Abstract
The spliceosome discriminates against suboptimal substrates, both during assembly and catalysis, thereby enhancing specificity during pre-mRNA splicing. Central to such fidelity mechanisms are a conserved subset of the DEAD- and DEAH-box ATPases, which belong to a superfamily of proteins that mediate RNP rearrangements in almost all RNA-dependent processes in the cell. Through an investigation of the mechanisms contributing to the specificity of 5′ splice site cleavage, two related reports, one from our lab and the other from the Cheng lab, have provided insights into fidelity mechanisms utilized by the spliceosome. In our work, we found evidence for a kinetic proofreading mechanism in splicing in which the DEAH-box ATPase Prp16 discriminates against substrates undergoing slow 5′ splice site cleavage. Additionally, our study revealed that discriminated substrates are discarded through a general spliceosome disassembly pathway, mediated by another DEAH-box ATPase Prp43. In their work, Tseng et al. described the underlying molecular events through which Prp16 discriminates against a splicing substrate during 5′ splice site cleavage. Here, we present a synthesis of these two studies and, additionally, provide the first biochemical evidence for discrimination of a suboptimal splicing substrate just prior to 5′ splice site cleavage. Together, these findings support a general mechanism for a ubiquitous superfamily of ATPases in enhancing specificity during RNA-dependent processes in the cell.
Fidelity Mechanisms During Pre-mRNA Splicing
Primary transcripts of eukaryotic genes are interrupted by non-coding sequences (introns) that must be removed prior to translation of the coding sequences (exons). Intron removal and exon joining, known as pre-mRNA splicing, is catalyzed by a dynamic ribonucleoprotein complex called the spliceosome, which is composed of five snRNAs and roughly 80 conserved proteins.Citation1 The spliceosome, unlike classical enzymes, does not include a pre-formed catalytic center; instead, the spliceosome assembles on each pre-mRNA co-transcriptionallyCitation2 and then rearranges to configure the catalytic core. The catalytic core promotes splicing catalysis in two sequential phosphoryl transfer reactions: 5′ splice site cleavage and exon ligation. While the spliceosome must assemble and catalyze intron removal with sufficient speed to compete effectively with RNA exportCitation3-Citation8or nuclear turnover,Citation9-Citation12 the spliceosome must also function with sufficient specificity to prevent errors during gene expression. Given that introns are 10‒100 times longer than exons and are defined by only minimal sequence elements, establishing fidelity during splicing for the accurate and precise removal of introns is a daunting, yet essential, challenge to the cell. Indeed, the spliceosome has evolved fidelity mechanisms that allow discrimination against suboptimal substrates not only during the early stages of intron recognition but also during the later stages of splicing catalysis.
The components of the spliceosome have been demonstrated to enhance fidelity during splicing through both thermodynamic and kinetic mechanisms. In the thermodynamic mechanism, best understood during the catalytic stages of splicing, the spliceosome preferentially sequesters suboptimal substrates in a stable, non-productive conformation that is in equilibrium with the catalytic conformations of the spliceosome.Citation13 In the kinetic mechanism, also known as kinetic proofreading, the spliceosome actively rejects suboptimal substrates through branches in the splicing pathway that compete with productive splicing.Citation13,Citation14These rejection steps are ATP-dependent and mediated by members of the DEAD- and DEAH/RHA-box families (“DEAD/H-box ATPases”) belonging to the superfamily 2 (SF2) RNA helicases.Citation15 In splicing, eight of these DEAD/H-box ATPases mediate specific ribonucleoprotein (RNP) rearrangements to promote splicing of an optimal substrate. Additionally, at least five of them—Prp5, Prp28, Prp16, Prp22 and Prp43Citation8,Citation16-Citation19—have been shown to increase specificity of splicing by rejecting suboptimal substrates. For example, while Prp16, a bona fide RNA helicase,Citation20 promotes splicing of an optimal substrate by mediating repositioning of splicing intermediates for exon ligation,Citation21,Citation22 Prp16 also antagonizes substrates containing suboptimal branch sites.Citation17,Citation23,Citation24 These DEAD/H-box ATPases have been proposed to establish fidelity in splicing by a kinetic proofreading mechanism in which the DEAD/H-box ATPase promotes an optimal substrate by acting after a productive step and by rejecting a suboptimal substrate by acting before the productive step and consequently shunting the substrate down a competitive pathway (). However, it has remained unclear why DEAD/H-box ATPases act before a productive step for suboptimal substrates and after the productive step for optimal substrates—how DEAD/H-box ATPases antagonize suboptimal substrates while promoting optimal substrates. Additionally, the mechanism underlying a discard pathway that leads to elimination of rejected substrates has remained poorly understood.
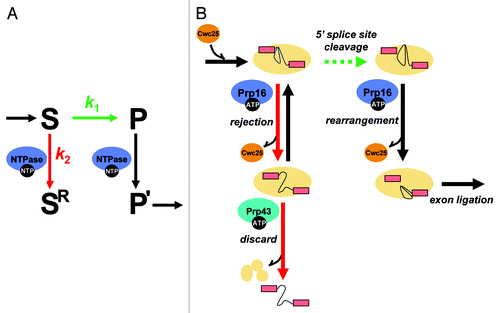
Figure 1. An ATP-dependent framework for kinetic proofreading by DEAD/H-box ATPases. (A) A general framework for kinetic proofreading by NTPases. In the kinetic proofreading scheme, an NTPase increases the specificity of a step (S → P, shown in green) by competing with it (S → SR, shown in red). In this scheme, k1 represents the rate of proceeding on-pathway, such as through 5′ splice site cleavage during pre-mRNA splicing, while k2 represents the rate of rejecting a substrate. Specificity of the step under inspection (S → P) is enhanced when the ratio of k1/k2 is higher for an optimal substrate than a suboptimal substrate. In this scheme, a proofreading NTPase not only antagonizes a suboptimal substrate (S → SR) but also promotes an optimal substrate (P → P’, shown in black), if the NTPase functions after, rather than before, the step under inspection. (B) A molecular framework by which the DEAH-box ATPase Prp16 establishes specificity during 5′ splice site cleavage. For an optimal substrate, Prp16 promotes splicing by enabling rearrangements after 5′ splice site cleavage that dissociate Cwc25, allowing repositioning of the reaction intermediates for exon ligation. For a suboptimal substrate, Prp16 antagonizes splicing, rejecting the substrate, by enabling rearrangements before 5′ splice site cleavage that similarly dissociate Cwc25 but also prime the spliceosome for discard of the substrate by the DEAH-box ATPase Prp43. In the kinetic proofreading model, specificity of 5′ splice site cleavage can be established if k5′ splice site cleavage (optimal substrate) > k5′ splice site cleavge (suboptimal substrate) and/or if krejection(suboptimal substrate) > krejection(optimal substrate). Our data provided evidence for the former case and that this proofreading NTPase functions, minimally, as a timer, setting a standard for the rate of 5′ splice site cleavage.
The DEAH-box ATPase Prp16 Discriminates Against Slow Substrates
In a kinetic proofreading model, a DEAD/H-box ATPase could compete with a productive step in splicing by one of two non-mutually exclusive mechanisms. In the first case, a DEAD/H-box ATPase would enhance specificity by sensing the authenticity of a substrate. For example, in the case of a suboptimal substrate that generally deviates from the consensus and, therefore, decreases the stability of binding to the spliceosome due to lack of proper fit, a DEAD/H-box ATPase could act preferentially before a productive step. For an optimal substrate, in contrast, proper fit of the substrate in the spliceosome could impart increased stability and, therefore, could delay the activity of DEAD/H-box ATPase until after the productive step, when the substrate association with the spliceosome would weaken thereby activating the DEAD/H-box ATPase. Consistent with this idea, the activities of DEAD/H-box ATPases have been found to be sensitive to substrate stability.Citation25-Citation28 In the second model, a DEAD/H-box ATPase would act by sensing the passage of time, enforcing a fixed, temporal window in which the substrate is permitted to proceed. In contrast to the first model where the activity of a DEAD/H-box ATPase could be modulated by a substrate, in the second model, at least in the extreme case, the DEAD/H-box ATPase would function at a constant rate, whereas the rate of splicing would vary between an optimal and a suboptimal substrate.Citation14 Because these models are not mutually exclusive, both could contribute to specificity.
While it remains to be tested whether any of the spliceosomal DEAD/H-box ATPases functions as “sensors” of substrate authenticity, we established a correlation between a slower rate of splicing and increased rejection mediated by a DEAD/H-box ATPase, strongly suggesting that DEAD/H-box ATPases generally antagonize slowly splicing substrates. Specifically, we provided evidence that Prp16 antagonizes substrates that undergo 5′ splice site cleavage slowly.Citation29
These investigations of the role of Prp16 in fidelity were inspired by earlier studies from our lab on the fidelity of exon ligation. Previously, we showed that the DEAH-box ATPase Prp22 rejects suboptimal substrates prior to exon ligation, implicating a role for Prp22 in promoting the specificity of exon ligation. Given a well-established role for Prp22 in promoting the release of mRNA from the spliceosome after exon ligation in the case of optimal substrates,Citation30,Citation31 our data suggested a model wherein Prp22 establishes the fidelity of exon ligation by acting either before exon ligation in the case of a suboptimal substrate or after exon ligation in the case of an optimal substrate. These findings also suggested that the fidelity of 5′ splice site cleavage may similarly require a DEAD/H box ATPase.
Given that Prp16 functions immediately after 5′ splice site cleavage when splicing an optimal substrate,Citation21 we hypothesized that Prp16 might function before 5′ splice site cleavage to antagonize suboptimal substrates. Indeed, while genetic studies have clearly established a role for Prp16 in rejecting suboptimal substrates containing mutations at the branch site consensus sequence during 5′ splice site cleavage (for example, mutation of the branch site UACUAAC from A to C, “brC substrate”),Citation17,Citation23,Citation24 it was unclear at what stage these substrates are rejected. Additionally, in vitro, substrates containing mutations at the branch site either showed strong defects earlier in spliceosome assemblyCitation32 or appeared to escape proofreading by Prp16 (see below),Citation33 precluding a detailed biochemical investigation of the proofreading mechanism during 5′ splice site cleavage.
To test for a role for Prp16 in competing with 5′ splice site cleavage to reject suboptimal substrates, we developed an in vitro assay in which we stalled spliceosomes reversibly just before 5′ splice site cleavage and then tested whether the defective spliceosomes were capable of undergoing splicing when the competing ATP-dependent activity of Prp16 is compromised. To reversibly stall the spliceosomes prior to 5′ splice site cleavage, we substituted sulfur for the pro-Sp non-bridging oxygen at position U80 of U6 snRNA to yield U6/sU80 (Sp), given that this modification has been shown before to disrupt Mg2+ binding just before 5′ splice site cleavage.Citation34 Because 5′ splice site cleavage in U6/sU80 (Sp) spliceosomes was rescued with more thiophilic metals such as Mn2+ or Cd2+, the pro-Sp oxygen of U6-U80 was interpreted to play a direct role in binding a Mg2+ ion important for 5′ splice site cleavage. Suggesting a direct catalytic role for this metal-ligand interaction, crystal structures of a group II intron revealed that a Mg2+ ion at the substrate binding pocket binds to a backbone oxygen atom analogous to that of the pro-Sp oxygen of U80 in U6 snRNA.Citation35,Citation36 Indeed, recent findings indicate that this ligand of U80 interacts directly with the 5′ splice site, as well as the 3′ splice site, establishing a direct role for this divalent-metal ligand in catalysis.Citation37
In our test for a proofreading activity, we indeed observed an ATP- and Prp16-dependent pathway that competes with 5′ splice site cleavage and that rejects defective spliceosomes.Citation29 Specifically, when we incubated stalled and partially purified U6/sU80 (Sp) spliceosomes in ATP and Mg2+, Mn2+ or Cd2+, we observed the expected metal rescue behavior: 5′ splice site cleavage remained repressed in Mg2+ but became rescued in Mn2+ or Cd.Citation2 However, in the absence of ATP, or in the presence of ATPase-defective variants of Prp16, we observed 5′ splice site cleavage in Mg2+, in addition to Mn2+ and Cd2+. Thus, the ATP-dependent activity of Prp16 specifically represses splicing in suboptimal spliceosomes lacking the critical interaction between a divalent metal ion and the pro-Sp non-bridging oxygen atom of U80 in U6 snRNA. While Prp16 failed to repress 5′ splice site cleavage in optimal spliceosomes having this critical metal-ligand interaction restored, Prp16 promoted splicing in these spliceosomes after 5′ splice site cleavage by triggering rearrangements necessary for exon ligation, validating the specificity of Prp16-mediated rejection for suboptimal substrates.Citation29 Importantly, while Prp16 is not required to bind the spliceosomes until after 5′ splice site cleavage when splicing an optimal substrate,Citation21 we observed that Prp16 can bind spliceosomes prior to 5′splice site cleavage when splicing is suboptimal, as predicted by its role prior to 5′ cleavage.Citation29 Together with previous experiments showing a role for Prp16 in antagonizing suboptimal branch site substratesCitation17,Citation23,Citation24 and in rearranging spliceosomes to the second catalytic conformation,Citation21,Citation22 our data emphasize the dual roles for Prp16 in splicing.
How does Prp16 promote splicing of an optimal substrate after 5′ splice site cleavage, while rejecting a suboptimal substrate before 5′ splice site cleavage? A strong prediction of the timer model of kinetic proofreading for 5′ splice site cleavage is that the rate of 5′ splice site cleavage of a suboptimal substrate is significantly slower than that of an optimal substrate. Consistent with this prediction, we observed that U6/sU80 (Sp) spliceosomes catalyzed 5′ splice site cleavage 10-fold slower in the suboptimal Mg2+ conditions as compared with the optimal Mn2+ conditions. Thus, a slower rate of 5′ splice site cleavage correlates with sensitivity to the ATP- and Prp16-dependent rejection pathway. Another prediction of the timer model is that the rate of Prp16-dependent rejection will be faster than the rate of 5′ splice site cleavage for suboptimal spliceosomes and slower than that for optimal spliceosomes, a prediction which remains to be tested. Nevertheless, our results suggest that DEAD/H-box ATPases in general discriminate against slow substrates during pre-mRNA splicing.
Molecular Events Underlying Fidelity Mechanisms Mediated by Spliceosomal DEAD/H-box ATPases
What is the molecular basis of fidelity mechanisms promoted by spliceosomal DEAD/H-box ATPases? While the detailed mechanisms by which spliceosomal DEAD/H-box ATPases function remain poorly understood, recent biochemical studies provide important clues to the molecular events mediated by a number of spliceosomal DEAD/H-box ATPases.Citation38-Citation46 With regards to Prp16 function, Tseng et al.Citation33 investigated protein dynamics during 5′ splice site cleavage, and provided important insights into the molecular events underlying Prp16-mediated proofreading of 5′ splice site cleavage. They observed that Prp16 promotes the release of two splicing factors essential for 5′ splice site cleavage, Yju2 and Cwc25, after 5′ splice site cleavage.Citation33 This observation is consistent with the findings from a proteomic study that spliceosomes stalled at the 5′ splice site cleavage stage retain Yju2 and Cwc25, while spliceosomes stalled at the stage of exon ligation lacked both Yju2 and Cwc25.Citation42 Additionally, when spliceosomes are assembled on a suboptimal brC substrate, Tseng et al. observed that Prp16 becomes essential for 5′ splice site cleavage, consistent with our observation that Prp16 can associate with pre-mRNA prior to 5′ splice site cleavage. Further, Tseng et al. observed binding of Prp16 and Cwc25 to the suboptimal brC pre-mRNA, but only in the absence of ATP, implying that ATP mediates dissociation of these factors from the brC pre-mRNA. Together, these data suggest a broad molecular framework for the timer model of kinetic proofreading in which Prp16 promotes an optimal substrate that proceeds rapidly by enabling rearrangements after 5′ splice site cleavage that dissociate Yju2 and Cwc25, allowing repositioning of the reaction intermediates for exon ligation and, in which Prp16 antagonizes a suboptimal substrate that splices slowly by enabling rearrangements before 5′ splice site cleavage that also dissociate Yju2 and Cwc25 ().
However, the observation of Tseng et al. that Prp16 promotes splicing of the suboptimal brC substrate appeared to be contradictory to a number of previous genetic studies.Citation17,Citation23,Citation24,Citation47 For example, Burgess and Guthrie observed that, in vivo, for splicing reporters containing mutations at the branch site, including the brC mutation, the efficiency of splicing is higher in a mutant prp16 background compared with that in a wild-type PRP16 background, thus providing the first evidence for a role for DEAD/H-box ATPases in pre-mRNA splicing fidelity—specifically, evidence that wild-type Prp16 antagonizes substrates with branch site mutations.Citation17 Tseng et al., however, failed to observe in vitro improved splicing efficiency for the brC substrate in the presence of mutated Prp16 and, therefore, failed to observe evidence for Prp16-mediated proofreading. Instead, they observed that Prp16, whether wild-type or mutated, promoted 5′ splice site cleavage of the brC substrate. Importantly, Tseng et al. found that Prp16 promoted splicing of the brC substrate by improving the association of Cwc25 with the brC substrate, an interaction they observed is weakened by the brC mutation.Citation33
In an effort to resolve these apparently opposing observations, we considered that the weakened binding of Cwc25 to the mutated branch site provides a potential path for reconciling the published data. Specifically, in the in vitro system of Tseng et al., the binding of Cwc25 to the mutated branch site may be rate limiting for 5′ splice site cleavage, reflecting an independent fidelity mechanismCitation47 and, thereby, mask a subsequent proofreading step in which the mutated brC substrate is antagonized by the ATP-dependent activity of Prp16. Further, we hypothesized that we could circumvent this limitation by accumulating spliceosomes assembled on the brC substrate after stable binding of Cwc25 but before 5′ splice site cleavage, thereby allowing a test for Prp16-dependent proofreading of the brC substrate in vitro.
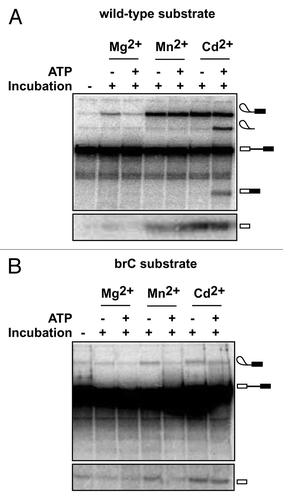
To test this prediction, we stalled and accumulated spliceosomes at the 5′ splice site cleavage stage on either wild-type or brC substrates in yeast extract reconstituted with the U6/sU80 (Sp) modification, as described above. Then, we affinity purified the stalled spliceosomes to remove ATP and further incubated the stalled spliceosomes in Mg2+, Mn2+ or Cd2+, to test for metal rescue, and with or without ATP, to test for ATP- and Prp16-dependent repression of 5′ splice site cleavage.Citation29 As previously observed for U6/sU80 (Sp) spliceosomes assembled on a wild-type substrate, 5′ splice site cleavage in the absence of ATP was observed in Mg2+, Mn2+ and Cd2+, whereas 5′ splice site cleavage in the presence of ATP was observed in Mn2+ and Cd2+ but not in Mg2+, reflecting rejection by Prp16 of the suboptimal Mg2+-bound spliceosomes ().Citation29 With U6/sU80 (Sp) spliceosomes assembled on a brC substrate, splicing was not observed in Mg2+, indicating that disruption of the metal-ligand interaction involving U80 was not tolerated in the context of the brC mutation. More importantly, in the absence of ATP 5′ splice site cleavage in these spliceosomes was well above background levels in both Mn2+ and Cd2+ (). In contrast, in the presence of ATP 5′ splice site cleavage was observed only in Cd2+ and not in Mn2+, indicating that ATP reduced the efficiency of splicing by at least 2-fold in Mn2+. Thus, in Mn2+ the ATP-dependent activity of Prp16 competes with 5′ splice site cleavage specifically for the brC substrate but not the wild-type substrate. While in the context of a modified spliceosome, this result demonstrates in vitro for the first time that Prp16 can discriminate a suboptimal substrate from an optimal substrate prior to 5′ splice site cleavage.
Figure 2. ATP-dependent discrimination of a suboptimal substrate containing a mutation at the branch site prior to 5′ splice site cleavage. (A) An ATP-dependent activity, reflecting the action of Prp16, competes with 5′ splice site cleavage and rejects spliceosomes containing the U6/sU80 (Sp) modification in Mg2+ (ref. Citation29). Spliceosomes were assembled on wild-type ACT1 pre-mRNA in extracts reconstituted with U6 containing the sU80 (Sp) modification, affinity-purified using TAP-tagged Prp19—an integral component of catalytically activated spliceosomes,Citation55 and then incubated under the indicated conditions and assayed for splicing, as described.Citation29 Two different regions of the same gel are shown. (B) ATP-dependent discrimination of a suboptimal substrate containing an A to C mutation of the branch site nucleophile. Spliceosomes were assembled as in A except using an ACT1 pre-mRNA containing the branch site A to C mutation (brC). The efficiency of 5′ splice site cleavage for the brC substrate in Mn2+ was reduced to background levels in the presence of ATP; specifically, ATP reduced 5′ splice site cleavage efficiency, calculated as the ratio of lariat intermediate/(lariat intermediate + pre-mRNA), by a factor 2.00 ± 0.04, with the error representing the standard deviation for two independent experiments. Two different regions of the same gel are shown.
A General Spliceosome Disassembly Pathway that Discards Suboptimal Substrates, in Addition to the Excised Intron
While proofreading of on-pathway splicing steps is necessary for discrimination against suboptimal substrates, it is not sufficient for the discard of rejected substrates from the spliceosome for turnover. Indeed, the rejection step mediated by DEAD/H-box ATPase Prp16, and perhaps others, appears reversible,Citation29 necessitating a downstream discard pathway that results not only in discard of the rejected substrates but also recycling of stalled snRNP components for further rounds of splicing. While nuclear surveillance pathways can compete with splicing and eliminate suboptimal pre-mRNA stalled on spliceosomes,Citation9-Citation12 accumulating evidence indicate that rejected pre-mRNA and splicing intermediates are alternatively released through a spliceosome disassembly pathway, exported to the cytoplasm, and turned over by cytoplasmic mRNA surveillance pathways.Citation7,Citation8,Citation14
Recent studies implicate a central role for the DEAH-box ATPase, Prp43, in releasing suboptimal substrates stalled in spliceosomal complexes, in addition to its canonical function in discarding the excised intron and disassembling spliceosomes for recycling. Specifically, when spliceosomes are stalled due to assembly defects resulting from mutations in PRP38 or PRP8 splicing factors, mutations in PRP43 or its cofactor NTR1/SPP382 suppress spliceosome assembly defects—the first hint of a role for Prp43 in disassembling stalled spliceosomes.Citation48 Through in vitro experiments, we were able to establish such a discard role by observing directly that Prp43 is required for release of pre-mRNA rejected by Prp16Citation29 and for release of splicing intermediates rejected prior to 3′ splice site cleavage.Citation8 Through an analysis of spliceosomes stalled at different stages of the splicing cycle, a recent study from the Cheng lab emphasizes the importance of the disassembly pathway mediated by Prp43, and provides insights into the mechanism by which the discard pathway eliminates slowly splicing substrates specifically.Citation49 Notably, Prp43-mediated disassembly of stalled spliceosomal complexes promotes fidelity, at least during 3′ splice site cleavage,Citation8 demonstrating the importance of coupling discard to rejection.
The Prp43-mediated disassembly of rejected spliceosomal complexes parallels the disassembly pathway of the spliceosome at the end of a canonical splicing reaction. Interestingly, this general spliceosome disassembly pathway seems to function analogously to a general disassembly pathway of the ribosome in which ribosomes stalled prematurely due to defects in translation elongation undergo disassembly parallel to the canonical disassembly pathway at the end of translation.Citation14 Given that Prp43 acts as a general splicing terminator, and its activity is modulated by cofactors,Citation50-Citation52 Prp43 represents a potential regulatory link between cellular or extra-cellular cues and pre-mRNA splicing. Consistent with a potential role for Prp43 in regulating splicing at the intermediate stage, a genome-wide analysis in S. cerevisiae identified endogenous, de-branched lariat intermediates,Citation53 indicating a discard of splicing intermediates before exon ligation that could be regulated to modulate gene expression. Further, in S. pombe, Prp43 is required to terminate splicing of the TER1 transcript at the intermediate stage to promote biogenesis of telomerase RNA, which corresponds to a 5′ exon.Citation54
Summary
To summarize, key advances in in vitro splicing experiments have provided important insights into the mechanism by which spliceosomes ensure fidelity during pre-mRNA splicing. These experiments have reinforced a central role for DEAD/H-box ATPases in pre-mRNA splicing fidelity and have revealed a two-tiered strategy in antagonizing and discarding slowly splicing suboptimal substrates from the spliceosome. Prp16, the first splicing fidelity factor discovered, continues to play a leading role in these advances. A complete understanding of the mechanism of splicing fidelity awaits further breakthroughs that will permit the most rigorous tests of the kinetic proofreading model.
| Abbreviations: | ||
| Prp | = | Pre-mRNA processing |
| ATPase | = | Adenosinetriphosphatase |
| snRNA | = | small nuclear RNA |
Acknowledgments
We thank D. Semlow and S. Fica for critically reading the manuscript, and S. Fica for reagents. This work was supported by a grant to J.P.S. from the US. National Institutes of Health (R01GM062264).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Wahl MC, Will CL, Lührmann R. The spliceosome: design principles of a dynamic RNP machine. Cell 2009; 136:701 - 18; http://dx.doi.org/10.1016/j.cell.2009.02.009; PMID: 19239890
- Perales R, Bentley D. “Cotranscriptionality”: the transcription elongation complex as a nexus for nuclear transactions. Mol Cell 2009; 36:178 - 91; http://dx.doi.org/10.1016/j.molcel.2009.09.018; PMID: 19854129
- Legrain P, Rosbash M. Some cis- and trans-acting mutants for splicing target pre-mRNA to the cytoplasm. Cell 1989; 57:573 - 83; http://dx.doi.org/10.1016/0092-8674(89)90127-X; PMID: 2655924
- Rain JC, Legrain P. In vivo commitment to splicing in yeast involves the nucleotide upstream from the branch site conserved sequence and the Mud2 protein. EMBO J 1997; 16:1759 - 71; http://dx.doi.org/10.1093/emboj/16.7.1759; PMID: 9130720
- Rutz B, Séraphin B. A dual role for BBP/ScSF1 in nuclear pre-mRNA retention and splicing. EMBO J 2000; 19:1873 - 86; http://dx.doi.org/10.1093/emboj/19.8.1873; PMID: 10775271
- Jaillon O, Bouhouche K, Gout JF, Aury JM, Noel B, Saudemont B, et al. Translational control of intron splicing in eukaryotes. Nature 2008; 451:359 - 62; http://dx.doi.org/10.1038/nature06495; PMID: 18202663
- Hilleren PJ, Parker R. Cytoplasmic degradation of splice-defective pre-mRNAs and intermediates. Mol Cell 2003; 12:1453 - 65; http://dx.doi.org/10.1016/S1097-2765(03)00488-X; PMID: 14690599
- Mayas RM, Maita H, Semlow DR, Staley JP. Spliceosome discards intermediates via the DEAH box ATPase Prp43p. Proc Natl Acad Sci USA 2010; 107:10020 - 5; http://dx.doi.org/10.1073/pnas.0906022107; PMID: 20463285
- Bousquet-Antonelli C, Presutti C, Tollervey D. Identification of a regulated pathway for nuclear pre-mRNA turnover. Cell 2000; 102:765 - 75; http://dx.doi.org/10.1016/S0092-8674(00)00065-9; PMID: 11030620
- Lemieux C, Marguerat S, Lafontaine J, Barbezier N, Bähler J, Bachand F. A Pre-mRNA degradation pathway that selectively targets intron-containing genes requires the nuclear poly(A)-binding protein. Mol Cell 2011; 44:108 - 19; http://dx.doi.org/10.1016/j.molcel.2011.06.035; PMID: 21981922
- Davidson L, Kerr A, West S. Co-transcriptional degradation of aberrant pre-mRNA by Xrn2. EMBO J 2012; 31:2566 - 78; http://dx.doi.org/10.1038/emboj.2012.101; PMID: 22522706
- Egecioglu DE, Kawashima TR, Chanfreau GF. Quality control of MATa1 splicing and exon skipping by nuclear RNA degradation. Nucleic Acids Res 2012; 40:1787 - 96; http://dx.doi.org/10.1093/nar/gkr864; PMID: 22021379
- Smith DJ, Query CC, Konarska MM. “Nought may endure but mutability”: spliceosome dynamics and the regulation of splicing. Mol Cell 2008; 30:657 - 66; http://dx.doi.org/10.1016/j.molcel.2008.04.013; PMID: 18570869
- Semlow DR, Staley JP. Staying on message: ensuring fidelity in pre-mRNA splicing. Trends Biochem Sci 2012; 37:263 - 73; http://dx.doi.org/10.1016/j.tibs.2012.04.001; PMID: 22564363
- Jankowsky E. RNA helicases at work: binding and rearranging. Trends Biochem Sci 2011; 36:19 - 29; http://dx.doi.org/10.1016/j.tibs.2010.07.008; PMID: 20813532
- Xu YZ, Query CC. Competition between the ATPase Prp5 and branch region-U2 snRNA pairing modulates the fidelity of spliceosome assembly. Mol Cell 2007; 28:838 - 49; http://dx.doi.org/10.1016/j.molcel.2007.09.022; PMID: 18082608
- Burgess SM, Guthrie C. A mechanism to enhance mRNA splicing fidelity: the RNA-dependent ATPase Prp16 governs usage of a discard pathway for aberrant lariat intermediates. Cell 1993; 73:1377 - 91; http://dx.doi.org/10.1016/0092-8674(93)90363-U; PMID: 8324826
- Mayas RM, Maita H, Staley JP. Exon ligation is proofread by the DExD/H-box ATPase Prp22p. Nat Struct Mol Biol 2006; 13:482 - 90; http://dx.doi.org/10.1038/nsmb1093; PMID: 16680161
- Yang F, Wang XY, Zhang ZM, Pu J, Fan YJ, Zhou J, et al. Splicing proofreading at 5′ splice sites by ATPase Prp28p. Nucleic Acids Res 2013; 41:4660 - 70; http://dx.doi.org/10.1093/nar/gkt149; PMID: 23462954
- Wang Y, Wagner JD, Guthrie C. The DEAH-box splicing factor Prp16 unwinds RNA duplexes in vitro. Curr Biol 1998; 8:441 - 51; http://dx.doi.org/10.1016/S0960-9822(98)70178-2; PMID: 9550699
- Schwer B, Guthrie C. PRP16 is an RNA-dependent ATPase that interacts transiently with the spliceosome. Nature 1991; 349:494 - 9; http://dx.doi.org/10.1038/349494a0; PMID: 1825134
- Schwer B, Guthrie C. A conformational rearrangement in the spliceosome is dependent on PRP16 and ATP hydrolysis. EMBO J 1992; 11:5033 - 9; PMID: 1464325
- Query CC, Konarska MM. Suppression of multiple substrate mutations by spliceosomal prp8 alleles suggests functional correlations with ribosomal ambiguity mutants. Mol Cell 2004; 14:343 - 54; http://dx.doi.org/10.1016/S1097-2765(04)00217-5; PMID: 15125837
- Villa T, Guthrie C. The Isy1p component of the NineTeen complex interacts with the ATPase Prp16p to regulate the fidelity of pre-mRNA splicing. Genes Dev 2005; 19:1894 - 904; http://dx.doi.org/10.1101/gad.1336305; PMID: 16103217
- Bhaskaran H, Russell R. Kinetic redistribution of native and misfolded RNAs by a DEAD-box chaperone. Nature 2007; 449:1014 - 8; http://dx.doi.org/10.1038/nature06235; PMID: 17960235
- Rogers GW Jr., Richter NJ, Merrick WC. Biochemical and kinetic characterization of the RNA helicase activity of eukaryotic initiation factor 4A. J Biol Chem 1999; 274:12236 - 44; http://dx.doi.org/10.1074/jbc.274.18.12236; PMID: 10212190
- Tanaka N, Schwer B. Characterization of the NTPase, RNA-binding, and RNA helicase activities of the DEAH-box splicing factor Prp22. Biochemistry 2005; 44:9795 - 803; http://dx.doi.org/10.1021/bi050407m; PMID: 16008364
- Pisareva VP, Pisarev AV, Komar AA, Hellen CU, Pestova TV. Translation initiation on mammalian mRNAs with structured 5’UTRs requires DExH-box protein DHX29. Cell 2008; 135:1237 - 50; http://dx.doi.org/10.1016/j.cell.2008.10.037; PMID: 19109895
- Koodathingal P, Novak T, Piccirilli JA, Staley JP. The DEAH box ATPases Prp16 and Prp43 cooperate to proofread 5′ splice site cleavage during pre-mRNA splicing. Mol Cell 2010; 39:385 - 95; http://dx.doi.org/10.1016/j.molcel.2010.07.014; PMID: 20705241
- Schwer B, Gross CH. Prp22, a DExH-box RNA helicase, plays two distinct roles in yeast pre-mRNA splicing. EMBO J 1998; 17:2086 - 94; http://dx.doi.org/10.1093/emboj/17.7.2086; PMID: 9524130
- Wagner JD, Jankowsky E, Company M, Pyle AM, Abelson JN. The DEAH-box protein PRP22 is an ATPase that mediates ATP-dependent mRNA release from the spliceosome and unwinds RNA duplexes. EMBO J 1998; 17:2926 - 37; http://dx.doi.org/10.1093/emboj/17.10.2926; PMID: 9582286
- Vijayraghavan U, Parker R, Tamm J, Iimura Y, Rossi J, Abelson J, et al. Mutations in conserved intron sequences affect multiple steps in the yeast splicing pathway, particularly assembly of the spliceosome. EMBO J 1986; 5:1683 - 95; PMID: 3017708
- Tseng CK, Liu HL, Cheng SC. DEAH-box ATPase Prp16 has dual roles in remodeling of the spliceosome in catalytic steps. RNA 2011; 17:145 - 54; http://dx.doi.org/10.1261/rna.2459611; PMID: 21098140
- Yean SL, Wuenschell G, Termini J, Lin RJ. Metal-ion coordination by U6 small nuclear RNA contributes to catalysis in the spliceosome. Nature 2000; 408:881 - 4; http://dx.doi.org/10.1038/35048617; PMID: 11130730
- Toor N, Keating KS, Taylor SD, Pyle AM. Crystal structure of a self-spliced group II intron. Science 2008; 320:77 - 82; http://dx.doi.org/10.1126/science.1153803; PMID: 18388288
- Marcia M, Pyle AM. Visualizing group II intron catalysis through the stages of splicing. Cell 2012; 151:497 - 507; http://dx.doi.org/10.1016/j.cell.2012.09.033; PMID: 23101623
- Fica SM, Tuttle N, Novak T, Li N-S, Lu J, Koodathingal P, et al. Direct evidence that RNA catalyzes nuclear pre-mRNA splicing; submitted.
- Warkocki Z, Odenwälder P, Schmitzová J, Platzmann F, Stark H, Urlaub H, et al. Reconstitution of both steps of Saccharomyces cerevisiae splicing with purified spliceosomal components. Nat Struct Mol Biol 2009; 16:1237 - 43; http://dx.doi.org/10.1038/nsmb.1729; PMID: 19935684
- Perriman R, Ares M Jr.. Invariant U2 snRNA nucleotides form a stem loop to recognize the intron early in splicing. Mol Cell 2010; 38:416 - 27; http://dx.doi.org/10.1016/j.molcel.2010.02.036; PMID: 20471947
- Hahn D, Kudla G, Tollervey D, Beggs JD. Brr2p-mediated conformational rearrangements in the spliceosome during activation and substrate repositioning. Genes Dev 2012; 26:2408 - 21; http://dx.doi.org/10.1101/gad.199307.112; PMID: 23124065
- Mozaffari-Jovin S, Santos KF, Hsiao HH, Will CL, Urlaub H, Wahl MC, et al. The Prp8 RNase H-like domain inhibits Brr2-mediated U4/U6 snRNA unwinding by blocking Brr2 loading onto the U4 snRNA. Genes Dev 2012; 26:2422 - 34; http://dx.doi.org/10.1101/gad.200949.112; PMID: 23124066
- Lardelli RM, Thompson JX, Yates JR 3rd, Stevens SW. Release of SF3 from the intron branchpoint activates the first step of pre-mRNA splicing. RNA 2010; 16:516 - 28; http://dx.doi.org/10.1261/rna.2030510; PMID: 20089683
- Schwer B. A conformational rearrangement in the spliceosome sets the stage for Prp22-dependent mRNA release. Mol Cell 2008; 30:743 - 54; http://dx.doi.org/10.1016/j.molcel.2008.05.003; PMID: 18570877
- Chen JY, Stands L, Staley JP, Jackups RR Jr., Latus LJ, Chang TH. Specific alterations of U1-C protein or U1 small nuclear RNA can eliminate the requirement of Prp28p, an essential DEAD box splicing factor. Mol Cell 2001; 7:227 - 32; http://dx.doi.org/10.1016/S1097-2765(01)00170-8; PMID: 11172727
- Shen H, Zheng X, Shen J, Zhang L, Zhao R, Green MR. Distinct activities of the DExD/H-box splicing factor hUAP56 facilitate stepwise assembly of the spliceosome. Genes Dev 2008; 22:1796 - 803; http://dx.doi.org/10.1101/gad.1657308; PMID: 18593880
- Kistler AL, Guthrie C. Deletion of MUD2, the yeast homolog of U2AF65, can bypass the requirement for sub2, an essential spliceosomal ATPase. Genes Dev 2001; 15:42 - 9; http://dx.doi.org/10.1101/gad.851301; PMID: 11156604
- Horowitz DS. The splice is right: guarantors of fidelity in pre-mRNA splicing. RNA 2011; 17:551 - 4; http://dx.doi.org/10.1261/rna.2577511; PMID: 21357751
- Pandit S, Lynn B, Rymond BC. Inhibition of a spliceosome turnover pathway suppresses splicing defects. Proc Natl Acad Sci USA 2006; 103:13700 - 5; http://dx.doi.org/10.1073/pnas.0603188103; PMID: 16945917
- Chen HC, Tseng CK, Tsai RT, Chung CS, Cheng SC. Link of NTR-mediated spliceosome disassembly with DEAH-box ATPases Prp2, Prp16, and Prp22. Mol Cell Biol 2013; 33:514 - 25; http://dx.doi.org/10.1128/MCB.01093-12; PMID: 23166295
- Tsai RT, Fu RH, Yeh FL, Tseng CK, Lin YC, Huang YH, et al. Spliceosome disassembly catalyzed by Prp43 and its associated components Ntr1 and Ntr2. Genes Dev 2005; 19:2991 - 3003; http://dx.doi.org/10.1101/gad.1377405; PMID: 16357217
- Boon KL, Auchynnikava T, Edwalds-Gilbert G, Barrass JD, Droop AP, Dez C, et al. Yeast ntr1/spp382 mediates prp43 function in postspliceosomes. Mol Cell Biol 2006; 26:6016 - 23; http://dx.doi.org/10.1128/MCB.02347-05; PMID: 16880513
- Tanaka N, Aronova A, Schwer B. Ntr1 activates the Prp43 helicase to trigger release of lariat-intron from the spliceosome. Genes Dev 2007; 21:2312 - 25; http://dx.doi.org/10.1101/gad.1580507; PMID: 17875666
- Harigaya Y, Parker R. Global analysis of mRNA decay intermediates in Saccharomyces cerevisiae. Proc Natl Acad Sci USA 2012; 109:11764 - 9; http://dx.doi.org/10.1073/pnas.1119741109; PMID: 22752303
- Kannan R, Hartnett S, Voelker RB, Berglund JA, Staley JP, Baumann P. Intronic sequence elements impede exon ligation and trigger a discard pathway that yields functional telomerase RNA in fission yeast. Genes Dev 2013; 27:627 - 38; http://dx.doi.org/10.1101/gad.212738.112; PMID: 23468430
- Chan SP, Kao DI, Tsai WY, Cheng SC. The Prp19p-associated complex in spliceosome activation. Science 2003; 302:279 - 82; http://dx.doi.org/10.1126/science.1086602; PMID: 12970570