administration. Data are reported as means ± SD. Lower panel: immunoblotting assessment of PARP cleavage in RKO cells treated with ADR alone or in combination with Z-VAD-fmk. l.c., loading control. (D) RKO cells treated as in Panel C were co-cultured with immature DCs for 24 h, followed by the cytofluorometric assessment of CD86. Data for one representative experiment are reported as the percentage of CD86+ DCs ± SD (E) Total and membrane proteins were extracted from equal amounts of RKO cells exposed to 2 μM ADR for 24 h and immunoblotted with a calreticulin (CRT)-specific antibody.
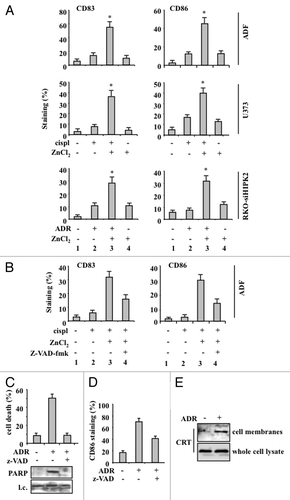
Next, we evaluated whether DCs could be activated upon exposure to wtp53-expressing RKO cells undergoing apoptosis in response to ADR. The ability of ADR to promote the activation of p53 in RKO cells is extensively documented in the literature.Citation4 ADR indeed triggered the apoptotic demise of RKO cells, as evidenced by TUNEL positivity as well as by the cleavage of the caspase substrate poly(ADP-ribose) polymerase (PARP), a process that was efficiently inhibited by Z-VAD-fmk (). Upon co-culture, RKO cells treated with ADR induced the activation of DCs, which also was (at least to some extent) inhibited by Z-VAD-fmk (). Immunoblotting analyses of plasma membrane proteins purified from ADR-treated and control RKO cells revealed a significant increase in local abundance of CRT upon ADR treatment (). This increase did not reflect the upregulation of CRT, as demonstrated by the immunoblotting of whole-cell protein extracts (). Altogether, these data demonstrate that p53-proficient cancer cells killed by chemotherapy, at odds with their p53-functionally deficient counterparts, may expose CRT in response to chemotherapy (see below), hence promoting the maturation of DCs.
ZnCl2 promotes CRT exposure, which is required for DC activation by dying chemoresistant cancer cells
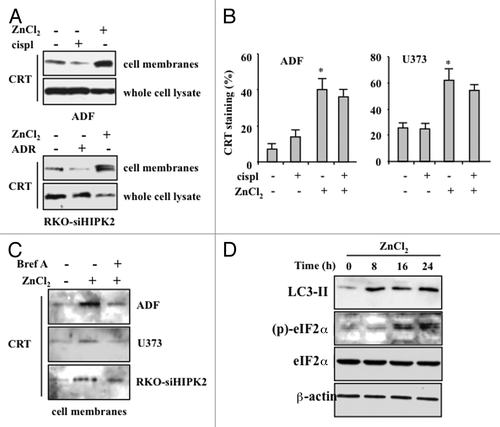
As the maturation of DCs elicited by cancer cells succumbing to chemotherapy in the presence of ZnCl2 dependent only in part on the extent of cell death, we investigated how ZnCl2 could affect the immunogenicity of this process. To this aim, we explored the effects of ZnCl2 on the exposure of CRT on the surface of cancer cells, since this event has previously been shown to elicit potent anticancer immune responses.Citation10-Citation12,Citation20 Immunoblotting analyses of purified plasma membrane proteins revealed a significant increase in the amount of cell-surface CRT in cancer cells treated with chemotherapy plus ZnCl2, while cells receiving chemotherapy alone exhibited decreased CRT levels on their surface (). Of note, this was not a consequence of a global upregulation of CRT (). A similar effect could also be documented by flow cytometry, revealing that living cancer cells exposed to ZnCl2, alone or in the presence of chemotherapy, exhibit increased amounts of CRT of their surface (). Brefeldin A (BrefA), an inhibitor of anterograde protein transport from the ER to the Golgi apparatusCitation21 that has previously been shown to inhibit CRT exposure,Citation12 limited the ZnCl2-induced translocation of CRT on the plasma membrane of chemoresistant cancer cells (). Importantly, ZnCl2-induced CRT translocation correlated with the phosphorylation of eIF2α (), a protein that is generally phosphorylated in the course of ER stress owing to the activity of stress-inducible kinases such as eIF2α kinase 3 (EIF2AK3, best known as PERK).Citation22 This finding is in agreement with the results of previous studies showing that the ER stress-induced phosphorylation of eIF2α correlates with CRT exposure.Citation23 We also found that eIF2α phosphorylation as promoted by ZnCl2 is preceded by the activation of autophagy, as indicated by the appearance of the lipidated form of LC3 (LC3-II) (). This is also in line with previous reports showing that pre-mortem autophagy is fundamental for the immunogenicity of cell death.Citation24,Citation25
Figure 4. ZnCl2 induces calreticulin exposure on the membrane of chemoresistant cancer cells. (A) Human glioblastoma ADF cells and colorectal carcinoma RKO-HIPK2i cells were treated with 100 μM ZnCl2, 4 μg/mL cisplatin (cispl) or 2 μM adriamycin (ADR), as indicated, for 16 h. Thereafter, total and membrane proteins were extracted from equal amounts of RKO cells and immunoblotted with a calreticulin (CRT)-specific antibody. (B) CRT exposure by living human glioblastoma U373 cells exposed to 100 μM ZnCl2 and 4 μg/mL cisp, alone or in combination, was verified by flow cytometry. Data, which are representative of two independent experiments, are reported as means ± SD (C) ADF, U373 and RKO-HIPK2i cells were treated with 100 μM ZnCl2 alone or in the presence of 10 μM brefeldin A (Bref A), for 16 h. Membrane proteins extracted from equal amounts of cells were assayed by immunoblotting with an anti-CRT antibody. (D) ADF cells were treated with 100 μM ZnCl2 for the indicated time (h), followed by the extraction of total proteins from equal amounts of cells and immunoblotting with antibodies specific for the indicated proteins. β actin levels were monitored as a loading control.
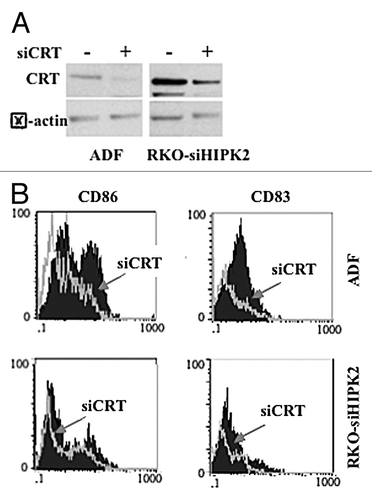
To evaluate the role of CRT exposure in the activation of DCs as triggered by chemoresistant cancer cells succumbing to chemotherapy in the presence of ZnCl2, we co-cultured immature DCs with ADF cells treated with chemotherapy plus ZnCl2 in the presence of BrefA. We found that DC activation in this setting is significantly impaired by the pre-treatment of ADF cells with BrefA (). Similar results were obtained with RKO-siHIPK2 (data not shown). To obtain further insights in this process, we depleted ADF and RKO-siHIPK2 cells of CRT with specific small-interfering RNAs (siRNAs) (). This intervention efficiently inhibited the ability of ADF and RKO-siHIPK2 cells treated with ZnCl2 plus ADR and cisplatin, respectively, to promote DC activation, as shown by flow cytometry upon staining of DCs with anti-CD83 and anti-CD86 antibodies (). Altogether, these data demonstrate that ZnCl2 induces the exposure of CRT on the surface of malignant cells, which is a pre-requisite for DC activation in response to their demise.
Figure 5. The inhibition of calreticulin exposure by brefeldin A impairs dendritic cell activation by chemoresistant cancer cells succumbing to chemotherapy in the presence of ZnCl2. (A and B) Human glioblastoma ADF cells were treated with 100 μM ZnCl2 alone or together with 10 μM brefeldin A (BrefA) for 6 h, followed by the administration of 4 μg/mL cisplatin (cispl) for additional 16 h. Thereafter, cancer cells were co-cultured with immature dendritic cells (DCs) for 24 h and the DC maturation markers CD83 and CD86 were monitored by flow cytometry. Data from one representative experiment are reported as staining profiles (A), isotype controls in black) or mean percentage of CD83+ and CD86+ DCs ± SD; *p = 0.001 (B).
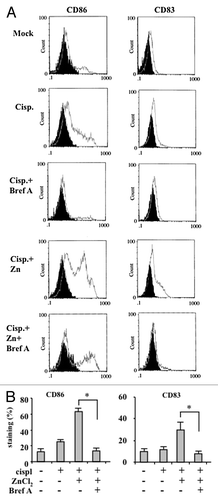
Figure 6. Calreticulin depletion in cancer cells impairs their capacity to activate dendritic cells in the course of immunogenic cell death. (A) Human glioblastoma ADF cells and colorectal carcinoma RKO-HIPK2i cells were transfected with a control siRNA (-) or a siRNA specific for calreticulin (siCRT; +). 36 h after transfection, cells were assayed by immunoblotting for calreticulin (CRT) expression levels. β actin was monitored as a loading control. (B) Control ADF and RKO-HIPK2i cells (empty gray histograms) or ADF and RKO-HIPK2i subjected to CRT depletion as in Panel A (black-filled hystograms) were treated with 100 μM ZnCl2 for 6 h, followed by the administration of 4 μg/mL cisplatin (cispl; in ADF cells) or 2 μM adriamycin (ADR; in RKO-HIPK2i cells) for 16 h cancer cells were co-cultured with immature dendritic cells (DCs) for 24 h and the DC maturation markers CD83 and CD86 were monitored by flow cytometry. Data from one representative experiment are reported as staining profiles.
Discussion
The 2 ideal goals of anticancer therapy are the induction of a strong cytotoxic responses among malignant cells and the stimulation of host tumor-specific response, cooperating in the achievement of clinically relevant effects. Here we show that both these goals can be obtained in chemoresistant, p53-functionally deficient cell lines by combining ZnCl2 with chemotherapeutic agents such as cisplatin and ADR. Indeed, such a combinatorial regimen turned out to reactivate p53 and promote the exposure of CRT on the cell surface in the course of cell death, resulting in the activation of DCs.
ZnCl2 is an essential microelement that participates in the activity of approximately 300 enzymes and is involved in the regulation of over 2000 transcription factors.Citation26 We have previously demonstrated that ZnCl2 can reactivate mutant or misfolded p53 in cancer cells,Citation4-Citation7 in line with previous findings on the importance of ZnCl2 for p53 folding and stability.Citation27,Citation28 Thus, the co-administration of ZnCl2 and chemotherapeutic agents such as cisplatin and ADR promotes the reestablishment of wt p53 conformation, in turn rescuing p53 oncosuppressor activity and mediating prominent antineoplastic effects, in vivo.Citation4-Citation7 In agreement with these findings, here we report that ZnCl2 reactivates non-functional wtp53 in glioblastoma ADF cells, thereby restoring their chemosensitivity. Although the molecular mechanisms of p53 reactivation in ADF cells remain to be elucidated, we and others have demonstrated that ZnCl2 supplementation may be useful, at in some circumstances, to improve the therapeutic efficacy of anticancer drugs.
The apoptotic response of cancer cells to chemotherapy leads to tumor eradication if it is accompanied by an efficient immune response. According to some authors, this may represent the sole successful approach to anticancer therapy.Citation5,Citation20 DCs not only play a pivotal role in the eradication of apoptotic cancer cells, but also are capable of cross-presenting tumor-associated antigens to cytotoxic T cells, hence promoting the initiation of tumor-specific immune responses.Citation9 For these reasons, the activation of DCs by dying tumor cells can counteract cancer-driven immunosuppression and eventually improve disease outcome.Citation29 Here we found that chemoresistant p53-functionally deficient cancer cells undergo apoptosis only when chemotherapy is combined with ZnCl2, and that such an apoptotic response is able to trigger DC maturation. The pre-treatment of cancer cells with the pan-caspase inhibitor Z-VAD-fmk inhibited their demise in response to chemotherapy and ZnCl2, yet impaired DC maturation only to partial extents. This indicates that the degree of cell death is not sufficient the major determinant of the immunogenicity of the process. Moreover, both cancer cells expressing mt variants of p53 (upon exposure to chemotherapy plus ZnCl2) and p53-proficient malignant cells (succumbing to chemotherapy alone) were able to induce the maturation of DCs, underscoring a functional role for p53 in immunogenic apoptosis that warrant further investigation.
The immunogenicity of cell death requires the translocation to the cell surface of proteins that are normally found in the ER lumen, which belong to so-called “damage-associated molecular patterns” (DAMPs).Citation30 One of the most important DAMPs is calreticulin, which translocates from the ER to the plasma membrane in response to ER stress. This process stimulates the activation of DCs, resulting in enhanced antigen uptake, processing and presentation.Citation10 CRT exposure usually ensues the ER stress-associated phosphorylation of eIF2α, which is also linked to the induction of autophagy.Citation12,Citation23,Citation31 Interestingly, pre-mortem autophagy is an important determinant of the immunogenicity of chemotherapy-induced cell death.Citation24,Citation25 We have previously reported that the immunogenicity of dying PEL cells is not completely dependent on the degree of cell death. Rather, in this context DC activation appeared to critically rely on the exposure of CRT, HSP70 and HSP90 on the cell surface.Citation13,Citation14 In line with these findings, here we reported that ZnCl2 induces the exposure of CRT on chemoresistant cancer cells, correlating with eIF2α phosphorylation and autophagy induction. The siRNA-mediated knockdown of CRT as well as the inhibition of CRT exposure by BrefA strongly impaired the activation of DC in response to dying cancer cells, indicating that the translocation of CRT on the outer leaflet of the plasma membrane is the key event for immunogenic cell death as triggered by chemotherapy plus ZnCl2.
In conclusion, our data suggest that ZnCl2 may exacerbate the cytotoxic effects of chemotherapy in chemoresistant p53-functionally deficient cells and promote the exposure of CRT in the course of cell death, hence rendering it immunogenic. ZnCl2 might therefore have a heavy impact on the crosstalk between malignant cells and the immune system, both as it increases the immunogenic potential of cell death (as shown in this report) and as it counteracts mechanisms of immunosuppression that are set in place by tumor cells (as we have previously reported).Citation31-Citation33 Future studies will generate additional insights into the potential application of ZnCl2 in anticancer therapy.
Materials and Methods
Cell culture and reagents
Human ADF and U373MG glioblastoma cells, wt human colon carcinoma RKO cells and RKO cells stably depleted of HIPK2 (RKO-HIPK2i cells)Citation34 were routinely maintained in RPMI-1640 medium (Life-Technology-Invitrogen) containing 10% heat-inactivated fetal calf serum (FCS), 100 units/mL penicillin/streptomycin, and 2 mM l-glutamine, in 5% CO2 humidified incubators at 37°C. The following reagents were used in this study at the indicated concentrations: ZnCl2 (Sigma), dissolved in dH2O2, 100 μM; cisplatin (Pharmachemie B.V., Harleem, Holland), 4 μg/mL; ADR (EBEWE Pharma, Italy, S.r.l.), 2 μg/mL; BrefA (Cell Signaling) 10 μM; PFT (ENZO Life Sciences, Lausen Switzerland), 30 μM; Z-VAD-fmk (R&D Systems), 40 μM. Appropriate volumes of PBS or DMSO were used to generate negative control conditions.
Clonogenic assays and assessment of apoptosis
For long-term clonogenic assays, cells were pre-treated with ZnCl2 for 16 h and then subjected to a 2 h pulse of cisplatin. Upon treatment, cells were washed, trypsinized, and counted, and equal cell numbers were seeded in drug-free medium in 60-mm petri dishes. Colonies were stained with crystal violet 14 d later and quantified. For TUNEL assays, 4 × 104 cells were cytospun on a slide and fixed in 4% paraformaldheyde for 30 min at room temperature, as previously reported.Citation35 After rinsing with PBS, samples were permeabilized in 0.01% Triton X-100 in sodium citrate for 2 min. Samples were then washed with PBS and incubated in the TUNEL reaction mix for 1 h at 37°C, according to the manufacturer’s instructions (Roche, Germany). Cells were counterstained with Hoechst 33342 before assessment of TUNEL positivity on a fluorescence microscope (Zeiss).
RNA isolation and RT-PCR
Total RNA was isolated by using the TRizol reagent (Invitrogen). cDNA was syntesized from 2 μg of total RNA with a MuLV reverse transcriptase-based kit (Applied Biosystems). Semi-quantitative RT-PCR was performed by means of the Hot-Master Taq polymerase (Eppendorf) with 2 μL cDNA template and gene-specific oligonucleotides under conditions of linear amplification. The housekeeping gene coding for β actin was monitored as an internal standard. mRNA expression levels were quantified by densitometric procedures.
Immunoblotting
Whole-cell lysates were prepared for immunoblotting analyses by means of a lysis buffer containing 50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 150 mM KCl, 1 mM dithiothreitol, 5 mM EDTA, pH 8.0, 1% Nonidet P-40 plus a commercial mix of protease and phosphatase inhibitors (Sigma Chemical Company). Membrane proteins were extracted from 5 × 106 cells by using the Mem-PER Eukaryotic Membrane Protein Extraction Reagent (Thermo Scientific, Pierce Biotechnology). Samples were denatured in SDS-based sample buffer and proteins were separated on SDS-PAGE precast gels (BioRad), followed by transfer to polyvinylidene difluoride (PVDF) membranes (Millipore). Membranes were blocked with 5% nonfat dry milk in PBS and incubated with primary antibodies specific for CRT (PA3–900, Thermo Scientific), LC3B (L7543, Sigma), phosphorylated eIF2α (Ser15) (Cell Signaling), total eIF2α (Cell Signaling), PARP (Millipore) and β actin (Calbiochem). Secondary antibodies conjugated to horseradish peroxidase (BioRad) were used to detect primary antibodies by chemoluminescence (ECL kit, Amersham Corporation).
RNA interference
siRNAs specific for human CRT (Santa Cruz Biotechnology) were transfected by using the proprietary siRNA transfection kit, according to the manufacturer’s instructions. 36 h after transfection, CRT downregulation was monitored by immunoblotting.
Generation of monocyte-derived DCs and DC maturation
To generate monocyte-derived DCs, human peripheral blood mononuclear cells obtained from healthy donors (under informed consent) were isolated by Fycoll-Paque gradient centrifugation (Pharmacia, Uppsala, Sweden). CD14+ monocytes were positively selected using anti-CD14 antibody-conjugated magnetic microbeads (Miltenyi Biotec, Auburn, Calif.). To generate immature DCs, purified monocytes were then cultured in 12-well plates for 6 d, at a density of 106 cells/3 mL RPMI 1640 containing 10% FCS, 2 mM l-glutamine, 100 U/mL penicillin G, 100 mg/mL streptomycin, 50 ng/mL recombinant human granulocyte macrophage colony-stimulating factor (GM-CSF) and 20 ng/mL interleukin-4 (IL-4) (Miltenyi Biotec). Cytokines were replenished every other day along with 20% fresh medium.
Cytofluorometric analyses of DC phenotypes
Tumor cells were treated with ZnCl2 for 6 h prior to the administration of cisplatin or ADR for additional 16 h. Thereafter, tumor cells were washed and co-cultured with immature DCs for 24 h. The phenotype of DCs was monitored by flow cytometry upon staining with phycoerytrin (PE)-conjugated anti-CD83 and fluorescein isothiocianate (FITC)-conjugated anti-CD86 antibodies (Becton Dickinson, San Diego, CA) for 30 min at 4°C and two washes in PBS. DCs were gated according to FSC and SSC parameters. Appropriate isotype-matched control antibodies were included in the assessments and 5000 viable DCs were acquired in each experiment. Acquisition was performed on a EPICS XL flow cytometer (Coulter, Hialeah, FL).
Statistical analyses
Unless otherwise specified, ach experiment was performed at least 3 times, and data are presented as means ± SD. Statistical significance was determined using the Student’s t-test. p values ≤ 0.05 were considered as statistically significant.
| Abbreviations: | ||
| ADR | = | adriamycin |
| BrefA | = | brefeldin A |
| CRT | = | calreticulin |
| DAMP | = | damage-associated molecular pattern |
| DC | = | dendritic cell |
| eIF2α | = | eukaryotic initiation factor 2α |
| ER | = | endoplasmic reticulum |
| HIPK2 | = | homeodomain-interacting protein kinase 2 |
| mt | = | mutant |
| PARP | = | poly (ADP-ribose) polymerase |
| PFT | = | pifithryn α |
| wt | = | wild-type |
| ZnCl2 | = | zinc dichloride |
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
This study was supported by grants from the Italian Association for Cancer Research (AIRC, IG 11377 to GD and IG 10265 to AF). We thank Gianluca Bossi for critical discussion and Sandro Valia, Valeria Conte, Marina Peddis, and Donatella D’Eliseo for technical assistance.
Citation: Garufi A, Di Renzo L, Granato M, Faggioni A, D’Orazi G, Cirone M. Zinc supplementation is required for the cytotoxic and immunogenic effects of chemotherapy in chemoresistant p53-functionally deficient cells. OncoImmunology 2013; 2:e26198; 10.4161/onci.26198
Related Research Data
References
- Vacchelli E, Senovilla L, Eggermont A, Fridman WH, Galon J, Zitvogel L, Kroemer G, Galluzzi L. Trial watch: Chemotherapy with immunogenic cell death inducers. Oncoimmunology 2013; 2:e23510; http://dx.doi.org/10.4161/onci.23510; PMID: 23687621
- Brown CJ, Lain S, Verma CS, Fersht AR, Lane DP. Awakening guardian angels: drugging the p53 pathway. Nat Rev Cancer 2009; 9:862 - 73; http://dx.doi.org/10.1038/nrc2763; PMID: 19935675
- Selivanova G, Wiman KG. Reactivation of mutant p53: molecular mechanisms and therapeutic potential. Oncogene 2007; 26:2243 - 54; http://dx.doi.org/10.1038/sj.onc.1210295; PMID: 17401433
- Puca R, Nardinocchi L, Gal H, Rechavi G, Amariglio N, Domany E, Notterman DA, Scarsella M, Leonetti C, Sacchi A, et al. Reversible dysfunction of wild-type p53 following homeodomain-interacting protein kinase-2 knockdown. Cancer Res 2008; 68:3707 - 14; http://dx.doi.org/10.1158/0008-5472.CAN-07-6776; PMID: 18483253
- Nardinocchi L, Puca R, Sacchi A, Rechavi G, Givol D, D’Orazi G. Targeting hypoxia in cancer cells by restoring homeodomain interacting protein-kinase 2 and p53 activity and suppressing HIF-1alpha. PLoS One 2009; 4:e6819; http://dx.doi.org/10.1371/journal.pone.0006819; PMID: 19714248
- Puca R, Nardinocchi L, Porru M, Simon AJ, Rechavi G, Leonetti C, Givol D, D’Orazi G. Restoring p53 active conformation by zinc increases the response of mutant p53 tumor cells to anticancer drugs. Cell Cycle 2011; 10:1679 - 89; http://dx.doi.org/10.4161/cc.10.10.15642; PMID: 21508668
- Margalit O, Simon AJ, Yakubov E, Puca R, Yosepovich A, Avivi C, Jacob-Hirsch J, Gelernter I, Harmelin A, Barshack I, et al. Zinc supplementation augments in vivo antitumor effect of chemotherapy by restoring p53 function. Int J Cancer 2012; 131:E562 - 8; http://dx.doi.org/10.1002/ijc.26441; PMID: 21932419
- Zitvogel L, Casares N, Péquignot MO, Chaput N, Albert ML, Kroemer G. Immune response against dying tumor cells. Adv Immunol 2004; 84:131 - 79; http://dx.doi.org/10.1016/S0065-2776(04)84004-5; PMID: 15246252
- Ma Y, Aymeric L, Locher C, Kroemer G, Zitvogel L. The dendritic cell-tumor cross-talk in cancer. Curr Opin Immunol 2011; 23:146 - 52; http://dx.doi.org/10.1016/j.coi.2010.09.008; PMID: 20970973
- Obeid M, Tesniere A, Ghiringhelli F, Fimia GM, Apetoh L, Perfettini JL, Castedo M, Mignot G, Panaretakis T, Casares N, et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat Med 2007; 13:54 - 61; http://dx.doi.org/10.1038/nm1523; PMID: 17187072
- Garg AD, Krysko DV, Verfaillie T, Kaczmarek A, Ferreira GB, Marysael T, Rubio N, Firczuk M, Mathieu C, Roebroek AJ, et al. A novel pathway combining calreticulin exposure and ATP secretion in immunogenic cancer cell death. EMBO J 2012; 31:1062 - 79; http://dx.doi.org/10.1038/emboj.2011.497; PMID: 22252128
- Panaretakis T, Kepp O, Brockmeier U, Tesniere A, Bjorklund AC, Chapman DC, Durchschlag M, Joza N, Pierron G, van Endert P, et al. Mechanisms of pre-apoptotic calreticulin exposure in immunogenic cell death. EMBO J 2009; 28:578 - 90; http://dx.doi.org/10.1038/emboj.2009.1; PMID: 19165151
- Cirone M, Di Renzo L, Lotti LV, Conte V, Trivedi P, Santarelli R, Gonnella R, Frati L, Faggioni A. Primary effusion lymphoma cell death induced by bortezomib and AG 490 activates dendritic cells through CD91. PLoS One 2012; 7:e31732; http://dx.doi.org/10.1371/journal.pone.0031732; PMID: 22412839
- Cirone M, Di Renzo L, Lotti LV, Conte V, Trivedi P, Santarelli R, Gonnella R, Frati L, Faggioni A. Activation of dendritic cells by tumor cell death. Oncoimmunology 2012; 1:1218 - 9; http://dx.doi.org/10.4161/onci.20428; PMID: 23170286
- Biroccio A, Bufalo DD, Ricca A, D’Angelo C, D’Orazi G, Sacchi A, Soddu S, Zupi G. Increase of BCNU sensitivity by wt-p53 gene therapy in glioblastoma lines depends on the administration schedule. Gene Ther 1999; 6:1064 - 72; http://dx.doi.org/10.1038/sj.gt.3300935; PMID: 10455409
- D’Avenia P, Porrello A, Berardo M, Angelo MD, Soddu S, Arcangeli G, Sacchi A, D’Orazi G. Tp53-gene transfer induces hypersensitivity to low doses of X-rays in glioblastoma cells: a strategy to convert a radio-resistant phenotype into a radiosensitive one. Cancer Lett 2006; 231:102 - 12; http://dx.doi.org/10.1016/j.canlet.2005.01.033; PMID: 16356835
- Komarov PG, Komarova EA, Kondratov RV, Christov-Tselkov K, Coon JS, Chernov MV, Gudkov AV. A chemical inhibitor of p53 that protects mice from the side effects of cancer therapy. Science 1999; 285:1733 - 7; http://dx.doi.org/10.1126/science.285.5434.1733; PMID: 10481009
- Steinman RM, Turley S, Mellman I, Inaba K. The induction of tolerance by dendritic cells that have captured apoptotic cells. J Exp Med 2000; 191:411 - 6; http://dx.doi.org/10.1084/jem.191.3.411; PMID: 10662786
- Blachère NE, Darnell RB, Albert ML. Apoptotic cells deliver processed antigen to dendritic cells for cross-presentation. PLoS Biol 2005; 3:e185; http://dx.doi.org/10.1371/journal.pbio.0030185; PMID: 15839733
- Zitvogel L, Kepp O, Senovilla L, Menger L, Chaput N, Kroemer G. Immunogenic tumor cell death for optimal anticancer therapy: the calreticulin exposure pathway. Clin Cancer Res 2010; 16:3100 - 4; http://dx.doi.org/10.1158/1078-0432.CCR-09-2891; PMID: 20421432
- Lippincott-Schwartz J, Yuan LC, Bonifacino JS, Klausner RD. Rapid redistribution of Golgi proteins into the ER in cells treated with brefeldin A: evidence for membrane cycling from Golgi to ER. Cell 1989; 56:801 - 13; http://dx.doi.org/10.1016/0092-8674(89)90685-5; PMID: 2647301
- Zhang K, Kaufman RJ. Signaling the unfolded protein response from the endoplasmic reticulum. J Biol Chem 2004; 279:25935 - 8; http://dx.doi.org/10.1074/jbc.R400008200; PMID: 15070890
- Panaretakis T, Joza N, Modjtahedi N, Tesniere A, Vitale I, Durchschlag M, Fimia GM, Kepp O, Piacentini M, Froehlich KU, et al. The co-translocation of ERp57 and calreticulin determines the immunogenicity of cell death. Cell Death Differ 2008; 15:1499 - 509; http://dx.doi.org/10.1038/cdd.2008.67; PMID: 18464797
- Martins I, Michaud M, Sukkurwala AQ, Adjemian S, Ma Y, Shen S, Kepp O, Menger L, Vacchelli E, Galluzzi L, et al. Premortem autophagy determines the immunogenicity of chemotherapy-induced cancer cell death. Autophagy 2012; 8:413 - 5; http://dx.doi.org/10.4161/auto.19009; PMID: 22361584
- Garg AD, Martin S, Golab J, Agostinis P. Danger signalling during cancer cell death: origins, plasticity and regulation. Cell Death Differ 2013; [Epub ahead of print] http://dx.doi.org/10.1038/cdd.2013.48; PMID: 23686135
- Beyersmann D, Haase H. Functions of zinc in signaling, proliferation and differentiation of mammalian cells. Biometals 2001; 14:331 - 41; http://dx.doi.org/10.1023/A:1012905406548; PMID: 11831463
- Hainaut P, Milner J. A structural role for metal ions in the “wild-type” conformation of the tumor suppressor protein p53. Cancer Res 1993; 53:1739 - 42; PMID: 8467489
- Méplan C, Richard M-J, Hainaut P. Metalloregulation of the tumor suppressor protein p53: zinc mediates the renaturation of p53 after exposure to metal chelators in vitro and in intact cells. Oncogene 2000; 19:5227 - 36; http://dx.doi.org/10.1038/sj.onc.1203907; PMID: 11077439
- Petersen TR, Dickgreber N, Hermans IF. Tumor antigen presentation by dendritic cells. Crit Rev Immunol 2010; 30:345 - 86; http://dx.doi.org/10.1615/CritRevImmunol.v30.i4.30; PMID: 20666707
- Tesniere A, Panaretakis T, Kepp O, Apetoh L, Ghiringhelli F, Zitvogel L, Kroemer G. Molecular characteristics of immunogenic cancer cell death. Cell Death Differ 2008; 15:3 - 12; http://dx.doi.org/10.1038/sj.cdd.4402269; PMID: 18007663
- Cirone M, Lucania G, Aleandri S, Borgia G, Trivedi P, Cuomo L, Frati L, Faggioni A. Suppression of dendritic cell differentiation through cytokines released by Primary Effusion Lymphoma cells. Immunol Lett 2008; 120:37 - 41; http://dx.doi.org/10.1016/j.imlet.2008.06.011; PMID: 18680763
- Cirone M, Di Renzo L, Trivedi P, Lucania G, Borgia G, Frati L, Faggioni A. Dendritic cell differentiation blocked by primary effusion lymphoma-released factors is partially restored by inhibition of P38 MAPK. Int J Immunopathol Pharmacol 2010; 23:1079 - 86; PMID: 21244757
- Garufi A, Pistritto G, Ceci C, Di Renzo L, Santarelli R, Faggioni A, Cirone M, D’Orazi G. Targeting COX-2/PGE(2) pathway in HIPK2 knockdown cancer cells: impact on dendritic cell maturation. PLoS One 2012; 7:e48342; http://dx.doi.org/10.1371/journal.pone.0048342; PMID: 23144866
- Di Stefano V, Rinaldo C, Sacchi A, Soddu S, D’Orazi G. Homeodomain-interacting protein kinase-2 activity and p53 phosphorylation are critical events for cisplatin-mediated apoptosis. Exp Cell Res 2004; 293:311 - 20; http://dx.doi.org/10.1016/j.yexcr.2003.09.032; PMID: 14729469
- Puca R, Nardinocchi L, Starace G, Rechavi G, Sacchi A, Givol D, D’Orazi G. Nox1 is involved in p53 deacetylation and suppression of its transcriptional activity and apoptosis. Free Radic Biol Med 2010; 48:1338 - 46; http://dx.doi.org/10.1016/j.freeradbiomed.2010.02.015; PMID: 20171273