Abstract
Survivin-3B (S-3B), an alternative splice isoform of survivin, plays a key role in tumorigenesis. S-3B promotes the escape of malignant cells from immune recognition by blocking the cytotoxicity of natural killer (NK) cells. Such an effect reflects the ability of S-3B to interfere with the assembly of the so-called “death-inducing signaling complex” upon the interaction of FAS with its ligand (FASL). S-3B also inhibits the activation of caspase-6, thus increasing the resistance of neoplastic cells to granzyme B and various chemotherapeutics.
Tumors develop along a cascade of steps that include (as an early event) the dysregulation of alternative splicing. Such an aberrant splicing results in the synthesis of new polypeptides, some of which are cancer-specific. One example of these cancer-specific proteins is survivin-3B (S-3B), which originates as a splicing variant of survivin (baculoviral IAP repeat containing 5, BIRC5).Citation1 Conversely, survivin and 3 other alternative isoforms, namely, survivin-ΔEx3, survivin-2B, and survivin-2α are expressed in both neoplastic and normal cells. It is therefore important to understand whether S-3B plays a role in oncogenesis and tumor progression.
The S-3B-coding mRNA includes the 4 first exons of BIRC5 plus 69 nucleotides from intron 3, a new coding sequence (named exon 3B) that contains a stop codon.Citation2 Similar to survivin (but not other survivin isoforms) S-3B harbors a complete BIR domain. Thus, S-3B mediates anti-apoptotic functions and has been involved in the resistance of malignant cells to cisplatinCitation3 and 5-fluorouracil.Citation4
The cancer-specific expression profile of S-3B prompted us to focus on its effects on tumorigenesis. Normally, the immune system is programmed to eliminate abnormal cells that may become malignant. The elimination of such pre-cancerous cells is mainly attributable to CD8+ T lymphocytes and natural killer (NK) cells. Only when neoplastic cells resist the cytotoxic activity of immune effectors, they can proliferate unrestrained and generate tumors, a process that is known as immune escape. In the course of tumor progression, immune cells of multiple types infiltrate malignant lesions. Some of these cells exert tumor-supporting functions, while others—including NK cells—mediate robust antitumor effects.Citation5
To get insights into the functions of S-3B, we first tested the effect of its overexpression on a non-tumorigenic cell line. We observed that inoculation of originally non-tumorigenic cells that had been engineered to overexpress S-3B in nude mice, which have no T lymphocytes but harbor high NK-cell activity, promoted the development of malignant lesions. We therefore hypothesized that S-3B could inhibit the antitumor activity of NK cells. To test this hypothesis, we downregulated S-3B in vivo by injecting specific small-interfering RNAs (siRNAs) into neoplastic lesions generated by a highly tumorigenic cell line, an intervention that considerably reduced tumor growth. The impact of S-3B on the cytotoxic activity of NK cells was confirmed as the effects of S-3B-targeting siRNAs completely disappeared in NK cell-depleted nude mice. In vitro, S-3B was shown to interact with pro-caspase-8, hence preventing its proteolytic maturation upon the interaction of FAS with its ligand (FASL). Owing to the ability to inhibit the formation of the so-called death-inducing signaling complex (DISC), S-3B completely blocked the ability of NK cells to provoke the apoptotic demise of cancer cells. NK cells can also eliminate unwanted cells via the granzyme B/perforin system. This mechanism operates through mitochondrial depolarization, cytochrome c release and the activation of caspases-9, -3, -6 and -7. Since intrinsic apoptosis is also the cell death subroutine whereby most anticancer agents exert their activity, we decided to explore the effects of S-3B on the response of neoplastic cells to staurosporine and 5-fluorouracil. Upon exposure to these treatments, cells expressing high levels of S-3B not only failed to die, but also were able to divide and form colonies in clonogenic assays. In contrast, cells lacking S-3B died massively in response to apoptotic stimuli. By studying the mechanisms underlying intrinsic apoptosis as triggered in our model by staurosporine and 5-fluorouracil, we observed that cancer cells underwent mitochondrial depolarization followed by the activation of caspases-9 and -3 irrespective of S-3B expression levels. Rather, S-3B was involved in the events downstream of caspase-3 activation, notably as it inhibited the cleavage and activation of pro-caspase-6. Thus, S-3B sequestered pro-caspase-6 upon physical interaction, thus impeding its activation by active caspase-3.
Taken together, these data suggest that combining S-3B-targeting interventions with chemotherapy could result in a superior efficacy by improving both the activity of immune cells and the direct cytotoxicity of antineoplastic agents. In support of this hypothesis, we demonstrated that the association of S-3B-depleting siRNAs and 5-fluorouracil improves anticancer responses in mice.
As S-3B appeared to provide cancer cells with improved protection as compared with survivin, through different mechanisms, we studied in detail the sequence of S-3B. S-3B shares the first 113 amino acids with survivin but contains 7 amino acids at the C-terminus that differ from the corresponding residues of survivin. We showed that this short polypeptide, which we named LEO sequence, is required for the interaction of S-3B with its targets.
In conclusion, S-3B turned out to play a critical role in cancer initiation, progression and dissemination (). This protein is not the sole responsible for oncogenesis, but some evidence indicates that the expression level of S-3B might be related to tumor progressionCitation1 and poor disease outcome.Citation6,Citation7 The importance of S-3B in the resistance of cancer cells to immune effectors (especially NK cells) and chemotherapy, coupled to its tumor-specific expression pattern, should encourage the development of S-3B-targeting therapies, for example siRNA-based approaches or inhibitors of the LEO domain.
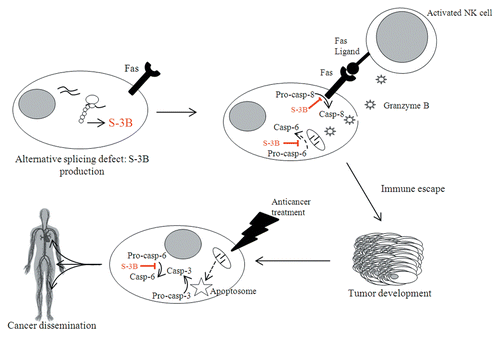
Figure 1. Involvement of survivin-3B in cancer initiation, progression, and dissemination. The defects in alternative splicing that normally accompany oncogenesis can result in the production of survivin-3B (S-3B). Pre-malignant cells are normally detected by the immune system, in particular by natural killer (NK) cells. Upon such a recognition, activated NK cells attempt to eliminate target cells by triggering FAS-dependent cell death and by secreting granzyme B. However, S-3B inhibits both the extrinsic and the intrinsic pathways of apoptosis. In this way, pre-malignant cells can escape elimination by the immune system and generate neoplastic lesions. In addition, S-3B inhibits the apoptotic response of cancer cells to chemotherapy and perhaps favors their metastatic dissemination. Dashed lines summarize several events occurring upstream of the represented process.
| Abbreviations: | ||
| BIR | = | baculovirus IAP repeat |
| DISC | = | death-inducing signaling complex |
| NK | = | natural killer |
| S-3B | = | survivin-3B |
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Citation: Végran F, Boidot R. Survivin-3B promotes chemoresistance and immune escape by inhibiting caspase-8 and -6 in cancer cells OncoImmunology 2013; 2:e26328; 10.4161/onci.26328
Related Research Data
References
- Végran F, Mary R, Gibeaud A, Mirjolet C, Collin B, Oudot A, Charon-Barra C, Arnould L, Lizard-Nacol S, Boidot R.. Survivin-3B potentiates immune escape in cancer but also inhibits the toxicity of cancer chemotherapy. Forthcoming http://dx.doi.org/10.1158/0008-5472.CAN-13-0036
- Badran A, Yoshida A, Ishikawa K, Goi T, Yamaguchi A, Ueda T, Inuzuka M. Identification of a novel splice variant of the human anti-apoptopsis gene survivin. Biochem Biophys Res Commun 2004; 314:902 - 7; http://dx.doi.org/10.1016/j.bbrc.2003.12.178; PMID: 14741722
- Knauer SK, Bier C, Schlag P, Fritzmann J, Dietmaier W, Rödel F, Klein-Hitpass L, Kovács AF, Döring C, Hansmann ML, et al. The survivin isoform survivin-3B is cytoprotective and can function as a chromosomal passenger complex protein. Cell Cycle 2007; 6:1502 - 9; http://dx.doi.org/10.4161/cc.6.12.4305; PMID: 17582222
- Sawai K, Goi T, Hirono Y, Katayama K, Yamaguchi A. Survivin-3B gene decreases the invasion-inhibitory effect of colon cancer cells with 5-fluorouracil. Oncol Res 2010; 18:541 - 7; http://dx.doi.org/10.3727/096504010X12767359113848; PMID: 20939430
- Fregni G, Perier A, Avril MF, Caignard A. NK cells sense tumors, course of disease and treatments: Consequences for NK-based therapies. Oncoimmunology 2012; 1:38 - 47; http://dx.doi.org/10.4161/onci.1.1.18312; PMID: 22720210
- Boidot R, Végran F, Lizard-Nacol S. Predictive value of survivin alternative transcript expression in locally advanced breast cancer patients treated with neoadjuvant chemotherapy. Int J Mol Med 2009; 23:285 - 91; PMID: 19148555
- Végran F, Boidot R, Bonnetain F, Cadouot M, Chevrier S, Lizard-Nacol S. Apoptosis gene signature of Survivin and its splice variant expression in breast carcinoma. Endocr Relat Cancer 2011; 18:783 - 92; http://dx.doi.org/10.1530/ERC-11-0105; PMID: 21878572