Abstract
Tumor-associated macrophages play a central role in tumor progression and metastasis. Macrophages can also promote the resistance of malignant cells to chemotherapy by stimulating the upregulation of cytidine deaminase, an intracellular enzyme that catabolizes the active form of gemcitabine. Targeting macrophage-dependent chemoresistance may reduce tumor-associated morbidity and mortality.
In the last decade, the role of the tumor microenvironment in the progression of stroma-rich pancreatic ductal adenocarcinoma (PDA) lesions has been increasingly understood.Citation1 Early studies of hematological malignancies suggested that tumor-associated macrophages (TAMs) can promote chemoresistance by directly interacting with malignant cells within the tumor microenvironment.Citation2 It is now clear that TAMs can influence the response of cancer cells to chemotherapy in the context of a process known as environment-mediated drug resistance.Citation3,Citation4 Gemcitabine (dFdC), the chemotherapeutic agent most often employed for the treatment of PDA is metabolized intracellularly by deoxycytidine kinase (DCK) to the active metabolites dFdCDP and dFdCTP. Several mechanisms may be responsible for gemcitabine resistance:Citation1 the loss of import via nucleoside transporters;Citation2 the downregulation of DCK, the rate-limiting enzyme in the formation of bioactive gemcitabine metabolitesCitation3 the upregulation of cytidine deaminase (CDA), the enzyme that metabolizes gemcitabine to one of its inactive forms (dFdU);Citation4 the upregulation of ribonucleotide reductases, which promote DNA synthesis by catalyzing the formation of deoxyribonucleotides from ribonucleotides; andCitation5 the inhibition of caspases, the enzymes that execute the apoptotic demise of cancer cells as induced by gemcitabine.Citation5
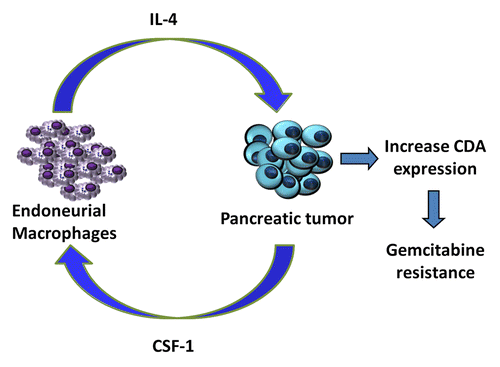
We have recently shown that TAMs stimulate chemoresistance by promoting the expression of CDA, the enzyme responsible for the inactivation of gemcitabine, by cancer cells (). Using TAM-conditioned medium, we observed a significant reduction in caspase-3 activation in malignant cells exposed to gemcitabine. The co-culture of TAMs and cancer cells had similar effect on gemcitabine-induced apoptosis than the exposure of the latter to TAM-conditioned medium, suggesting that one or more soluble factors secreted by TAMs mediate(s) this effect. In line with this finding, macrophage-depleted mice (be they Ccr2−/− animals or animals treated with clodronate-loaded liposomes) were more sensitive to the antineoplastic effects of gemcitabine than their wild-type counterparts. Similarly, in transgenic mice that spontaneously develop pancreatic carcinomas (owing to mutations in Kras and Trp53 expressed under the control of a pancreatic cell-specific promoter) the anticancer activity of gemcitabine was significantly improved by the co-administration of the colony-stimulating factor 1 receptor (CSF1R) antagonist GW2580.
Figure 1. Paracrine loop between endoneurial macrophages and pancreatic ductal adenocarcinoma cells. Colony-stimulating factor 1 (CSF1) secreted by pancreatic cancer cells attracts endoneurial macrophages (EMΦs) that express the CSF1 receptor (CSF1R). EMΦs recruited to the tumor site are activated by cancer cells and stimulate them to express high levels of cytidine deaminase (CDA). By catabolizing the bioactive form of gemcitabine, CDA exerts consistent chemoprotective effects, hence reducing the susceptibility of cancer cells to chemotherapy.
Real-time PCR analyses were used to evaluate the regulation of the multiple proteins involved in gemcitabine transport and metabolism, includingCitation1 gemcitabine transporters such as concentrative nucleoside transporter 1 (CNT1, official name SLC28A1) and equilibrative nucleoside transporter 1 (ENT1, official name SLC29A1);Citation2 DCK;Citation3 various ribonucleotide reductases, including ribonucleotide reductase M1 (RRM1) and RRM2; andCitation4 CDA. These analyses demonstrated that TAM-conditioned medium induce a 75-fold increase in the expression levels of CDA observed in the presence of gemcitabine. The immunohistochemical assessment of CDA levels in vivo revealed that tumors growing in wild-type mice significantly upregulate CDA in response to gemcitabine, whereas neoplasms developing in Ccr2−/− mice (which lack macrophages) do so to limited extents. Macrophages depletion also resulted in increased caspase-3 activation (a marker of apoptotic cells death) and decreased Ki67 immunoreactivity (which is indicative of reduced proliferation rates) in gemcitabine-treated neoplastic lesions, correlating with limited CDA expression levels. Finally, we demonstrated that pancreatic cancer cells depleted of CDA by means of a specific small-interfering RNA were less sensitive to chemoprotective effects of TAM-conditioned medium.
Our results demonstrate that paracrine signals delivered by the tumor microenvironment can be harnessed by cancer cells to maintain growth in spite of cytotoxic insults. Thus, tumor growth, progression and chemoresistance are supported by the tumor microenvironment, which provides a network of interactions between malignant cells and their stroma. A recent study revealed that the efficacy of anticancer drugs is markedly reduced when cancer cells are co-cultured with stromal cells.Citation6 Macrophages, which account for a significant fraction of the tumor stroma, are recruited and activated by signals emitted by cancer cells.Citation7
The first hint on the chemoprotective activity of macrophages was obtained in vitro, in breast cancer cells exposed to 6-thioguanine.Citation8 Cathepsin-expressing macrophages can also blunt the efficacy of paclitaxel against breast carcinoma cells, both in co-culture experiments and in vivo.Citation9 The authors of these studies suggested that macrophages promote chemotherapy resistance by impacting on cell-mediated antitumor immune response. Conversely, our findings point to the upregulation of intracellular proteins involved in the metabolism of chemotherapeutic agents as a major determinant of the chemoprotective activity of macrophages. Several factors released by macrophages may increase the resistance of cancer cells to gemcitabine. Data from a phosphorylation array covering 71 distinct tyrosine kinase receptors (Raybio, #AAH-PRTK-G1) performed on PANC-1 pancreatic carcinoma cells upon exposure to macrophage-condition medium revealed a significant increase in the phosphorylation levels of Janus kinase 1 (JAK1) and JAK3 (Gil and collaborators, unpublished data). Both JAK1 and JAK3 are activated by multiple cytokines, including interleukin-4 (IL-4), which is abundantly secreted by TAMs ().Citation10
In summary, our study demonstrates for the first time that macrophages can increase the resistance of cancer cells to chemotherapy through the upregulation of CDA, an intracellular enzyme which catabolizes the active form of gemcitabine. The clinical significance of our findings is substantial, since targeting the chemoprotective effects of TAMs may improve the efficacy of chemotherapy, hence decreasing disease morbidity and prolonging the survival of cancer patients.
Acknowledgments
The authors would like to thank their collaborators Noam Weizman, Yakov Krelin, Ayelet Shabtay-Orbach and Yoav Binenbaum from The Laboratory for Applied Cancer Research, Department of Otolaryngology Head and Neck Surgery Rambam Medical Center, the Technion Israel Institute of Technology, Haifa, Israel and Richard J Wong from the Department of Surgery Memorial Sloan Kettering Cancer Center, New York, NY.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Citation: Amit M, Gil Z. Macrophages increase the resistance of pancreatic adenocarcinoma cells to gemcitabine by upregulating cytidine deaminase. OncoImmunology 2013; 2:e27231; 10.4161/onci.27231
References
- Andre F, Berrada N, Desmedt C. Implication of tumor microenvironment in the resistance to chemotherapy in breast cancer patients. Curr Opin Oncol 2010; 22:547 - 51; http://dx.doi.org/10.1097/CCO.0b013e32833fb384; PMID: 20842030
- Zheng Y, Cai Z, Wang S, Zhang X, Qian J, Hong S, Li H, Wang M, Yang J, Yi Q. Macrophages are an abundant component of myeloma microenvironment and protect myeloma cells from chemotherapy drug-induced apoptosis. Blood 2009; 114:3625 - 8; http://dx.doi.org/10.1182/blood-2009-05-220285; PMID: 19710503
- De Palma M, Lewis CE. Cancer: Macrophages limit chemotherapy. Nature 2011; 472:303 - 4; http://dx.doi.org/10.1038/472303a; PMID: 21512566
- Mitchem JB, Brennan DJ, Knolhoff BL, Belt BA, Zhu Y, Sanford DE, Belaygorod L, Carpenter D, Collins L, Piwnica-Worms D, et al. Targeting tumor-infiltrating macrophages decreases tumor-initiating cells, relieves immunosuppression, and improves chemotherapeutic responses. Cancer Res 2013; 73:1128 - 41; http://dx.doi.org/10.1158/0008-5472.CAN-12-2731; PMID: 23221383
- Morgan MA, Parsels LA, Parsels JD, Mesiwala AK, Maybaum J, Lawrence TS. Role of checkpoint kinase 1 in preventing premature mitosis in response to gemcitabine. Cancer Res 2005; 65:6835 - 42; http://dx.doi.org/10.1158/0008-5472.CAN-04-2246; PMID: 16061666
- Straussman R, Morikawa T, Shee K, Barzily-Rokni M, Qian ZR, Du J, Davis A, Mongare MM, Gould J, Frederick DT, et al. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature 2012; 487:500 - 4; http://dx.doi.org/10.1038/nature11183; PMID: 22763439
- Gil Z, Cavel O, Kelly K, Brader P, Rein A, Gao SP, Carlson DL, Shah JP, Fong Y, Wong RJ. Paracrine regulation of pancreatic cancer cell invasion by peripheral nerves. J Natl Cancer Inst 2010; 102:107 - 18; http://dx.doi.org/10.1093/jnci/djp456; PMID: 20068194
- Yamashina K, Miller BE, Heppner GH. Macrophage-mediated induction of drug-resistant variants in a mouse mammary tumor cell line. Cancer Res 1986; 46:2396 - 401; PMID: 3084067
- Shree T, Olson OC, Elie BT, Kester JC, Garfall AL, Simpson K, Bell-McGuinn KM, Zabor EC, Brogi E, Joyce JA. Macrophages and cathepsin proteases blunt chemotherapeutic response in breast cancer. Genes Dev 2011; 25:2465 - 79; http://dx.doi.org/10.1101/gad.180331.111; PMID: 22156207
- Dijkgraaf EM, Heusinkveld M, Tummers B, Vogelpoel LT, Goedemans R, Jha V, Nortier JW, Welters MJ, Kroep JR, van der Burg SH. Chemotherapy alters monocyte differentiation to favor generation of cancer-supporting M2 macrophages in the tumor microenvironment. Cancer Res 2013; 73:2480 - 92; http://dx.doi.org/10.1158/0008-5472.CAN-12-3542; PMID: 23436796