Abstract
Inflammatory hepatocellular adenomas (IHCAs) are benign liver lesions that can be characterized histologically by the presence of an inflammatory infiltrate and at the molecular level by the overexpression of acute phase inflammatory response genes. Recurrent somatic mutations of the interleukin-6 (IL-6) signal transducer (IL6ST) locus, encoding the critical component of the IL-6 signal transduction machinery gp130, are present in 60% of IHCAs and in a subset (2%) of hepatocellular carcinoma (HCCs). By screening of 256 human hepatic adenoma specimens (the largest genetic analysis of IL6ST performed to date in this setting), we identified 24 distinct somatic IL6ST mutations among 66 mutant adenomas. The functional analysis of nine different gp130 mutants expressed in hepatic cancer cell lines consistently revealed the constitutive and IL-6-independent activation of the JAK/STAT signaling pathway. We further demonstrated that the signaling activity of mutant gp130 in IHCA remains responsive to suppressor of cytokine signaling 3 (SOCS3), a physiological gp130 inhibitor. Specifically, cells expressing a double mutant variant of gp130 with a disrupted SOCS3-binding site at residue 759 (Y186/Y759F) displayed a hyperactivation of signal transducer and activator of transcription 3 (STAT3) as compared with cells expressing the endogenous IHCA-associated Y186 gp130 mutant. Notably, we identified that constitutive signaling via gp130 in IHCA requires the Janus kinase family member JAK1, but not JAK2 or tyrosine kinase 2. In support of this notion, AG490, a tyrosine kinase inhibitor that selectively blocks JAK2, had no effect on gp130 activity. In stark contrast, we showed that ruxolitinib, a JAK1/JAK2-selective tyrosine kinase inhibitor used to treat patients with myelofibrosis, dramatically impaired JAK1-STAT signaling downstream of all IHCA-associated gp130 mutants. In conclusion, our findings provide a rationale for the use of JAK1 inhibitors for the treatment of HCAs expressing mutant gp130 as well as a subset of HCCs that bear similar mutations.
Keywords: :
Introduction
Hepatocellular adenomas (HCAs) are rare benign liver tumors that usually develop in young females following the use of contraceptives.Citation1 HCAs are heterogeneous and 4 major molecular subgroups have been defined so far, based on both genotypic and phenotypic features.Citation2-Citation4 Inflammatory hepatocellular adenomas (IHCAs) account for 45–60% of HCAs, hence representing the most common subtype of this disease. IHCAs are characterized; (1) genetically by the overexpression of acute phase inflammatory genes, and (2) immunophenotypically by the presence of a constituent inflammatory infiltrate. This particular subtype of HCA occurs most frequently in women on oral contraceptives, but has also been associated with obesity and alcohol abuse.Citation3 We have previously identified a select panel of IHCA-driving mutations. In particular: (1) 60% of IHCAs harbor activating somatic mutations in the interleukin (IL)-6 signal transducer (IL6ST) locus, gene coding for gp130, the co-receptor and signal transducer of the IL-6 receptor (IL6R)Citation5; (2) 5% harbor somatic mutations in signal transducer and activator of transcription 3 (STAT3);Citation6 and (3) 5% harbor somatic mutations in the GNAS complex locus (GNAS) encoding, among other proteins, the G-protein α subunit.Citation1,Citation7-Citation9 Notably, all these IHCA-associated mutations promote the constitutive activation of STAT3.
All IL-6 family cytokines, namely, IL-6, IL-11, IL-27, ciliary neurotrophic factor (CNTF), cardiotrophin 1 (CTF1), CTF1-like cytokine factor 1 (CLCF1), leukemia inhibitory factor (LIF), oncostatin M (OSM) and neuropoietin (NPN), activate the gp130-JAK-STAT signaling pathway. IL-6 and IL-11 are the only of these cytokines that exclusively transduce a signal via gp130 homodimers. Generally, IL-6 binds to its specific α receptor, IL6R (also known as gp80) to generate heterodimeric IL6/IL6R complexes that can associate with a gp130 homodimer, resulting in a hexameric structure that activates the signaling functions of gp130. Gp130 activation induces the phosphorylation of JAK kinase 1 (JAK1) and JAK2, which in turn phosphorylate STAT3 at Y705, triggering its homodimerization, nuclear translocation, DNA binding and the transactivation of pro-inflammatory STAT3 target genes.Citation10
STAT transcription factors are directly phosphorylated and activated by JAK tyrosine kinases, which play a pivotal role in the development of both solid and hematopoietic tumors.Citation11-Citation13 The JAK-STAT signaling pathway can be triggered either by the activation of oncogenes or via the paracrine or autocrine production of cytokines whose receptors signal to JAK family members, which in turn phosphorylate and activate STAT transcription factors.Citation14 In this context, IHCA-associated IL6ST mutations that functionally activate gp130 are thought to lead to the constitutive activation of JAK and STAT3, resulting in a sustained inflammatory response that promote the formation of adenomas. Thus, new inhibitors that selectively inhibit JAK kinasesCitation15-Citation18 may represent attractive therapeutics for this class of adenomas, which are relatively benign but can progress to hepatocellular carcinoma (HCC). Moreover, 1 to 2% of HCCs harbor activating mutations in IL6ST, implying that JAK kinases might represent a candidate target for the therapy of this subset of hepatic malignancies as well.Citation5,Citation19,Citation20
To assess the mechanisms of activation of the JAK-STAT signaling axis in IHCAs bearing IL6ST mutations, we evaluated the functional and biochemical properties of nine distinct gp130-activating mutations identified by the screening of a large panel of 256 HCA patient samples. Further, we assessed the therapeutic potential of disabling the JAK-STAT signaling pathways in IHCA bearing expressing mutant gp130.
Results
Spectrum of IL6ST mutations in IHCA
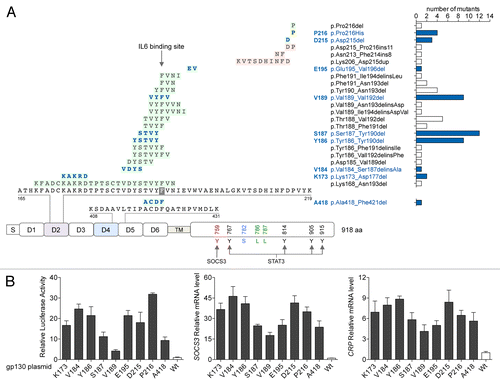
Among 256 HCA patient samples screened for genetic abnormalities, we identified 66 heterozygous, somatic IL6ST mutations. Thus, IL6ST mutations were found in 25.7% of all HCA samples tested, and—of particular interest—were exclusively identified among IHCA specimens, accounting of 56.9% of our HCA patient cohort. In addition, we found that 13% of IHCA specimens (15) exhibited activating mutations in both IL6ST and the third exon of CTNNB1, which encodes β catenin (Table S1). In contrast, we confirmed that IL6ST mutations are mutually exclusive with mutations in HNF1A (coding for HNF1 homeobox A), STAT3 and GNAS, which are recurrent in benign HCAs.Citation4,Citation6,Citation7 The spectrum of IL6ST mutations that we identified included 20 distinct in-frame deletions, 1 missense substitution and 3 in-frame insertion or deletions. Almost all of these mutations affected the D2 domain of gp130, which is directly involved in IL-6 binding ( and Table S1). At this hot spot, 2 amino acids, Y190 and F191, are essential for the interaction between gp130 and IL-6. Most of the in-frame deletions affected these residues.
Figure 1. Gain-of-function mutations of gp130 in IHCA. (A) Spectrum of somatic mutations affecting interleukin-6 (IL-6) signal transducer (IL6ST) in human inflammatory hepatocellular adenoma (IHCA) samples (n = 256). DNA sequencing of IL6ST of was performed to identify the resultant alterations in gp130, including in-frame deletions (in green), insertions or deletions (in pink) and amino acid substitutions (in yellow) occurring in the different domains of the protein (S, signal peptide; D1-D6, extracellular domains; TM, transmembrane domain). Right, occurrence of the different mutants with their official nomenclature. Mutants reproduced by site-directed mutagenesis (for functional analysis) are in blue. (B) Plasmids engineered to express either IHCA-associated gp130 mutants or wild-type (WT) gp130 were co-transfected into Hep3B cells (n = 3) along with a STAT3-driven luciferase (Luc) reporter. STAT3 activation (left) was measured by luciferase activity 6 h after serum starvation. Shown are the means ± SD luciferase activity. Quantitative PCR was also used to examine the effects of expressing mutant gp130 on SOCS3 (center) or CRP (right) mRNA expression levels in comparison to WT gp130. Shown is the mean ± SD of the normalized mRNA levels in mutants relative to WT gp130 controls (1-fold).
The IL-6/IL6R/gp130 hexamer is held together by 10 2-fold related interfaces of which 5 are unique (sites I, IIa, IIb, IIIa, and IIIb).Citation21 Within site IIa, g130 F191 (F169 in ProteinDataBank) is conserved and crucial for all cytokine interactions,Citation22-Citation24 contributing 25% of the total buried surfaces area (Fig. S1). To determine the effects of the IL6ST in-frame deletions on the quaternary structure of the complex, we took advantage of the availability of the IL-6/IL6R/gp130 crystal structure.Citation21 In particular, we deleted these residues in silico and performed 200 rounds of energy minimizations of the resulting model. In our final model, deletion of gp130 residues 187–190 (165–168 in ProteinDataBank) appeared to affect cytokine interaction substantially as moiety F191 no longer stacked against the side of IL-6 R27 but rather localized near IL-6 Y31 (Fig. S1). This contrasts with prior structural and calorimetric data suggesting that the disruption of site IIa does not affect IL-6/IL-6R interactions but abrogates hexamer assembly, particularly considering that gp130 assembles as a loop of 5 codependent interfaces (I → IIa/IIb → IIIa → IIIb → I).Citation21
We also identified one tumor harboring an atypical in-frame deletion of 4 residues localized to the D4 domain of gp130 ( and Table S1). The structure of the entire extracellular domain of gp130 allows for modeling the effects of the A418-F421 deletion (A396-F399 in ProteinDataBank) on the IL-6/IL6R/gp130 hexamer (Fig. S2). These amino acids reside on a loop, and in our energy-minimized model we found that this deletion shortens the loop without significantly changing the structure of the overall hexamer. It is tempting to speculate that this region might be important for the flexibility of extracellular domains of gp130. Thus, mutations that decrease such a flexibility might favor gp130 dimerization.
To clarify the mechanism(s) by which the JAK-STAT signaling axis is activated in IHCA cells expressing mutant gp130, we generated (by site-directed site mutagenesis) recombinant variants of gp130 harboring nine of the IL6ST genetic alterations that we identified, including 8 in-frame small deletions targeting the D2 and D4 domains (of 1 to 5 amino acids in length) as well as the P216H point substitution (). The overexpression of these mutants in 3 different hepatic cancer cell lines (Hep3B, HepG2, and Huh7 cells) cotransfected with a STAT3-luciferase reporter demonstrated that all nine mutants drive constitutive, IL-6-independent STAT3 activation ( and Fig. S3), as anticipated. All gp130 mutants that we tested exhibited a similar ability to activate STAT3, except A418 (deletion in the D4 domain) and V189 (deletion in the D2 domain), which were the least efficient in this sense (). Finally, in contrast to wild-type gp130, all gp130 mutants induced high levels of suppressor of cytokine signaling 3 (SOCS3) and C-reactive protein, pentraxin-related (CRP) mRNAs (), 2 acute-phase inflammatory genes transcriptionally regulated by STAT3. Thus, all nine IHCA-associated mutations of gp130 induced the constitutive, cytokine-independent activation of STAT3.
The activity of IHCA-associated gp130 mutants is attenuated by SOCS3
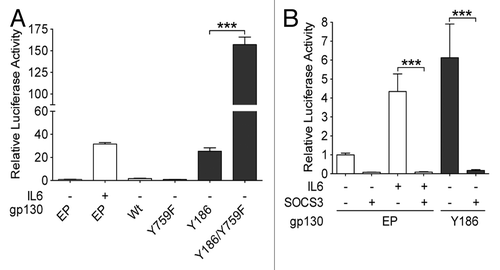
SOCS3 is induced as a direct target of STAT3 and modulates JAK signaling upon interaction with gp130 Y759.Citation25 In order to test whether gp130 mutants remain under the control of SOCS3, we selected Y186, the gp130 mutant most frequently associated with IHCA, and generated a double mutant (Y186/Y759F). We found that the Y186/Y759F gp130 double mutant exhibits an increased ability to drive STAT3 activation than the Y186 simple mutant (). To confirm this observation, we further tested the effect of exogenous SOCS3 expression on STAT3 activity by co-expressing the Y186 gp130 mutant and wild-type SOCS3 in Hep3B cells. Cells forced to constitutively express SOCS3 together with the gp130 mutant displayed a marked reduction in STAT3 activation as compared with cells expressing the gp130 mutant only (). Thus, the activation of the JAK-STAT signaling axis by IHCA-associated gp130 mutants remains under the control of SOCS3. In line with notion, the Y186/Y759F double mutation produced a superactive gp130 variant.
Figure 2. Signaling activity of IHCA gp130 mutants is attenuated by SOCS3. (A and B) Plasmids engineered to express either inflammatory hepatocellular adenoma (IHCA)-associated gp130 mutants (black bars) or wild-type (WT) gp130 were transfected into Hep3B cells (n = 3) together with a STAT3-driven luciferase (Luc) reporter construct (pSIEM-Luc). STAT3 activation was measured by luciferase activity 6 h after serum starvation. Shown are the means ± SD luciferase activity. (A) STAT3 activity following the co-transfection of Hep3B cells with the STAT3-luciferase reporter and a control empty plasmid (EP) or constructs coding withWT gp130, Y186 gp130 mutant, Y759F gp130 mutant, or Y186/Y759F gp130 double mutant. (B) Hep3B cells were co-transfected with the STAT3-luciferase reporter and a vector expressing the Y186 gp130 mutant or the corresponding EP along with a SOCS3 expression construct (+) or the corresponding EP (−). Data shown are the mean luciferase activities ± SD relative to pSIEM-Luc alone (EP control) following serum starvation (6 h). Where indicated, cells were treated for the final 3 h with 100 ng/mL interleukin-6 (IL-6). Statistical significance was determined by 2-tailed Student t test; ***P < 0.001.
Interaction of gp130 mutants with wild-type gp130, IL6R, and OSMR
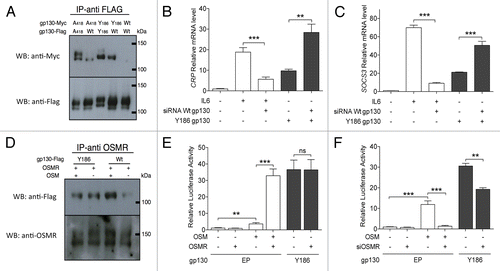
IHCA cells most often bear heterozygous IL6ST mutations, implying that both the mutant and the wild-type allele are expressed.Citation5 Thus, we assessed the effects of IHCA-associated gp130 mutations on gp130 dimerization, as well as on its interactions with IL6R and the OSM receptor (OSMR), two IL-6 receptor family members that are expressed by IHCA cells. As previously shown,Citation5 wild-type gp130 do not homodimerize in the absence of IL-6. Conversely, IHCA-associated gp130 mutants are fully capable of homodimerizing as well as of heterodimerizing with wild-type gp130 in the absence of IL-6 (). Furthermore, the selective depletion of wild-type gp130 by means of specific small-interfering RNAs (siRNAs) significantly augmented the expression levels of CRP and SOCS3, reflecting a burst in STAT3 activation driven by mutant gp130 (). Thus, our findings suggest that wild-type gp130 antagonizes the activity of IHCA-associated gp130 mutants via heterodimerization.
Figure 3. Interaction of IHCA gp130 mutants with wild-type gp130 and OSMR. (A) MYC- or Flag-tagged constructs expressing either wild-type (WT) gp130 or the inflammatory hepatocellular adenoma (IHCA)-associated gp130 mutants A418 or Y186 were co-transfected (1:1) into Hep3B cells. Gp130 hetero- and homo-dimerization in the absence of interleukin-6 (IL-6) was assessed by immunoprecipitation using the anti-Flag antibodies followed by immunoblotting. (B and C) Hep3B cells (n = 3) were co-transfected with a control vector or a plasmid expressing the Y186 gp130 mutant and a control siRNA (−) or a siRNA specific for WT gp130 (+) and quantitative PCR was used to examine the effects on transcription of STAT3 target genes. Shown is the mean ± SD expression level of endogenous CRP (B) and SOCS3 (C) transcripts in cell expressing gp130 mutants relative to WT gp130 (1-fold). On hundred ng/mL IL-6 was added to the culture medium where indicated. (D) The dimerization potential of the oncostatin M receptor (OSMR) with WT or mutant gp130 in the absence of OSM was assessed in Hep3B cells expressing Flag-tagged WT or mutant gp130. Shown are the immunoblots of immunoprecipitates obtained with anti-OSMR antibodies in the presence or in the absence of 100 ng/mL OSM. (E) Hep3B cells were co-transfected with the STAT3-luciferase reporter and a control vector (EP) or plasmids encoding the Y186 gp130 mutant or OSMR, as indicated. One hundred ng/mL OSM was added when indicated. Shown are the mean ± SD luciferase activities determined from triplicate co-transfections. (F) The knockdown of OSMR in Hep3B cells (n = 3) impairs STAT3 activity a driven by the Y186 gp130 mutant or treatment with 100 ng/mL OSM. Shown are the mean luciferase activities ± SD relative to cells transfected with the EP only. Results were confirmed using a second siRNA (data not shown). Statistical significance was determined by 2-tailed Student t test; **P < 0.01; ***P < 0.001.
We also assessed the effects of IHCA-associated gp130 mutations on its interactions with the gp80 subunit of IL6R. Interestingly, although the silencing of gp80 dramatically inhibited IL-6-induced STAT3 activity in the presence of wild-type gp130, this was not the case when cells were engineered to express IHCA-associated gp130 mutants (Fig. S4). However, the transgene-driven overexpression of gp80 did slightly decrease the activation of STAT3 in cells expressing either the Y186 or A418 gp130 mutant (Fig. S4), consistent with a limited role for gp80 in the constitutive activation of gp130 mutants.
Gp130 also binds to the OSMR, an interaction that is required for OSMR signaling. As anticipated, the interaction between wild-type gp130 and OSMR in Hep3B cells required OSM, whereas gp130 mutants formed heterodimers with OSMR even in the absence of its ligand (). The overexpression of OSMR did not affect STAT3 activity as stimulated by IHCA-associated gp130 mutants (). However, the siRNA-mediated knockdown of OSMR partially decreased the transcriptional activity of STAT3 as induced by gp130 mutants in both HepG2 (; P < 0.01) and Huh7 cells (Fig. S5). These findings suggest that ligand-independent interactions between gp130 mutants and the OSMR contribute to the activation of STAT3 in IHCAs.
The ability of IHCA-associated gp130 mutants to activate STAT3 is impaired by JAK1 inhibitors
JAK1, JAK2 and tyrosine kinase 2 (TYK2) are members of the Janus kinase family that operate downstream of the IL6R/gp130 complex and hence potentially serve to activate STAT3.Citation26 Interestingly, in human HCC HepG2 cells, the knockdown of JAK2 or TYK2 had no effect on the transcription activity of STAT3 as driven by IHCA-associated gp130 mutants or IL-6 administration (). In stark contrast, JAK1 silencing led to a dramatic decrease in STAT3 activation in 3 different HCC cell lines engineered to express IHCA-associated gp130 mutants (; Fig. S6). Thus, the activation of STAT3 by IHCA-associated gp130 mutants is dependent upon JAK1 but not JAK2 or TYK2.
Figure 4. Activity of IHCA gp130 mutants is selectively dependent upon JAK1. (A) Two different siRNAs silencing JAK1 (siJAK1#1 and #2) and two different siRNAs silencing JAK2 (siJAK2#1 and #2) or TYK2 (siTYK2#1 and #2) were tested for their ability to interfere with STAT3 activity in HepG2 cells co-transfected with a STAT3-luciferase reporter and an empty plasmid (EP)- or a plasmid coding for the Y186 gp30 mutant (n = 3). Where indicated, cells were treated for 3 h with 100 ng/mL interleukin-6 (IL-6). Shown are the mean luciferase activity ± SD relative to cells receiving the EP and a non-targeting control siRNA (siCTL). (B) Hep3B cells transfected with a plasmid encoding the Y186 gp130 mutant (filled circles) or the corresponding EP and then treated with 100 ng/mL IL-6 were exposed for 16 h to increasing concentrations of the JAK1 inhibitor ruxolitinib (n = 3). The data shown are the mean luciferase activities ± SD relative to Hep3B cells not treated with the inhibitor. (C) Hep3B cells transfected with constructs coding for nine different IHCA gp130 mutants (K173, V184, Y186, S187, V189, E195, D215, P216 and A418) or the corresponding EP were treated with 100 ng/mL IL-6 and exposed for 18 h to increasing concentrations of ruxolitinib. Shown are the mean luciferase activities ± SD from triplicate assessments relative to transfected cells not treated with the inhibitor. Statistical significance was determined by 2-tailed Student t test; **P < 0.01; ***P < 0.001.
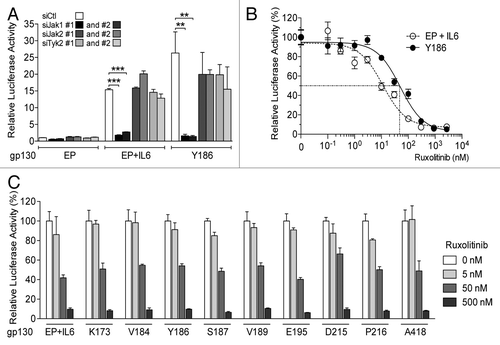
In view of these findings, we tested the effects of ruxolitinib (INCB01842), one of the first oral JAK1 and JAK2 inhibitors with clinical activity,Citation27,Citation28 on the ability of IHCA-associated gp130 mutants to activate STAT3. The treatment of Hep3B cells with ruxolitinib impaired the capacity of all nine IHCA-associated gp130 mutants to signal to STAT3 in a dose-dependent fashion, with an IC50 of ~50 nM (; Fig. S7). The sensitivity of the Y186 gp130 mutant to ruxolitinib was demonstrated in two additional HCC cell lines, namely HepG2 and Huh7 cells (Fig. S7). In accord with the results of our knockdown studies, the treatment of cells expressing IHCA-associated gp130 mutants with AG490, a tyrosine kinase inhibitor that selectively blocks JAK2,Citation18 had no effect on STAT3-driven luciferase reporter activity. Similarly, AG490 blocked only in part the activation of STAT3 by IL-6 (Fig. S8). Thus, the physiological activation of STAT3 by IL-6 is sensitive to high concentrations of JAK2 inhibitors whereas gp130 mutants appear to signal to STAT3 in a JAK2-independent fashion. We tested 2 additional classes of drugs known to inhibit STAT3, curcumin and SRC kinase inhibitors.Citation29,Citation30 We found that all these inhibitors block STAT3 activation as driven by IHCA-associated gp130 mutants, yet low doses of ruxolitinib were more efficient in doing so (Fig. S8B and C).
Discussion
Here, we identified a spectrum of IL6ST mutations in a large cohort of human IHCAs and we investigated the mechanisms by which the JAK/STAT signaling pathway is hyperactivated by the IHCA-associated gp130 mutants. Using this strategy, we established a central role for mutant gp130 in the activation of the JAK/STAT pathway, which is a hallmark of these benign liver tumors.Citation5 These data reinforce the notion that hepatocytes themselves are among the inflammatory cells involved in the development of IHCA, together with Kuppfer cells and other liver-infiltrating bone marrow-derived cells.Citation31
The spectrum of IL6ST mutations associated to IHCA appears to be the result of spontaneous mutagenesis. These genetic alterations may drive an inflammatory response that stimulates the benign proliferation of hepatocytes. Most of the IHCA-associated IL6ST mutations target the IL-6-binding site, which is located in the D2 domain of the protein. These residues have recently been involved in hydrophobic D2-D3 interactions that induce the ligand-independent activation of gp130.Citation32 Here, we report a new rare mutation that targets 4 residues at the D4-D5 junction of gp130, a flexible region that is important for the dimerization of gp130 extracellular domains. All IHCA-associated IL6ST mutations cause the constitutive activation of gp130, which is associated with the spontaneous formation of gp130 homodimers. Physiologically, the activation of the JAK/STAT signaling axis by IL-6 in hepatocytes requires the formation of a IL-6/IL6R/gp130 hexameric complex.Citation33 In contrast, gp130 mutants fully activate this signal transduction pathway independently of IL-6 or IL6R.
Although IHCA-associated gp130 mutants remain under the control of SOCS3, this checkpoint appears to be insufficient to inhibit the constitutive activation of STAT3 observed in this setting. We also demonstrated that wild-type gp130 normally is in an inactive basal conformation that can be converted into a constitutively active one by gain-of-function mutations of the D2 domain or the D4-D5 junction. These findings support the notion that the IL-6-binding site and the D4-D5 junction of g130 may function as intrinsic inhibitory domains that need to be modified by the binding of IL-6/IL6R to activate the JAK/STAT signaling cascade.
In the canonical IL-6/STAT3 signaling axis, JAK1, JAK2, and TYK2 kinases function downstream of gp130 and are required for STAT3 activation. In contrast, we provided evidence that the activity of IHCA-associated gp130 mutant only relies on JAK1 in various HCC cell lines. Accordingly, STAT3 activation as driven by gp130 mutants is strongly inhibited by ruxolitinib, a JAK1 inhibitor, but is insensitive to AG490, an inhibitor of JAK2. Consequently, the canonical activation of this signal transduction cascade by paracrine or autocrine signals is not equivalent to the activation of the same pathway by an oncogenic event that give rise to constitutively active gp130.
We have previously shown that ~5% of IHCAs, specifically those lacking gp130 mutations, bear somatic mutations in the gene coding for STAT3, which promote its activating homodimerization.Citation6 Although activating STAT3 and IL6ST mutations result in similar phenotypes, there are striking differences in the mechanism whereby the JAK/STAT axis is activated in these tumors. For example, IHCAs bearing activating STAT3 mutations are independent of JAK2, only partially rely on JAK1, are resistant to ruxolitinib but are sensitive to SRC kinase inhibitors.Citation6 Thus, understanding the specifics of the regulatory circuits that operate in these adenomas is required for implementing effectively tailored therapies.
In the clinical practice, the accepted treatment for HCA bearing IL6ST mutations is surgical resection, mostly due to the risk of malignant transformation or hemorrhage when lesions are larger than 5 cm.Citation1 However major hepatectomy, even in the normal liver, is invasive and accompanied by significant morbidity. In addition, multiple HCAs are frequently difficult to treat, as complete resection is difficult to perform. Adenomas are benign tumors with few genetics alterations that drive tumorigenesis. Thus, these lesions should be highly sensitive to the inhibition of oncogenic drivers. A recent study has demonstrated that an anti-gp130 antibody, B-P4, can block the constitutive activity of mutant gp130.Citation34 Of particular interest, B-P4, which targets the D4 domain of gp130, has been shown to block the ability of IL-11 (another inflammatory cytokine overexpressed in IHCAs), but not that of IL-6, to mediate the activation of gp130.Citation5 B-P4 has been used only in pre-clinical models and data on its clinical safety and efficacy are lacking. In contrast, ruxolitinib is well known for its safety and efficacy in the treatment of myelofibrosis, a clonal benign hematological disease characterized by JAK/STAT activation.
In conclusion, we propose a clinical trial to test ruxolitinib for the treatment of IHCAs bearing IL6ST mutations. This therapeutic approach could be particularly indicated to treat the occurrence of multiple adenomas or adenomatosis with large nodules that cannot be easily resected by surgery with minimal risk. Moreover, because the therapeutic options for advanced HCC are limited, ruxolitinib should be also evaluated in the rare cases of HCCs expressing gp130 mutants.
Materials And Methods
Tumors and patients
A series of 256 human HCA samples were previously collected (according to French ethical guidelines) and characterized.Citation2,Citation3 Patients with HCA were predominantly female (87.8%) with a mean age of 40 y. Of these, 45.3% were diagnosed with IHCA, as defined by the presence of an inflammatory tumor infiltrate (analyzed by immunostaining) and by the overexpression of the acute phase inflammatory response markers SAA and CRP analyzed (detected by qRT-PCR, with a fold-change of differential gene expression between tumor and normal liver tissues > 5). All patients gave informed consent according to French law and in accordance with the Declaration of Helsinki. The ethical committee of Saint Louis Hospital approved the study. Malignant and non-malignant liver samples were frozen immediately after surgery or biopsy and stored at −80 °C. Tissues samples were also preserved in 10% formalin, paraffin-embedded and stained with hematein-eosin and masson trichrome. The diagnosis of HCA was performed using established histological criteria and immunohistochemistry for the classification as previously described.Citation3,Citation35,Citation36
Quantitative RT-PCR
Total RNA was extracted using the RNeasy kit (Qiagen) according to the manufacturer's protocol. Single-stranded cDNA synthesis and qRT-PCR was performed as describedCitation5 using pre-designed TaqMan primers and probe sets from Applied Biosystems (catalog # Hs00269575_s1 and Hs00265044_m1 for SOCS3 and CRP, respectively). The ribosomal 18S RNA (R18S) was used to normalize expression data and the 2−∆∆CT method was applied.
DNA sequencing
DNA sequencing was performed as previously described to screen for IL6ST mutations.Citation5 All mutations were validated by sequencing a second independent PCR product on both strands. In all cases, the somatic origin of the mutation found in the tumor was verified by sequencing the adjacent, non-malignant liver tissue.
Generation of gp130 mutants
A full-length IL6ST open reading frame cloned into the pORF9 expression vector was purchased from InvivoGen (catalog #porf-hil6st). Mutagenesis reactions were performed using the QuickChange XL site-directed mutagenesis kit (Stratagene) using primers described in Table S2. All constructs were verified by sequencing.
Cell culture
Human HCC Hep3B, Huh7, and HepG2 cells (ATCC) were grown in Dulbecco-modified essential medium (DMEM) supplemented with 10% fetal calf serum (FCS), 100 units/mL penicillin and 0.1 mg/mL streptomycin. For transfections, cells were plated 16 h earlier to produce monolayers that were 60% confluent and transfections were performed using either Lipofectamine™ LTX (plasmid alone) or Lipofectamine™ 2000 (plasmid and siRNA) according to the manufacturer’s instructions (Invitrogen). Transfection efficiency was monitored by measuring the level of either wild-type or mutated IL6ST mRNA using qRT-PCR. Transfection efficiency was comparable in wild-type and mutant gp130-transfected cells. For luciferase assays, Hep3B, Huh7 and HepG2 cells were co-transfected with 1 μg of a STAT3 luciferase reporter (pSIEM-Luc) expressing a firefly-luciferase reporter gene that contains three copies of a consensus STAT3-binding site linked to a minimal thymidine kinase promoter (kindly provided by Dr. H. Gascan, Institut National de la Santé et de la Recherche Médicale UMR564, Angers, France)Citation37 together with 1 µg of a plasmid coding for wild-type or mutant gp130. Forty-eight hr after transfection, cells were starved in a serum-free medium for 3 h, and then were left untreated or treated with 100 ng/mL IL-6 or OSM for 3 h. Cells were then lysed and the luciferase activity determined according to the manufacturer’s recommendations (Promega). Luciferase activity was normalized to total protein concentration. All analyses were performed in triplicate.
RNA interference
In experiments with siRNA-mediated knockdown, cells were co-transfected with plasmids (see above) and 5 nM siRNA. The efficiency of knockdown was monitored by measuring levels of targeted mRNA using qRT-PCR. The efficiency of siRNA-mediated knockdown was invariably greater than 80%. All siRNAs (Table S3) were purchased from Applied Biosystems, with the sole exception of the control siRNA (Block-IT Alexa Fluor Red) which was purchased from Invitrogen.
Pharmacological treatments
Cells were exposed, in serum-free medium, to Tyrphostin AG490 (Sigma France) for 16 h; SRC Inhibitor-1 (Sigma France), SRC Inhibitor-5 (JS Res Chemical Trading, Germany) or PP2 (Sigma) for 10 h; Ruxolitinib (INCB-018424, JS Res Chemical Trading, Germany) for 16 h or curcumin (Sigma) for 9 h. In the last 3 h of treatment, cells were optionally stimulated with 100 ng/mL IL-6, as indicated.
Immunoprecipitation and immunonoblotting
For dimerization assays cell lysates were incubated with Immobilized Protein G agarose (Pierce) and anti-flag (Cell signaling Technology, 1:50) or anti-OSMR antibodies (Santa Cruz, 1:200) at 4 °C overnight, prior to immunoprecipitation and immunoblotting studies. Immunoblotting was performed as previously describedCitation5 using anti-flag (Cell Signaling Technology, 1:1000), anti-MYC (Cell Signaling Technology, 1:1000) or anti-OSMR (Santa Cruz, 1:200) antibodies. The of β actin (Sigma, 1:3000) was monitored to ensure equal lane loading.
| Abbreviations: | ||
| CRP | = | C-reactive protein, pentraxin-related |
| HCA | = | hepatocellular adenoma |
| HCC | = | hepatocellular carcinoma |
| IHCA | = | inflammatory HCA |
| IL-6 | = | interleukin-6 |
| IL6R | = | IL-6 receptor |
| IL6ST | = | IL-6 signal transducer |
| OSM | = | oncostatin M |
| OSMR | = | OSM receptor |
| STAT3 | = | signal transducer and activator of transcription 3 |
| SOCS3 | = | suppressor of cytokine signaling 3 |
| TYK2 | = | tyrosine kinase 2 |
Additional material
Download Zip (1.1 MB)Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We warmly thank Mohamed Amessou and Karine Bacri for their participation to this work, and Hugues Gascan who kindly provided the pSIEM-Luc plasmid. We also thank Charles Balabaud, Laurence Chiche, and Jean-Frédéric Blanc (Bordeaux hospital), Alexis Laurent (Créteil hospital), Eric Savier (AP-HP, department of digestive surgery, Groupe Hospitalier Pitié-Salpêtrière), Anne de Muret (Pathology department, CHRU de Tours), Centre des Collections Biologiques Hospitalières de Montpellier (CCBH-M, F-34285 Montpellier, France), all the participants to the GENTHEP network (Génétique des tumeurs hépatiques), and the center de resource national des tumeurs hépatiques (CRB). This work was supported by the association pour la recherche Contre le Cancer (ARC, grant n°3194), Institut National de la Santé et de la Recherche Médicale (Réseaux de recherche clinique et réseaux de recherche en santé des populations) and the BioIntelligence collaborative program. C.P. and J-C.N. were supported by a fellowship from the MENRT, ARC, and INCa.
Citation: Poussin K, Pilati C, Couchy G, Calderaro J, Bioulac-Sage P, Bacq Y, Paradis V, Leteurtre E, Sturm N, Ramos J, et al. Biochemical and functional analyses of gp130 mutants unveil JAK1 as a novel therapeutic target in human inflammatory hepatocellular adenoma. OncoImmunology 2013; 2:e27090; 10.4161/onci.27090
References
- Nault JC, Bioulac-Sage P, Zucman-Rossi J. Hepatocellular benign tumors-from molecular classification to personalized clinical care. Gastroenterology 2013; 144:888 - 902; http://dx.doi.org/10.1053/j.gastro.2013.02.032; PMID: 23485860
- Zucman-Rossi J, Jeannot E, Nhieu JT, Scoazec JY, Guettier C, Rebouissou S, Bacq Y, Leteurtre E, Paradis V, Michalak S, et al. Genotype-phenotype correlation in hepatocellular adenoma: new classification and relationship with HCC. Hepatology 2006; 43:515 - 24; http://dx.doi.org/10.1002/hep.21068; PMID: 16496320
- Bioulac-Sage P, Rebouissou S, Thomas C, Blanc JF, Saric J, Sa Cunha A, Rullier A, Cubel G, Couchy G, Imbeaud S, et al. Hepatocellular adenoma subtype classification using molecular markers and immunohistochemistry. Hepatology 2007; 46:740 - 8; http://dx.doi.org/10.1002/hep.21743; PMID: 17663417
- Bluteau O, Jeannot E, Bioulac-Sage P, Marqués JM, Blanc JF, Bui H, Beaudoin JC, Franco D, Balabaud C, Laurent-Puig P, et al. Bi-allelic inactivation of TCF1 in hepatic adenomas. Nat Genet 2002; 32:312 - 5; http://dx.doi.org/10.1038/ng1001; PMID: 12355088
- Rebouissou S, Amessou M, Couchy G, Poussin K, Imbeaud S, Pilati C, Izard T, Balabaud C, Bioulac-Sage P, Zucman-Rossi J. Frequent in-frame somatic deletions activate gp130 in inflammatory hepatocellular tumours. Nature 2009; 457:200 - 4; http://dx.doi.org/10.1038/nature07475; PMID: 19020503
- Pilati C, Amessou M, Bihl MP, Balabaud C, Nhieu JT, Paradis V, Nault JC, Izard T, Bioulac-Sage P, Couchy G, et al. Somatic mutations activating STAT3 in human inflammatory hepatocellular adenomas. J Exp Med 2011; 208:1359 - 66; http://dx.doi.org/10.1084/jem.20110283; PMID: 21690253
- Nault JC, Fabre M, Couchy G, Pilati C, Jeannot E, Tran Van Nhieu J, Saint-Paul MC, De Muret A, Redon MJ, Buffet C, et al. GNAS-activating mutations define a rare subgroup of inflammatory liver tumors characterized by STAT3 activation. J Hepatol 2012; 56:184 - 91; http://dx.doi.org/10.1016/j.jhep.2011.07.018; PMID: 21835143
- Calderaro J, Labrune P, Morcrette G, Rebouissou S, Franco D, Prévot S, Quaglia A, Bedossa P, Libbrecht L, Terracciano L, et al. Molecular characterization of hepatocellular adenomas developed in patients with glycogen storage disease type I. J Hepatol 2013; 58:350 - 7; http://dx.doi.org/10.1016/j.jhep.2012.09.030; PMID: 23046672
- Chun YS, Calderaro J, Zucman-Rossi J. Synchronous hepatocellular carcinoma and Castleman’s disease: the role of the interleukin-6-signaling pathway. Hepatology 2012; 56:392 - 3; http://dx.doi.org/10.1002/hep.25857; PMID: 22611056
- Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer 2009; 9:798 - 809; http://dx.doi.org/10.1038/nrc2734; PMID: 19851315
- Grivennikov S, Karin E, Terzic J, Mucida D, Yu GY, Vallabhapurapu S, Scheller J, Rose-John S, Cheroutre H, Eckmann L, et al. IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell 2009; 15:103 - 13; http://dx.doi.org/10.1016/j.ccr.2009.01.001; PMID: 19185845
- Matsumoto S, Hara T, Mitsuyama K, Yamamoto M, Tsuruta O, Sata M, Scheller J, Rose-John S, Kado S, Takada T. Essential roles of IL-6 trans-signaling in colonic epithelial cells, induced by the IL-6/soluble-IL-6 receptor derived from lamina propria macrophages, on the development of colitis-associated premalignant cancer in a murine model. J Immunol 2010; 184:1543 - 51; http://dx.doi.org/10.4049/jimmunol.0801217; PMID: 20042582
- Tebbutt NC, Giraud AS, Inglese M, Jenkins B, Waring P, Clay FJ, Malki S, Alderman BM, Grail D, Hollande F, et al. Reciprocal regulation of gastrointestinal homeostasis by SHP2 and STAT-mediated trefoil gene activation in gp130 mutant mice. Nat Med 2002; 8:1089 - 97; http://dx.doi.org/10.1038/nm763; PMID: 12219085
- Benekli M, Baer MR, Baumann H, Wetzler M. Signal transducer and activator of transcription proteins in leukemias. Blood 2003; 101:2940 - 54; http://dx.doi.org/10.1182/blood-2002-04-1204; PMID: 12480704
- Kunnumakkara AB, Anand P, Aggarwal BB. Curcumin inhibits proliferation, invasion, angiogenesis and metastasis of different cancers through interaction with multiple cell signaling proteins. Cancer Lett 2008; 269:199 - 225; http://dx.doi.org/10.1016/j.canlet.2008.03.009; PMID: 18479807
- Li F, Fernandez PP, Rajendran P, Hui KM, Sethi G. Diosgenin, a steroidal saponin, inhibits STAT3 signaling pathway leading to suppression of proliferation and chemosensitization of human hepatocellular carcinoma cells. Cancer Lett 2010; 292:197 - 207; http://dx.doi.org/10.1016/j.canlet.2009.12.003; PMID: 20053498
- Michaud-Levesque J, Bousquet-Gagnon N, Béliveau R. Quercetin abrogates IL-6/STAT3 signaling and inhibits glioblastoma cell line growth and migration. Exp Cell Res 2012; 318:925 - 35; http://dx.doi.org/10.1016/j.yexcr.2012.02.017; PMID: 22394507
- Miyamoto N, Sugita K, Goi K, Inukai T, Lijima K, Tezuka T, Kojika S, Nakamura M, Kagami K, Nakazawa S. The JAK2 inhibitor AG490 predominantly abrogates the growth of human B-precursor leukemic cells with 11q23 translocation or Philadelphia chromosome. Leukemia: official journal of the Leukemia Society of America. Leukemia Research Fund, UK 2001; 15:1758 - 68; http://dx.doi.org/10.1038/sj.leu.2402260
- Guichard C, Amaddeo G, Imbeaud S, Ladeiro Y, Pelletier L, Maad IB, Calderaro J, Bioulac-Sage P, Letexier M, Degos F, et al. Integrated analysis of somatic mutations and focal copy-number changes identifies key genes and pathways in hepatocellular carcinoma. Nat Genet 2012; 44:694 - 8; http://dx.doi.org/10.1038/ng.2256; PMID: 22561517
- Amaddeo G, Guichard C, Imbeaud S, Zucman-Rossi J. Next-generation sequencing identified new oncogenes and tumor suppressor genes in human hepatic tumors. Oncoimmunology 2012; 1:1612 - 3; http://dx.doi.org/10.4161/onci.21480; PMID: 23264911
- Boulanger MJ, Chow DC, Brevnova EE, Garcia KC. Hexameric structure and assembly of the interleukin-6/IL-6 alpha-receptor/gp130 complex. Science 2003; 300:2101 - 4; http://dx.doi.org/10.1126/science.1083901; PMID: 12829785
- Bravo J, Staunton D, Heath JK, Jones EY. Crystal structure of a cytokine-binding region of gp130. EMBO J 1998; 17:1665 - 74; http://dx.doi.org/10.1093/emboj/17.6.1665; PMID: 9501088
- Timmermann A, Pflanz S, Grötzinger J, Küster A, Kurth I, Pitard V, Heinrich PC, Müller-Newen G. Different epitopes are required for gp130 activation by interleukin-6, oncostatin M and leukemia inhibitory factor. FEBS Lett 2000; 468:120 - 4; http://dx.doi.org/10.1016/S0014-5793(00)01205-9; PMID: 10692570
- Pflanz S, Kurth I, Grötzinger J, Heinrich PC, Müller-Newen G. Two different epitopes of the signal transducer gp130 sequentially cooperate on IL-6-induced receptor activation. J Immunol 2000; 165:7042 - 9; PMID: 11120832
- Schmitz J, Weissenbach M, Haan S, Heinrich PC, Schaper F. SOCS3 exerts its inhibitory function on interleukin-6 signal transduction through the SHP2 recruitment site of gp130. J Biol Chem 2000; 275:12848 - 56; http://dx.doi.org/10.1074/jbc.275.17.12848; PMID: 10777583
- Mohr A, Chatain N, Domoszlai T, Rinis N, Sommerauer M, Vogt M, Müller-Newen G. Dynamics and non-canonical aspects of JAK/STAT signalling. Eur J Cell Biol 2012; 91:524 - 32; http://dx.doi.org/10.1016/j.ejcb.2011.09.005; PMID: 22018664
- Quintás-Cardama A, Vaddi K, Liu P, Manshouri T, Li J, Scherle PA, Caulder E, Wen X, Li Y, Waeltz P, et al. Preclinical characterization of the selective JAK1/2 inhibitor INCB018424: therapeutic implications for the treatment of myeloproliferative neoplasms. Blood 2010; 115:3109 - 17; http://dx.doi.org/10.1182/blood-2009-04-214957; PMID: 20130243
- Harrison C, Kiladjian JJ, Al-Ali HK, Gisslinger H, Waltzman R, Stalbovskaya V, McQuitty M, Hunter DS, Levy R, Knoops L, Cervantes F, Vannucchi AM, Barbui T, Barosi G. JAK Inhibition with Ruxolitinib versus Best Available Therapy for Myelofibrosis. N Engl J Med 2012; 366:787 98; http://dx.doi.org/10.1056/NEJMc1203704; PMID: 22621633
- Edwards J. Src kinase inhibitors: an emerging therapeutic treatment option for prostate cancer. Expert Opin Investig Drugs 2010; 19:605 - 14; http://dx.doi.org/10.1517/13543781003789388; PMID: 20367532
- Milacic V, Banerjee S, Landis-Piwowar KR, Sarkar FH, Majumdar AP, Dou QP. Curcumin inhibits the proteasome activity in human colon cancer cells in vitro and in vivo. Cancer Res 2008; 68:7283 - 92; http://dx.doi.org/10.1158/0008-5472.CAN-07-6246; PMID: 18794115
- Naugler WE, Sakurai T, Kim S, Maeda S, Kim K, Elsharkawy AM, Karin M. Gender disparity in liver cancer due to sex differences in MyD88-dependent IL-6 production. Science 2007; 317:121 - 4; http://dx.doi.org/10.1126/science.1140485; PMID: 17615358
- Schütt A, Zacharias M, Schneider N, Horn S, Grötzinger J, Rose-John S, Schmidt-Arras D. gp130 activation is regulated by D2-D3 interdomain connectivity. Biochem J 2013; 450:487 - 96; http://dx.doi.org/10.1042/BJ20121660; PMID: 23294003
- Streetz KL, Luedde T, Manns MP, Trautwein C. Interleukin 6 and liver regeneration. Gut 2000; 47:309 - 12; http://dx.doi.org/10.1136/gut.47.2.309; PMID: 10896929
- Sommer J, Effenberger T, Volpi E, Waetzig GH, Bernhardt M, Suthaus J, Garbers C, Rose-John S, Floss DM, Scheller J. Constitutively active mutant gp130 receptor protein from inflammatory hepatocellular adenoma is inhibited by an anti-gp130 antibody that specifically neutralizes interleukin 11 signaling. J Biol Chem 2012; 287:13743 - 51; http://dx.doi.org/10.1074/jbc.M111.349167; PMID: 22523320
- Bioulac-Sage P, Cubel G, Balabaud C, Zucman-Rossi J. Revisiting the pathology of resected benign hepatocellular nodules using new immunohistochemical markers. Semin Liver Dis 2011; 31:91 - 103; http://dx.doi.org/10.1055/s-0031-1272837; PMID: 21344354
- Bioulac-Sage P, Laumonier H, Couchy G, Le Bail B, Sa Cunha A, Rullier A, Laurent C, Blanc JF, Cubel G, Trillaud H, et al. Hepatocellular adenoma management and phenotypic classification: the Bordeaux experience. Hepatology 2009; 50:481 - 9; http://dx.doi.org/10.1002/hep.22995; PMID: 19585623
- Coqueret O, Gascan H. Functional interaction of STAT3 transcription factor with the cell cycle inhibitor p21WAF1/CIP1/SDI1. J Biol Chem 2000; 275:18794 - 800; http://dx.doi.org/10.1074/jbc.M001601200; PMID: 10764767