
Abstract
Recent evidence suggests that host immune cells contribute to the development of malignant pleural effusion (MPE), a common manifestation of metastatic cancer. We have identified such cells, predominantly mononuclear myeloid cells, recruited by tumor-orchestrated inflammatory chemokines. Moreover, targeting of these inflammation-associated mediators modified the disease course of MPE in mice.
Malignant pleural effusion (MPE) is a lethal clinical condition that affects a large portion of patients with different cancer types, predominantly those suffering from lung and breast adenocarcinomas.Citation1 The appearance of MPE in a cancer patient defines Stage IV disease, distinguished from early-stage disease by surgical incurability, decreased survival, and poor quality of life.Citation2 Current treatments are invasive, cause adverse effects, and do not combat the etiology of the condition.Citation3 In this setting, devising novel therapeutic modalities for patients with MPE is a task of high priority, which prerequisites understanding its biologic basis.
To this end, it is long known that pleural metastatic tumors block the lymphatic evacuation tracts that normally serve to drain the constantly produced pleural fluid, thereby causing accumulation of pleural fluid in the pleural space.Citation4 However, newer evidence from immune-intact mouse models and observations from humans, indicate that lymphatic obstruction is not enough and that enhanced pleural fluid production is absolutely required for MPE.Citation5 It has also become clear that pleural tumor cells initiate a complex inflammatory crosstalk with resident cells (pleural macrophages and lymphocytes, pleural mesothelial cells, and endothelial cells of juxtapleural capillaries) as well as with bone-marrow- and lymphatic system-accrued cells (mononuclear cells, neutrophils, lymphocytes) in the pleural microenvironment, which culminates in sharp elevations of inflammatory and vasoactive mediator levels in the pleural space. This ‘Molotov’ cocktail of tumor- and host-originated inflammatory signals (representative examples are tumor-derived tumor necrosis factor and host-cell-originated interleukin-5) renders pleural blood vessels leaky to plasma proteins and directly leads to MPE formation.Citation5-Citation7
We had described early on that in novel mouse models of MPE developed by our group, as well as in human MPE, monocytes/macrophages are a predominant cell population. In addition, we had identified that most of these cells in MPE are newly recruited from the bone marrow and the bloodstream and expressed CD45, CD11b, and Gr1, among other myeloid cell markers.Citation6-Citation8 In 2008, we discovered why and how these monocytes were trafficked to the pleural space: tumor cells secreted high levels of C-C-motif chemokine ligand 2 (CCL2) into the pleural space generating chemotactic gradients for myeloid cells along the whole systemic circulation.Citation9 Tumor CCL2 expression was required and sufficient for MPE precipitation, and exerted multiple pathogenesis-promoting effects on MPE, including myeloid cell recruitment, induction of vascular leakiness, and endothelial proliferation, among others.Citation9 However, we had no clinically-relevant means to antagonize tumor-derived CCL2 at that time and the clinical relevance of our findings was uncertain.
This changed when Centocor (currently with Janssen R&D, Oncology Discovery Research, Spring House, PA) introduced monoclonal antibodies directed against murine CCL2 and its ortholog CCL12 into preclinical trials. These agents allowed us to continue our experimental endeavors with CCL2 using established and newly developed mouse models of MPE triggered by mouse and human tumor cells, to find that anti-CCL2 treatment had identical effects with genetic ablation of CCL2 expression.Citation10 For this, we innoculated syngeneic lung and colon adenocarcinoma cells into the pleural space of C57BL/6 mice and started intraperitoneal treatment with the neutralizing antibodies. In both models, chemokine blockade limited pleural fluid accumulation by suppressing tumor effects on the host vasculature and immune system. Intriguingly, CCL2 and CCL12 showed redundancy and blockade of either or both had identical inhibitory effects on mononuclear cell accrual and MPE development. It is worth noting that to definitively prove this, we developed an innovative experimental setup for monitoring the process of inflammatory cell recruitment in the pleural space. Specifically, we generated chimeric mice transplanted with luminescent bone marrow cells, a model suitable for real-time monitoring of cancer-related inflammation by bioluminescent imaging. In addition to integral determinations of MPE-related inflammation, we also attempted to analyze the cell types that composed the inflammatory infiltrates of our mice. Interestingly, although CCL2 and CCL12 are mononuclear cell chemoattractants, we observed an overall decrease of different immune cell types, indicating that these chemokines orchestrate the recruitment of additional immune cell types (i.e., neutrophils and others). We have corroborated this since the publication of our above-referenced article, and have identified additional bone marrow cells that respond to CCL2 gradients with pleural-directed migration.
As an attempt to translate our work closer to the human setting, we developed a new animal model of MPE by delivering intrapleural human A549 lung adenocarcinoma cells to immunocompromized mice and proceeded with intraperitoneal injections of mouse CCL2 neutralizing antibody. This setup, would allow us to study the contributions of host-originated CCL2, since in this model, tumor-derived CCL2 is human and cannot be neutralized by our antibody. Strikingly, CCL2 blockade showed significant efficacy, lending hope that this approach could also benefit human cancer patients with MPE. In addition, these experiments showed that host-cell-originated CCL2 also contributes to MPE pathobiology. The host cell type(s) that elaborate the chemokine remain to be determined, but macrophages and mesothelial cells are likely candidates.
Many questions regarding CCL2 functions in cancer remain, and are the objects of ongoing work in our and other laboratories. First of all, why do some tumors produce high levels of the chemokine while others do not? Is the cancer genome or the cellular origin of tumors to blame? The literature and recent unpublished results from our laboratory suggest that both factors are important: tumor cells need to possess an appropriate tissue-specific epigenetic gene expression profile as well as harbor defined mutations in order to switch to the CCL2-expressing phenotype. Second, are there other mononuclear chemokines that supplement CCL2 functions, and do these relate to the recruitment of other inflammatory cell populations (e.g., neutrophils) to human MPE? Identification of the most important players in this game would allow careful tailoring of targeted therapies to effectively block multiple inflammatory axes in MPE. Third and most important: why are CCL2 effects specific to MPE and only marginal with solid tumors? What aspects of the malignant phenotype are especially key to MPE development and are also especially promoted by tumor-originated CCL2? There is a pressing need to answer these, and other questions to allow the translation of the findings discussed herein (summarized in ) to clinical trials of anti-CCL2 monoclonal antibodies in patients with metastatic MPE.
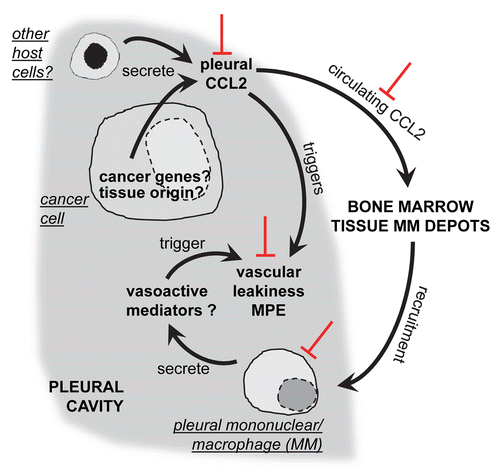
Figure 1. Role of C-C-motif chemokine ligand 2 (CCL2) in malignant pleural effusion. Tumor cells, as well as other host cells, secrete the C-C-motif chemokine ligand 2 (CCL2) into the pleural cavity, contributing to elevated intrapleural levels of the chemokine relative to the bloodstream. Local CCL2 levels directly cause vascular leakiness, possibly facilitating the influx of inflammatory cells. Overflow CCL2 circulates generating systemic chemotactic gradients that function to recruit mononuclear myeloid cells from the bone marrow (or other tissue depots) to the pleural space. Pleural mononuclear/macrophage (MM) cells, in turn, release proinflammatory and vasoactive mediators to further promote MPE development. Red inhibitory symbols indicate the effects of anti-CCL2 antibody therapy. Question marks indicate areas of future research.
| Abbreviations: | ||
| BRAF | = | v-raf murine sarcoma viral oncogene homolog B |
| CCL | = | C-C-motif chemokine ligand |
| EGFR | = | epidermal growth factor receptor |
| KRAS | = | Kirsten rat sarcoma viral oncogene homolog |
| MPE | = | malignant pleural effusion |
| NF | = | nuclear factor |
| TNF | = | tumor necrosis factor |
| VEGF | = | vascular endothelial growth factor |
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by the European Research Council and the European Respiratory Society.
References
- Ryu JS, Ryu HJ, Lee SN, Memon A, Lee SK, Nam HS, Kim HJ, Lee KH, Cho JH, Hwang SS. Prognostic impact of minimal pleural effusion in non-small-cell lung cancer. J Clin Oncol 2014; 32:960 - 7; http://dx.doi.org/10.1200/JCO.2013.50.5453; PMID: 24550423
- Postmus PE, Brambilla E, Chansky K, Crowley J, Goldstraw P, Patz EF Jr., Yokomise H, International Association for the Study of Lung Cancer International Staging Committee, Cancer Research and Biostatistics, Observers to the Committee, Participating Institutions. The IASLC Lung Cancer Staging Project: proposals for revision of the M descriptors in the forthcoming (seventh) edition of the TNM classification of lung cancer. J Thorac Oncol 2007; 2:686 - 93; http://dx.doi.org/10.1097/JTO.0b013e31811f4703; PMID: 17762334
- Maskell NA. Treatment options for malignant pleural effusions: patient preference does matter. JAMA 2012; 307:2432 - 3; http://dx.doi.org/10.1001/jama.2012.5543; PMID: 22609878
- Marazioti A, Blackwell TS, Stathopoulos GT. The lymphatic system in malignant pleural effusion. Drain or immune switch?. Am J Respir Crit Care Med 2014; 189:626 - 7; http://dx.doi.org/10.1164/rccm.201401-0140ED; PMID: 24628311
- Stathopoulos GT, Kalomenidis I. Malignant pleural effusion: tumor-host interactions unleashed. Am J Respir Crit Care Med 2012; 186:487 - 92; http://dx.doi.org/10.1164/rccm.201203-0465PP; PMID: 22652027
- Stathopoulos GT, Kollintza A, Moschos C, Psallidas I, Sherrill TP, Pitsinos EN, Vassiliou S, Karatza M, Papiris SA, Graf D, et al. Tumor necrosis factor-alpha promotes malignant pleural effusion. Cancer Res 2007; 67:9825 - 34; http://dx.doi.org/10.1158/0008-5472.CAN-07-1064; PMID: 17942913
- Stathopoulos GT, Sherrill TP, Karabela SP, Goleniewska K, Kalomenidis I, Roussos C, Fingleton B, Yull FE, Peebles RS Jr., Blackwell TS. Host-derived interleukin-5 promotes adenocarcinoma-induced malignant pleural effusion. Am J Respir Crit Care Med 2010; 182:1273 - 81; http://dx.doi.org/10.1164/rccm.201001-0001OC; PMID: 20595227
- Stathopoulos GT, Zhu Z, Everhart MB, Kalomenidis I, Lawson WE, Bilaceroglu S, Peterson TE, Mitchell D, Yull FE, Light RW, et al. Nuclear factor-kappaB affects tumor progression in a mouse model of malignant pleural effusion. Am J Respir Cell Mol Biol 2006; 34:142 - 50; http://dx.doi.org/10.1165/rcmb.2005-0130OC; PMID: 16210694
- Stathopoulos GT, Psallidas I, Moustaki A, Moschos C, Kollintza A, Karabela S, Porfyridis I, Vassiliou S, Karatza M, Zhou Z, et al. A central role for tumor-derived monocyte chemoattractant protein-1 in malignant pleural effusion. J Natl Cancer Inst 2008; 100:1464 - 76; http://dx.doi.org/10.1093/jnci/djn325; PMID: 18840818
- Marazioti A, Kairi CA, Spella M, Giannou AD, Magkouta S, Giopanou I, Papaleonidopoulos V, Kalomenidis I, Snyder LA, Kardamakis D, et al. Beneficial impact of CCL2 and CCL12 neutralization on experimental malignant pleural effusion. PLoS One 2013; 8:e71207; http://dx.doi.org/10.1371/journal.pone.0071207; PMID: 23967166