Abstract
Peptide-pulsed T2 cells are routinely used to study T-cell activation by MHC-restricted peptides derived from tumor-associated antigens (TAAs). Nevertheless, the capacity of T2 cells to present antigenic epitopes remains to be precisely quantified, primarily due to the detection limits imposed by available methods. Since naturally-processed TAA-derived epitopes have been shown to be displayed at levels as low as 10–150 copies per cell, highly sensitive detection and quantification techniques are essential to assess the natural degree of T-cell sensitivity. Here, we report the use of soluble, high-affinity T-cell receptors (TCRs) coupled with single-molecule fluorescence microscopy to quantify three reported TAA-derived epitopes on peptide-pulsed T2 cells, dissecting the relationship between concentration of exogenous peptide, number of epitopes presented, and activation of epitope-specific T cells. Our findings indicate that peptide concentrations in the low nanomolar range are required for T2 cells to present TAAs in extents that are comparable to those of malignant cells.
Introduction
The activation of cytotoxic T cells (CTLs) is mediated by the recognition of antigenic peptides displayed on the surface of antigen-presenting cells (APCs) in association with MHC class I molecules. The process of antigen recognition is controlled by the T-cell receptor (TCR) and is essential for the maintenance of protective immunity against infectious disease as well as (pre)malignant conditions. Consequently, methodologies that facilitate the investigation of TCR-dependent antigen recognition and subsequent CTL activation not only are vital for the development of new immunotherapeutic strategies, but also are essential tools for experimental immunologists. The use of T2 cells to probe antigen recognition by CTLs is well-established and relies on the induction of T2 cells to display MHC class I molecules associated with exogenously administered peptides. T2 cells are deficient in a peptide transporter involved in antigen processing (TAP) and therefore fail to correctly translocate endogenous (processed) peptides to the site of MHC loading in the endoplasmic reticulum/Golgi apparatus.Citation1 Thus, peptide-pulsed T2 cells can be used to monitor the CTL response to an exogenous antigen of interest in a non-competitive environment. Despite the widespread use of peptide-pulsed T2 cells, a correlation between the concentration of exogenous peptides used for pulsing and the number of epitopes presented on the cell surface has not yet been provided. This is relevant especially when CTL activation by tumor-associated antigen (TAA)-derived peptides is assessed, as these antigenic epitopes may be displayed in amounts as low as 10–150 copies per APC.Citation2,Citation3
Epitopes presented on the surface of APCs can be quantified by means of soluble proteins, including antibody fragments, that specifically bind to the peptide/MHC complex of interest.Citation4 However, one of the major drawbacks of this approach is the generally low binding affinity of antibody fragments, posing a significant problem for the detection and quantification of TAA epitopes presented in limited amounts. While improvements in binding affinity have been achieved upon oligomerization, these appear to be beneficial only for antigens that are presented on the cell surface at high densities.Citation5 The use of soluble, monoclonal TCRs (mTCRs) genetically engineered to possess an extremely high affinity for cognate antigens (in the picomolar range) not only offers a new solution for the quantification of epitopes presented in limited quantities,Citation3,Citation6,Citation7 but also is able to resolve information at the single-cell level. In addition, the prolonged binding half-life of high-affinity mTCRs reduces the need for rapid experimental assessments.
Here, we used high affinity mTCRs to investigate the presentation of reported TAA-derived epitopes on T2 cells. First, single-molecule microscopy was used to detect and quantify the presentation of 3 distinct epitopes. Second, the influence of peptide concentration on epitope presentation by T2 cells was examined and compared with natural presentation levels. Finally, the potency of the T-cell response to various amounts of presented epitopes was analyzed. Our findings demonstrate that peptide concentrations in the nanomolar range lead to the presentation of a number of epitopes that is comparable to that observed on the surface of tumor cells and that these concentrations are suitable to investigate the natural level of T-cell sensitivity to TAAs. These observations apply to three subclasses of TAAs: cancer-testis antigens, such as NY-ESO-1, overexpressed TAAs, such as Wilms’ tumor 1 (WT1) and telomerase reverse transcriptase (TERT), and differentiation-associated TAAs, such as premelanosome protein (PMEL, best known as gp100).
Results
Quantification of epitopes presented on peptide-pulsed T2 cells
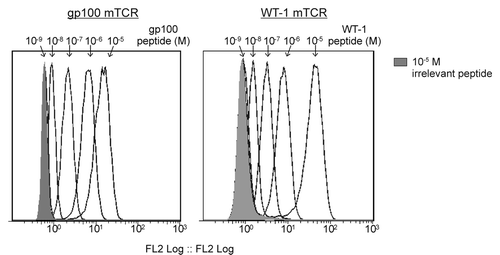
Soluble, biotinylated mTCRs were used to detect MHC-restricted epitopes on the surface of peptide-pulsed T2 cells. Each mTCR is antigen specific and exhibits a binding affinity in the low picomolar range. mTCRs specific for NY-ESO-1Citation2 and TERTCitation8 have previously been shown by flow cytometry to bind T2 cells pulsed with cognate antigenic peptides. Here we show that this is also the case for gp100- and WT1-specific mTCRs (). In summary, all four mTCRs employed in this study bind their cognate epitopes in a manner that depends on peptide concentration, indicating that, for any given peptide, T2 cell presentation can be modulated in a predictable manner by altering the concentration of exogenous peptides.
Figure 1. Flow cytometry-based detection of epitope-bound monoclonal TCRs on peptide-pulsed T2 cells. T2 cells were pulsed with the indicated HLA-A2-restricted peptides in concentrations ranging from 10−5 to 10−9 M. Epitope presentation was detected using a high-affinity biotinylated monoclonal T-cell receptor (mTCR) specific for gp100 (left panel) or Wilms’ tumor 1 (WT1, right panel) and flow cytometry, upon staining with phycoerythrin (PE)-conjugated streptavidin. A control measurement was made using T2 cell pulsed with 10−5 M of an irrelevant peptide (shaded gray).
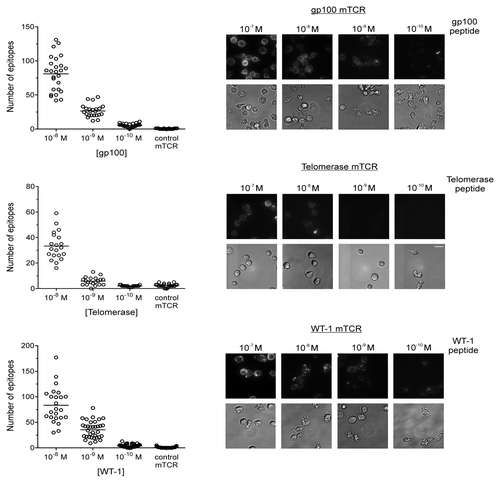
The detection of epitope-bound mTCRs using single-molecule fluorescence microscopy rather than flow cytometry has a number of advantages, including the possibility to resolve the number of epitopes presented by individual cells as well as an improved detection sensitivity.Citation2 Indeed, Purbhoo et al. provided microscopy data indicating that very low numbers of NY-ESO-1-derived epitopes (in the range of 10–50 per cell) could be detected on T2 cells pulsed with exogenous peptide concentrations ranging from 10−9 to 10−11 M.Citation2 To investigate whether this observation could be extended to other reported TAA-derived epitopes, epitope-bound gp100-, TERT- and WT1-targeting mTCRs were quantified by single-molecule fluorescence microscopy on T2 cells pulsed with nanomolar amounts of the corresponding peptides. We observed that T2 cells pulsed with peptide in concentrations of 10−8 to 10−10 M do present low numbers of epitopes, on average 4–80 copies per cell (). Since each mTCR displays a high affinity for its cognate antigen and the half-life of binding to antigenic epitopes presented on the cell surface is approximately 20 h (data not shown), each fluorescent signal is assumed to correspond to one single epitope and all epitopes are assumed to be bound by an mTCR. We also observed an increasingly large variation in the number of epitopes presented by individual cells as the concentration of peptide used for pulsing T2 cells is raised. In particular, at 10−8 M some T2 cells individually displayed over 100 epitopes, while others displayed fewer than 20 ().
Figure 2. Quantification of epitopes on T2 cells using monoclonal TCRs and single-molecule microscopy. Peptides derived from the tumor-associated antigens (TAAs) gp100, telomerase reverse transcriptase (TERT), and Wilms’ tumor 1 (WT1) were used to pulse T2 cells in concentrations ranging from 10−7 to 10−10 M. Epitopes were bound by high affinity, biotinylated monoclonal T-cell receptor (mTCRs) and visualized by microscopy upon staining with phycoerythrin (PE)-conjugated streptavidin. Solid bars depict the number of epitopes presented by individual T2 cells (means). At concentrations of 10−7 M, the number of epitopes was incompatible with accurate counting. In each case a background measurement was made using a high-affinity mTCR specific for an irrelevant, HIV-1-derived antigen (Gag77–85), using a peptide concentration of 10−5M. The Y-axis on each graph has been optimized to best represent the distribution. Representative phase contrast and fluorescence images are shown for gp100 (upper panel), TERT (middle panel), and WT1 (lower panel). Fluorescence images are three-dimensional reconstructions of individual planes. The brightness/contrast of individual phase contrast and fluorescence images was adjusted to optimize epitope visualization. Scale bar = 20 µm.
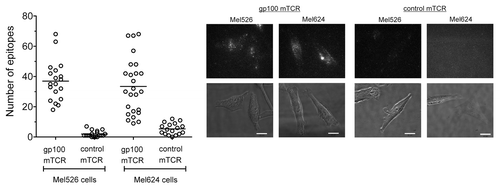
To link the number of epitopes observed on pulsed T2 cells with the number naturally presented on malignant cells, the gp100-derived epitope was quantified on two melanoma cell lines, Mel526 and Mel624 cells. The data presented in demonstrate that, on average, Mel526 and Mel624 cells present 37 and 34 epitopes per cell, respectively, consistent with the range detected on T2 cells pulsed with nanomolar amounts of a gp100-derived peptide (). These data are also in line with the previously reported amounts of NY-ESO epitopes presented on melanoma cell lines, averaging ~25 and ~45 epitopes per cell for SK-Mel37 and Mel624 cells, respectively.Citation2 Furthermore, we observed a similarly wide distribution in the number of gp100-derived epitopes presented on individual melanoma cells () and T2 cells pulsed with nanomolar amounts of gp100-derived peptides (). In particular, T2 cells pulsed with 10−9 M of the gp100-derived peptide presented 12–47 gp100 epitopes per cell (), while Mel526 and Mel624 cells naturally presented 18–68 and 9–68 gp100 epitopes per cell, respectively ().
Figure 3. Quantification of epitopes on Mel526 and Mel624 melanoma cells. A gp100-specific biotinylated monoclonal T-cell receptor (mTCR) was allowed to bind to gp100-derived epitopes naturally presented on melanoma Mel526 and Mel624 cells, and visualized by microscopy upon staining with phycoerythrin (PE)-conjugated streptavidin. Solid bars depict the number of epitopes presented by individual tumor cells (means). A background control measurement was made using a high-affinity mTCR specific for an irrelevant, HIV-1-derived antigen (Gag77–85). Representative phase contrast and fluorescence images for the 2 cell lines are shown. Fluorescence images are three-dimensional reconstructions of individual planes. The brightness/contrast of fluorescence images was adjusted to optimize epitope visualization. Scale bar = 20 µm.
Activation of T cells by peptide-pulsed T2 cells
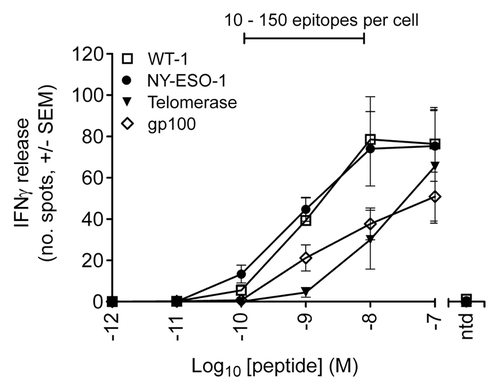
To correlate the number of epitopes presented on the cell surface of T2 cells with their ability to activate T cells, interferon γ (IFNγ) release was monitored from T cells transduced with wild-type TCRs specific for NY-ESO-1-, gp100-, TERT-, or WT1-derived peptides, upon exposure to T2 cells previously pulsed with 10−7 to 10−12 M of the corresponding peptides. We observed that, regardless of the TAA specificity, partial T-cell activation is induced in response to T2 cells pulsed with 10−8 M to 10−10 M TAA-derived peptides ().
Figure 4. T-cell response to peptide pulsed T2 cells as determined by interferon γ release. ELIPOST assays were used to measure the level of T-cell activation, assessed in terms of interferon γ (IFNγ) release, in response to T2 cells presenting varying levels of epitopes. CD8+ and CD4+ T cells were isolated from peripheral blood mononuclear cells and transduced with wild-type TCRs specific for NY-ESO-1-, gp100-, telomerase reverse transcriptase (TERT)-, and Wilms’ tumor 1 (WT1)-derived peptides. T2 cells were pulsed with antigenic peptides in concentrations ranging from 10−7 to 10−12 M. A control was performed in each case using non-transduced (ntd) T cells and a peptide concentration of 10−7 M. The bar above the graph indicates the peptide concentration range in which physiological numbers of epitopes are presented.
Discussion
The combination of high affinity mTCRs and single-molecule fluorescence microscopy provides an extremely sensitive method for the detection and quantification of MHC-restricted epitopes at the single cell level, not only on T2 cells and cancer cell lines, as demonstrated here, but also potentially on many other antigen-presenting cells, including dendritic cells and primary tissue cells.
Since pulsing T2 cells with reported TAA-derived peptides in low nanomolar concentrations (10−10 –10−8 M) results in the presentation of a physiological amount of epitopes, the corresponding T-cell response is likely to reflect the natural level of T-cell sensitivity to the specific epitope under investigation. This observation has important implications for scientists using peptide-pulsed T2 cells to determine T-cell sensitivity to TAAs. Peptide pulsing is routinely performed using peptide concentrations in the micromolar range.Citation9,Citation10 However, on the basis of the data presented here, such high concentrations are associated with the risk of producing T2 cells that present an unnaturally elevated number of epitopes. In turn this not only may result in an inaccurate assessment of T-cell sensitivity to naturally presented TAAs, but also may give false indications on the suitability of specific T cells or TCRs for immunotherapeutic applications.
For the four different TAAs investigated here, T cells expressing wild-type TCRs against a specific peptide/MHC complex respond only to partial extents to physiological numbers of epitopes presented on the surface of T2 cells. Given the wide variation in the number of epitopes presented by individual T2 cells, which is exacerbated with increasing peptide concentrations (), these results point to a mechanism whereby the potency of T-cell responses vary depending on the number of antigenic epitopes encountered, with a minimum number of epitopes being required to provoke T-cell activation. Furthermore, these observations indicate how suboptimal levels of antigen presentation, such as those that occur upon the downregulation of MHC class I molecules by cancer cells or when TAA-specific T cells exhibit a weak affinity for cognate antigens, may prevent productive T-cell activation, allowing malignant cells to evade immune detection, one of the key obstacles against achieving greater success with some anticancer immunotherapies.Citation11
The results presented herein demonstrate the potential utility of high-affinity mTCRs as diagnostic tools to detect and quantify the presentation of TAA-derived epitopes on the cell surface. Furthermore, new bi-specific therapeutic reagents comprising a CD3-specific single-chain variable fragments fused to the high-affinity soluble mTCRs used here are able to elicit a potent immune response despite low levels of antigen presentation and the weak affinity of TAA-specific T cells. Provided that comprehensive preclinical assessments are undertaken and show reassuring safety and efficacy results, these reagents offer a new immunotherapeutic strategy against cancer.Citation3,Citation12
Materials and Methods
Peptides and cell lines
Synthetic peptides corresponding to NY-ESO-1157–165 (SLLMWITQV), gp100280–288 (YLEPGPVTV), TERT540–548 (ILAKFLHWL), WT1126–134 (RLFPNAPYV) and HIV-1-derived Gag77–85 (SLYNTVATL) were obtained from Peptide Protein Research and dissolved in dimethylsulfoxide (DMSO) at 4 mg/mL prior to use. The C-terminal residues of NY-ESO-1157–165 and gp100280–288, as well as the residues at position 2 and 9 of WT1126–134 peptide, have been mutated to improve the stability of peptide/MHC complexes. Mel526 and Mel624 cells were obtained from Thymed and maintained in standard culture conditions.
Protein production
Soluble mTCRs with picomolar affinity for cognate antigens were produced as previously described.Citation6,Citation7 Briefly, the DNA encoding the α and β chains of affinity-enhanced TCRs were isolated from peptide-specific T-cell lines by PCR. The resulting TCR chain-coding sequences were then expressed in Escherichia coli and soluble disulfide-linked heterodimeric mTCRs were purified by anion exchange and size exclusion chromatography. mTCRs were biotinylated on the β chain and purified as previously described.Citation2
Peptide pulsing and flow cytometry
One million T2 cells were pulsed with a serial dilution of each TAA-derived peptide, ranging from 10−5 to 10−11 M, for 90 min at 37°C. Cells were then stained as described previously.Citation2 Briefly, cells were incubated with 5 µg/mL high-affinity biotinylated mTCRs at in PBS supplemented with 0.5% bovine serum albumin (BSA) for 30 min at room temperature (RT), followed by a 20 min incubation at RT with 10 µg/mL phycoerythrin (PE)-conjugated streptavidin (BD Bioscience). Samples were acquired on an FC500 flow cytometer (Beckman Coulter) and plated onto a multi-well glass chamber for microscopic acquisitions. Flow cytometry data files were analyzed with the FloJo software version 7.6.
Microscopy
T2 cells or melanoma cells were plated on glass coverslip chambers and stained with 5 µg/mL high-affinity biotinylated mTCRs in PBS supplemented with 0.5% BSA, 400 nM CaCl2 and 400 nM MgCl2 for 30 min at RT. Then, samples were incubated with for 20 min at RT with 10 µg/mL PE-conjugated streptavidin (BD Bioscience). Phase-contrast and PE-dependent fluorescence images were acquired as previously describedCitation2 using a 200M/Universal Imaging system with a 63 × objective (Carl Zeiss Inc.). Z-stack fluorescent images were taken (27 individual planes, 0.7 µm apart) to cover the entire 3D extension of the cell. The fluorescent spots corresponding to PE-labeled mTCRs bound to peptide/MHC complexes on each Z-stack were summed up to obtain total counts per individual cell. In each experiment epitopes were quantified on more than 18 individual cells.
Lentivirus manufacture
A T150 flask of semi-confluent HEK293T cells was transfected with 15 µg of a lentiviral vector encoding the TCR of interest, along with a total of 43 µg of 3 packaging plasmids,Citation13 using the Express-In Transfection Reagent (Open Biosystems/Thermo-Scientific). Supernatants were collected 24 and 48 h later and were concentrated by centrifugation at 10,000 g for 16 h at 4°C. Cell pellets were then resuspended in 2 mL of R10 culture medium and snap frozen on dry ice until required.
T-cell transduction
Peripheral blood mononuclear cells (PBMCs) were isolated from venous blood freshly drawn from healthy individuals. CD8+ and CD4+ T cells were isolated using untouched CD8+ and CD4+ isolation kits (Invitrogen/Life Technologies), as per manufacturer’s instructions, and incubated overnight in R10 culture medium containing 50 U/mL human recombinant IL-2 (Peprotech) and Dynabeads CTS antiCD3/CD28 beads (Invitrogen/Life Technologies) at a bead-to-cell ratio of 3 (1 × 106 cells/mL, 1 mL per well of a 24-well culture plate). Following overnight stimulation, T cells were transduced by addition of 1 mL crude lentiviral supernatant. T cells were then fed by addition of fresh medium (with IL-2) every other day, and beads were removed on day 5. After 12 d, cells were cryopreserved at 2.51 × 106 cells/mL until required. T-cell transduction efficiency was determined by flow cytometry following incubation with PC7-conjugate anti-CD8 antibodies (BD PharMingen) and either Vβ-specific antibodies (Beckman Coulter) or appropriate PE-conjugated tetramers.
ELISPOT assays
The response of transduced T cells to peptide-pulsed T2 cells was determined by IFNγ-specific ELISPOT kits (BD PharMingen), as per manufacturer’s recommendations. Briefly, T2 cells were pulsed with TAA-derived peptides in concentrations ranging from 10−7 to 10−12 M for 90 min, and subsequently plated out at a concentration of 3 × 104 cells per well. TCR-transduced T cells were defrosted and allowed to recover in R10 medium for 2 h, before washing and plating at 5 × 103 viable transduced cells/well. Plates then were incubated overnight (37°C and 5% CO2), developed according to manufacturer’s instructions and evaluated on a Immunospot Series 4 ELISPOT Analyzer (CTL).
| Abbreviations: | ||
| APC | = | antigen-presenting cell |
| CTL | = | cytotoxic T lymphocyte |
| mTCR | = | monoclonal TCR |
| TAA | = | tumor-associated antigen |
| TCR | = | T-cell receptor |
Disclosure of Potential Conflicts of Interest
GB, SJP, DHS, NJH and BKJ are employees of Immunocore Ltd. ABG and BKJ are employees of Adaptimmune Ltd.
Acknowledgments
The authors would like to thank all members of the protein engineering group at Immunocore for their invaluable assistance in these studies; and Joanne Oates for assistance in manuscript preparation. Funding was provided by Immunocore Ltd.
Citation: Bossi G, Gerry AB, Paston SJ, Sutton DH, Hassan NJ, Jakobsen BK. Examining the presentation of tumor associated antigens on peptide pulsed T2 cells. OncoImmunology 2013; 2:e26840; 10.4161/onci.26840
Related Research Data
References
- Hosken NA, Bevan MJ. Defective presentation of endogenous antigen by a cell line expressing class I molecules. Science 1990; 248:367 - 70; http://dx.doi.org/10.1126/science.2326647; PMID: 2326647
- Purbhoo MA, Sutton DH, Brewer JE, Mullings RE, Hill ME, Mahon TM, Karbach J, Jäger E, Cameron BJ, Lissin N, et al. Quantifying and imaging NY-ESO-1/LAGE-1-derived epitopes on tumor cells using high affinity T cell receptors. J Immunol 2006; 176:7308 - 16; PMID: 16751374
- Liddy N, Bossi G, Adams KJ, Lissina A, Mahon TM, Hassan NJ, Gavarret J, Bianchi FC, Pumphrey NJ, Ladell K, et al. Monoclonal TCR-redirected tumor cell killing. Nat Med 2012; 18:980 - 7; http://dx.doi.org/10.1038/nm.2764; PMID: 22561687
- Cohen CJ, Hoffmann N, Farago M, Hoogenboom HR, Eisenbach L, Reiter Y. Direct detection and quantitation of a distinct T-cell epitope derived from tumor-specific epithelial cell-associated mucin using human recombinant antibodies endowed with the antigen-specific, major histocompatibility complex-restricted specificity of T cells. Cancer Res 2002; 62:5835 - 44; PMID: 12384546
- Michaeli Y, Denkberg G, Sinik K, Lantzy L, Chih-Sheng C, Beauverd C, Ziv T, Romero P, Reiter Y. Expression hierarchy of T cell epitopes from melanoma differentiation antigens: unexpected high level presentation of tyrosinase-HLA-A2 Complexes revealed by peptide-specific, MHC-restricted, TCR-like antibodies. J Immunol 2009; 182:6328 - 41; http://dx.doi.org/10.4049/jimmunol.0801898; PMID: 19414786
- Boulter JM, Glick M, Todorov PT, Baston E, Sami M, Rizkallah P, Jakobsen BK. Stable, soluble T-cell receptor molecules for crystallization and therapeutics. Protein Eng 2003; 16:707 - 11; http://dx.doi.org/10.1093/protein/gzg087; PMID: 14560057
- Li Y, Moysey R, Molloy PE, Vuidepot AL, Mahon T, Baston E, Dunn S, Liddy N, Jacob J, Jakobsen BK, et al. Directed evolution of human T-cell receptors with picomolar affinities by phage display. Nat Biotechnol 2005; 23:349 - 54; http://dx.doi.org/10.1038/nbt1070; PMID: 15723046
- Purbhoo MA, Li Y, Sutton DH, Brewer JE, Gostick E, Bossi G, Laugel B, Moysey R, Baston E, Liddy N, et al. The HLA A*0201-restricted hTERT(540-548) peptide is not detected on tumor cells by a CTL clone or a high-affinity T-cell receptor. Mol Cancer Ther 2007; 6:2081 - 91; http://dx.doi.org/10.1158/1535-7163.MCT-07-0092; PMID: 17620437
- Ge L, Hoa NT, Cornforth AN, Bota DA, Mai A, Kim DI, Chiou SK, Hickey MJ, Kruse CA, Jadus MR. Glioma big potassium channel expression in human cancers and possible T cell epitopes for their immunotherapy. J Immunol 2012; 189:2625 - 34; http://dx.doi.org/10.4049/jimmunol.1102965; PMID: 22844111
- Mahnke YD, Devevre E, Baumgaertner P, Matter M, Rufer N, Romero P, Speiser DE. Human melanoma-specific CD8(+) T-cells from metastases are capable of antigen-specific degranulation and cytolysis directly ex vivo. Oncoimmunology 2012; 1:467 - 530; http://dx.doi.org/10.4161/onci.19856; PMID: 22754765
- Devaud C, John LB, Westwood JA, Darcy PK, Kershaw MH. Immune modulation of the tumor microenvironment for enhancing cancer immunotherapy. Oncoimmunology 2013; 2:e25961; http://dx.doi.org/10.4161/onci.25961; PMID: 24083084
- Oates J, Jakobsen BK. ImmTACs: Novel bi-specific agents for targeted cancer therapy. Oncoimmunology 2013; 2:e22891; http://dx.doi.org/10.4161/onci.22891; PMID: 23525668
- Dull T, Zufferey R, Kelly M, Mandel RJ, Nguyen M, Trono D, Naldini L. A third-generation lentivirus vector with a conditional packaging system. J Virol 1998; 72:8463 - 71; PMID: 9765382