Abstract
Neurons have highly developed Ca2+ signaling systems responsible for regulating a large number of neural functions such as the control of brain rhythms, information processing and the changes in synaptic plasticity that underpin learning and memory. The tonic excitatory drive, which is activated by the ascending arousal system, is particularly important for processes such as sensory perception, cognition and consciousness. The Ca2+ signaling pathway is a key component of this arousal system that regulates the neuronal excitability responsible for controlling the neural brain rhythms required for information processing and cognition. Dysregulation of the Ca2+ signaling pathway responsible for many of these neuronal processes has been implicated in the development of some of the major neural diseases in man such as Alzheimer disease, bipolar disorder and schizophrenia. Various treatments, which are known to act by reducing the activity of Ca2+ signaling, have proved successful in alleviating the symptoms of some of these neural diseases.
Introduction
Calcium (Ca2+) signaling plays a central role in regulating multiple neuronal processes such as transmitter release from presynaptic endings and multiple postsynaptic processes including the regulation of neuronal excitability and the changes in synaptic plasticity responsible for learning and memory. Many of the major neural disease in man are caused by alterations in these various Ca2+-sensitive processes. The regulation of excitability might be of particular importance as it is responsible for controlling the neural brain rhythms responsible for information processing and cognition. It will be argued that alterations in the mechanisms responsible for this Ca2+-dependent tonic excitatory drive that regulates neuronal excitability might play an important role in the development of diseases such as Alzheimer disease, bipolar disorder and schizophrenia.
Tonic Excitatory Drive
The brain can switch rapidly between sleep and consciousness by the activation of an ascending arousal system that consists of groups of neurons that project their axons throughout the brain where transmitters such as orexin, acetylcholine (ACh), norepinephrine (NE), 5-hydroxytryptamine (5-HT), histamine and dopamine (DA) are released on to the excitatory and inhibitory neurons that constitute the functional neural circuits. The cholinergic system, which projects diffusely throughout the cortex and hippocampus, controls processes such as sensory perception, cognition and consciousness by activating this tonic excitatory drive.Citation1 ACh functions as an arousal-promoting neuromodulator that can switch the brain from a sleep to an awake mode of firing that is characterized by the appearance of gamma rhythms. Such arousal transmitters act on receptors that are coupled to various signaling pathways () that induce the tonic excitatory drive that enables these neurons to process information during the wake state. For example, ACh acts through M1 receptors that are coupled to the Ins(1,4,5)P3/Ca2+ signaling pathway, which functions to regulate the excitability of both hippocampal and dentate gyrus neurons.Citation2,Citation3
Figure 1. Tonic excitatory drive and control of neuronal rhythms. The ascending arousal system releases transmitters such as orexin, acetylcholine (ACh), 5-hydroxytryptamine (5-HT), norepinephrine (NE) and dopamine (DA) that activate signaling systems that control neural rhythms that occur during the sleep/wake cycle. The tonic excitatory drive mechanisms depend on membrane depolarization that results from closing the KV7.2/KV7.3 channels responsible for the M current, the opening of the Ca2+-activated non-specific cation channel (ICAN) and the hyperpolarizing-activated cyclic nucleotide-gated (HCN) channel responsible for the Ih current.
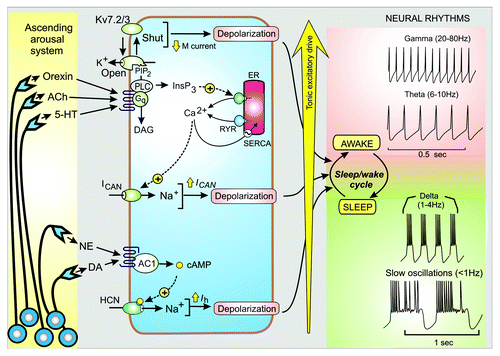
Variations in the activity of this arousal system, which is translated into variations in the tonic excitatory drive, determines the hierarchy of the neural rhythms with the lowest frequencies occurring during sleep (i.e the slow waves and delta rhythms) that then switch to the higher frequency theta and gamma rhythms of the wake state (). In effect, the variable tonic excitatory drive induced by the ascending arousal system functions as a rhythm rheostat capable of generating the panoply of neural rhythms that occur during the course of the sleep/wake cycle. Alterations in the frequency of the gamma rhythms have been implicated in both schizophrenia and Alzheimer disease.Citation4-Citation6 Thus, there is an imperative to understand the nature of the tonic excitatory drive and how it might be modified in neural diseases.
A variety of signaling mechanisms are used to control the ion channels that provide the tonic excitatory drive that depolarizes the target neurons to regulate their oscillatory outputs (). Transmitters such as orexin, ACh and 5-HT act on receptors that are coupled to the phosphoinositide signaling pathway, which has multiple outputs. A particularly important output depends on the ability of phosphatidylinositol 4,5-bisphosphate (PtdIns(4,5)P2) to regulate Kv7.2 and Kv7.3 ion channels responsible for the M current.Citation7 Under resting conditions, PtdIns(4,5)P2 accumulates and keeps the channels open, but when this lipid is hydrolysed by the transmitters released by the ascending arousal system, the channel closes. Switching off this M current depolarizes the membrane to increase neuronal activity.
There are other K+ channels that can modulate the characteristics of neuronal rhythms. In particular, the Ca2+-sensitive K+ channels, such as the large conductance (BK) channels and the small conductance (SK) channels, have an important role in determining rhythm frequency by regulating the interval between the spikes. The contribution of these two channels depends on the nature of the activating Ca2+ signal. The BK channels are often closely associated with the voltage-operated Ca2+ channels (VOCs), which enables their low-affinity Ca2+-binding sites to respond to the high levels of Ca2+ found near the opening of these Ca2+ entry channels. In this way, the BK channels function primarily to facilitate the repolarization of the individual action potentials. By contrast, the SK channels, which have calmodulin (CaM) as a Ca2+ sensor, respond to more global elevations such as those that occur when Ca2+ is released from internal stores by Ins(1,4,5)P3 receptors or ryanodine receptors (RYRs). Such global elevations in Ca2+ may also act to enhance membrane depolarization by stimulating an inward Ca2+ current (ICAN).Citation2,Citation8
Norepinephrine and dopamine also contribute to the tonic excitatory drive by acting through the cyclic AMP signaling pathway to enhancing the activity of the hyperpolarizing-activated cyclic nucleotide-gated (HCN) channel responsible for the Ih current ().
Schizophrenia
Schizophrenia is a severe psychiatric condition characterized by both positive (hallucinations and paranoia) and negative symptoms (poor attention, decline in social interactions and lack of motivation).Citation9 Some of these symptoms may be linked to subtle changes in the brain rhythms, which are driven by the tonic excitatory drive and are responsible for processes such as perception, consciousness and memory. With regard to the latter, memory formation depends on pyramidal neurons in different regions of the cortex firing in a sustained and synchronous manner in the gamma frequency range of approximately 40 Hz. It is these rhythms that are known to be impaired in schizophrenia.Citation4,Citation5 The essential components of the network oscillator that generates this gamma rhythm are the inhibitory GABAergic interneurons and the excitatory pyramidal glutamatergic neurons.Citation10 As part of the oscillatory cycle, the glutamatergic neurons release glutamate to excite the inhibitory interneurons to release GABA, which then feeds back to inhibit the pyramidal neurons thereby setting up a positive and negative feedback loop to generate the gamma rhythms (). Many of the susceptibility genes and pharmacological inducers that have been linked to schizophrenia seem to be associated with defects in these fast spiking GABAergic inhibitory neurons that result in either a decrease of NMDA receptor (NMDAR) activity or a change in the tonic excitatory drive (). Such changes in the function of these inhibitory neurons interfere with their role in the network oscillator resulting in changes in the gamma and theta rhythms that are a feature of schizophrenia.
Figure 2. Phenotypic changes and compensatory responses of inhibitory GABAergic interneurons in schizophrenia. Many of the schizophrenia susceptibility genes and pharmacological inducers are associated with the fast spiking GABAergic inhibitory neurons where they act either to decrease the activity of the NMDA receptor (NMDAR) or they play a role in the tonic excitatory drive. All these changes in the activity of these GABAergic neurons interferes with their role in the network oscillator resulting in alterations of the gamma rhythms that are a feature of schizophrenia. Some of the changes are compensatory responses (green arrows) to the genetic modifications.
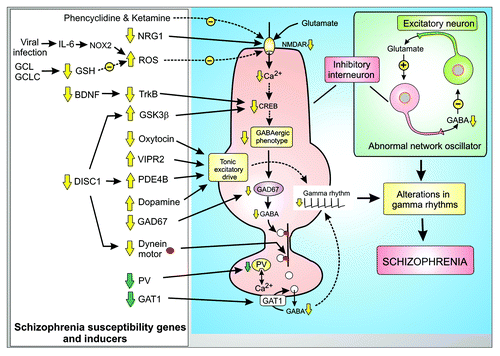
The network oscillator that generates gamma rhythms depends on the excitatory neurons releasing glutamate to activate the inhibitory GABAergic interneurons. The latter then respond to glutamate to release GABA that then inhibits the excitatory neurons (). A defect in the ability of the NMDARs on the inhibitory interneurons to respond to this glutamate is the basis of the glutamate hypothesis of schizophrenia.Citation11,Citation12 This hypothesis is supported by the fact that administering NMDAR antagonists such as ketamine and phencyclidine (PCP) can induce schizophrenic symptoms in healthy adults. One of the functions of the NMDARs is to generate the repetitive Ca2+ signals that stimulate the transcription factor CREB responsible for maintaining the phenotype of these GABAergic inhibitory interneurons. Hypofunction of the NMDARs results in a reduction in the expression of the glutamic acid decarboxylase 67 (GAD67), which is one of the key components of the phenotype in that it synthesizes the inhibitory transmitter GABACitation13 (). This GABA is packaged into vesicles that are transported to the presynaptic terminals where they are released as part of the network oscillatory mechanism that generates the gamma rhythms. A reduction in the level of GAD67 and the resulting decreased availability of GABA is one of the most characteristic features of schizophrenia. The transport of GABA containing vesicles down the axon to the presynaptic ending is mediated by the dynein motor, which travels down microtubules (). One of the genes mutated in schizophrenia is disrupted in schizophrenia 1 (DISC1), which functions as a dynein adaptor. The mutations in DISC1 will thus disrupt the transport of GABA vesicles.
Some of the phenotypic changes that occur in the interneurons seem to be compensatory responses caused by this primary defect in the expression of GAD67.Citation13 This may explain the decline in the level of the Ca2+ buffer parvalbumin (PV), which modulates the Ca2+-dependent release of GABA. Another compensatory mechanism depends on a decrease in the expression of the GABA membrane transporter 1 (GAT1), which is located in the presynaptic ending where it functions to return GABA back into the interneuron. A reduction in this removal mechanism will help to enhance the activity of the reduced amount of GABA being released in schizophrenia.
In summary, hypofunction of the NMDARs and the resulting reduction of Ca2+ signaling sets in train a complex series of transcriptional and compensatory events that remodel the GABAergic phenotype. A reduction in the release of the inhibitory neurotransmitter GABA may explain the gamma rhythm changes that occur in schizophrenia. Identifying the reason for the decrease in NMDA receptor function may help to understand the biochemical basis for schizophrenia. The working hypothesis summarized in attempts to integrate much of the genetic (e.g., neuregulin-1, DISC1 and VIPR2) and biochemical susceptibility factors, such as brain-derived neurotrophic factor (BDNF) and glutathione (GSH) into a unifying concept based on the Ca2+-dependent remodelling of the GABAergic phenotype and the changes in the tonic excitatory drive responsible for schizophrenia.
NMDAR hypofunction and redox signaling in schizophrenia
The fast spiking inhibitory interneurons in the cortex seem to be particularly sensitive to the redox state of the brain. Hypofunction of the NMDAR has been linked to a change in redox signaling, which is used normally as a mechanism to modulate the activity of this channel. The NMDAR channel, which consists of NR1 and NR2A subunits, is physically linked to neuronal nitric oxide synthase (nNOS) in the post-synaptic density through the scaffolding protein PSD95. The nNOS is thus ideally positioned to respond to the pulses of Ca2+ entering through the NMDA receptor to generate the NO that then interacts rapidly with O2−• to form peroxynitrite (ONOO-), which is very much more reactive than the two parent molecules. The ONOO- then nitrosylates the NR2A subunit at Cys-399 to inhibit channel open probability and thus reduces the ability of the NMDA receptor to gate Ca2+ as part of a negative feedback loop to guard against excessive Ca2+ entry. An abnormal increase in the oxidation state of these inhibitory neurons may be responsible for the onset of schizophrenia.Citation14,Citation15
Dysregulation of the redox signaling pathway may provide an explanation for the developmental origins of schizophrenia because there appears to be a link between maternal viral infection during gestation and the incidence of this disease in the offspring. In rat models that reproduce this phenomenon, the developmental defects induced by such inflammatory responses are caused by activation of redox signaling. During viral infections, there is an increase in interleukin-6 (IL-6), which has a prominent role in activating the redox signaling pathway.Citation14 IL-6 acts through the JAK/STAT signaling pathway to increase the expression of Nox2, which is one of the NADPH oxidases responsible for generating superoxide (O2−•) (). Both the superoxide (O2−•) and the hydrogen peroxide (H2O2), which is formed from O2−• by superoxide dismutase (SOD), react with nitric oxide (NO) to form ONOO- that then nitrosylates the NR2A subunit at Cys 399 to reduce the open probability of the NMDAR. This reduction in the ability of the NMDA receptor to generate a Ca2+ signal may have a direct bearing on the cognitive defects in schizophrenia. In addition, the reduction in Ca2+ signaling can explain the phenotypic remodelling of the GABAergic phenotype as described earlier.
The role of enhanced oxidation in driving these phenotypic alterations of the inhibitory interneurons is also consistent with studies on the changes in the antioxidant mechanisms that have been described in neurodegenerative disordersCitation16 including schizophrenia. The denitrosylation reaction, which functions to reverse the nitrosylation reaction that reduces the activity of the NMDAR, is performed by two main antioxidants: glutathione (GSH) and thioredoxin (TRX). In schizophrenia, gene polymorphisms have been described in the enzymes responsible for the synthesis of GSH such as the GCL catalytic subunit (GCLC) and a GCL modifier subunit (GCLM) that combine to form the glutamate cysteine ligase (GCL) responsible for one of the steps of GSH synthesis.Citation17 Such defects in GSH synthesis may explain the decreased GSH levels found in schizophrenia ().
NMDA receptor expression in schizophrenia
One of the prominent gene mutations, which have been linked to schizophrenia, are found in the neuregulin-1 gene that encodes NRG1 that acts through the ErbB3 signaling pathway to maintain the GABAergic phenotype.Citation9 It controls the expression of various NMDAR signaling functions such as NMDAR itself, the scaffolding protein PSD95 that links the NMDAR to nNOS and the nicotinic acetylcholine receptor (nAChR). ACh can facilitate the release of glutamate at presynaptic endings by acting through ionotropic nAChRs.Citation18 In patients with schizophrenia, the expression of nAChRs, which is regulated by this NRG1/ErbB pathway, is reduced and this could contribute to hypofunction of glutamatergic signaling in the interneurons. Mutations in NRG1 decrease its activity and may thus contribute to hypofunction of the NMDAR and a remodelling of the GABAergic phenotype. A reduction in activity of the NRG1/ ErbB3 signaling pathway also inhibits the Src kinase that is a regulator of the NMDAR.Citation19
Brain-derived neurotrophic factor (BDNF) and TrkB receptor dysfunction in schizophrenia
The expression of brain-derived neurotrophic factor (BDNF) and its receptor TrkB are reduced in schizophrenia and this may have a major impact on the function of the inhibitory interneurons particularly during early development. The signaling pathways induced by TrkB receptors control the transcription factor CREB, which is the same transcription factor that is activated by Ca2+ to maintain the GABAergic phenotype as described earlier (). The TrkB receptors also activate the PI 3-kinase signaling pathway, which regulates the activity of glycogen synthesis kinase 3β (GSK3β) that controls the transcription factor β-catenin. The activity of GSK3β is regulated by disrupted in schizophrenia (DISC1), which is one of the prominent genes mutated in schizophrenia.Citation20 The mutated form of DISC1 is incapable of inhibiting GSK3 β resulting in a more pronounced inhibition of transcription thus contributing to a decline of the GABAergic phenotype and inhibitory interneuron function.
Modulation of the tonic excitatory drive of GABAergic interneurons and schizophrenia
The neuronal oscillator that generates the gamma rhythm depends on the tonic excitatory drive (see above) that functions to reduce the membrane potential of the participating neurons to a level that enables them to oscillate at gamma frequencies (). This tonic drive is present in both the inhibitory GABAergic and excitatory glutamatergic neurons. In the case of the GABAergic interneurons, transmitters such as dopamine, vasoactive intestinal peptide (VIP) and oxytocin, which use cyclic AMP or Ins(1,4,5)P3/Ca2+ signaling pathways to regulate this tonic excitatory drive, have been implicated in schizophrenia ().
Schizophrenic-like symptoms occur in response to drugs such as the amphetamines that release dopamine, whereas drugs that inhibit the D2 receptor are antipsychotic. These pharmacological actions would thus predict that schizophrenic symptoms may arise through excessive activation of the D2 receptor resulting in a reduction in the level of cyclic AMP and a decrease in the tonic drive. This inhibition of cyclic AMP signaling alters the input resistance by reducing the activity of the HCN1 channels that provides an inward Na+ current (). Since the activity of HCN1 is regulated by cyclic AMP, it could also explain how alterations in the activity of the dopamine D2 and vasoactive intestinal peptide receptor 2 (VIPR2) receptors may contribute to schizophrenia. It is somewhat surprisingly to find that duplications of VIPR2, which confers a significant risk for schizophrenia,Citation21 has the opposite effect on cyclic AMP levels to those just described for dopamine. VIP acting on VIPR2 enhances the cyclic AMP signaling pathway to increase the tonic drive. It would seem that changes in the cyclic AMP signaling pathway can either enhance or reduce the tonic excitatory drive and this would have repercussions on the generation of the gamma rhythms resulting in schizophrenia. Such contradictory effects on the tonic excitatory drive can be reconciled by the fact that gamma rhythms may be increased or reduced in different schizophrenia syndromes.Citation22
An important role for cyclic AMP is evident from the fact that alterations in the activity of the phosphodiesterase 4B (PDE4B), which hydrolyses cyclic AMP (), have been identified in schizophrenia. SNPs associated with the gene that codes for PDE4B have been described in schizophrenia. In addition, the gene for DISC1, which is mutated in schizophrenia, has also been shown to interact with PDE4B to alter the metabolism of cyclic AMP. A decrease in oxytocin, which acts through the Ins(1,4,5)P3/Ca2+ signaling pathway and may thus play a role in regulating the tonic excitatory drive (), has also been implicated in schizophrenia and other mental disorders such as autism.Citation23
Bipolar Disorder
The underlying causes of bipolar disorder (BD), which is characterized by extreme mood swings between mania and depression, are still a mystery. Early attempts to understand BD focused on possible defects in neurotransmitters such as 5-HT, NE, dopamine and ACh. Organophosphates, which inhibit the acetylcholinesterase that hydrolyses ACh, cause depression. On the other hand, inhibition of muscarinic M1 receptors by scopolamine has the opposite effect by inducing symptoms of mania. Such M1 receptors stimulate phosphoinositide hydrolysis, which induces the Ins(1,4,5)P3/Ca2+ and DAG/protein kinase C signaling pathways (). Dopamine, which acts through the cyclic AMP signaling pathway, also has a marked effect on mood. Drugs such as haloperidol that decreases dopaminergic transmission are antimanic. It seems that low cyclic AMP levels promote mania whereas elevated levels induce depression. Since these transmitters have a major role in regulating the tonic excitatory drive (), it seems that changes in mood might arise from alterations in the downstream signaling pathways that modulate neural excitability. Alterations in these signaling pathways may thus play a role in BD.
Figure 3. Signaling pathways in bipolar disorder (BD). Control of mood seems to depend on a number of interacting signaling mechanisms. Acetylcholine (ACh) acting through muscarinic M1 receptors (M1R) stimulates the hydrolysis of phosphatidylinositol 4,5-bisphosphate (PtdIns(4,5)P2) to generate inositol 1,4,5-trisphospahate (Ins(1,4,5)P3) and diacylglycerol (DAG). The Ins(1,4,5)P3 releases Ca2+ whereas DAG activates protein kinase C (PKC). Both Ca2+ and PKC may influence mood by modulating membrane excitability. The DAG is inactivated following its phosphorylation by DAG kinase (DGKH), whereas Ins(1,4,5)P3 is recycled back to inositol through a sequential series of dephosphorylation reactions with the final step being performed by an inositol monophosphatase (IMPase), which is sensitive to lithium. The same IMPase hydrolyzes the InsP1 formed by the inositol synthase, which is inhibited by valproate. Other neurotransmitters such as dopamine and norepinephrine (NE) seem to act through adenylyl cyclase (AC) to form cyclic AMP, which acts through protein kinase A (PKA) to phosphorylate the transcription factor CREB to control expression of brain-derived neurotrophic factor (BDNF) and Bcl-2 that function to promote neurogenesis and to inhibit apoptosis, respectively. Bcl-2 may inhibit apoptosis by reducing the release of Ca2+ by the Ins(1,4,5)P3 receptor (Ins(1,4,5)P3R). The transcriptional activity of CREB is inhibited by glycogen synthase kinase 3β (GSK-3β), which is inhibited by lithium.
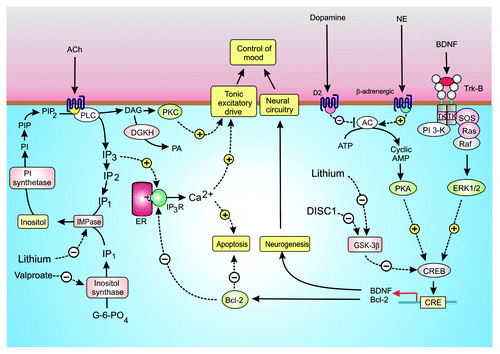
There is considerable evidence to link changes in mood to dysfunctional signaling mechanisms that control neurogenesis.Citation24 Neurogenesis is responsible for the differentiation and survival of new neurons necessary to maintain the neural circuitry in the adult brain.Citation25 Much interest is now focused on the idea that BD may be caused by a decline in the action of neurotrophins, such as BDNF, which regulate this process of neurogenesis.Citation25 Patients with depression have low serum levels of BDNF. Some of the most effective mood stabilizing drugs, such as lithium (Li+) and valproate, act by modulating downstream intracellular signaling pathways that regulate both neurogenesis and membrane excitability (). For example, Li+ is a potent inhibitor of enzymes such as GSK-3β and inositol monophosphatase (IMPase) that function in different but related signaling pathways that are related to each other through a number of feedback mechanisms.
GSK-3β inhibition and bipolar disorder
One of the primary intracellular targets of Li+ is GSK-3β, which regulates the activity of transcription factors (CREB and β-catenin) responsible for the expression of signaling components such as BDNF and Bcl-2, which control neurogenesis and survival respectively. Li+ promotes neurogenesis by inhibiting GSK-3β, which is a potent inhibitor of the transcription factor CREB.Citation20 This action of Li+ suggests that one of the causes of BD may be an overactive GSK-3β that suppresses the expression of both BDNF and Bcl-2. Such an interpretation is consistent with the fact that mutations in DISC1, which reduce its inhibitory effect on GSK-3β and is one of the prominent genes mutated in both schizophrenia and BD, results in a decrease in neurogenesis.Citation26
Single nucleotide polymorphisms (SNPs) of the Bcl-2 gene, which increase the risk of developing BD, are associated with elevated basal Ca2+ levels and enhanced Ins(1,4,5)P3-mediated cytosolic Ca2+ release.Citation27 Both Li+ and valproate can markedly enhance the level of Bcl-2.Citation28 These observations are of particular importance because one of the actions of Bcl-2 is to reversibly inhibit the ability of Ins(1,4,5)P3 to open the Ins(1,4,5)P3R channel to release Ca2+ from the ER.Citation29 The low level of Bcl-2 will enhance apoptosis and will also reduce its inhibitory effect on Ins(1,4,5)P3-induced Ca2+ release that may explain the increase in both resting and activated levels of Ca2+ that are a characteristic feature of BD.Citation30
Inositol depletion hypothesis of bipolar disorder
Another important Li+ target is the IMPase that hydrolyses IP1 to free inositol. By chocking off the supply of inositol, Li+ reduces the resynthesize of the PI necessary to provide the PtdIns(4,5)P2 required for the Ins(1,4,5)P3/Ca2+ signaling pathway ().Citation31 Valproate, which is another potent mood stabilizing drug, has a similar action in that it inhibits the inositol synthase responsible for the de novo synthesis of inositol from glucose 6-phosphate. Both Li+ and valproate act through inositol depletion to reduce the membrane phosphoinositides resulting in an increase in synapse formation between cultured hippocampal neurons.Citation32 The central feature of the inositol depletion hypothesis is that Li+ and valproate act to inhibit the supply of inositol required to maintain the inositol lipid signaling pathway. If this pathway is a target for Li+, it suggests that BD may be caused by hyperactivity of phosphoinositide signaling with enhanced activity of the Ins(1,4,5)P3/Ca2+ and DAG/protein kinase C pathways. This is consistent with finding that SNPs in the diacylglycerol kinase (DGKH) gene that metabolizes DAG () is a significant risk factor for BD.Citation33 An increase in the activity of the DAG/protein kinase C pathway may influence mood through its established action on membrane excitability. With regard to the Ins(1,4,5)P3/Ca2+ pathway, there are a number of observations indicating that the resting and activated levels of Ca2+ are elevated in BD.Citation30 The neuronal Ca2+ sensor-1 (NCS-1), which is known to be elevated in the prefrontal cortex in both BD and schizophrenia,Citation34 is known to enhance the activity of the Ins(1,4,5)P3RsCitation35 and this would contribute to an increase in the intracellular level of Ca2+. Increase NCS-1 activity may also influence membrane excitability by desensitizing the D2 receptors.Citation36 Such elevations of Ca2+ can activate apoptosis, which will contribute to a decrease in neurogenesis. This inhibitory effect on neurogenesis may be facilitated by Ca2+ stimulating Pyk2 to phosphorylate GSK-3β thus enhancing its inhibitory action on CREB.Citation37 The abnormal elevations in Ca2+ will enhance membrane excitability and this may distort the neural components of the circuits that control mood.
Alzheimer Disease
Alzheimer disease (AD) is a progressive neurodegenerative disorder caused by an increase in amyloid metabolism. The basis of the amyloid cascade hypothesis is that the disruption of neural activity and subsequent cell death results from the abnormal processing of β-amyloid (Aβ).Citation38 Mutations in some of the components of the amyloid pathway, such as the amyloid precursor protein (APP), ApoE4, presenilin-1 and presenilin-2 (PS1 and PS2) and SORL1 are responsible for autosomal-dominant early-onset familial Alzheimer disease (FAD). The APP is synthesized and transferred to the plasma membrane where it is processed by either the non-amyloidogenic or amyloidogenic pathways. In the non-amyloidogenic pathway, APP is processed without giving rise to the deleterious β amyloids. However, in the amyloidogenic pathway, APP is internalized and ends up in the late endosomes where it is hydrolysed by β-secretase (also known as BACE) that sheds the N-terminal sAPPβ region leaving the C-terminal fragment β (CTF β) in the membrane (). This CTF β is hydrolysed by the γ-secretase complex that contains the presenilin enzymes, either the PS1 or PS2 isoforms. This γ-secretase cleaves CTF β at two sites to yield either amyloid β 40 (Aβ40) or amyloid β 42 (Aβ42), which are then released to the inside of the vesicle, and the APP intracellular domain (AICD) that is released to the cytoplasm. The amyloids are transported and released to the surface via the constitutive secretory pathway. The calcium hypothesis of AD explores how activation of the amyloidgenic pathway remodels neuronal signaling pathways to reduce cognition and to promote neuronal cell death.
Figure 4. Calcium hypothesis of Alzheimer disease. Alzheimer disease (AD) is caused by an increase in the formation of the amyloid β peptides Aβ40 and Aβ42. The amyloid precursor protein (APP) enters the late endosome where it is cleaved by BACE and the γ-secretase complex to form Aβ40/42. The amyloid peptides are released where they form either fibrils and plaques or the oligomers that enhance Ca2+ signaling. The yellow arrows illustrate some of the changes in various Ca2+ signaling components that are responsible the for an overall increase in Ca2+ signaling that then induce dementia through the loss of memory and neuronal apoptosis. The white boxes highlight some of Ca2+-modifying agents that have been found to alleviate the symptoms of AD in mouse models
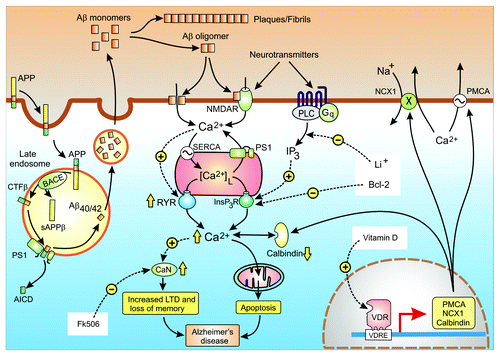
The basis of the Ca2+ hypothesis is that abnormal amyloid metabolism results in an upregulation of neuronal Ca2+ signaling to induce an initial decline in memory and then progresses to a later phase of apoptosis.Citation39-Citation42 When Ca2+ is measured in the spines and dendrites of cortical pyramidal neurons of transgenic mice, there was a higher than normal resting level in those neurons located close to amyloid deposits.Citation43 Similarly, the resting level of Ca2+ in the cortical neurons of 3xTg-AD animals was 247 nmol/L, which was twice that found in the non-Tg controls (110 nmol/L).Citation44 There are indications that this increase in Ca2+ signaling is caused by changes in both the entry of external Ca2+ and release from internal stores.
One mechanism for the upregulation of Ca2+ signaling is for the amyloids to enhance Ca2+ entry ().Citation43,Citation45-Citation47 The β-amyloid peptides, which aggregate to form complexes may enhance entry either directly by forming channels in the membraneCitation48 or by stimulating pre-existing channels such as the NMDARs.Citation49 The cellular prion protein (PrPC), which is tethered to the outside of the membrane through a glycosyl phosphatidylinositol (GPI) anchor, functions as an amyloid β receptor and may thus carry out some of these actions on Ca2+ entry at the plasma membrane.Citation50 Any Ca2+ that enters through these amyloid-dependent mechanisms will contribute to the remodelling of the Ca2+ signaling system that occurs during AD.
There also are indications that the amount of Ca2+ being released from the ER is also increased in AD.Citation51-Citation53 An increased expression of the ryanodine receptor (RYR), particularly the RYR3 isoform,Citation53 is one cause for this hypersensitivity of the internal release mechanism.Citation52,Citation54-Citation56 The increase in RYR expression results in a greater sensitivity to Ca2+ elevations during normal synaptic transmission.Citation57 In transgenic mice, entry of Ca2+ through the NMDA receptors triggered a greater release of Ca2+ from the RYRs in both the dendrites and spines through the process of Ca2+-induced Ca2+ release (CICR). Since one of the functions of these RYRs is to amplify the Ins(1,4,5)P3-mediated release of Ca2+, this remodelling of the ER release channels can markedly enhance neuronal Ca2+ signals.Citation52 Release of Ca2+ can also be altered by changing the Ca2+ content of the internal stores, which depends on the balance between the activity of the SERCA pump and the passive leak of Ca2+ back into the cytoplasm (). Presenilins have been shown to alter both activities. For example, the activity of the SERCA pump is enhanced by the presenilins.Citation58 The channels responsible for the passive leak remain to be properly characterized, but there is increasing evidence that presenilins may be leak channels.Citation45,Citation59 The mutated forms of PS1 that give rise to early-onset FAD reduced the passive leak resulting in enhanced Ca2+ signals.Citation59 The mutated PS1 can also interact with the Ins(1,4,5)P3R to enhance its sensitivity.Citation60,Citation61
Another important remodeling event associated with AD is a downregulation of the Ca2+ buffer calbindin D-28k (CB).Citation62 It has been known for some time that during normal aging there are gradual changes in certain Ca2+ signaling components that increase neuronal vulnerability to cell death stimuli. For example, there is a decline in the level of the Ca2+ buffer calbindin D-28k (CB) that normally functions to restrict the amplitude of Ca2+ signals.Citation63 A decline in this buffer may also increase the onset of AD because mice expressing mutant APP also display a decline in the level of CB especially in the dentate gyrus region of the hippocampus, which functions in learning and memory.Citation62 The upregulation of Ca2+ signaling may be responsible for the learning and memory deficits that occur early during the onset of AD.
Just how this upregulation of Ca2+ signaling results in the learning and memory deficits that occur during the onset of AD is still a mystery. It is known that Ca2+ is responsible for the synaptic plasticity responsible for learning and memory and one possibility is that persistent elevation in the resting level of Ca2+ may be responsible for the decline in cognition.Citation64,Citation65 High concentration spikes of Ca2+ activate the process of long-term potentiation (LTP) responsible for memory formation. In contrast, smaller elevation in Ca2+ activates a process of long-term depression (LTD) that can erase the information that is stored during the process of LTP. In other words, Ca2+ has two diametrically opposed actions: it can both form and erase memories. This Ca2+-dependent erasure of newly acquired memories depends on activation of the protein phosphatase calcineurin (CaN) (). The alteration in Ca2+ signaling will also contribute to the increase in apoptosis resulting in the neurodegeneration that characterizes the later stages of dementia.
Reversal of Ca2+-dependent neurodegeneration
The hypothesis that neurodegenerative diseases are caused by Ca2+dysregulation suggests that novel therapies designed to normalize Ca2+ signaling pathways could reverse the consequences of neurodegeneration. Such treatments must be fairly subtle and need to concentrate on rectifying the different defects, such as lowering the resting level of Ca2+, without interfering with the normal Ca2+ signaling pathways. There already is some evidence, based primarily on AD mouse models, that the deleterious effects of excess Ca2+ can be reversed by adjusting either the levels of Ca2+ or its downstream signaling events using treatments such as Li+, Bcl-2, FK506 and vitamin D ().
The risk of developing Alzheimer disease might be reduced by Li+, but how this occurs is not clear.Citation66 As described earlier, the action of Li+ in bipolar disorder may depend on its ability to reduce the activity of Ins(1,4,5)P3/Ca2+ signaling () and exactly the same mechanism could explain its protective effect in AD.
As indicated earlier, enhanced activity of the Ins(1,4,5)P3R is thought to contribute to the onset of AD and there are indications that lowering the activity of this channel by the anti-apoptotic factor Bcl-2 reduces the symptoms of AD. When expressed in a mouse model of AD, the anti-apoptotic factor Bcl-2 was able to improve cognition and prevented neuronal apoptosis.Citation67 This is consistent with the calcium hypothesis of AD because Bcl-2 is known to bind to the Ins(1,4,5)P3R to reversibly inhibit Ins(1,4,5)P3-dependent channel opening ().Citation29 If such a mechanism operates in neurons, a reduction in the release of Ca2+ from the internal store and the subsequent decline in the level of Ca2+ would support the notion that the upregulation of Ca2+ signaling is responsible for driving memory loss in AD.
As described earlier, the persistent elevated levels of Ca2+ stimulate CaN that is responsible for increasing the process of LTD (). The level of CaN was found to be elevated in aged rats and in an APP transgenic mouse model of AD that display defects in cognition.Citation68,Citation69 In the case of the transgenic mouse, the defects in cognition could be reversed by FK506, which is an inhibitor of CaN (). These inhibitory effects of Li+, Bcl-2 and FK506 on neurodegeneration not only support the idea that upregulation of Ca2+ may be responsible for the onset of AD but they also provide proof of concept that this debilitating neurodegenerative disease could be alleviated by treatments targeted at neuronal Ca2+ signaling pathways.
There are an increasing number of studies indicating that a deficiency in vitamin D may contribute to the onset of neurodegenerative diseases such as Alzheimer disease (AD) and Parkinson disease (PD).Citation70-Citation72 With regard to AD, the decline in cognition that occurs normally in older adults may also be linked to vitamin D deficiency.Citation73 Enhanced dietary vitamin D intake lowers the risk of developing AD in a study of older women.Citation74 Since both AD and PD seem to be caused by abnormal elevations in Ca2+, I shall develop the notion that the deleterious effect of vitamin D deficiency may be explained by an alteration in its normal role in regulating intracellular Ca2+ homeostasis.Citation75,Citation76
The brain possesses all the enzyme responsible for both vitamin D formation (vitamin D3 25-hydroxylase and 25-hydroxyvitamin D3–1α-hydroxylase) and degradation (vitamin D3 25-hydroxylase).Citation70,Citation77 Neurons also express the vitamin D receptor (VDR) and VDR polymorphisms have been associated with Parkinson disease,Citation76 age-related decline in cognition and the incidence of depressive symptomsCitation78 and is also a risk factor for AD.Citation72,Citation79 This VDR is also strongly expressed in the dopaminergic neurons in the substantia nigra, which are particularly vulnerable to Ca2+ stress in that they have to deal with repetitive surges in Ca2+ every few seconds.Citation80 The neurotoxic effect of 6-hydroxydopamine, which seems to depend on an increase in Ca2+ and reactive oxygen species (ROS), is reduced by vitamin D in rats.Citation81 Vitamin D may alleviate the deleterious effects of ROS by increasing expression of γ-glutamyl transpeptidase that synthesizes the redox buffer glutathione.Citation70
All the evidence outlined above indicates that vitamin D has a significant protective role in the brain by helping to maintain both Ca2+ and ROS homeostasis. Such an action is consistent with the fact that vitamin D can regulate the expression of those Ca2+ signaling toolkit components responsible for reducing Ca2+ levels (). For example, vitamin D stimulates the expression of the plasma membrane Ca2+ ATPase (PMCA), the Na+/Ca2+ exchanger (NCX) and Ca2+ buffers such as CB and parvalbumin.Citation82-Citation84 Neuronal levels of CB are known to be reduced in AD.Citation85 In addition to enhancing these mechanisms for lowering the level of Ca2+, vitamin D can curb the influx of external Ca2+ by reducing the expression of L-type voltage-sensitive channels, which are markedly elevated in rat hippocampal neurons.Citation86
In summary, any reduction in vitamin D levels will result in elevated neuronal Ca2+ levels and this could account for a number of neurodegenerative diseases such as AD and PD. A clinical trial is in progress to test whether vitamin D can alleviate some of the degenerative processes associated with ADCitation75 and there is every reason to suspect that it might prove efficacious in other neural diseases such as PD that are driven by a dysregulation of Ca2+ signaling.
Conclusion
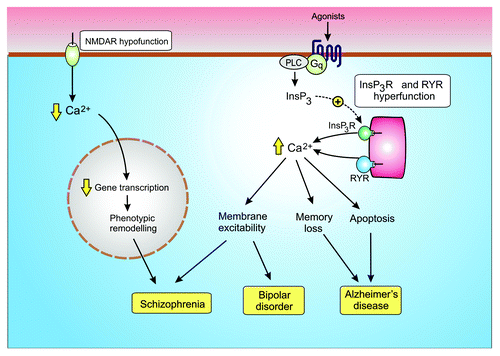
Neurons have sophisticated Ca2+ signaling systems that deliver the spatial and temporal Ca2+ signals necessary to control multiple neuronal functions. Fast acting voltage-sensitive Ca2+ entry mechanisms provide the primary Ca2+ signal for cell activation, whereas there are other Ca2+ signals that play a more modulatory role. For example, the NMDA receptors provide regular pulses of Ca2+ that maintain the phenotypic stability of the GABAergic interneurons. The Ins(1,4,5)P3/Ca2+ signaling pathway has an essential modulatory role in the ascending arousal system by maintaining the tonic excitatory drive responsible for regulating the activity of neuronal rhythms. Subtle alterations in the function of these modulatory pathways () contribute to some of the major neurodegenerative diseases such as Alzheimer disease, bipolar disorder and schizophrenia.
Figure 5. Dysregulation of modulatory Ca2+ signaling pathways contribute to neural diseases. Hypofunction of the NMDA receptor results in a reduction in the gene transcription necessary to maintain the GABAergic phenotype resulting in schizophrenia. Hyperfunction of the Ins(1,4,5)P3/Ca2+ signaling pathway has been linked to both bipolar disorder and Alzheimer disease.
| Abbreviations: | ||
| ACh | = | acetylcholine |
| AD | = | Alzheimer’s disease (AD) |
| APP | = | amyloid precursor protein |
| BD | = | bipolar disorder |
| BDNF | = | brain-derived neurotrophic factor |
| CB | = | calbindin D-28k |
| CaN | = | calcineurin |
| CaM | = | calmodulin |
| PrPC | = | cellular prion protein |
| CREB | = | cyclic AMP response element-binding protein |
| DGKH | = | diacylglycerol kinase |
| DISC1 | = | disrupted in schizophrenia 1 |
| DA | = | dopamine |
| FAD | = | familial Alzheimer’s disease |
| GCL | = | glutamate cysteine ligase |
| GCLC | = | GCL catalytic subunit |
| GCLM | = | GCL modifier subunit |
| GAD67 | = | glutamic acid decarboxylase 67 |
| GSH | = | glutathione |
| GSK3β | = | glycogen synthesis kinase 3β |
| H2O2 | = | hydrogen peroxide |
| 5-HT | = | 5-hydroxytryptamine |
| HCN | = | hyperpolarizing-activated cyclic nucleotide-gated |
| IMPase | = | inositol monophosphatase |
| IP3, inositol 1 | = | 4,5-trisphosphate |
| IL-6 | = | interleukin-6 |
| ICAN | = | inward Ca2+ current |
| LTD | = | long term depression |
| LTP | = | long term potentiation |
| NCX | = | Na+/Ca2+ exchanger |
| NCS-1 | = | neuronal Ca2+ sensor-1 |
| nNOS | = | neuronal nitric oxide synthase |
| NAChR | = | nicotinic acetylcholine receptor |
| NO | = | nitric oxide |
| NE | = | norepinephrine, PD, Parkinson’s disease |
| PV | = | parvalbumin |
| ONOO- | = | peroxynitrite |
| PIP2 | = | phosphatidylinositol 4,5-bisphosphate |
| PDE48 | = | phosphodiesterase 4B |
| PMCA | = | plasma membrane Ca2+ ATPase |
| PS1 and PS2 | = | presenilin-1 and presenilin-2 |
| ROS | = | reactive oxygen species |
| RYRs | = | ryanodine receptors |
| SNPs | = | single nucleotide polymorphisms |
| O2− | = | superoxide |
| SOD | = | superoxide dismutase |
| VIP | = | vasoactive intestinal peptide receptor |
| VIPR | = | vasoactive intestinal peptide receptor, VDR, vitamin D receptor |
| VOCs | = | voltage-operated Ca2+ channels |
References
- Lawrence JJ. Cholinergic control of GABA release: emerging parallels between neocortex and hippocampus. Trends Neurosci 2008; 31:317 - 27; http://dx.doi.org/10.1016/j.tins.2008.03.008; PMID: 18556072
- Fisahn A, Yamada M, Duttaroy A, Gan J-W, Deng C-X, McBain CJ, et al. Muscarinic induction of hippocampal gamma oscillations requires coupling of the M1 receptor to two mixed cation currents. Neuron 2002; 33:615 - 24; http://dx.doi.org/10.1016/S0896-6273(02)00587-1; PMID: 11856534
- Chiang PH, Yeh WC, Lee CT, Weng JY, Huang YY, Lien CC. M(1)-like muscarinic acetylcholine receptors regulate fast-spiking interneuron excitability in rat dentate gyrus. Neuroscience 2010; 169:39 - 51; http://dx.doi.org/10.1016/j.neuroscience.2010.04.051; PMID: 20433901
- Lee K-H, Williams LM, Breakspear M, Gordon E. Synchronous gamma activity: a review and contribution to an integrative neuroscience model of schizophrenia. Brain Res Brain Res Rev 2003; 41:57 - 78; http://dx.doi.org/10.1016/S0165-0173(02)00220-5; PMID: 12505648
- Uhlhaas PJ, Singer W. Abnormal neural oscillations and synchrony in schizophrenia. Nat Rev Neurosci 2010; 11:100 - 13; http://dx.doi.org/10.1038/nrn2774; PMID: 20087360
- Verret L, Mann EO, Hang GB, Barth AMI, Cobos I, Ho K, et al. Inhibitory interneuron deficit links altered network activity and cognitive dysfunction in Alzheimer model. Cell 2012; 149:708 - 21; http://dx.doi.org/10.1016/j.cell.2012.02.046; PMID: 22541439
- Delmas P, Brown DA. Pathways modulating neural KCNQ/M (Kv7) potassium channels. Nat Rev Neurosci 2005; 6:850 - 62; http://dx.doi.org/10.1038/nrn1785; PMID: 16261179
- Congar P, Leinekugel X, Ben-Ari Y, Crépel V. A long-lasting calcium-activated nonselective cationic current is generated by synaptic stimulation or exogenous activation of group I metabotropic glutamate receptors in CA1 pyramidal neurons. J Neurosci 1997; 17:5366 - 79; PMID: 9204921
- Ross CA, Margolis RL, Reading SAJ, Pletnikov M, Coyle JT. Neurobiology of schizophrenia. Neuron 2006; 52:139 - 53; http://dx.doi.org/10.1016/j.neuron.2006.09.015; PMID: 17015232
- Curley AA, Lewis DA. Cortical basket cell dysfunction in schizophrenia. J Physiol 2012; 590:715 - 24; PMID: 22219337
- Coyle JT. Glutamate and schizophrenia: beyond the dopamine hypothesis. Cell Mol Neurobiol 2006; 26:365 - 84; http://dx.doi.org/10.1007/s10571-006-9062-8; PMID: 16773445
- Kantrowitz JT, Javitt DC. N-methyl-d-aspartate (NMDA) receptor dysfunction or dysregulation: the final common pathway on the road to schizophrenia?. Brain Res Bull 2010; 83:108 - 21; http://dx.doi.org/10.1016/j.brainresbull.2010.04.006; PMID: 20417696
- Gonzalez-Burgos G, Lewis DA. GABA neurons and the mechanisms of network oscillations: implications for understanding cortical dysfunction in schizophrenia. Schizophr Bull 2008; 34:944 - 61; http://dx.doi.org/10.1093/schbul/sbn070; PMID: 18586694
- Behrens MM, Sejnowski TJ. Does schizophrenia arise from oxidative dysregulation of parvalbumin-interneurons in the developing cortex?. Neuropharmacology 2009; 57:193 - 200; http://dx.doi.org/10.1016/j.neuropharm.2009.06.002; PMID: 19523965
- Do KQ, Cabungcal JH, Frank A, Steullet P, Cuenod M. Redox dysregulation, neurodevelopment, and schizophrenia. Curr Opin Neurobiol 2009; 19:220 - 30; http://dx.doi.org/10.1016/j.conb.2009.05.001; PMID: 19481443
- Hardingham GE, Bading H. Synaptic versus extrasynaptic NMDA receptor signalling: implications for neurodegenerative disorders. Nat Rev Neurosci 2010; 11:682 - 96; http://dx.doi.org/10.1038/nrn2911; PMID: 20842175
- Berk M, Ng F, Dean O, Dodd S, Bush AI. Glutathione: a novel treatment target in psychiatry. Trends Pharmacol Sci 2008; 29:346 - 51; http://dx.doi.org/10.1016/j.tips.2008.05.001; PMID: 18538422
- Sharma G, Vijayaraghavan S. Modulation of presynaptic store calcium induces release of glutamate and postsynaptic firing. Neuron 2003; 38:929 - 39; http://dx.doi.org/10.1016/S0896-6273(03)00322-2; PMID: 12818178
- Pitcher GM, Kalia LV, Ng D, Goodfellow NM, Yee KT, Lambe EK, et al. Schizophrenia susceptibility pathway neuregulin 1-ErbB4 suppresses Src upregulation of NMDA receptors. Nat Med 2011; 17:470 - 8; http://dx.doi.org/10.1038/nm.2315; PMID: 21441918
- Li X, Jope RS. Is glycogen synthase kinase-3 a central modulator in mood regulation?. Neuropsychopharmacology 2010; 35:2143 - 54; http://dx.doi.org/10.1038/npp.2010.105; PMID: 20668436
- Vacic V, McCarthy S, Malhotra D, Murray F, Chou H-H, Peoples A, et al. Duplications of the neuropeptide receptor gene VIPR2 confer significant risk for schizophrenia. Nature 2011; 471:499 - 503; http://dx.doi.org/10.1038/nature09884; PMID: 21346763
- Lee K-H, Williams LM, Breakspear M, Gordon E. Synchronous gamma activity: a review and contribution to an integrative neuroscience model of schizophrenia. Brain Res Brain Res Rev 2003; 41:57 - 78; http://dx.doi.org/10.1016/S0165-0173(02)00220-5; PMID: 12505648
- Meyer-Lindenberg A, Domes G, Kirsch P, Heinrichs M. Oxytocin and vasopressin in the human brain: social neuropeptides for translational medicine. Nat Rev Neurosci 2011; 12:524 - 38; http://dx.doi.org/10.1038/nrn3044; PMID: 21852800
- Machado-Vieira R, Manji HK, Zarate CA Jr.. The role of lithium in the treatment of bipolar disorder: convergent evidence for neurotrophic effects as a unifying hypothesis. Bipolar Disord 2009; 11:Suppl 2 92 - 109; http://dx.doi.org/10.1111/j.1399-5618.2009.00714.x; PMID: 19538689
- Quiroz JA, Machado-Vieira R, Zarate CA Jr., Manji HK. Novel insights into lithium’s mechanism of action: neurotrophic and neuroprotective effects. Neuropsychobiology 2010; 62:50 - 60; http://dx.doi.org/10.1159/000314310; PMID: 20453535
- Mao Y, Ge X, Frank CL, Madison JM, Koehler AN, Doud MK, et al. Disrupted in schizophrenia 1 regulates neuronal progenitor proliferation via modulation of GSK3β/β-catenin signaling. Cell 2009; 136:1017 - 31; http://dx.doi.org/10.1016/j.cell.2008.12.044; PMID: 19303846
- Machado-Vieira R, Pivovarova NB, Stanika RI, Yuan P, Wang Y, Zhou R, et al. The Bcl-2 gene polymorphism rs956572AA increases inositol 1,4,5-trisphosphate receptor-mediated endoplasmic reticulum calcium release in subjects with bipolar disorder. Biol Psychiatry 2011; 69:344 - 52; http://dx.doi.org/10.1016/j.biopsych.2010.10.019; PMID: 21167476
- Chen G, Zeng WZ, Yuan PX, Huang LD, Jiang YM, Zhao ZH, et al. The mood-stabilizing agents lithium and valproate robustly increase the levels of the neuroprotective protein bcl-2 in the CNS. J Neurochem 1999; 72:879 - 82; http://dx.doi.org/10.1046/j.1471-4159.1999.720879.x; PMID: 9930766
- Rong YP, Distelhorst CW. Bcl-2 protein family members: versatile regulators of calcium signaling in cell survival and apoptosis. Annu Rev Physiol 2008; 70:73 - 91; http://dx.doi.org/10.1146/annurev.physiol.70.021507.105852; PMID: 17680735
- Warsh JJ, Andreopoulos S, Li PP. Role of intracellular calcium signaling in the pathophysiology and pharmacotherapy of bipolar disorder: current status. Clin Neurosci Res 2004; 4:201 - 13; http://dx.doi.org/10.1016/j.cnr.2004.09.012
- Berridge MJ, Downes CP, Hanley MR. Neural and developmental actions of lithium: a unifying hypothesis. Cell 1989; 59:411 - 9; http://dx.doi.org/10.1016/0092-8674(89)90026-3; PMID: 2553271
- Kim HJ, Thayer SA. Lithium increases synapse formation between hippocampal neurons by depleting phosphoinositides. Mol Pharmacol 2009; 75:1021 - 30; http://dx.doi.org/10.1124/mol.108.052357; PMID: 19188338
- Baum AE, Akula N, Cabanero M, Cardona I, Corona W, Klemens B, et al. A genome-wide association study implicates diacylglycerol kinase eta (DGKH) and several other genes in the etiology of bipolar disorder. Mol Psychiatry 2008; 13:197 - 207; http://dx.doi.org/10.1038/sj.mp.4002012; PMID: 17486107
- Koh PO, Undie AS, Kabbani N, Levenson R, Goldman-Rakic PS, Lidow MS. Up-regulation of neuronal calcium sensor-1 (NCS-1) in the prefrontal cortex of schizophrenic and bipolar patients. Proc Natl Acad Sci U S A 2003; 100:313 - 7; http://dx.doi.org/10.1073/pnas.232693499; PMID: 12496348
- Schlecker C, Boehmerle W, Jeromin A, DeGray B, Varshney A, Sharma Y, et al. Neuronal calcium sensor-1 enhancement of InsP3 receptor activity is inhibited by therapeutic levels of lithium. J Clin Invest 2006; 116:1668 - 74; http://dx.doi.org/10.1172/JCI22466; PMID: 16691292
- Kabbani N, Negyessy L, Lin RW, Goldman-Rakic P, Levenson R. Interaction with neuronal calcium sensor NCS-1 mediates desensitization of the D2 dopamine receptor. J Neurosci 2002; 22:8476 - 86; PMID: 12351722
- Sayas CL, Ariaens A, Ponsioen B, Moolenaar WH. GSK-3 is activated by the tyrosine kinase Pyk2 during LPA1-mediated neurite retraction. Mol Biol Cell 2006; 17:1834 - 44; http://dx.doi.org/10.1091/mbc.E05-07-0688; PMID: 16452634
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 2002; 297:353 - 6; http://dx.doi.org/10.1126/science.1072994; PMID: 12130773
- Khachaturian ZS. Calcium, membranes, aging, and Alzheimer’s disease. Introduction and overview. Ann N Y Acad Sci 1989; 568:1 - 4; http://dx.doi.org/10.1111/j.1749-6632.1989.tb12485.x; PMID: 2629579
- LaFerla FM. Calcium dyshomeostasis and intracellular signalling in Alzheimer’s disease. Nat Rev Neurosci 2002; 3:862 - 72; http://dx.doi.org/10.1038/nrn960; PMID: 12415294
- Stutzmann GE. The pathogenesis of Alzheimers disease is it a lifelong “calciumopathy”?. Neuroscientist 2007; 13:546 - 59; http://dx.doi.org/10.1177/1073858407299730; PMID: 17901262
- Thibault O, Gant JC, Landfield PW. Expansion of the calcium hypothesis of brain aging and Alzheimer’s disease: minding the store. Aging Cell 2007; 6:307 - 17; http://dx.doi.org/10.1111/j.1474-9726.2007.00295.x; PMID: 17465978
- Kuchibhotla KV, Goldman ST, Lattarulo CR, Wu H-Y, Hyman BT, Bacskai BJA. Abeta plaques lead to aberrant regulation of calcium homeostasis in vivo resulting in structural and functional disruption of neuronal networks. Neuron 2008; 59:214 - 25; http://dx.doi.org/10.1016/j.neuron.2008.06.008; PMID: 18667150
- Lopez JR, Lyckman A, Oddo S, Laferla FM, Querfurth HW, Shtifman A. Increased intraneuronal resting [Ca2+] in adult Alzheimer’s disease mice. J Neurochem 2008; 105:262 - 71; http://dx.doi.org/10.1111/j.1471-4159.2007.05135.x; PMID: 18021291
- Bezprozvanny I, Mattson MP. Neuronal calcium mishandling and the pathogenesis of Alzheimer’s disease. Trends Neurosci 2008; 31:454 - 63; http://dx.doi.org/10.1016/j.tins.2008.06.005; PMID: 18675468
- Sanz-Blasco S, Valero RA, Rodríguez-Crespo I, Villalobos C, Núñez L. Mitochondrial Ca2+ overload underlies Abeta oligomers neurotoxicity providing an unexpected mechanism of neuroprotection by NSAIDs. PLoS One 2008; 3:e2718; http://dx.doi.org/10.1371/journal.pone.0002718; PMID: 18648507
- Shirwany NA, Payette D, Xie J, Guo Q. The amyloid beta ion channel hypothesis of Alzheimer’s disease. Neuropsychiatr Dis Treat 2007; 3:597 - 612; PMID: 19300589
- Demuro A, Smith M, Parker I. Single-channel Ca(2+) imaging implicates Aβ1-42 amyloid pores in Alzheimer’s disease pathology. J Cell Biol 2011; 195:515 - 24; http://dx.doi.org/10.1083/jcb.201104133; PMID: 22024165
- Ferreira IL, Bajouco LM, Mota SI, Auberson YP, Oliveira CR, Rego AC. Amyloid beta peptide 1-42 disturbs intracellular calcium homeostasis through activation of GluN2B-containing N-methyl-d-aspartate receptors in cortical cultures. Cell Calcium 2012; 51:95 - 106; http://dx.doi.org/10.1016/j.ceca.2011.11.008; PMID: 22177709
- Laurén J, Gimbel DA, Nygaard HB, Gilbert JW, Strittmatter SM. Cellular prion protein mediates impairment of synaptic plasticity by amyloid-β oligomers. Nature 2009; 457:1128 - 32; http://dx.doi.org/10.1038/nature07761; PMID: 19242475
- Guo Q, Furukawa K, Sopher BL, Pham DG, Xie J, Robinson N, et al. Alzheimer’s PS-1 mutation perturbs calcium homeostasis and sensitizes PC12 cells to death induced by amyloid beta-peptide. Neuroreport 1996; 8:379 - 83; http://dx.doi.org/10.1097/00001756-199612200-00074; PMID: 9051814
- Stutzmann GE, Smith I, Caccamo A, Oddo S, Laferla FM, Parker I. Enhanced ryanodine receptor recruitment contributes to Ca2+ disruptions in young, adult, and aged Alzheimer’s disease mice. J Neurosci 2006; 26:5180 - 9; http://dx.doi.org/10.1523/JNEUROSCI.0739-06.2006; PMID: 16687509
- Supnet C, Grant J, Kong H, Westaway D, Mayne M. Amyloid-β-(1-42) increases ryanodine receptor-3 expression and function in neurons of TgCRND8 mice. J Biol Chem 2006; 281:38440 - 7; http://dx.doi.org/10.1074/jbc.M606736200; PMID: 17050533
- Chan SL, Mayne M, Holden CP, Geiger JD, Mattson MP. Presenilin-1 mutations increase levels of ryanodine receptors and calcium release in PC12 cells and cortical neurons. J Biol Chem 2000; 275:18195 - 200; http://dx.doi.org/10.1074/jbc.M000040200; PMID: 10764737
- Smith IF, Hitt B, Green KN, Oddo S, LaFerla FM. Enhanced caffeine-induced Ca2+ release in the 3xTg-AD mouse model of Alzheimer’s disease. J Neurochem 2005; 94:1711 - 8; http://dx.doi.org/10.1111/j.1471-4159.2005.03332.x; PMID: 16156741
- Chakroborty S, Goussakov I, Miller MB, Stutzmann GE. Deviant ryanodine receptor-mediated calcium release resets synaptic homeostasis in presymptomatic 3xTg-AD mice. J Neurosci 2009; 29:9458 - 70; http://dx.doi.org/10.1523/JNEUROSCI.2047-09.2009; PMID: 19641109
- Goussakov I, Miller MB, Stutzmann GE. NMDA-mediated Ca(2+) influx drives aberrant ryanodine receptor activation in dendrites of young Alzheimer’s disease mice. J Neurosci 2010; 30:12128 - 37; http://dx.doi.org/10.1523/JNEUROSCI.2474-10.2010; PMID: 20826675
- Green KN, Demuro A, Akbari Y, Hitt BD, Smith IF, Parker I, et al. SERCA pump activity is physiologically regulated by presenilin and regulates amyloid β production. J Cell Biol 2008; 181:1107 - 16; http://dx.doi.org/10.1083/jcb.200706171; PMID: 18591429
- Tu H, Nelson O, Bezprozvanny A, Wang Z, Lee SF, Hao YH, et al. Presenilins form ER Ca2+ leak channels, a function disrupted by familial Alzheimer’s disease-linked mutations. Cell 2006; 126:981 - 93; http://dx.doi.org/10.1016/j.cell.2006.06.059; PMID: 16959576
- Cheung KH, Shineman D, Müller M, Cárdenas C, Mei L, Yang J, et al. Mechanism of Ca2+ disruption in Alzheimer’s disease by presenilin regulation of InsP3 receptor channel gating. Neuron 2008; 58:871 - 83; http://dx.doi.org/10.1016/j.neuron.2008.04.015; PMID: 18579078
- Müller M, Cárdenas C, Mei L, Cheung KH, Foskett JK. Constitutive cAMP response element binding protein (CREB) activation by Alzheimer’s disease presenilin-driven inositol trisphosphate receptor (InsP3R) Ca2+ signaling. Proc Natl Acad Sci U S A 2011; 108:13293 - 8; http://dx.doi.org/10.1073/pnas.1109297108; PMID: 21784978
- Palop JJ, Jones B, Kekonius L, Chin J, Yu G-Q, Raber J, et al. Neuronal depletion of calcium-dependent proteins in the dentate gyrus is tightly linked to Alzheimer’s disease-related cognitive deficits. Proc Natl Acad Sci U S A 2003; 100:9572 - 7; http://dx.doi.org/10.1073/pnas.1133381100; PMID: 12881482
- Geula C, Bu J, Nagykery N, Scinto LFM, Chan J, Joseph J, et al. Loss of calbindin-D28k from aging human cholinergic basal forebrain: relation to neuronal loss. J Comp Neurol 2003; 455:249 - 59; http://dx.doi.org/10.1002/cne.10475; PMID: 12454989
- Berridge MJ. Calcium hypothesis of Alzheimer’s disease. Pflugers Arch 2010; 459:441 - 9; http://dx.doi.org/10.1007/s00424-009-0736-1; PMID: 19795132
- Berridge MJ. Calcium signalling and Alzheimer’s disease. Neurochem Res 2011; 36:1149 - 56; http://dx.doi.org/10.1007/s11064-010-0371-4; PMID: 21184278
- Nunes PV, Forlenza OV, Gattaz WF. Lithium and risk for Alzheimer’s disease in elderly patients with bipolar disorder. Br J Psychiatry 2007; 190:359 - 60; http://dx.doi.org/10.1192/bjp.bp.106.029868; PMID: 17401045
- Rohn TT, Vyas V, Hernandez-Estrada T, Nichol KE, Christie L-A, Head E. Lack of pathology in a triple transgenic mouse model of Alzheimer’s disease after overexpression of the anti-apoptotic protein Bcl-2. J Neurosci 2008; 28:3051 - 9; http://dx.doi.org/10.1523/JNEUROSCI.5620-07.2008; PMID: 18354008
- Dineley KT, Hogan D, Zhang WR, Taglialatela G. Acute inhibition of calcineurin restores associative learning and memory in Tg2576 APP transgenic mice. Neurobiol Learn Mem 2007; 88:217 - 24; http://dx.doi.org/10.1016/j.nlm.2007.03.010; PMID: 17521929
- Foster TC, Sharrow KM, Masse JR, Norris CM, Kumar A. Calcineurin links Ca2+ dysregulation with brain aging. J Neurosci 2001; 21:4066 - 73; PMID: 11356894
- Garcion E, Wion-Barbot N, Montero-Menei CN, Berger F, Wion D. New clues about vitamin D functions in the nervous system. Trends Endocrinol Metab 2002; 13:100 - 5; http://dx.doi.org/10.1016/S1043-2760(01)00547-1; PMID: 11893522
- Tuohimaa P, Keisala T, Minasyan A, Cachat J, Kalueff A. Vitamin D, nervous system and aging. Psychoneuroendocrinology 2009; 34:Suppl 1 S278 - 86; http://dx.doi.org/10.1016/j.psyneuen.2009.07.003; PMID: 19660871
- Wang L, Hara K, Van Baaren JM, Price JC, Beecham GW, Gallins PJ, et al. Vitamin D receptor and Alzheimer’s disease: a genetic and functional study. Neurobiol Aging 2012; 33:1844.e1 - 9; http://dx.doi.org/10.1016/j.neurobiolaging.2011.12.038; PMID: 22306846
- Przybelski RJ, Binkley NC. Is vitamin D important for preserving cognition? A positive correlation of serum 25-hydroxyvitamin D concentration with cognitive function. Arch Biochem Biophys 2007; 460:202 - 5; http://dx.doi.org/10.1016/j.abb.2006.12.018; PMID: 17258168
- Annweiler C, Rolland Y, Schott AM, Blain H, Vellas B, Herrmann FR, et al. Higher vitamin D dietary intake is associated with lower risk of Alzheimer’s disease: A 7-year follow-up. J Gerontol A Biol Sci Med Sci 2012; In Press http://dx.doi.org/10.1093/gerona/gls107; PMID: 22503994
- Annweiler C, Beauchet O. Possibility of a new anti-alzheimer’s disease pharmaceutical composition combining memantine and vitamin D. Drugs Aging 2012; 29:81 - 91; http://dx.doi.org/10.2165/11597550-000000000-00000; PMID: 22233455
- Butler MW, Burt A, Edwards TL, Zuchner S, Scott WK, Martin ER, et al. Vitamin D receptor gene as a candidate gene for Parkinson disease. Ann Hum Genet 2011; 75:201 - 10; http://dx.doi.org/10.1111/j.1469-1809.2010.00631.x; PMID: 21309754
- Kesby JP, Eyles DW, Burne TH, McGrath JJ. The effects of vitamin D on brain development and adult brain function. Mol Cell Endocrinol 2011; 347:121 - 7; http://dx.doi.org/10.1016/j.mce.2011.05.014; PMID: 21664231
- Kuningas M, Mooijaart SP, Jolles J, Slagboom PE, Westendorp RG, van Heemst D. VDR gene variants associate with cognitive function and depressive symptoms in old age. Neurobiol Aging 2009; 30:466 - 73; http://dx.doi.org/10.1016/j.neurobiolaging.2007.07.001; PMID: 17714831
- Lehmann DJ, Refsum H, Warden DR, Medway C, Wilcock GK, Smith AD. The vitamin D receptor gene is associated with Alzheimer’s disease. Neurosci Lett 2011; 504:79 - 82; http://dx.doi.org/10.1016/j.neulet.2011.08.057; PMID: 21911036
- Chan CS, Gertler TS, Surmeier DJ. Calcium homeostasis, selective vulnerability and Parkinson’s disease. Trends Neurosci 2009; 32:249 - 56; http://dx.doi.org/10.1016/j.tins.2009.01.006; PMID: 19307031
- Wang JY, Wu J-N, Cherng T-L, Hoffer BJ, Chen H-H, Borlongan CV, et al. Vitamin D(3) attenuates 6-hydroxydopamine-induced neurotoxicity in rats. Brain Res 2001; 904:67 - 75; http://dx.doi.org/10.1016/S0006-8993(01)02450-7; PMID: 11516412
- de Viragh PA, Haglid KG, Celio MR. Parvalbumin increases in the caudate putamen of rats with vitamin D hypervitaminosis. Proc Natl Acad Sci U S A 1989; 86:3887 - 90; http://dx.doi.org/10.1073/pnas.86.10.3887; PMID: 2542952
- Wasserman RH. Vitamin D and the dual processes of intestinal calcium absorption. J Nutr 2004; 134:3137 - 9; PMID: 15514288
- Pérez AV, Picotto G, Carpentieri AR, Rivoira MA, Peralta López ME, Tolosa de Talamoni NG. Minireview on regulation of intestinal calcium absorption. Emphasis on molecular mechanisms of transcellular pathway. Digestion 2008; 77:22 - 34; PMID: 18277073
- Sutherland MK, Somerville MJ, Yoong LK, Bergeron C, Haussler MR, McLachlan DR. Reduction of vitamin D hormone receptor mRNA levels in Alzheimer as compared to Huntington hippocampus: correlation with calbindin-28k mRNA levels. Brain Res Mol Brain Res 1992; 13:239 - 50; http://dx.doi.org/10.1016/0169-328X(92)90032-7; PMID: 1317496
- Brewer LD, Porter NM, Kerr DS, Landfield PW, Thibault O. Chronic 1α,25-(OH)2 vitamin D3 treatment reduces Ca2+ -mediated hippocampal biomarkers of aging. Cell Calcium 2006; 40:277 - 86; http://dx.doi.org/10.1016/j.ceca.2006.04.001; PMID: 16780945