
Abstract
Established tumors are typified by an immunosuppresive microenvironment. Countering this naturally occurring phenomenon, emerging evidence suggests that radiation promotes a proimmunogenic milieu within the tumor capable of stimulating host cancer-specific immune responses. Three cryptic immunogenic components of cytotoxic-agent induced cell death—namely, calreticulin cell surface exposure, the release of high mobility group box 1 (HMGB1) protein, and the liberation of ATP—have been previously shown to be critical for dendritic cell (DC) activation and effector T-cell priming. Thus, these immune-mobilizing components commonly presage tumor rejection in response to treatment. We initially set out to address the hypothesis that radiation-induced immunogenic cell death (ICD) is dose-dependent. Next, we hypothesized that radiation would enhance chemotherapy-induced ICD when given concomitantly, as suggested by the favorable clinical outcomes observed in response to analogous concurrent chemoradiation regimens. Thus, we designed an in vitro assay to examine the 3 hallmark features of ICD at clinically relevant doses of radiation. We then tested the immunogenic-death inducing effects of radiation combined with carboplatin or paclitaxel, focusing on these combinations to mimic chemoradiation regimens actually used in clinical trials of early stage triple negative [NCT0128953/NYU-10–01969] and locally advanced [NYU-06209] breast cancer patients, respectively. Despite the obvious limitations of an in vitro model, radiotherapy produced both a dose-dependent induction and chemotherapeutic enhancement of ICD. These findings provide preliminary evidence that ICD stimulated by either high-dose radiotherapy alone, or concurrent chemoradiation regimens, may contribute to the establishment of a peritumoral proimmunogenic milieu.
Introduction
Clinically apparent tumors often exhibit immunosuppressive features conducive to unchecked growth of the primary tumor, as well as metastatic spread of the disease. Fortuitously, however, scientific evidence suggests that radiation may promote a type of tumor cell death that stimulates host antitumor immune responses attempting to gain parity with the immunosuppressive effects prevalent within the non-treated tumor microenvironment.Citation1-Citation3 This immunogenic type of cancer cell death effectively contributes to priming the host’s immune system to recognize and eliminate radiation exposed tumor cells, particularly after immune checkpoint blockade. Importantly, radiotherapy combined with immune checkpoint inhibitors, often stimulates an immune response that positively contributes to local tumor control and can result in memory that confers a selective ability to recognize and eliminate residual malignant cells, either localized outside the radiotherapy site or that emerge from dormancy at a later point in time.Citation4-Citation6
Founded by the seminal work of Zitvogel and Kroemer, 3 distinct ICD components essential for dendritic cell (DC) activation and immune priming have emerged. These include: 1) the cell surface translocation of calreticulin (CALR, better known as CRT), an endoplasmic reticulum (ER) resident chaperone protein and potent DC “eat me” signal; 2) the extracellular release of high mobility group box 1 (HMGB1), a DNA binding protein and toll-like receptor 4 (TLR-4) mediated DC activator; and 3) the liberation of adenosine-5′-triphosphate (ATP), a cell-cell signaling factor in the extracellular matrix (ECM) that serves to activate P2X7 purinergic receptors on DCs, triggering DC inflammasome activation, secretion of IL-1β, and subsequent priming of interferon-γ (IFNγ) producing CD8+ T cells.Citation7,Citation8 The cumulative effects of all 3 arms act to promote DC phagocytosis of tumor cells, thus facilitating DC processing of tumor-derived antigens and subsequent DC-associated cross-priming of CD8+ cytotoxic T lymphocytes.Citation7-Citation10 Using these phenotypic features of immunogenic death, some chemotherapeutic compounds have been demonstrated to induce ICD, particularly oxaliplatin.Citation11,Citation12
We developed an in vitro assay to test the effects of radiotherapy at clinically relevant doses alone or in combination with chemotherapeutic agents on each individual component of ICD.Citation38 Previously, using the TSA syngeneic murine model of mammary carcinoma, we reported that radiotherapy in combination with cytotoxic T lymphocyte associated protein 4 (CTLA-4) blockade induces an immune-mediated tumor-inhibitory effect outside the field of radiation, in other words an abscopal effect.Citation13,Citation14 Thus, we selected TSA cells for the development of an in vitro model to determine whether we can observe the hallmark features of ICD upon exposing the cells to radiation in a dose-dependent manner (i.e., with increasing doses of radiation) alone or in combination with chemotherapy. In vitro models are particularly advantageous due to the low cost and feasibility, permitting expedited screening of chemoradiotherapy regimens prior to validating their role in an in vivo setting. We report, herein, the immunogenic death signature displayed by TSA mammary cancer cells exposed to radiotherapy alone or in combination with the platinum agents oxaliplatin and carboplatin, or the taxane paclitaxel.
Results
Radiotherapy promotes ATP release
To rapidly quantify radiation-induced ATP release, TSA cells were transfected with a pGEN2.1 plasmid encoding firefly luciferase bound to the plasma membrane by virtue of a flanking folate receptor (FR) leader sequence and a glycophosphatidylinositol (GPI) anchor sequence, as previously described by Francesco di Virgilio (, top panel, Fig. S1).Citation15-Citation17 ATP is an intracellular molecule under homeostatic conditions in treatment-naïve TSA cells. However, ATP is readily released from stressed cells undergoing ICD.Citation3,Citation17 In our in vitro model, pericellular ATP, in the presence of oxygen, magnesium, and D-luciferin substrate, reacts with the extracellular localized, plasma membrane-bound luciferase (pMe-Luc) to produce photons, readily detectable and quantified with a luminometer, thereby serving as a surrogate for the presence of pericellular ATP (, bottom panel).
Figure 1. Ionizing radiation induces ATP release into the ECM. (A and B) TSA cells were stably transfected with a pGEN2.1 plasmid encoding a firefly luciferase reporter sequence flanked by a folate receptor (FR) leader sequence and glycophosphatidylinositol (GPI) anchor sequence (A, top panel). ATP remains intracellular when tumor cells are in their native condition, correlating with minimal luciferase activity. ATP release upon immunogenic cell death increases pericellular ATP that, in the presence of oxygen, magnesium, and D-luciferin, reacts with the external membrane-bound luciferase and produces photons that can be detected by luminometery (A, bottom panel). (B) The amount of luminescence detected from 2 x 104 cells per well (96-well plate) pGEN2.1-pMe-Luc transfected TSA cells after 24 h of exposure to increasing doses of ionizing radiation (IR) ranging from 0–20 Gray (Gy) reported as fold-change in relative luminescent units (RLU) in comparison to the luminescent signal detected from non-irradiated cells, normalized to 1. Shown are the mean RLU (n = 8 wells/group) ± SD.
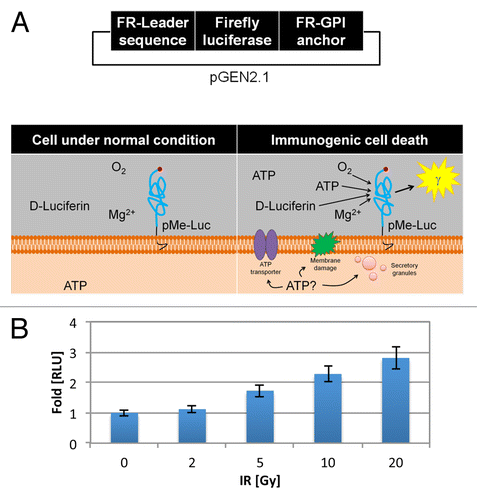
The amount of luminescence detected and reported as fold change in relative luminescent units (RLUs) after 24 h of exposure to increasing doses of ionizing radiation (IR) is shown (). We observed a radiation dose-dependent increase in RLUs from pGEN2.1-pMe-Luc transfected TSA cells exposed to IR reflecting higher levels of ATP in the ECM of irradiated cells. Specifically, irradiation with 0, 2, 5, 10, and 20 Gray (Gy) induced luciferase-reporter RLUs of 1 ± 0.10, 1.12 ± 0.11, 1.73 ± 0.2, 2.28 ± 0.25, and 2.81 ± 0.38, respectively.
Radiotherapy promotes CRT translocation
CRT is an ER resident chaperone protein that sequesters calcium and prevents misfolded proteins from leaving the ER.Citation18 However, CRT is also well known to serve as an “eat me” signal for DC phagocytosis upon translocation to the surface of dying tumor cells undergoing ICD.Citation19-Citation22 To gain insight into the amount of CRT cell surface translocation among dying TSA cells in response to IR, we stably transfected TSA cells with an engineered DNA construct from the pEZ-M02 vector that stably expresses CRT-HaloTag-KDEL fusion protein, a fluorescence-based reporter of cell surface localized CRT (, top panel, Fig. S2).Citation11
Figure 2. Radiotherapy promotes calreticulin surface translocation. (A–C) TSA cells were stably transfected with a pEZ-M02 plasmid encoding a calreticulin (CRT) fusion protein with the modular HaloTag® reporter,and endoplasmic reticulum targeting KDEL sequences (A, top panel) corresponding to the translation of a fusion CRT-HaloTag-KDEL protein (A, middle panel). CRT remains in the ER in non-stressed cells, whereas upon immunogenic cell death (ICD) CRT translocates to the cell surface (A, bottom panel). Externally localized CRT-HaloTag-KDEL is irreversible bound by membrane impermeable HaloTag® Alexa Fluor 488 ligand activating its fluorescent properties that can then be detected via fluorescence microscopy or flow cytometry (A, middle and bottom panels). (B and C) pEZ-M02-CRT-HaloTag-KDEL transfected TSA cells were treated for 24 h with the indicated dosage of ionizing radiation (IR, delivered at time 0 h) were exposed to the impermeable HaloTag® Alexa Fluor 488 ligand. The amount of green fluorescence indicative of cell surface CRT was detected via cytoflourimetric analysis. (B) Representative histograms at each dose of IR. (C) Mean fluorescent intensity (MFI) detected from cells irradiated with the indicated IR dosage vs. non-irradiated cells normalized to 1. Shown are the MFI (n = 10 000 cells/group) ± SD.
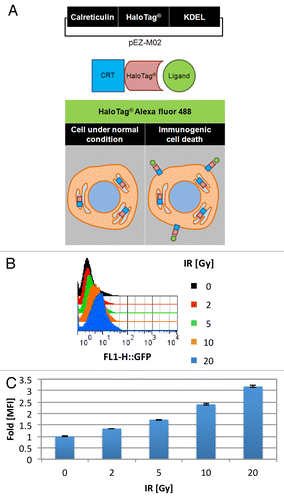
HaloTag® is a genetically engineered haloalkane dehalogenase designed to irreversibly bind to synthetic fluorescent ligands.Citation23 Under normal (i.e., non-stressed) conditions, the CRT-HaloTag-KDEL fusion protein, akin to native CRT, resides in the ER (). However, when cells undergo ICD, ER resident CRT, including CRT-HaloTag is translocated to the cell surface, whereby HaloTag® Alexa fluor 488, a cell membrane impermeable HaloTag® ligand, can selectively label ECM exposed CRT-HaloTag fusion protein (, bottom panel).Citation24 The fluorescent signal from the irreversibly bound ligand can then be detected via flow cytometry, whereby changes in the mean fluorescence intensity (MFI) under various conditions be ascertained.
To confirm the specificity of the impermeable HaloTag® Alexa Fluor 488 ligand, untreated cells were exposed to either the 5μM impermeable HaloTag® Alexa Fluor 488 ligand, or as a control, the cell permeable HaloTag® R110 direct ligand, and subject to fluorescence cytometric analysis. This control experiment documented that the system could detect compartmentalized CRT, as untreated TSA cells expressing CRT-Halotag-KDEL labeled with either HaloTag® Alexa fluor 488 (baseline control) or HaloTag® R110 direct was 1 ± 0.01 and 2.94 ± 0.10, respectively.
Before testing IR, we tested oxaliplatin, previously been shown to cause CRT surface-translocation tumor cells undergoing ICD, as a positive control for the assay.Citation24 Thus, to confirm the presence of CRT cell surface translocation in our system, CRT-HaloTag-KDEL engineered TSA cells were either untreated or treated with 500 µM oxaliplatin for 24 h.Citation24 Thereafter, the cells were exposed to 5 µM of HaloTag® Alexa Fluor 488 ligand and visualized via confocal microscopy. Similarly to previous reports, the oxaliplatin treated cells demonstrated significant fluorescence via confocal microscopy, whereas minimal fluorescence was observed in untreated cells (data not shown).Citation24 This pilot experiment confirmed that our system could selectively detect cell-surface exposed CRT.
To quantitatively determine cell surface CRT translocation in response to IR, TSA cells transfected with the CRT-HaloTag-KDEL reporter construct were first evaluated untreated (as a negative control) or treated for 24 h with 500 µM oxaliplatin (as a positive control) or 20 Gy of IR. Transfectants were subsequently stained with the cell membrane impermeable HaloTag® Alexa Fluor 488 ligand, and subjected to flow cytometry. The MFI fold change in the MFI of untreated cells relative to cells treated for 24 h with 500 µM oxaliplatin or 20 Gy IR increased from 1.17 ± 0.01 to 2.36 ± 0.03 and 2.07 ± 0.04, respectively. Of note, a small MFI fold difference was observed between untreated cells that were unexposed or exposed to 5 µM of HaloTag® Alexa Fluor 488 ligand, in which case the MFI was 1 ± 0.01 and 1.17 ± 0.01, respectively.
Interestingly, in a dose-dependent manner IR alone was capable of inducing CRT translocation to the cell surface. The amount of CRT externalization, as indicated by cell surface fluorescence detected via flow cytometry and reported as fold-change in MFI after 24 h of exposure to increasing doses of IR is shown in . The MFI detected from CRT-Halotag transfected TSA cells exposed to IR 0, 2, 5, 10, and 20 Gy was 1 ± 0.01, 1.34 ± 0.01, 1.73 ± 0.02, 2.40 ± 0.03, and 3.18 ± 0.05, respectively. Taken together, these data indicate that the majority of CRT is normally intracellular under homeostatic conditions. However, when cells are exposed to increasing doses of IR, significant amounts of CRT are translocated to the cell surface.
Radiotherapy promotes HMGB1 release
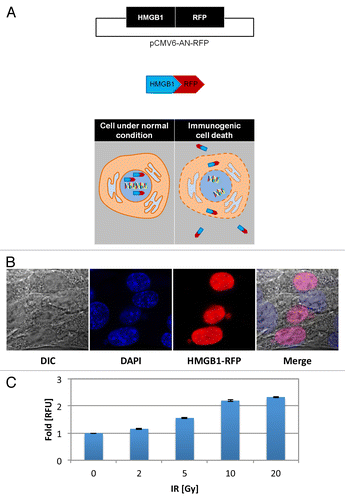
To rapidly determine the degree to which IR triggers another ICD hallmark, HMGB1 release from dying tumor cells, TSA cells were transfected with a different fluorescent reporter, a pCMV6-AN-RFP plasmid encoding a HMGB1 with a red fluorescent protein (RFP) C-terminal tag (, top panel, refer to Fig. S3). HMGB1 has been previously shown to reside in the nucleus under native conditions.Citation25 Thus, in HMGB1-RFP transfected TSA cells, we anticipated the presence of fused HMGB1-RFP protein in the nucleus of untreated cells (, bottom panel), a subcellular localization verified via confocal microscopy (). Conversely, released HMGB1-RFP protein was anticipated in the conditioned medium of cells exposed to IR (, bottom panel) validated via fluorescence cytometry (exλ530 nM and emλ593 nm) and reported as fold-change in relative fluorescent units (RFUs) in comparison to untreated controls (). After 72 h of exposure to increasing IR doses, increasing fluorescence was detected in the conditioned media of irradiated cells (relative to untreated controls) reflecting higher levels of HMGB1-RFP release. Specifically, irradiation with 0, 2, 5, 10, and 20 Gy resulted in RFUs of 1 ± 0.01, 1.16 ± 0.02, 1.56 ± 0.02, 2.20 ± 0.03, and 2.33 ± 0.02, respectively.
Figure 3. Radiation therapy promotes HMGB1 release. (A–C) TSA cells were stably transfected with a pCMV6-AN-RFP plasmid (A, top panel) comprising the high mobility group box 1 (HMGB1) protein coding open-reading frame sequence fused with a C-terminal red fluorescent protein (RFP) tag (A, middle panel). Under native conditions, HMGB1 remains in the nucleus, whereas HMGB1 is released from cells undergoing ICD, detectable in the conditioned medium of cultured HMGB1-RFP TSA cells via fluorimetry (A, bottom panel). (B) Chimeric HMGB1-RFP protein can be detected in the nucleus of untreated cells via confocal fluorescence microscopy. (C) Transfected cells were exposed to ionizing radiation (IR) ranging from 0–20 Gray (Gy), as indicated. Released HMGB1-RFP was detected in the conditioned medium 72 h after treatment via cytofluorimetric analysis and reported as fold change in relative fluorescent units (RFU) in comparison to untreated control cells, normalized to 1. Shown are the MFI (n = 6 wells/group) ± SD.
Platinum and radiotherapy enhance TSA cytotoxicity
We next sought to address the combined effects of platinums and radiation on markers of ICD. To this end, we selected oxaliplatin (a known inducer of ICD) and carboplatin, both drugs that are currently used in the clinic. Due to cell line specific pharmacodynamic differences, we first set out to understand the cytotoxic profiles of the platinum compounds on TSA cells.
To ascertain the short term cytotoxic effects of the platinum compounds, we treated TSA cells with the platinums for 48 h and determined cell viability via a methylthiazolyldiphenyl-tetrazolium bromide (MTT) colorimetric assay.Citation26,Citation27 MTT powder is a yellow water-soluble tetrazolium salt that is catalyzed to a purple formazan chromogen in the presence of dehydrogenases and reductases. This reaction readily occurs in living cells, whereby the absorbance of this purple solution directly corresponds to cell viability and can be measured with a spectrophotometer at λ490 nm. Utilizing this cell viability assay, we found that 100 µM of oxaliplatin or carboplatin, reduced the percent cell viability (relative to untreated controls) from 100 ± 10.73% to 16.19 ± 0.99% and 10.16 ± 1.21%, respectively.
We also evaluated the persistent growth-inhibiting effects of platinum therapy on TSA cells following 48h treatment by colony forming assay to further assess the long-term survival (2 wk) of individual cells and their ability to spawn clonal descendants following treatment.Citation28 In cells treated with 1 µM of oxaliplatin or carboplatin, the percent of colonies formed after 2 wk incubation was reduced (relative to the untreated control) from 100 ± 2.63% to 63.49 ± 6.69% and 71.75 ± 4.79%, respectively (). Taken together, our results indicate that TSA cells are comparably sensitive to oxaliplatin and carboplatin as measured by both the short-term MTT viability assay and the long-term clonal growth assay.
Figure 4. Cytotoxicity induced by platinum and ionizing radiation combinatorial treatment in TSA cells. (A and B) The differential cytotoxic effects of platinum compounds ± ionizing radiation (IR) at a dosage of 2 Gray (Gy) was evaluated via colony forming assay. TSA cells (200 cells/well in a 6-well plate; n = 6) were exposed to increasing doses of the indicated platinum agent [0–5 μM] for 48 h ± IR [2 Gy], delivered at time 0 h. After incubating the cells for 10 d, the colonies formed were fixed, stained, and subsequently counted. (A) The percent colonies formed are displayed, normalized to untreated cells (100% colony formation). (B) Corresponding images of crystal violet stained colonies. Shown are the mean colony formation ± SD. Statistical analyses were performed using a paired Student’s t test; P values < 0.05 were considered statistically significant.
![Figure 4. Cytotoxicity induced by platinum and ionizing radiation combinatorial treatment in TSA cells. (A and B) The differential cytotoxic effects of platinum compounds ± ionizing radiation (IR) at a dosage of 2 Gray (Gy) was evaluated via colony forming assay. TSA cells (200 cells/well in a 6-well plate; n = 6) were exposed to increasing doses of the indicated platinum agent [0–5 μM] for 48 h ± IR [2 Gy], delivered at time 0 h. After incubating the cells for 10 d, the colonies formed were fixed, stained, and subsequently counted. (A) The percent colonies formed are displayed, normalized to untreated cells (100% colony formation). (B) Corresponding images of crystal violet stained colonies. Shown are the mean colony formation ± SD. Statistical analyses were performed using a paired Student’s t test; P values < 0.05 were considered statistically significant.](/cms/asset/4be5b0dc-2d44-4a17-b9e3-2d09bf090c35/koni_a_10928518_f0004.gif)
Since IR therapeutic activity on solid tumors is a cumulative effect of cancer cell death occurring over several rounds of cell division following irradiation, we determined the combined growth-inhibiting effects of platinum and radiotherapy via the long-term colony forming assay, the gold-standard for determining radiation induced cell death.Citation3,Citation28,Citation29 We found that in TSA cells the fraction of surviving (SF) colonies after IR 2 Gy (SF2, a measurement of radiosensitivity) was reduced from 100 ± 2.63% in the control unexposed cells, to 58.41 ± 5.74% (). Of particular interest, the SF2 in TSA cells was further reduced when either platinum was added. For instance, when TSA cells were irradiated with 2 Gy and exposed to 1 µM of oxaliplatin or carboplatin for 48 h, the percent of colonies formed was further reduced from 58.41 ± 5.74% to 29.84 ± 2.90% and 24.76 ± 10.08%, respectively (). Collectively, these results indicate that the combination of IR and platinum have an enhanced cytotoxic effect on TSA cells.
Platinum and radiotherapy induce ATP release from dying tumor cells
To address the degree to which chemotherapy and radiotherapy combined to elicit the ICD hallmark of ATP release, the amount of luminescence from the extracellular anchored luciferase reporter (pMe-Luc) is shown in after 24 h of exposure to increasing doses of oxaliplatin, a known inducer of ATP release.Citation30 pMe-Luc expressing TSA cells exposed to 1, 5, and 10 µM oxaliplatin, resulted in an RLU fold increase (relative to untreated control levels) from 1 ± 0.09 to 1.27 ± 0.12, 1.87 ± 0.19, and 1.56 ± 0.18, respectively.
Figure 5. Pericellular ATP in platinum and ionizing radiation treated TSA cells. (A and B) TSA cells transfected with pGEN2.1 vector encoding plasma membrane localized luciferase (pMe-Luc) were used to assay the effects of platinum ± ionizing radiation (IR) on pericellular ATP concentrations. The amount of luminescence detected from 2 x 104 pGEN2.1-pMe-Luc transfected TSA cells/well (96-well plate) in the presence of D-luciferin. (A) Luminescence detected after 24 h of exposure to increasing doses of oxaliplatin (0–10 μM). (B) Luminescence detected after 24 h of exposure to increasing doses of the indicated platinum agent [0–5 μM] ± increasing doses of IR, ranging from 0–20 Gray (Gy) delivered at time 0 h. Values are reported as fold-change in relative luminescent units (RLU) in comparison to the luminescent signal detected from non-irradiated cells, normalized to 1. Shown are the means (n = 8 wells/group) ± SD. Statistical analyses were performed by paired Student’s t test; P values < 0.05 were considered statistically significant.
![Figure 5. Pericellular ATP in platinum and ionizing radiation treated TSA cells. (A and B) TSA cells transfected with pGEN2.1 vector encoding plasma membrane localized luciferase (pMe-Luc) were used to assay the effects of platinum ± ionizing radiation (IR) on pericellular ATP concentrations. The amount of luminescence detected from 2 x 104 pGEN2.1-pMe-Luc transfected TSA cells/well (96-well plate) in the presence of D-luciferin. (A) Luminescence detected after 24 h of exposure to increasing doses of oxaliplatin (0–10 μM). (B) Luminescence detected after 24 h of exposure to increasing doses of the indicated platinum agent [0–5 μM] ± increasing doses of IR, ranging from 0–20 Gray (Gy) delivered at time 0 h. Values are reported as fold-change in relative luminescent units (RLU) in comparison to the luminescent signal detected from non-irradiated cells, normalized to 1. Shown are the means (n = 8 wells/group) ± SD. Statistical analyses were performed by paired Student’s t test; P values < 0.05 were considered statistically significant.](/cms/asset/3f90bc36-761c-4034-9f55-57075abf8cfe/koni_a_10928518_f0005.gif)
Interestingly, we observed enhanced ATP release from cells exposed to IR when combined with oxaliplatin and parity of ATP release when IR was combined with carboplatin (). For example, untreated vs. either 5 µM oxaliplatin or carboplatin treated cells resulted in an RLU fold increase from 1 ± 0.08 to 1.43 ± 0.15 or 1.05 ± 0.15 respectively. Additionally, untreated or IR 20 Gy treated cells resulted in an RLU fold increase from 1 ± 0.08 to 2.61 ± 0.30. However, IR 20 Gy combined with 5 µM oxaliplatin significantly stimulated ATP release, as measured by a fold-change in RLU to 4.49 ± 0.68 (P value < 0.001). In contrast, carboplatin treatment, only modestly increased ATP release as shown by an insignificant RLU fold change of 2.47 ± 0.36. In summary, our results indicate that, at the doses of radiation tested IR-induced ATP-release is enhanced by oxaliplatin and conserved in the presence of carboplatin.
Platinum and radiotherapy cause CRT translocation in dying tumor cells
In order to delineate whether CRT translocation is platinum dose-dependent, TSA CRT-HaloTag-KDEL cells were treated with increasing doses of platinum and assayed 24h later. Interestingly, the degree of CRT translocation was dose-dependent in response to oxaliplatin, a known inducer of CRT translocation, whereas CRT translocation was not dose-dependent in response to carboplatin treatment. For example, the fold-change in MFI detected on CRT-HaloTag-KDEL TSA cells treated with 100µM oxaliplatin or carboplatin increased to 1.60 ± 0.04 and 1.47 ± 0.03, respectively from untreated controls levels of 1 ± 0.02. Additionally, the MFI fold-change in cells treated with 500 µM oxaliplatin further increased to 2.36 ± 0.03, while the MFI fold-change in cells treated with 500 µM carboplatin remained relatively stable at 1.23 ± 0.03 ().
Figure 6. Calreticulin translocation to the cell surface in platinum and ionizing radiation treated TSA cells. (A and B) Externalization of calreticulin (CRT) was monitored using pEZ-M02-CRT-Halotag-KDEL stably transfected TSA breast cancer cells treated for 24 h with platinum ± the indicated dosage of ionizing radiation (IR, delivered at time 0 h) exposed to the impermeable HaloTag® Alexa Fluor 488 ligand. The amount of green fluorescence indicative of cell surface CRT was detected via fluorescence cytometry. (A) Representative histograms from the indicated treatment. (B) Mean fluorescent intensity (MFI) detected from cells irradiated with the indicated platinum agent ± IR vs. non-irradiated cells normalized to 1. Shown are the MFI (n = 10 000 cells/group) ± SD. Statistical analysis was performed by paired Student’s t test; P values < 0.05 were considered statistically significant.
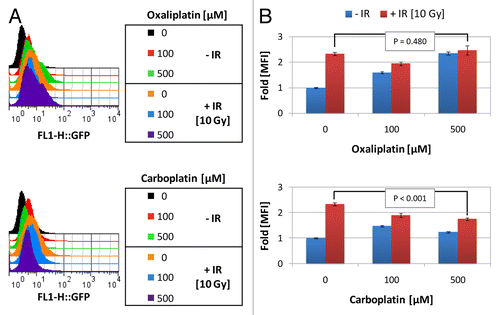
To determine whether concurrent platinum and radiotherapy could synergize to enhance CRT translocation in mammary carcinoma cells, we treated CRT-HaloTag-KDEL transfected TSA cells with various platinum and radiotherapy regimens. We found that upon the addition of IR, the amount of CRT translocation induced by the platinum agents remained elevated, but did not appear to significantly increase further. For instance, the MFI fold change in untreated vs. IR 10 Gy treated cells increased from 1 ± 0.02 to 2.34 ± 0.06. However, when IR 10 Gy was added to 100 µM of oxaliplatin or carboplatin the MFI fold change was 1.95 ± 0.07 and 1.89 ± 0.08, respectively, values only marginally higher than the chemotherapeutic agent alone (). Additionally, when IR 10 Gy was added to 500 µM of oxaliplatin or carboplatin the MFI fold change was 2.48 ± 0.17 and 1.75 ± 0.05, respectively, lower or nearly equivalent levels to the same dosage of IR alone and similar to that of the platinum agent only (). In summary, radiotherapy and oxaliplatin monotherapy induced CRT translocation at the dosages tested in a dose-dependent manner. That being said, IR did not further enhance platinum-induced CRT translocation, such that upon the addition of IR, the amount of CRT translocation in platinum treated cells remained relatively stable, albeit elevated.
Platinum and radiotherapy cause HMGB1 release from dying tumor cells
Considering that the kinetics of the reaction could impact the magnitude of the measured response, we next set out to ascertain the ideal timing of HMGB1 release in IR or platinum exposed tumor cells. To accomplish this, we used the RFP-tagged HMGB1 fusion protein to detect HMGB1 release into the surrounding media of dying cancer cells after treatment with IR or oxaliplatin, a known inducer of HMGB1 release, in a time and dose-dependent manner ().Citation11 The RFUs detected in the conditioned media of untreated controls barely changed over the time course of 24, 48, and 72 h, from 1 ± 0.16 to 1.16 ± 0.05, and 1.23 ± 0.04 fold respectively. Fluorescence from HMGB1-RFP TSA cells treated with 10 µM oxaliplatin similarly minimally changed over 24, 48, and 72 h, exhibiting slight increases in RFUs from 1.06 ± 0.03 to 1.37 ± 0.03, and 1.49 ± 0.02-fold, respectively, whereas cells treated with 100 µM oxaliplatin and incubated for 24, 48, and 72 h following treatment, the RFUs increased more substantially from 1 ± 0.16 in the controls to 1.51 ± 0.05, 2.49 ± 0.04, and 2.76 ± 0.21 fold, respectively. Similarly, HMGB1-RFP TSA cells treated with IR 2 Gy for 24, 48, and 72 h the fold change in RFUs increased from 1 ± 0.03 (untreated control) to 1.11 ± 0.04, 1.15 ± 0.03, and 1.5 ± 0.17, respectively, whereas cells treated with higher IR at 20 Gy for 24, 48, and 72 h exhibited a more robust increase in the RFUs at the later time points, from 1 ± 0.16 (untreated control) to 1.18 ± 0.05, 1.77 ± 0.03, and 2.95 ± 0.06-fold, respectively. In summary, both IR and oxaliplatin optimally induced HMGB1 release 72 h after treatment.
Figure 7. HMGB1 release from platinum and ionizing radiation treated TSA cells. (A and B) Red-fluorescence protein (RFP) tagged high mobility group box 1 (HMGB1) expressing TSA cells were used to analyze the combined effect of platinum and ionizing radiation (IR) on the release of HMGB1 from TSA mammary carcinoma cells. (A) HMGB1-RFP stably transfected TSA cells were exposed to oxaliplatin (0–100 μM) or IR (0–20 Gray) delivered at time 0 h. Released HMGB1-RFP was detected in the conditioned medium 24–72 h (as indicated) after treatment via fluorimetry and reported as a fold change in relative fluorescent units (RFUs) in comparison to untreated cells (24 h). (B) HMGB1-RFP was detected in the conditioned medium of transfected cells exposed for 72 h with increasing doses of platinum (0–20 μM) ± radiation therapy (RT) at a dosage of 2 Gray (Gy) delivered at time 0 h. The RFU is plotted relative to untreated control cells, normalized to 1. Shown are the mean (n = 6/group) ± SD. Statistical analyses were performed by paired Student’s t test: P values < 0.05 were considered statistically significant.
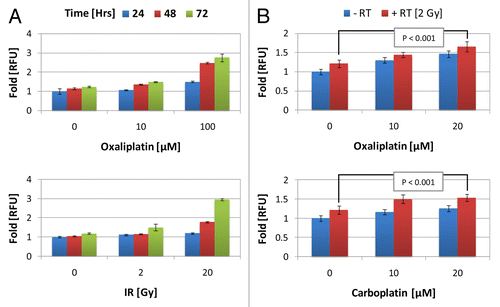
We next sought to determine whether other platinums could mediate HMGB1 release and to test whether combinatorial therapy with IR might further enhance liberation of HMGB1. To this end, we treated HMGB1-RFP transfected cells with oxaliplatin or carboplatin and assayed for HMGB1 release into the conditioned media 72 h later. As shown in , in TSA cells treated with 20 µM oxaliplatin or carboplatin, the fold change in RFUs increased from 1 ± 0.07 (untreated cells) to 1.44 ± 0.09 and 1.23 ± 0.08, respectively. Treatment with IR 2 Gy alone only affected a minor increase in RFUs from 1 ± 0.07 to 1.18 ± 0.10. However, when IR 2 Gy was added to oxaliplatin and carboplatin, enhanced HMGB1 release was observed as reflected by an increase in RFUs detected from 1.18 ± 0.1 (IR alone) to 1.62 ± 0.13 and 1.51 ± 0.09 in cells treated in combination with 20 µM oxaliplatin or carboplatin, respectively. Taken together, our results indicate that IR, oxaliplatin, and carboplatin could, in a dose-dependent fashion significantly increase HMGB1 release 72 h after treatment in our mammary carcinoma model. Moreover, IR combined with oxaliplatin or carboplatin may potentially enhance HMGB1 release from dying TSA cells.
Paclitaxel and radiotherapy elicit features of ICD from dying tumor cells
We sought out to test whether concurrent IR and paclitaxel were capable of eliciting features of ICD from dying tumor cells and determine whether the combined regimen could enhance TSA therapeutic cytotoxicity. For example, the clonal growth measure of radiosensitivity, SF2 in TSA cells was reduced from 100 ± 6.03% to 69.57 ± 3.33% in response to IR alone (). Nevertheless, the SF2 in TSA cells was further reduced when paclitaxel was added. For instance, when TSA cells were irradiated with 2 Gy and exposed to 50 nM of paclitaxel for 48 h, the percent of colonies formed was reduced further from 69.57 ± 3.33% to 31.36 ± 3.07% (). Of note, paclitaxel alone reduced the surviving fraction to 59.67 ± 3.42%. Hence, these results indicate that IR and paclitaxel enhance the cytotoxic effects on TSA cells.
Figure 8. Immunogenic cell death is enhanced in paclitaxel and ionizing radiation treated TSA cells. (A–D) The cytotoxic effects of paclitaxel (PTX) ± ionizing radiation (IR) were evaluated via colony forming assay and molecular markers of immunogenic cell death. (A) TSA cells were plated at 200 cells/well in a 6-well plate. After adherence, cells were exposed to 50 nM paclitaxel ± radiation therapy (RT) at a dosage of 2 Gray (Gy) delivered at time 0 h and evaluated via colony forming assay. After incubating the cells for 10 d, the colonies formed were fixed, stained, and counted. The percent colonies formed are displayed are displayed, normalized to untreated cells (100% colony formation). (B) To assay release of ATP, TSA cells stably transfected with a plasma membrane localized luciferase (pMe-Luc) plated at 2 x 104 cells per well (96-well plate) were exposed to 1 μM paclitaxel ± 2 Gy IR, delivered at time 0 h. The relative luminescent units (RLUs) detected 24 h later are shown in comparison to untreated cells, normalized to 1. Shown are the mean RLU (n = 8 wells/group) ± SD (C) To assay calreticulin (CRT) externalization, CRT-Halotag-KDEL stably transfected TSA cells were treated for 24 h with 100 nM paclitaxel ± 10 Gy IR delivered at time 0 h were exposed to the impermeable HaloTag® Alexa Fluor 488 ligand. The amount of green fluorescence indicative of cell surface CRT was detected via fluorescence cytometry and reported as fold change in mean fluorescent intensity (MFI) vs. untreated cell levels, normalized to 1. Shown are the MFI (n = 10 000 cells/group) ± SD (D) The release of high mobility group box 1 (HMGB1) protein was assayed using red fluorescence protein (RFP)-tagged HMGB1. TSA cells stably transfected with HMGB1-RFP stably were exposed to 1 μM paclitaxel ± 2 Gy IR, delivered at time 0 h. Released HMGB1-RFP was detected in the conditioned medium 24–72 h after treatment via fluorimetry and reported as fold change in relative fluorescent units (RFU) vs. untreated cell levels, normalized to 1. Shown are the mean RLU (n = 6 wells/group) ± SD.
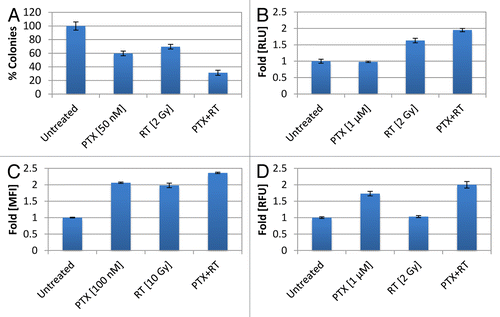
To determine whether IR and paclitaxel could intensify hallmarks of ICD, we tested the combined treatment regimen in our in vitro breast cancer model. Accordingly, we quantified the amount ATP release from dying pMe-Luc expressing TSA cells treated with the combined regimen and observed enhanced ATP release, as measured by increased luciferase activity (). For example, 1 µM paclitaxel treatment vs. untreated cells did not alter luciferase luminescence, as shown by a RLU fold-change from 1 ± 0.06 to 0.98 ± 0.02. IR 2 Gy treated cells exhibited a modest RLU fold increase from 1 ± 0.08 to 1.63 ± 0.07. However, IR 2 Gy plus 1 µM paclitaxel stimulated the highest RLU fold-change to 1.95 ± 0.05, indicating that IR and paclitaxel enhanced the release of ATP. We also quantified the amount of CRT translocation to the cell surface using the CRT-HaloTag-KDEL transfected TSA cells and found that the combination of IR and paclitaxel enhanced CRT externalization. For instance, 100 nM paclitaxel only treated cells exhibited a MFI increase from 1 ± 0.01 (untreated cells) to 2.06 ± 0.02-fold and IR 10 Gy only affected a MFI fold-increase to 1.98 ± 0.07. However, the combination exerted an enhanced effect, displaying a MFI fold-increase to 2.36 ± 0.02. Finally, using our HMGB1-RFP transfected TSA cells, we observed an enhanced effect with the combinatorial regimen of paclitaxel and IR with respect to HMGB1 release. For example, 1 µM paclitaxel caused a MFI fold-change from 1 ± 0.025 (untreated cells) to 1.73 ± 0.07, whereas, IR 2 Gy barely altered the RFUs detected to 1.03 ± 0.03. Nevertheless, the combined regimen markedly increased the RFUs to 2 ± 0.10-fold control levels, reflecting higher levels of HMGB1 release. Taken together, these results indicate that paclitaxel and IR combinatorial therapies have the potential to enhance each arm of ICD.
Discussion
The central dogma of traditional radiobiology states that the cytotoxic effects of IR on tumor cells are primarily due to the production of DNA double strand breaks followed by cell death, either via apoptosis, necrosis, autophagy, mitotic catastrophy, or replicative senescence.Citation3 This traditional view of radiation-induced cell death is rapidly evolving to take into account the contribution of the tumor microenvironment and the host’s antitumor immunity, particularly in the modern era following the advent of immune-checkpoint inhibitors applied as a means to overcome the immunosuppressive effects of established tumors.Citation1,Citation3,Citation5,Citation31 Importantly, the recruitment of the host’s immune system as a contributor of the “in field” response to radiotherapy can result in immune memory, an advantageous systemic effect that transcends the localized nature of this treatment modality.Citation4
Here, we showed that clinically relevant doses of IR effectively induce the signals for each individual component of ICD, in a dose-dependent manner from 2–20 Gy (–). Still, IR alone may not be sufficient to produce clinically observable effects. In vivo and in the clinic, IR-produced pro-immunogenic effects are often masked by the overwhelming immunosuppressive microenvironment that characterizes established cancers.Citation5,Citation32 Nevertheless, when some barriers of established immunosuppression are removed, for instance by adding immune checkpoint inhibitors (such as anti-CTLA4 or anti-PD1) to local radiation therapy, the pro-immunogenic effects of IR are leveraged and foster immune-mediated tumor rejection.Citation6,Citation13,Citation14,Citation33
An alternative approach to achieve immunogenic cell death is the concurrent use of IR and some chemotherapy agents. In such settings, IR may “reposition” some of the drugs used, a mechanism suggested by Zitvogel and colleagues.Citation34,Citation35 We originally introduced the concept that some of the success of concurrent chemo-radiation treatment regimens could derive from the contribution of ICD, a clinically relevant biological phenotype more likely to be achieved when the modalities are used concomitantly rather than sequentially.Citation36
We previously reported the results of a multicenter trial of neoadjuvant paclitaxel and radiotherapy given concurrently to treat locally advanced breast cancer patients. A 34% pathologic complete response rate, measured by histological examination of the breast and resected lymph nodes from the surgical specimens after mastectomy, was demonstrated in a cohort of 105 locally advanced breast cancer patients.Citation37 Since pathological response to chemo-radiation was significantly associated with 5 y survival probability, we hypothesized that ICD may at least in part contribute to the successful outcomes seen in these patients. Likewise, we hypothesized that ICD could partially contribute to the success of concurrent platinum-based chemo-radiation regimens, as witnessed in numerous clinical trials.Citation36 Therefore, we focused here on testing IR in combination with platinums or paclitaxel, to mimic these clinical conditions and test mechanistically whether the single vs. combined therapies manifest differences in ICD. Our in vitro assay confirmed our hypothesis, in that we observed enhanced features of ICD when TSA cells were treated concurrently with IR and chemotherapy.
Although the range of our in vitro assay is limited and the findings presented herein may be cell line-dependent, the hallmark features of ICD were demonstrated upon exposing TSA cells to various combinations of IR with platinums or paclitaxel. This preliminary evidence suggests that IR in a therapeutic dose-range effectively induces ICD. In addition, when combined with drugs commonly used in the clinic (platinums and taxanes), IR stimulates an optimized repertoire of pro-immunogenic signals from dying tumor cells that may facilitate host anticancer immune responses and could, at least in part, explain the enhanced local and systemic clinical benefits of combinatorial regimens. If the predictive role of this assay is further validated by subsequent experiments in vivo, this mammary cancer in vitro system could represent a convenient screening tool for elucidating optimal therapeutic combinations suitable for clinical testing.
Materials and Methods
Reagents and materials
Halotag® R110DirectTM cell permeable ligand, Halotag® Alexa Fluor 488 cell impermeable ligand, and firefly luciferin potassium salt were purchased from Promega. oxaliplatin, cisplatin, and carboplatin were purchased from Sigma-Aldrich and dissolved in dimethyl sulfoxide (DMSO) at 10 mM, 100 mM, and 25 mM concentrations, respectively.
Cell culture
TSA mammary carcinoma cells were propagated in DMEM GlutaMAXTM (Gibco) containing 4.5 g/L D-glucose and supplemented with 10% (v/v) fetal bovine serum (HyClone Laboratories). Prior to transfection or experimentation, TSA cells tested negative for Mycoplasma using a CELLshipper Mycoplasma Detection Kit (Bionique). All TSA derived clones were propagated and assayed in phenol-free DMEM supplemented with 10% (v/v) fetal bovine serum (HyClone Laboratories), 4.5 g/L D-glucose, L-glutamine, and 25 mM HEPES (Gibco). Additionally, all TSA derived clones were selected and maintained in 400 µg/mL G418 (Gibco). All cell lines were grown in a humidified incubator (ThermoScientific) at 37 °C and a 5% CO2 atmosphere.
Radiation treatment
Cells were exposed to ionizing radiation with a Varian 2300 C/D linear accelerator (Varian Medical Systems) using a bolus set to 1.5 cm tissue equivalent material at 6 MV energy and a 600 cGy/minute dosing rate.
Colony-formation assay
TSA cells were seeded into 6-well plates at 200 cells/well with DMEM GlutaMAXTM (Gibco) supplemented with 10% (v/v) fetal bovine serum (HyClone Laboratories). After complete cell adherence, TSA cells were exposed to ionizing radiation with or without 50 nM paclitaxel in replicates of 6. After 48 h, the medium was removed, fresh growth medium was added, and the cells were kept in culture undisturbed for 10–12 d, during which time the surviving cells spawned colonies. Colonies were fixed with 70% ethanol for 10 min and subsequently washed with PBS. Colonies with > 50 cells were visualized and counted after staining for 20 min with 0.1% crystal violet (Sigma-Aldrich) dissolved in distilled water.
Establishment of a TSA derived clone that stably expresses a plasma membrane targeted luciferase
For pericellular ATP detection, TSA cells were transfected using TurboFectin 8.0 (Origene) with a pGEN2.1 CMV expression vector (GenScript) containing an engineered firefly luciferase-folate receptor chimeric construct (refer to Fig. S3) in accordance to the manufacturer’s instructions (GenScript). Stable pGEN2.1-pMe-Luc clones were selected by means of G418. Pericellular ATP detection was determined by measuring photons using a Lumistar Galaxy luminometer (BMG LABTECH GmbH) from the enzymatic reaction of the plasma membrane luciferase in the presence of luciferin substrate solution comprising 3 mM D-luciferin (Sigma-Aldrich), 15 mM MgSO4, (Sigma-Aldrich), and 30 mM HEPES buffer (Gibco).
Establishment of a TSA derived clone that stably expresses a HaloTag-CRT-KDEL chimeric protein
For CRT analysis, Turbofectin 8.0 (Origene) was used to transfect TSA cells with a pEZ-M02 expression vector containing the Mus musculus CRT-HaloTag-KDEL fusion construct (refer to Fig. S1) following the manufacturer’s instructions (GeneCopoeia). Stable clones were selected by means of G418 selection (Gibco). To validate and detect CRT-HaloTag-KDEL endoplasmic reticulum localization, cloned cells were incubated overnight with 5 µM R110DirectTM ligand (Promega), diluted in phenol free medium, according to the manufacturers instructions. CRT-HaloTag-KDEL bound R110DirectTM ligand, a small molecule that crosses the cell membrane and labels total HaloTag® fusion protein, was visualized on an EVOS fluorescence microscope (Advanced Microscopy Group) at exλ470 nm/emλ525 nm. Positive clones were chosen for subsequent experiments. To visualize CRT-HaloTag-KDEL membranous localization, cloned cells were incubated with the 5 µM of the cell membrane impermeable HaloTag® Alexa Fluor 488 ligand (Promega) for 30 min and processed according to the manufacturers instructions. CRT-HaloTag-KDEL bound HaloTag® Alexa Fluor 488 ligand was visualized on an EVOS fluorescence microscope (Advanced Microscopy Group) at exλ470 nm/emλ525 nm.
Establishment of a TSA derived clone that stably expresses red fluorescence protein (RFP) tagged HMGB1
For HMGB1 analysis, TSA cells were transfected using TurboFectin 8.0 (Origene) with a PrecisionShuttle mammalian pCMV6-AC-RFP expression vector containing a Mus musculus HMGB1 construct (refer to Fig. S2) and a C-terminal RFP tag (Origene) following the manufacturers instructions. Stable clones were selected by means of G418 selection (Gibco). To validate and detect HMGB1-RFP nuclear localization, cloned cells were visualized on an EVOS fluorescence microscope (Advanced Microscopy Group) at exλ531 nm/emλ593 nm. Positive clones were chosen for subsequent experiments.
Luminescence-based detection of pericellular ATP via analysis of pMe-Luciferase expressing TSA cells
TSA cells stably transfected with pGEN2.1-pMe-Luc were seeded in 96-well plates, at a density of 2 x 104 cells per well in 50 µL of DMEM phenol free medium. The following day, cells were left either untreated or exposed to IR with or without drug, to a total of 100 µL of medium per well, and allowed to incubate for 24 h. ATP-dependent luminescence was detected on a LUMIstar Galaxy microplate luminometer (BMG LABTECH GmbH) upon the addition of 100 µL of luciferin substrate solution containing PBS dissolved D-luciferin, HEPES buffer (Gibco), and MgSO4 (Sigma-Aldrich), as described above. Luminescence was detected for 10s at a gain of 127 immediately after addition of luciferin substrate solution. The luminescence detected upon analysis of untreated cells was normalized to 1 for all experiments and the relative luminescence units for experimental groups are shown.
Cytofluorimetric detection of cell surface CRT via analysis of CRT-HaloTag-KDEL expressing TSA cells
TSA cells stably transfected with CRT-HaloTag-KDEL were seeded in 6-well plates at a density of 2.5 x 105 cells/well. The following day cells were treated with IR (or chemotherapeutic control agent), as described above and allowed to incubate for 24 h. CRT cell surface expression was detected with the impermeable Halotag® Alexa488 according to the manufacturer’s instructions. Per treatment group, 104 transfected cells were analyzed on the FACSCalibur (BD Biosciences) flow cytometer with CellQuest software (Becton Dickinson).
Detection of extracellular HMGB1 from stably expressing HMGB1-RFP TSA cells
Stably transfected HMGB1-RFP transfected TSA cells were seeded in replicates (n = 6) in 6-well plates, at a density of 1 x 105 cells per well. The following day, cells were IR treated and allowed to incubate for the indicated length of times (24, 48, or 72 h). Conditioned media was collected, floating cells were removed via centrifugation, and released HMGB1-RFP was detected on the Flexstation 3 Spectrophotometer (Molecular Devices) at excitation λ530 nM and emission λ593 nM. Fluorescence signal detected in the medium of untreated cells was normalized to 1 for all experiments and relative fluorescence units for experimental groups are shown.
Statistical analysis
The means ± the standard deviation (S.D.) were calculated and presented for each data point. Statistical analyses were performed using a paired Student’s t test. For all experiments, P values < 0.05 were considered statistically significant.
| Abbreviations: | ||
| CRT | = | calreticulin |
| CTLA4 | = | cytotoxic T lymphocyte associated protein 4 |
| DC | = | dendritic cell |
| ECM | = | extracellular matrix |
| GPI | = | glycophosphatidylinositol |
| HMGB1 | = | high mobility group box 1 |
| ICD | = | immunogenic cell death |
| IR | = | ionizing radiation |
| MTT | = | methylthiazolyldiphenyl-tetrazolium bromide |
| RT | = | radiation therapy |
| TLR-4 | = | toll-like receptor 4 |
Additional material
Download Zip (80.4 KB)Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
E.B.G. is supported by the Chemotherapy Foundation Competitive Research Fellowship made possible by the Joyce and Irving Goldman Family Foundation. S.CF., S.D., and M.H.B-H. were supported by the Department of Defense Breast Cancer Research Program BC100481. S.C.F and S.D. are supported by Breast Cancer Research Foundation 13-A0-00-001870-01. S.C.F. is supported by NCI RO1CA161891-01.
Citation: Golden EB, Francis D, Pellicciotta I, Demaria S, Helen Barcellos-Hoff M, Formenti SC. Radiation fosters dose-dependent and chemotherapy-induced immunogenic cell death. OncoImmunology 2014; 3:e28518; 10.4161/onci.28518
Related Research Data
References
- Kroemer G, Galluzzi L, Kepp O, Zitvogel L. Immunogenic cell death in cancer therapy. Annu Rev Immunol 2013; 31:51 - 72; http://dx.doi.org/10.1146/annurev-immunol-032712-100008; PMID: 23157435
- Kroemer G, Zitvogel L. Abscopal but desirable: The contribution of immune responses to the efficacy of radiotherapy. Oncoimmunology 2012; 1:407 - 8; http://dx.doi.org/10.4161/onci.20074; PMID: 22754758
- Golden EB, Pellicciotta I, Demaria S, Barcellos-Hoff MH, Formenti SC. The convergence of radiation and immunogenic cell death signaling pathways. Front Oncol 2012; 2:88; http://dx.doi.org/10.3389/fonc.2012.00088; PMID: 22891162
- Formenti SC, Demaria S. Systemic effects of local radiotherapy. Lancet Oncol 2009; 10:718 - 26; http://dx.doi.org/10.1016/S1470-2045(09)70082-8; PMID: 19573801
- Formenti SC, Demaria S. Combining radiotherapy and cancer immunotherapy: a paradigm shift. J Natl Cancer Inst 2013; 105:256 - 65; http://dx.doi.org/10.1093/jnci/djs629; PMID: 23291374
- Golden EB, Demaria S, Schiff PB, Chachoua A, Formenti SC. An Abscopal Response to Radiation and Ipilimumab in a Patient with Metastatic Non-Small Cell Lung Cancer. Cancer Immunol Res 2013; 1:365 - 72; http://dx.doi.org/10.1158/2326-6066.CIR-13-0115; PMID: 24563870
- Ma Y, Kepp O, Ghiringhelli F, Apetoh L, Aymeric L, Locher C, Tesniere A, Martins I, Ly A, Haynes NM, et al. Chemotherapy and radiotherapy: cryptic anticancer vaccines. Semin Immunol 2010; 22:113 - 24; http://dx.doi.org/10.1016/j.smim.2010.03.001; PMID: 20403709
- Kroemer G, Adjemian S, Michaud M, Martins I, Kepp O, Sukkurwala AQ, Menger L, Vacchelli E, Ma Y, Zitvogel L. Contributions of immunogenic cell death to the efficacy of anticancer chemotherapy. Bull Mem Acad R Med Belg 2011; 166:130 - 8, discussion 139-40; PMID: 22375493
- Ma Y, Conforti R, Aymeric L, Locher C, Kepp O, Kroemer G, Zitvogel L. How to improve the immunogenicity of chemotherapy and radiotherapy. Cancer Metastasis Rev 2011; 30:71 - 82; http://dx.doi.org/10.1007/s10555-011-9283-2; PMID: 21298323
- Locher C, Conforti R, Aymeric L, Ma Y, Yamazaki T, Rusakiewicz S, Tesnière A, Ghiringhelli F, Apetoh L, Morel Y, et al. Desirable cell death during anticancer chemotherapy. Ann N Y Acad Sci 2010; 1209:99 - 108; http://dx.doi.org/10.1111/j.1749-6632.2010.05763.x; PMID: 20958322
- Tesniere A, Schlemmer F, Boige V, Kepp O, Martins I, Ghiringhelli F, Aymeric L, Michaud M, Apetoh L, Barault L, et al. Immunogenic death of colon cancer cells treated with oxaliplatin. Oncogene 2010; 29:482 - 91; http://dx.doi.org/10.1038/onc.2009.356; PMID: 19881547
- Obeid M, Panaretakis T, Joza N, Tufi R, Tesniere A, van Endert P, Zitvogel L, Kroemer G. Calreticulin exposure is required for the immunogenicity of gamma-irradiation and UVC light-induced apoptosis. Cell Death Differ 2007; 14:1848 - 50; http://dx.doi.org/10.1038/sj.cdd.4402201; PMID: 17657249
- Dewan MZ, Galloway AE, Kawashima N, Dewyngaert JK, Babb JS, Formenti SC, Demaria S. Fractionated but not single-dose radiotherapy induces an immune-mediated abscopal effect when combined with anti-CTLA-4 antibody. Clin Cancer Res 2009; 15:5379 - 88; http://dx.doi.org/10.1158/1078-0432.CCR-09-0265; PMID: 19706802
- Demaria S, Ng B, Devitt ML, Babb JS, Kawashima N, Liebes L, Formenti SC. Ionizing radiation inhibition of distant untreated tumors (abscopal effect) is immune mediated. Int J Radiat Oncol Biol Phys 2004; 58:862 - 70; http://dx.doi.org/10.1016/j.ijrobp.2003.09.012; PMID: 14967443
- Pellegatti P, Falzoni S, Pinton P, Rizzuto R, Di Virgilio F. A novel recombinant plasma membrane-targeted luciferase reveals a new pathway for ATP secretion. Mol Biol Cell 2005; 16:3659 - 65; http://dx.doi.org/10.1091/mbc.E05-03-0222; PMID: 15944221
- Pellegatti P, Raffaghello L, Bianchi G, Piccardi F, Pistoia V, Di Virgilio F. Increased level of extracellular ATP at tumor sites: in vivo imaging with plasma membrane luciferase. PLoS One 2008; 3:e2599; http://dx.doi.org/10.1371/journal.pone.0002599; PMID: 18612415
- Michaud M, Martins I, Sukkurwala AQ, Adjemian S, Ma Y, Pellegatti P, Shen S, Kepp O, Scoazec M, Mignot G, et al. Autophagy-dependent anticancer immune responses induced by chemotherapeutic agents in mice. Science 2011; 334:1573 - 7; http://dx.doi.org/10.1126/science.1208347; PMID: 22174255
- Michalak M, Mariani P, Opas M. Calreticulin, a multifunctional Ca2+ binding chaperone of the endoplasmic reticulum. Biochem Cell Biol 1998; 76:779 - 85; http://dx.doi.org/10.1139/bcb-76-5-779; PMID: 10353711
- Tesniere A, Panaretakis T, Kepp O, Apetoh L, Ghiringhelli F, Zitvogel L, Kroemer G. Molecular characteristics of immunogenic cancer cell death. Cell Death Differ 2008; 15:3 - 12; http://dx.doi.org/10.1038/sj.cdd.4402269; PMID: 18007663
- Chaput N, De Botton S, Obeid M, Apetoh L, Ghiringhelli F, Panaretakis T, Flament C, Zitvogel L, Kroemer G. Molecular determinants of immunogenic cell death: surface exposure of calreticulin makes the difference. J Mol Med (Berl) 2007; 85:1069 - 76; http://dx.doi.org/10.1007/s00109-007-0214-1; PMID: 17891368
- Obeid M, Tesniere A, Ghiringhelli F, Fimia GM, Apetoh L, Perfettini JL, Castedo M, Mignot G, Panaretakis T, Casares N, et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat Med 2007; 13:54 - 61; http://dx.doi.org/10.1038/nm1523; PMID: 17187072
- Zitvogel L, Kepp O, Senovilla L, Menger L, Chaput N, Kroemer G. Immunogenic tumor cell death for optimal anticancer therapy: the calreticulin exposure pathway. Clin Cancer Res 2010; 16:3100 - 4; http://dx.doi.org/10.1158/1078-0432.CCR-09-2891; PMID: 20421432
- Los GV, Wood K. The HaloTag: a novel technology for cell imaging and protein analysis. Methods Mol Biol 2007; 356:195 - 208; PMID: 16988404
- Martins I, Kepp O, Schlemmer F, Adjemian S, Tailler M, Shen S, Michaud M, Menger L, Gdoura A, Tajeddine N, et al. Restoration of the immunogenicity of cisplatin-induced cancer cell death by endoplasmic reticulum stress. Oncogene 2011; 30:1147 - 58; http://dx.doi.org/10.1038/onc.2010.500; PMID: 21151176
- Apetoh L, Ghiringhelli F, Tesniere A, Criollo A, Ortiz C, Lidereau R, Mariette C, Chaput N, Mira JP, Delaloge S, et al. The interaction between HMGB1 and TLR4 dictates the outcome of anticancer chemotherapy and radiotherapy. Immunol Rev 2007; 220:47 - 59; http://dx.doi.org/10.1111/j.1600-065X.2007.00573.x; PMID: 17979839
- Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods 1983; 65:55 - 63; http://dx.doi.org/10.1016/0022-1759(83)90303-4; PMID: 6606682
- Alley MC, Scudiero DA, Monks A, Hursey ML, Czerwinski MJ, Fine DL, Abbott BJ, Mayo JG, Shoemaker RH, Boyd MR. Feasibility of drug screening with panels of human tumor cell lines using a microculture tetrazolium assay. Cancer Res 1988; 48:589 - 601; PMID: 3335022
- Henriksson E, Kjellén E, Wahlberg P, Wennerberg J, Kjellström JH. Differences in estimates of cisplatin-induced cell kill in vitro between colorimetric and cell count/colony assays. In Vitro Cell Dev Biol Anim 2006; 42:320 - 3; PMID: 17316066
- Puck TT, Marcus PI. Action of x-rays on mammalian cells. J Exp Med 1956; 103:653 - 66; http://dx.doi.org/10.1084/jem.103.5.653; PMID: 13319584
- Martins I, Tesniere A, Kepp O, Michaud M, Schlemmer F, Senovilla L, Séror C, Métivier D, Perfettini JL, Zitvogel L, et al. Chemotherapy induces ATP release from tumor cells. Cell Cycle 2009; 8:3723 - 8; http://dx.doi.org/10.4161/cc.8.22.10026; PMID: 19855167
- Barcellos-Hoff MH, Cordes N. Resistance to radio- and chemotherapy and the tumour microenvironment. Int J Radiat Biol 2009; 85:920 - 2; http://dx.doi.org/10.3109/09553000903274043; PMID: 19895267
- Russell JS, Brown JM. The irradiated tumor microenvironment: role of tumor-associated macrophages in vascular recovery. Front Physiol 2013; 4:157; http://dx.doi.org/10.3389/fphys.2013.00157; PMID: 23882218
- Postow MA, Callahan MK, Barker CA, Yamada Y, Yuan J, Kitano S, Mu Z, Rasalan T, Adamow M, Ritter E, et al. Immunologic correlates of the abscopal effect in a patient with melanoma. N Engl J Med 2012; 366:925 - 31; http://dx.doi.org/10.1056/NEJMoa1112824; PMID: 22397654
- Sistigu A, Viaud S, Chaput N, Bracci L, Proietti E, Zitvogel L. Immunomodulatory effects of cyclophosphamide and implementations for vaccine design. Semin Immunopathol 2011; 33:369 - 83; http://dx.doi.org/10.1007/s00281-011-0245-0; PMID: 21611872
- Menger L, Vacchelli E, Adjemian S, Martins I, Ma Y, Shen S, Yamazaki T, Sukkurwala AQ, Michaud M, Mignot G, et al. Cardiac glycosides exert anticancer effects by inducing immunogenic cell death. Sci Transl Med 2012; 4:43ra99; http://dx.doi.org/10.1126/scitranslmed.3003807; PMID: 22814852
- Formenti SC, Demaria S. Effects of chemoradiation on tumor-host interactions: the immunologic side. J Clin Oncol 2008; 26:1562 - 3, author reply 1563; http://dx.doi.org/10.1200/JCO.2007.15.5499; PMID: 18349411
- Formenti SC, Volm M, Skinner KA, Spicer D, Cohen D, Perez E, Bettini AC, Groshen S, Gee C, Florentine B, et al. Preoperative twice-weekly paclitaxel with concurrent radiation therapy followed by surgery and postoperative doxorubicin-based chemotherapy in locally advanced breast cancer: a phase I/II trial. J Clin Oncol 2003; 21:864 - 70; http://dx.doi.org/10.1200/JCO.2003.06.132; PMID: 12610186
- Golden EB, Formenti SC; 2013 July 29. Assays and methods for assessing immunogenic cell death, efficacy of anti-cancer therapy and identifying potentially efficacious therapies. United States provisional patent Serial No. 61/859,351