
Abstract
Anti-PD-1/PD-L1 antibodies are emerging as promising anticancer therapeutics. Interestingly, elevated response rates to these agents are mostly documented among patients with tumors that bear high level of somatic mutations, like melanoma or non-small cell lung carcinoma. We herein formulate the hypothesis that high levels of mutational heterogeneity in the tumor could be the key for the success of immune checkpoint-targeting therapies.
Introduction
Recently, promising results have been obtained in patients with advanced cancers like melanoma, renal cell carcinoma (RCC), or non-small cell lung carcinoma (NSCLC) with immune checkpoint blockers, including monoclonal antibodies targeting cytotoxic T lymphocyte-associated protein 4 (CTLA4), programmed cell death 1 (PDCD1, best known as PD-1), or its main ligand CD174 (best known as PD-L1), resulting in remarkable and long-lasting clinical responses. Interestingly, Phase I efficacy data on anti-PD-1/PD-L1 agents point to a higher response rate among patients with melanoma and NSCLC, 2 tumor types associated with the highest levels of somatic mutations that develop upon exposure to UV light and carcinogens (tobacco smoke), respectively. Also, preliminary Phase IA data on the anti-PD-L1 antibody MPDL3280A suggest an association between smoking status and response in NSCLC patients.Citation1 These observations may suggest that the mutational heterogeneity of the tumor could be the key of the success of immune checkpoint-targeting therapies. To evaluate this hypothesis, we looked at the overall response rate (ORR) to PD-1/PD-L1-targeting agents among patients affected by solid tumors and compared it to mutational load reported in the literature for neoplasms of the same type. Herein, we report the results of this analysis, provide a rationale to explain how the accumulation of somatic mutations in tumors could improve the response to immune checkpoint blockers and propose future directions to improve the development of these new immunotherapies.
Response Rate to PD-1/PD-L1-Targeting Agents Across Solid Tumors
The PD-1/PD-L1 pathway mediates one major immune checkpoint.Citation7 PD-1/PD-L1 are a receptor/ligand pair that delivers inhibitory signals to activated T cells. Malignant cells are able to avoid immune destruction by diverting such an immune checkpoint.Citation7 Thus, anti-PD-1/PD-L1 monoclonal antibodies often restore effective antitumor immune responses.
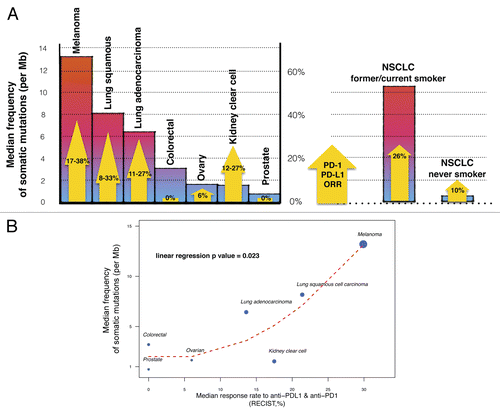
Although anti-PD-1/PD-L1 monoclonal antibodies have been tested across multiple solid tumors, they were first investigated in melanoma and RCC patients, mostly because of renown sensitivity of these malignancies to immunotherapy. Indeed, PD-1/PD-L1-targeting agents are associated with significant response rates in these clinical settingsCitation1, Citation2, Citation3, Citation4, Citation5, Citation6, as shown in : ORRs up to 38% (44/117) or 27% (9/33) have been reported in melanoma patients treated with the anti-PD-1 antibody lambrolizumab or RCC patients treated with nivolumab (another PD-1-targeting antibody), respectivelyCitation2,Citation3. Nonetheless, comparable response rates were later on observed in subjects with neoplasms expected to be poorly immunogenic. For instance, the ORR observed in NSCLC patients treated with MDPL3280A and nivolumab is 23% (12/53) and 18% (14/76), respectivelyCitation1,Citation2. Individuals bearing both the squamous and non-squamous NSCLC subtype seems to equally benefit from PD-1/PD-L1-targeting agents: ORRs of 8–33% and 11–27% were observed among squamous and non-squamous NSCLC patients, respectivelyCitation1,Citation2,Citation4 (). This suggests that the immunogenicity of tumors, as evaluated by their sensitivity to interleukin (IL)-2 or interferon (IFN)α is not a good surrogate marker for the efficacy of anti-PD-1/PD-L1 antibodies. Interestingly, patients affected by other solid tumors exhibit null or very low ORRs to anti-PD-1/PD-L1 agents: 0% in colorectal cancer patients (0/19 with nivolumab, 0/18 with lambrolizumab), 0% in prostate cancer (0/13 with nivolumab) and 6% in ovarian carcinoma (1/17 with BMS-936559)Citation2,Citation3,Citation4. The expression of PD-L1 on cancer cells has been suggested to constitute a predictive marker of clinical efficacy for anti-PD-1/PD-L1 agents.Citation1,Citation2,Citation8 Nevertheless, the method to quantify PD-L1 positivity is still debated and available data show a significant amount of responses among PD-L1-negative tumors. The expression of PD-L1 by malignant cells may therefore not constitute such an exclusive and critical predictive factor and other tumor characteristics may be as important for its sensitivity to anti-PD-1/PD-L1 agents. The remarkable ORRs observed upon the administration of anti-PD-1/PD-L1 antibodies to NSCLC patients raise the question on how NSCLC, melanoma and RCC differ from other solid tumors, which are associated with limited responses to these immunotherapeutics. Recent Phase I data on the treatment of NSCLC patients with MPDL3280A revealed a higher ORR of 26% (11/43) among former/current smokers than among never smokers (10%, 1/10)Citation1. These preliminary results in the setting of NSCLC and the ORRs documented among melanoma patients suggest that carcinogens (tobacco smoke or UV light) may play a key role in the susceptibility of tumors to PD-1/PD-L1 blockers, perhaps through their ability to promote mutagenesis. We thus formulated the hypothesis that the specific mutational profile of NSCLC, melanoma and RCC could underlie, at least in part, their ability to respond to checkpoints blockers.
Table 1. PD-1 and PD-L1 overall response rates across solid tumors
Mutational Heterogeneity Across Solid Tumors
To support this contention we looked at mutations rates that had been previously reported in the literature for various solid tumors.Citation9,Citation10,Citation11 Somatic mutations may be the consequence of defective DNA repair, the infidelity of the DNA replication machinery or the exposure to mutagens. Recent advances in the sequencing technology nowadays allow for the identification of a large panel of somatic mutations across different types of cancer and offer the possibility to validate predictive models in humans. The frequency of somatic mutations has been shown to be highly variable across cancer types, ranging from 0.001 to more than 400 per megabase (Mb).Citation10 The highest mutational rate is observed in melanoma (median of 13.2 mutations per Mb) and in NSCLC (8.17 and 6.43 mutations per Mb for the squamous and non-squamous subtype, respectively), in which mutations are known to be secondary to UV light and tobacco smoke exposure, respectively. As expected, the mutation rate is radically different between NSCLC patients that are former/current smoker and their counterparts who never smoked. The median of somatic mutations per Mb is indeed 10.5 for the former and 0.6 for the latter. On the contrary, relative low median rates of somatic mutations are observed in tumor types exhibiting limited responses to anti-PD-1/PD-L1 agents: 3.2 mutations per Mb for colorectal carcinoma, 1.53 mutations per Mb for RCC, 1.65 mutations per Mb for ovarian carcinoma and 0.73 mutations per Mb for prostate cancer. As a hypothesis-generating case, we observed a significant association between the mutational load of tumors and their susceptibility to PD-1/PD-L1-targeting agents ().
Figure 1. Link between mutational heterogeneity and response to immune checkpoint blockers. (A) Mutational heterogeneity of tumors and overall response rates (ORRs) to PD-1/PD-L1-targeting agents. Colored bars indicate the median frequency of somatic mutations per megabase (Mb) reported for patients affected by different solid tumors. Yellow arrows represent the ORRs of these patients to anti-PD-1/PD-L1 antibodies, as detailed in . NSCLC, non-small cell lung carcinoma. (B) Correlation between median frequency of somatic mutations and ORR to PD-1/PD-L1-targeting agents in solid tumors. Dot size is proportional to the number of patients in which the efficacy of anti-PD-1/PD-L1 antibodies was tested. The red dashed line represents the LOESS regression curve. The P value is derived from a linear univariate model (median somatic mutation frequency ~ORR to anti-PD-1/PD-L1 agents).
Defining which Antitumor Immune Responses are Stimulated by Immune Checkpoint Blockers
In the past decades, cancer vaccine specialists have been focusing on the identification of antigens selectively expressed by various solid tumors to developed efficient anticancer vaccines. Generally, tumor-specific T cells can recognize antigens resulting from oncogenic proteins, differentiation-associated proteins, overexpressed or aberrantly expressed proteins, proteins coded by oncogenic viruses, and cancer-testis proteins.Citation12 All these tumor-associated antigens are usually shared by different patients affected by the same neoplasm and sometimes even by distinct tumor types. Unfortunately, the results of clinical trials that have tested anticancer vaccines so far are deceiving, with null or very low ORRs. Now that immune checkpoint blockers are showing encouraging results, the future of anticancer immunotherapy looks more promising. Indeed, checkpoint-targeting agents represent, together with the adoptive transfer of tumor-infiltrating lymphocytes, the only immunotherapeutics that have been assocaited with high ORR and long-term responses. Compared with peptide-based vaccines or the adoptive transfer of engineered T cells with a narrow specificity, immune checkpoint blockers activate immune responses targeting a broad spectrum of antigens. Interestingly, patients responding to checkpoint-targeting agents show modest reactivity to known tumor-associated antigens, suggesting they are not strongly implicated in the immune response elicited by these agents.Citation13,Citation14 In fact, the tumor antigens that drive efficient T cells responses in patients who obtain clinical benefits from immune checkpoint blockers have not been clearly identified so far. The exome-guided immunomonitoring of patients treated with checkpoint-targeting agents has recently revealed that tumors are producing a large panel of neo-antigens that can drive immune responses.Citation15 These neo-antigens originate from the so-called tumor “mutanome,” the ensemble of tumor-specific (and hence most often patient-specific) mutations accumulated in the course of oncogenesis and tumor progression, which can involve both oncogenes and passenger genes. Moreover, mutations can result in neo-epitopes that are characterized by an improved MHC-binding profile, resulting in superior presentation to T cells. New technologies based on next-generation sequencing or peptide/MHC multimers are being developed to identify the numerous potential T-cell epitopes resulting from neo-antigens and to create optimal polytopic vaccines.Citation16-Citation18 However, since neo-antigens most often are tumor-specific, it seems particularly complex nowadays to employ these technologies to develop personalized treatments at a large scale.
Mutational Heterogeneity and Immune Response
Elevated mutation frequencies are sometimes attributable to the exposure to well known carcinogens, such as UV radiation in the case of melanoma and tobacco smoke in the case of lung cancer. As UV rays form pyrimidine dimers, melanoma-associated mutations show a high prevalence of C- > T transitions on the untranscribed, as compared with the transcribed, strand.Citation19 The mutational signature linked to NSCLC is dominated by C- > A transitions, which are associated with the exposure to the polycyclic hydrocarbons found in tobacco smoke.Citation20 Indeed, whole genome and transcriptome sequencing revealed a much higher number of mutations per Mb in NSCLC patients who were or had been tobacco smokers (mutations per Mb: median 10.5, range 4.9–17.6) than in never smokers (mutations per Mb: median 0.6, range 0.6–0.9), with a possible dose-response relationship between the amount and duration of smoking and the frequency of mutations.Citation21 Such an extreme mutational heterogeneity (be it intrinsic or induced by mutagens) represents a fundamental problem for targeted therapies, as it is responsible for the evolution of cancer cells exhibiting multiple molecular subtypes that exhibit variable susceptibility to treatment, hence facilitating resistance. As any mutation can potentially produce neo-antigens, the levels of mutagenesis may correlate with the degree of immunogenicity. Also, contrarily to targeted therapies, the immune system is highly adaptable, implying that a high mutational heterogeneity could play a key role in the tumor susceptibility to checkpoint blockers by eliciting a wide immune response to neo-antigens and therefore improving efficacy in particular against individual subclonal heterogeneity. Unlike anticancer vaccines, which may allow for the outgrowth of antigen-loss tumor variants, these neo-antigens could help to shape the antitumor immune response toward an improved fitness. High amounts of neo-antigens may also induce a beneficial effect by broadening the repertoire of responding T cells. Moreover, high levels of mutational heterogeneity may also promote immunogenicity by facilitating the interaction between tumor-derived epitopes and MHC class I and II molecules, resulting in improved presentation to T cells. The immune system is known to shape the tumor immunogenicity through immunoediting, reflecting the constant selection pressure exerted by the immune system on malignant cells.Citation22 On the other side, tumor’s mutational heterogeneity could reciprocally improve anticancer immune responses by widening the T-cell repertoire. In this setting, checkpoint blockers could reinstate therapeutically relevant immune responses against a hidden repertoire of neo-antigens.
Direct Effects of Carcinogens on the Immune System
Besides producing neo-antigens, carcinogens like UV rays or tobacco smoke have direct effects on the immune system. In addition to their carcinogenic potential, UV rays exert immunosuppressive effects by impairing the function of antigen-presenting cells, by promoting clonal anergy, by stimulating the accumulation of regulatory T cells or by favoring the secretion of immunosuppressive cytokines.Citation23 Detrimental mutations induced by tobacco carcinogens are also expected to facilitates the escape of cancer cells from the immune system through different mechanisms: they reduce immune recognition by favoring the loss of MHC class I proteins, by limiting the expression of anti-apoptotic proteins and by promoting the establishing of an immunosuppressive microenvironment characterized by high levels of IL-10, transforming growth factor β1, (TGFβ1) and indoleamine 2–3-dioxygenase 1 (IDO1).Citation22,Citation24 Finally, smoking may also help to maintain an inflammatory environment in the lungs, resulting in the IFNγ-driven expression of PD-L1.Citation25
Treatment-Induced Mutational Changes
It has been reported that melanoma cell clones becoming resistant to BRAF inhibitors harbor a different mutation profile upon exposure to these therapeutic agents. BRAF inhibitors may therefore contribute to mutational heterogeneity. Since approximately 50% of metastatic melanomas harbor BRAF V600 mutations, many melanoma patients enrolled in Phase I clinical trials have previously received BRAF inhibitors. Likewise, many NSCLC or RCC patients treated with PD-1/PD-L1-targeting agents in the context of Phase I clinical studies had previously been treated with a tyrosine kinase inhibitor (TKI), which may have directly shaped the tumor mutational pattern through drug selective pressure. These drug-induced mutations may play a key role at modeling the clinical response to PD-1/PD-L1-targeting agents. Additionally, these mutations that emerge under drug selection can directly affect the expression of immune checkpoint-related molecules on cancer cells, as it has been reported for melanoma in BRAF inhibitor resistant clones manifesting increased expression levels of PD-L1.
Conclusions and Future Directions
Our analysis is based on a limited amount of data. Moreover, the efficacy observed in Phase I clinical trials is of course very preliminary, and ORR data from Phase II and III studies are extremely expected. Nonetheless, we believe that our analysis has the merit to generate a new hypothesis to explain the responses to checkpoint inhibitors observed so far among patients with solid tumors, notably that mutagen-induced tumors appear to exhibit an improved response to these immunotherapeutic agents. This may indirectly suggest that targeted anticancer agents such as TKIs can affect the immunogenicity of tumors as they impose a selective pressure, a notion that should be taken into consideration for the design of future clinical trials involving immune checkpoint blockers. Moreover, if high levels of mutational heterogeneity increase the tumor immunogenicity, it will be interesting to evaluate the clinical activity of PD-1/PD-L1 agents in DNA mismatch repair (MM)-deficient tumors, such as microsatellite instability (MSI)+ colorectal carcinoma as well as BRCA1 and 2 neoplasms (breast cancer 1and 2, early onset), all of which display severe genomic instability. Other tumors that present foci of localized hypermutations as observed in subsets of breast, pancreatic, lung and hematological cancers, could also be good candidates for immune checkpoint inhibitors, a possibility that should be evaluated in future clinical trials.
| Abbreviations: | ||
| BRCA1 | = | breast cancer 1, early onset |
| BRCA2 breast cancer 1 | = | early onset |
| CTLA4 | = | cytotoxic T lymphocyte-associated protein 4 |
| IDO1 | = | indoleamine 2,3-dioxygenase 1 |
| IFN | = | interferon |
| IL | = | interleukin |
| MMR | = | mismatch repair |
| MSI | = | microsatellite instability |
| NSCLC | = | non-small cell lung carcinoma |
| ORR | = | overall response rate |
| PDCD1 | = | programmed cell death 1 |
| PD-L1 | = | PD-1 ligand 1 |
| RCC | = | renal cell carcinoma |
| TGFβ1 | = | transforming growth factor β1 |
| TKI | = | tyrosine kinase inhibitor |
Disclosure of Potential Conflicts of Interest
Drs Champiat, Ferté, and Lebel-Binay have declared no conflict of interest. Dr Eggermont has received consulting fee or honorarium from BMS, GSK, MedImmune, and MSD. Dr Soria has received consulting fee or honorarium from Genentech.
Citation: Champiat S, Ferté C, Lebel-Binay S, Eggermont A, Soria J. Exomics and immunogenics: Bridging mutational load and immune checkpoints efficacy. OncoImmunology 2014; 3:e27817; 10.4161/onci.27817
References
- Soria JC, et al. Clinical activity, safety and biomarkers of PD-L1 blockade in non-small cell lung cancer (NSCLC): Additional analyses from a clinical study of the engineered antibody MPDL3280A (anti-PDL1). ECCO ESMO Congress 2013; abstract 3408.
- Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med 2012; 366:2443 - 54; http://dx.doi.org/10.1056/NEJMoa1200690; PMID: 22658127
- Hamid O, Robert C, Daud A, Hodi FS, Hwu W-J, Kefford R, Wolchok JD, Hersey P, Joseph RW, Weber JS, et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med 2013; 369:134 - 44; http://dx.doi.org/10.1056/NEJMoa1305133; PMID: 23724846
- Brahmer JR, Tykodi SS, Chow LQM, Hwu W-J, Topalian SL, Hwu P, Drake CG, Camacho LH, Kauh J, Odunsi K, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med 2012; 366:2455 - 65; http://dx.doi.org/10.1056/NEJMoa1200694; PMID: 22658128
- Daniel C. Cho, Jeffrey Alan Sosman, Mario Sznol, Michael S. Gordon, Antoine Hollebecque, Omid Hamid, David F. McDermott, Jean-Pierre Delord, Ina Park Rhee, Ahmad Mokatri,, et al. Clinical activity, safety, and biomarkers of MPDL3280A, an engineered PD-L1 antibody in patients with metastatic renal cell carcinoma (mRCC). J Clin Oncol 31, 2013 (suppl; abstr 4505)
- Omid Hamid, Jeffrey Alan Sosman, Donald P. Lawrence, Ryan J. Sullivan, Nageatte Ibrahim, Harriet M. Kluger, Peter D. Boasberg, Keith Flaherty, Patrick Hwu, Marcus Ballinger, et al. Clinical activity, safety, and biomarkers of MPDL3280A, an engineered PD-L1 antibody in patients with locally advanced or metastatic melanoma (mM). J Clin Oncol 31, 2013 (suppl; abstr 9010)
- Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer 2012; 12:252 - 64; http://dx.doi.org/10.1038/nrc3239; PMID: 22437870
- Chen DS, Irving BA, Hodi FS. Molecular pathways: next-generation immunotherapy--inhibiting programmed death-ligand 1 and programmed death-1. Clin Cancer Res 2012; 18:6580 - 7; http://dx.doi.org/10.1158/1078-0432.CCR-12-1362; PMID: 23087408
- Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Sivachenko A, Carter SL, Stewart C, Mermel CH, Roberts SA, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013; 499:214 - 8; http://dx.doi.org/10.1038/nature12213; PMID: 23770567
- Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SAJR, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Børresen-Dale A-L, et al, Australian Pancreatic Cancer Genome Initiative. ICGC Breast Cancer Consortium; ICGC MMML-Seq Consortium; ICGC PedBrain. Signatures of mutational processes in human cancer. Nature 2013; 500:415 - 21; http://dx.doi.org/10.1038/nature12477; PMID: 23945592
- Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA Jr., Kinzler KW. Cancer genome landscapes. Science 2013; 339:1546 - 58; http://dx.doi.org/10.1126/science.1235122; PMID: 23539594
- Buonaguro L, Petrizzo A, Tornesello ML, Buonaguro FM. Translating tumor antigens into cancer vaccines. Clin Vaccine Immunol 2011; 18:23 - 34; http://dx.doi.org/10.1128/CVI.00286-10; PMID: 21048000
- Yuan J, Ginsberg B, Page D, Li Y, Rasalan T, Gallardo HF, Xu Y, Adams S, Bhardwaj N, Busam K, et al. CTLA-4 blockade increases antigen-specific CD8(+) T cells in prevaccinated patients with melanoma: three cases. Cancer Immunol Immunother 2011; 60:1137 - 46; http://dx.doi.org/10.1007/s00262-011-1011-9; PMID: 21465316
- Yuan J, Adamow M, Ginsberg BA, Rasalan TS, Ritter E, Gallardo HF, Xu Y, Pogoriler E, Terzulli SL, Kuk D, et al. Integrated NY-ESO-1 antibody and CD8+ T-cell responses correlate with clinical benefit in advanced melanoma patients treated with ipilimumab. Proc Natl Acad Sci USA 2011; 108:16723 - 8; http://dx.doi.org/10.1073/pnas.1110814108; PMID: 21933959
- van Rooij N, van Buuren MM, Philips D, Velds A, Toebes M, Heemskerk B, van Dijk LJA, Behjati S, Hilkmann H, El Atmioui D, et al. Tumor exome analysis reveals neoantigen-specific T-cell reactivity in an ipilimumab-responsive melanoma. J Clin Oncol 2013; 31:e439 - 42; http://dx.doi.org/10.1200/JCO.2012.47.7521; PMID: 24043743
- Hadrup SR, Bakker AH, Shu CJ, Andersen RS, van Veluw J, Hombrink P, Castermans E, Thor Straten P, Blank C, Haanen JB, et al. Parallel detection of antigen-specific T-cell responses by multidimensional encoding of MHC multimers. Nat Methods 2009; 6:520 - 6; http://dx.doi.org/10.1038/nmeth.1345; PMID: 19543285
- Andersen RS, Kvistborg P, Frøsig TM, Pedersen NW, Lyngaa R, Bakker AH, Shu CJ. Straten Pt, Schumacher TN, Hadrup SR. Parallel detection of antigen-specific T cell responses by combinatorial encoding of MHC multimers. Nat Protoc 2012; 7:891 - 902; http://dx.doi.org/10.1038/nprot.2012.037; PMID: 22498709
- Castle JC, Kreiter S, Diekmann J, Löwer M, van de Roemer N, de Graaf J, Selmi A, Diken M, Boegel S, Paret C, et al. Exploiting the mutanome for tumor vaccination. Cancer Res 2012; 72:1081 - 91; http://dx.doi.org/10.1158/0008-5472.CAN-11-3722; PMID: 22237626
- Pfeifer GP, You Y-H, Besaratinia A. Mutations induced by ultraviolet light. Mutat Res 2005; 571:19 - 31; http://dx.doi.org/10.1016/j.mrfmmm.2004.06.057; PMID: 15748635
- Pleasance ED, Stephens PJ, O’Meara S, McBride DJ, Meynert A, Jones D, Lin M-L, Beare D, Lau KW, Greenman C, et al. A small-cell lung cancer genome with complex signatures of tobacco exposure. Nature 2010; 463:184 - 90; http://dx.doi.org/10.1038/nature08629; PMID: 20016488
- Govindan R, Ding L, Griffith M, Subramanian J, Dees ND, Kanchi KL, Maher CA, Fulton R, Fulton L, Wallis J, et al. Genomic landscape of non-small cell lung cancer in smokers and never-smokers. Cell 2012; 150:1121 - 34; http://dx.doi.org/10.1016/j.cell.2012.08.024; PMID: 22980976
- Chow MT, Möller A, Smyth MJ. Inflammation and immune surveillance in cancer. Semin Cancer Biol 2012; 22:23 - 32; http://dx.doi.org/10.1016/j.semcancer.2011.12.004; PMID: 22210181
- Ullrich SE. Mechanisms underlying UV-induced immune suppression. Mutat Res 2005; 571:185 - 205; http://dx.doi.org/10.1016/j.mrfmmm.2004.06.059; PMID: 15748647
- Garrido F, Cabrera T, Aptsiauri N. “Hard” and “soft” lesions underlying the HLA class I alterations in cancer cells: implications for immunotherapy. Int J Cancer 2010; 127:249 - 56; PMID: 20178101
- Taube JM, Anders RA, Young GD, Xu H, Sharma R, McMiller TL, Chen S, Klein AP, Pardoll DM, Topalian SL, et al. Colocalization of inflammatory response with B7-h1 expression in human melanocytic lesions supports an adaptive resistance mechanism of immune escape. Sci Transl Med 2012; 4:127ra37; http://dx.doi.org/10.1126/scitranslmed.3003689; PMID: 22461641