
Abstract
The RNA chaperone Hfq is a global post-transcriptional regulator in bacteria. Here, we used RNAseq to analyze RNA populations from the legume symbiont Sinorhizobium meliloti that were co-immunoprecipitated (CoIP-RNA) with a FLAG-tagged Hfq in five growth/stress conditions. Hfq-bound transcripts (1315) were largely identified in stressed bacteria and derived from small RNAs (sRNAs), both trans-encoded (6.4%) and antisense (asRNAs; 6.3%), and mRNAs (86%). Pull-down with Hfq recovered a small proportion of annotated S. meliloti sRNAs (14% of trans-sRNAs and 2% of asRNAs) suggesting a discrete impact of this protein in sRNA pathways. Nonetheless, Hfq selectively stabilized CoIP-enriched sRNAs, anticipating that these interactions are functionally significant. Transcription of 26 Hfq-bound sRNAs was predicted to occur from promoters recognized by the major stress σ factors σE2 or σH1/2. Recovery rates of sRNAs in each of the CoIP–RNA libraries suggest a large impact of Hfq-assisted riboregulation in S. meliloti osmoadaptation. Hfq directly targeted 18% of the predicted S. meliloti mRNAs, which encode functionally diverse proteins involved in transport and metabolism, σE2-dependent stress responses, quorum sensing, flagella biosynthesis, ribosome, and membrane assembly or symbiotic nitrogen fixation. Canonical targeting of the 5′ regions of two of the ABC transporter mRNAs by the homologous Hfq-binding AbcR1 and AbcR2 sRNAs leading to inhibition of protein synthesis was confirmed in vivo. We therefore provide a comprehensive resource for the systems-level deciphering of hitherto unexplored S. meliloti stress and symbiotic post-transcriptional regulons and the identification of Hfq-dependent sRNA–mRNA regulatory pairs.
Introduction
Deep sequencing of cDNA libraries (RNAseq) has uncovered an unexpected complexity in the structure and regulation of prokaryotic transcriptomes.Citation1 A significant proportion of the bacterial transcriptional output consists of small untranslated RNA molecules (sRNAs) that mostly act as post-transcriptional regulators of gene expression to fine-tune the cellular responses to changing environments.Citation2 A large fraction of these sRNAs modulate translation and/or turnover rates of trans-encoded target mRNAs by short and discontinuous antisense interactions, which are commonly assisted by the widespread RNA chaperone Hfq.Citation2-Citation4
The impact of Hfq in the cell physiology has been addressed by reverse genetics approaches in bacterial species representing widely diverse lifestyles (for a review, see ref. Citation5). In the vast majority of cases, loss of Hfq compromises bacterial fitness to the environment altering a wide range of phenotypes, e.g., growth kinetics, cell morphology, motility, adaptation to abiotic stresses, host colonization, or virulence. High-throughput profiling of Hfq RNA ligands in some of these bacteria has revealed that the RNA-binding capacity of this protein extends beyond its association with trans-encoded sRNAs (trans-sRNAs) and their mRNA partners to target transcripts expressed antisense to protein coding genes (i.e., cis-encoded antisense RNAs; asRNA) and even tRNAs.Citation6-Citation13 Moreover, post-transcriptional regulation of certain Hfq-associated mRNAs can be exerted by the protein itself in a sRNA-independent manner involving translational repression or regulation of mRNA decay dependent on polyadenylation.Citation14,Citation15
The soil-dwelling α-proteobacterium Sinorhizobium meliloti belongs to the group of microorganisms collectively referred to as rhizobia that fix nitrogen in endosymbiosis with legume plants. Rhizobia induce the de novo formation of nodules on the roots of their cognate legumes, where invading bacteria are accommodated intracellularly upon a profound morphological differentiation to their nitrogen-fixing competent form, the bacteroids.Citation16 Successful transition of rhizobia from a competitive free-living state in soil to their final intracellular residence within nodules involves extensive transcriptional reprogramming of gene expression that has been intensively investigated.Citation16 In S. meliloti, transcriptional control of adaptive responses to changing environments can be exerted by a large set of transcription factors (8.7% of the S. meliloti protein-coding genes) and 15 alternative RNA polymerase holoenzymes (σ factors), of which 11 belong to the extracytoplasmic function (ECF) subfamily.Citation17 RpoH1/2 (σH1/2 homologous to E. coli heat shock σ32) and the ECF σ factor RpoE2 (σE2, functionally analogous to the enterobacterial global stress regulator RpoS) have been shown to be major regulators of the general (stationary phase), heat shock, and hyperosmotic stress responses.Citation18-Citation23 However, the impact of post-transcriptional regulation in free-living and symbiotic gene expression has remained largely unexplored.
Random chemical mutagenesis screenings in the legume symbiont Azorhizobium caulinodans early identified Hfq as an activator of translation of one of the master regulators of nitrogen-fixation, NifA.Citation24 Such a positive contribution of Hfq to the post-transcriptional regulation of nifA has also been observed in other free-living and symbiotic α-proteobacterial diazotrophs.Citation25,Citation26 More recently, a series of studies on S. meliloti revealed a much broader regulatory role of Hfq in rhizobia. Deletion of the chromosomal S. meliloti hfq gene resulted in delayed growth, alterations in the metabolism of certain nitrogen sources, changes in the profiles of quorum sensing (QS) signals, reduced swarming and swimming motilities, and increased vulnerability to oxidative, heat, or pH stresses in free-living bacteria.Citation27-Citation31 This pleiotropic phenotype is partially explained by the large Hfq-dependent changes in gene expression observed in cultured bacteria. Remarkably, the S. meliloti hfq mutants evidenced massive misregulation of transport systems, which anticipates a major role of Hfq in controlling the metabolic versatility of this bacterium.Citation28,Citation29,Citation31,Citation32 Not unexpectedly, this global influence of Hfq on S. meliloti physiology was also found to compromise an array of symbiotic traits, including competitiveness for infection, nodule development, intracellular survival of bacteroids, and efficiency of the nitrogen fixation process.Citation27-Citation30 Similarly, hfq mutants of animal and plant pathogens closely related to S. meliloti (e.g., Brucella sp. and Agrobacterium tumefaciens) have been also shown to be severely impaired in virulence, further evidencing a universal role of Hfq in the establishment and maintenance of chronic microbial infections on eukaryotic hosts.Citation33-Citation35
As reported for other bacteria, at least part of the rhizobial Hfq-dependent pathways are expected to be post-transcriptionally regulated by the concerted activity of Hfq and its cognate partner sRNAs. Several computational comparative genomics and high-throughput surveys (i.e., microarray hybridyzation and RNAseq) have revealed the complexity of the S. meliloti non-coding RNome.Citation23,Citation36-Citation40 The biological functions of the vast majority of the identified sRNAs remain to be elucidated, although a few have been already shown to bind Hfq.Citation29,Citation41,Citation42 In this work we have used a generic strategy relying on the combination of co-immunoprecipitation (CoIP) of an epitope-tagged Hfq and its associated RNA with RNAseq to generate an atlas of S. meliloti Hfq-bound transcripts. Our data reveal that major S. meliloti stress response and symbiotic gene networks are extensively subjected to Hfq-mediated post-transcriptional regulation.
Results
Identification of S. meliloti Hfq RNA targets
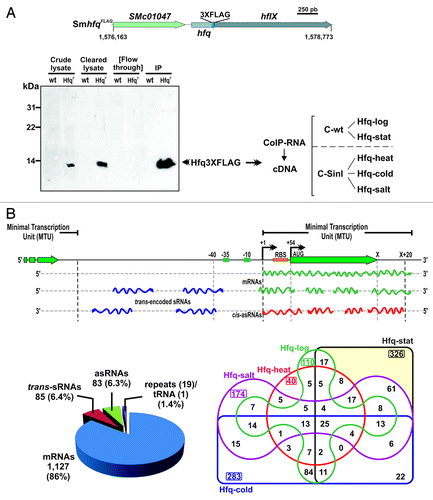
To comprehensively identify Hfq-bound transcripts in S. meliloti strain Sm2B3001, RNA was co-immunoprecipitated with the chromosomally encoded FLAG epitope-tagged Hfq variant (HfqFLAG) expressed by the Sm2B3001 derivative strain SmhfqFLAG. HfqFLAG was specifically recognized by monoclonal anti-FLAG antibodies in cell extracts of the recombinant strain and fully recovered in the procedure (). CoIP–RNA populations derived from bacteria grown under five different conditions (pools of two biological replicates per condition) were reverse transcribed to generate strand-specific cDNA libraries, henceforth referred to as Hfq-log (exponential growth phase TY cultures), Hfq-stat (stationary TY cultures), Hfq-cold (cold shock), Hfq-heat (heat shock), and Hfq-salt (osmotic upshift). Simultaneously, CoIP–RNA from exponential and stationary phase TY cultures of Sm2B3001 (wild-type) and from SmpWsinIFLAG (expressing a FLAG-tagged SinI protein, which has no known RNA-binding activity) upon cold, heat, and salt shocks, were pooled to obtain two additional cDNA libraries termed C-wt and C-SinI, respectively. Both strains, Sm2B3001 and SmpWsinIFLAG, encode an untagged Hfq and were used as controls to assess unspecific RNA binding to either the agarose beads used for CoIP or the FLAG tag itself.
Figure 1. Identification of Hfq-binding transcripts in S. meliloti. (A) Experimental setup. Chromosomal hfq region in S. meliloti with indication of tagging in the Sm2B3001 derivative strain SmhfqFLAG (coordinates according to the annotation of the reference S. meliloti 1021 genome). Below is a western blot probed with Anti-FLAG M2® antibodies showing the specific recognition and recovery of the FLAG-tagged Hfq in the different protein fractions from TY exponential cultures of SmhfqFLAG during CoIP. As a control the same fractions from the wild-type (wt) strain Sm2B3001 were similarly probed. CoIP–RNA was recovered from Hfq–RNA complexes (IP fraction) derived from Sm2B3001, SmpWsinIFLAG, and SmhfqFLAG strains grown to exponential and stationary phases in TY and upon cold, heat, and salt shocks. cDNA libraries for sequencing were generated from control and Hfq CoIP–RNA samples (see text for details). (B) Classification of Hfq-bound transcripts. A schematic of the different types of Hfq RNA species according to the MTU model described in Materials and Methods is shown on the upper panel. Lower panels show the number of Hfq-bound RNAs in both each category (left circle diagram) and cDNA library (right Venn diagram).
Illumina-based sequencing of all seven cDNA libraries delivered an average of approximately 4 000 000 reads per library of which 1 250 199 (C-wt library), 1 028 776 (Hfq-log), 1 155 531 (Hfq-stat), 906 528 (C-SinI), 1 178 600 (Hfq-cold), 326 838 (Hfq-heat), and 1 413 343 (Hfq-salt) unambiguously mapped to unique locations within the S. meliloti reference genome (Table S1). Read counts on annotated and putative novel S. meliloti features/genes were then computed from the sets of uniquely mapped reads. The relatively low number of such reads in the Hfq-heat library results from imperfect rRNA depletion during library preparation.
Enrichment of transcripts in each of the five Hfq CoIP–RNA libraries was calculated upon normalization of feature read counts to their control library pair, i.e., Hfq-log and Hfq-stat libraries were normalized independently with C-wt and so were Hfq-cold, Hfq-heat, and Hfq-salt with C-SinI. Transcripts represented by at least 30 reads and exceeding a fold change threshold of 5 in the experimental library with respect to the control were regarded as Hfq-bound (i.e., Hfq RNAs). Collectively, this analysis identified 1315 S. meliloti Hfq RNAs, which based on their genomic context with respect to a minimal transcription unit (MTU) model, were further cataloged as () (1) mRNAs, if matched the sense strand of protein-coding regions, including their experimentally determined or virtual 5′/3′ untranslated regions (UTR) (1127; 86% of the Hfq RNAs), (2) trans-sRNAs, if mapped to intergenic regions (IGRs) (85/6.4%), and (3) asRNAs, if encoded on the strand opposite to annotated mRNAs (83/6.3%). Additionally, 19 Hfq RNAs mapped to repeat regions and one corresponded to tRNA–Gly encoded by the SMc02206 gene.
Considering each growth condition independently, we found that only 8.3% (110) of the identified Hfq RNAs were recovered specifically from exponentially growing bacteria, whereas 24.8% (326) were likely expressed at the stationary phase alone (). Cold, heat, and salt shocks specifically contributed to the recovery of 21.5% (283), 3% (40), and 13.2% (174) of the Hfq RNAs, respectively. These data further support that Hfq functions as a global post-transcriptional regulator of bacterial responses to stress.
Hfq-associated sRNAs
About 13% of the transcripts enriched by Hfq CoIP were cataloged as either trans-sRNAs (85) or asRNAs (83) (, left panel). Of the 168 Hfq-bound sRNAs, 110 were already identified in previous RNAseq-based transcriptomics surveys (67 trans-sRNAs and 43 asRNAs),Citation23,Citation39 and of those, 20 trans-sRNAs had been shown to be conserved to varying extend in the Rhizobiales by bioinformatics methods.Citation43,Citation44 The same studies provided evidences of transcription for 29 more Hfq sRNAs (eight trans-sRNAs and 21 asRNAs) which, nonetheless, did not pass the established cutoff scores and were filtered out from the final lists of candidates. The remaining 29 Hfq-bound sRNAs represent newly discovered S. meliloti transcripts (10 trans-sRNAs and 19 asRNAs). Genomic distribution of the Hfq sRNA loci relative to the size of the three S. meliloti replicons evidenced a localization bias toward the symbiotic megaplasmid pSymA (, right panel). The coordinates of the Hfq sRNAs in the S. meliloti 1021 genome as well as their enrichment in each of the five CoIP cDNA libraries with respect to the controls are provided in Table S2.
Figure 2. Identification of Hfq-associated sRNAs. (A) Number of annotated and newly identified trans-sRNAs and asRNAs scored as Hfq-bound and their replicon distribution in S. meliloti. Latter values are relative to the sizes (Mb) of chromosome (Chr.) and megaplasmids pSymB and pSymA. (B) IGB visualization of the clone distribution over selected Hfq-dependent and -independent sRNAs. Shown are cDNA clusters corresponding to tmRNA (ssrA), SmelA060 (trans-sRNA), and SMc_Hfq_asRNA_11 (asRNA) in control and Hfq CoIP–RNA as indicated. The vertical axis indicates the number of sequencing reads that were obtained. Relevant genomic information for each sRNA is provided in the schematics below the plots. The asterisk indicates the tmRNA processing site. (C) Selective stabilization of CoIP-enriched sRNAs by Hfq. Northern analysis of Smr7C, SmelC289, and SmelC457 decay in Sm2B3001 (wt) and its hfq deletion mutant derivative (∆hfq) upon transcription arrest with rifampicin. Samples of exponentially growing bacteria in TY were withdrawn prior to or at the indicated time-points (in min) after antibiotic addition. Hybridization signal intensities were normalized to those of the 5S rRNA in each RNA sample to calculate the half-life of the transcripts (indicated in min). IGB plots for these sRNAs (as in B) are shown above each panel.
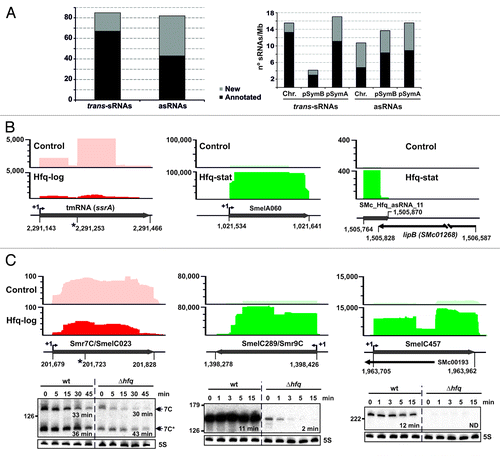
Upon normalization, the distribution of the sequencing reads from control and Hfq cDNA libraries over the S. meliloti 1021 genome was visualized using the integrated genome browser (IGB, Affymetrix). As an example, shows the clone distribution of cDNA sequences mapped to tmRNA, SmelA060, and SMc_Hfq_asRNA_11 sRNA loci. S. meliloti tmRNA has been shown to be transcribed as an unstable precursor that is processed into two abundant 82-nt and 214-nt RNA species.Citation45 Both mature tmRNA forms were represented in smaller numbers in the Hfq than in the control CoIP–RNA libraries and so scored as Hfq-independent transcripts. As expected, similar unspecific recovery was obtained for other housekeeping sRNAs, namely 6S, RNase P, and 4.5S RNAs (Fig. S1).
Conversely, SmelA060 and SMc_Hfq_asRNA_11 were significantly enriched by Hfq CoIP and are shown in as representatives of Hfq-dependent trans-sRNAs and asRNAs, respectively. SmelA060 is one of the most abundant among the identified Hfq trans-sRNAs, according to the number of cDNA sequences, particularly in the Hfq-stat library (> 100 000 reads) in which it is enriched by 624-fold in comparison to the controls (Table S2). Furthermore, the genomic positions determined by the borders of the cDNA cluster over the SmelA060 locus fully correlate with published 5′/3′ end mapping data of this sRNA.Citation23,Citation39 Remarkably, this is the case for most of the Hfq sRNAs listed in Table S2 (additional examples are provided in Fig. S1). SMc_Hfq_asRNA_11 exemplifies the identification of a novel chromosomal asRNA, highly enriched (~40-fold) by Hfq CoIP in lysates of bacteria grown to stationary phase, which is complementary to the 3′ region (including the putative 3′-UTR) of the lipB (SMc01268) mRNA (). This type seems to be predominant among the asRNAs identified in our study, but a number of Hfq RNAs antisense to the 5′-region or internal to mRNA coding sequences were also detected (Fig. S1).
As further experimental evidence to validate the transcripts listed in Table S2 as Hfq-binding sRNAs, we determined the Hfq-dependent decay of a new set of sRNAs (Smr7C, SmrC9, and SmelC457) upon transcriptional arrest of exponential cultures with rifampicin (). In S. meliloti, Smr7C (also known as SmelC023) exists in two forms, i.e., the primary 150-nt transcript and the 106-nt processed RNA, neither of which had been detected on northern blots of Hfq CoIP-RNA from S. meliloti 1021.Citation29 Accordingly, the enrichment pattern of Smr7C in the Hfq-log library with respect to the control resembled that of the housekeeping Hfq-independent sRNAs. In agreement with these findings, the half-life of both RNA species determined in the wild-type strain (33/36 min) was not significantly affected in the absence of Hfq (half-life of 30/43 min in the Δhfq genetic background) (). In contrast, the trans-sRNA Smr9C (also termed SmelC289) and the asRNA SmelC457 have been cataloged as Hfq-bound according to their enrichment in CoIP-RNA (specifically 14- and 10-fold, respectively, in the Hfq-log library; Table S2). Previous northern blot probing of Hfq CoIP–RNA derived from the S. meliloti reference strain 1021 anticipated that Smr9C is an Hfq-binding sRNA.Citation29 SmelC457 was identified in previous RNAseq-based surveys, but its expression had not been verified by Northern hybridization.Citation23,Citation39 Probing of total RNA with a 25-mer oligonucleotide targeting specifically the 5′ region of this sRNA yielded a signal for a small transcript, which size correlates with that inferred from RNAseq data. The half-life of these sRNAs in the wild-type Sm2B3001 background was approximately 11 min for SmrC9 and 12 min for SmelC457. Lack of Hfq strongly compromised stability of both transcripts; SmrC9 decayed with a more than 5-fold reduced half-life (2 min) whereas SmelC457 was undetectable in the Δhfq mutant (). Altogether, these data validate our approach as a reliable strategy to identify Hfq-dependent sRNAs at nucleotide-level resolution in S. meliloti strains.
Differential expression of the Hfq sRNAs
A common feature of the regulatory sRNAs is their expression under specific stress conditions. Overall, Hfq sRNAs were mostly recovered from bacteria subjected to an osmotic upshift, an observation that was particularly evident for the trans-sRNAs (). Of the 85 Hfq trans-sRNAs, 56 (66%) were identified in the Hfq-salt library, with 27 (32%) likely induced specifically by this abiotic stress. Similarly, up to 38 out of the 83 Hfq asRNAs (45%) were expressed in the presence of salt (25 were exclusive of the Hfq-salt library) but the cold shock also contributed significantly to extend this catalog of transcripts (29 Hfq asRNAs were only identified in the Hfq-cold library). To further compare the abundance of the Hfq sRNAs in the five CoIP-RNA libraries RPKM values (cDNA reads per kilobase transcript per million mapped reads) for each transcript were calculated (Table S2). RPKM-normalized data were plotted into heat maps provided in Figures S2 (trans-sRNAs) and S3 (asRNAs).
Figure 3. Expression profiling of the Hfq-dependent sRNAs. (A) Venn diagrams indicating the number of Hfq-bound trans-sRNAs and asRNAs identified in each CoIP–RNA library. (B) Northern analysis of the expression of selected Hfq-bound sRNAs. Total RNA was obtained from strain Sm2B3001 grown in the conditions of CoIP experiments. Growth in GMS medium was included as the reference for osmotic upshift induction. Sizes of each transcript as inferred from RNAseq data are indicated with a double arrowhead to the right. RPKM-normalized recovery rates of these sRNAs in each Hfq CoIP–RNA library were plotted under the blots according to the color scale shown in the middle.
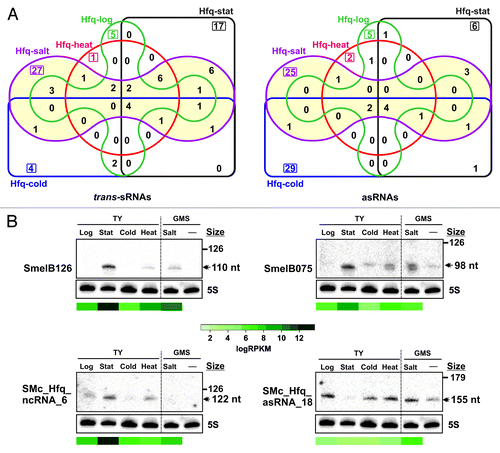
Sequence signatures recognized by the dual RpoH (H1 and/or H2) or the RpoE2 σ factors were predicted in the promoter regions of 14 (seven trans-sRNAs and seven asRNAs) and 12 (10 trans-sRNAs and 2 asRNAs) Hfq sRNAs, respectively, and thus, could explain the differential accumulation of these transcripts in our conditions (Table S2). The expression profiles of two of these sRNAs, SmelB126 (RpoE2 promoter) and SmelB075 (RpoH1/2 promoter), were assessed by northern blot hybridization (, upper panels). Both are pSymB-encoded trans-sRNAs identified by previous RNAseq surveys but their expression had not been confirmed by independent approaches.Citation23,Citation39 Nonetheless, reported microarray data provided evidence for RpoE2-dependent transcription of SmelB126.Citation23 For these experiments, total bacterial RNA was obtained from identical growth phases and stress conditions as applied for CoIP experiments. RNA from exponentially growing bacteria in glucose mannitol salt (GMS) medium was also probed as the reference of induction by salt stress. Oligonucleotide probes specific for each sRNA detected on gels small transcripts that accumulated differentially in the conditions tested. In both cases, the strongest hybridization signals were evident on RNA obtained from bacteria grown to stationary phase and subjected to heat and salt shocks (, upper panels). These expression profiles are in agreement with those expected from RpoE2- and RpoH-dependent genes. Furthermore, the sizes of the RNA species detected on gels and their relative abundance under each biological condition were in overall good correlation with RNAseq data (i.e., length of cDNA clusters and RPKM values). Probing of the same membranes for detection of the newly identified trans-sRNA SMc_Hfq_ncRNA_6 and asRNA SMc_Hfq_asRNA_18 further revealed such correlation (, lower panels).
Our data set thus provides insights into the expression profiles of the identified sRNAs, which collectively suggest a major impact of Hfq-dependent riboregulation on S. meliloti responses to high environmental salinity.
Hfq-dependent mRNAs
The most abundant class of RNAs enriched by Hfq CoIP derived from mRNAs (cistrons) (1127; 86%). According to the COG (Clusters of Orthologous Groups) database up to 30% of the proteins encoded by these Hfq mRNAs have no predictable function whereas the remaining ones could be putatively assigned to widely diverse functional categories; 48% are involved in transport and metabolism of small molecules, 12% in other cellular processes such as post-transcriptional modification, motility or signal transduction and 10% in the storage and processing of genetic information (). The full-length mRNA encoded by the conserved hfq–hflX tandem was found to be Hfq-bound (), further supporting that these genes are co-transcribed and autoregulated by Hfq as reported previously.Citation30 Table S3 lists all 1127 Hfq mRNAs identified in our study.
Figure 4. Functional distribution of the Hfq-bound mRNAs according to the COG database.
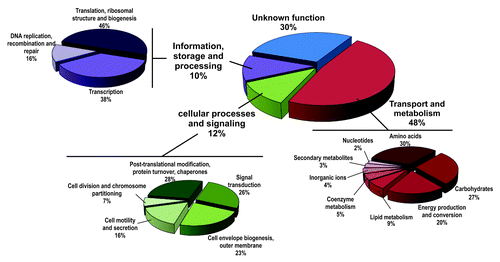
Figure 5. Enrichment patterns of Hfq-associated RNA species derived from mRNAs. (A) Recovery of mRNAs of major metabolic, stress, and symbiotic pathways. The clone distribution over the mRNAs is represented by an IGB stairstep diagram of fold enrichment in the indicated Hfq–CoIP RNA with respect to the control CoIP. The vertical axis indicates the enrichment factors. ORFs of the mRNAs are shown by arrows under each IGB plot. Numbers denote genomic positions of the recovered transcripts according to Table S3. TSSs are indicated if known. (B) Alternative distribution patterns of cDNA clusters over Hfq-bound mRNAs. Information has been diagrammed as in (A). Data correspond to the recovery of these three mRNAs in the Hfq-salt library.
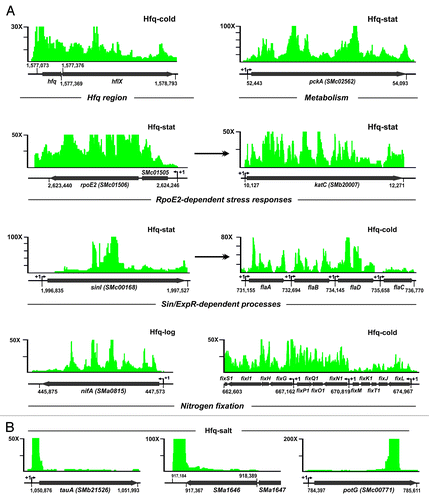
A compilation of published transcriptomic/proteomic data rendered a joint list of 940 S. meliloti protein-coding genes (i.e., approximately 15% of the annotated ORFs) reported as deregulated in S. meliloti hfq mutants.Citation28,Citation29,Citation31,Citation32 Despite of relevant differences in the experimental setups (i.e., biological conditions tested) preclude direct comparison of the mRNAs of the Hfq regulon and those scored as Hfq-associated in our study, an overlap of 322 genes was found between both data sets (Table S3). Almost half of these genes (158) encode transport proteins (mostly ABC transporter membrane complexes), cytoplasmic metabolic enzymes such as the phosphoenolpyruvate carboxykinase (PckA; ) or components of the electron transport chain, thus confirming a major impact of Hfq in the post-transcriptional control of S. meliloti uptake systems and energy production pathways.
A significant subset of the direct Hfq mRNA targets specify cellular functions related to stress responses known to be altered in S. meliloti strains lacking Hfq.Citation27-Citation31 For example, rpoH2 (SMc03873) and four out of the 11 ECF σ factor mRNAs (rpoE1 [SMc01419], rpoE2 [SMc01506], rpoE4 [SMc04051], and rpoE5 [SMb21484]) annotated in the S. meliloti genome were specifically recovered by Hfq CoIP. In particular, the mRNAs encoding RpoE2 and its anti-σ factor SMc01505 were highly enriched in the Hfq-stat library (56- and 10-fold, respectively) (). S. meliloti RpoE2 has been shown to activate the expression of more than 200 protein-coding genes under various stress conditions that include heat and salt shocks, and carbon or nitrogen starvation.Citation20,Citation21,Citation23 In our study, 127 mRNAs from genes currently assigned to the RpoE2 regulon were regarded as Hfq-associated (49 with recognizable RpoE2 promoter signatures; Table S3). These include rpoE5 and rpoH2 as well as mRNAs for determinants of oxidative stress resistance (sodC and katC) () or proteins operating under nutrient deprivation (ndiA-1, ndiA-2, and ndiB). The rpoE2, sodC (SMc02597), and katC (SMb20007) genes have been also found consistently downregulated in S. meliloti hfq mutants.Citation28,Citation32
Some other known S. meliloti hfq mutant phenotypes such as alteration of the N-acyl-homoserine lactones (AHLs) profile, reduced motility, or defects in root hair invasion, depend largely on functions controlled by the ExpR/Sin QS system. The sinI mRNA (SMc00168) encoding the S. meliloti AHLs synthase, which is upregulated in the absence of Hfq,Citation28 was enriched by ~11-fold in the Hfq-stat library (). Upon activation by AHLs, the LuxR-type regulator ExpR directly or indirectly controls the expression of at least 570 genes.Citation46-Citation49 Major outcomes of this regulatory activity are activation of galactoglucan (EPS II) production and repression of flagella biosynthesis. Hfq CoIP-RNA was enriched in 159 mRNAs from genes downstream of ExpR in the QS pathway (Table S3). This set of transcripts includes most of the mRNAs coding for structural elements of the flagellar apparatus () and some belonging to the wge cluster involved in the synthesis and secretion of EPS II. Consistent with both impaired motility and upregulation of sinI, flagellar genes have been reported to be downregulated in a S. meliloti hfq mutant.Citation28
It has also been shown that the abundance of S. meliloti fixK and nifA mRNAs, encoding the transcriptional activators of the symbiotic nitrogen fixation genes, is positively influenced by Hfq.Citation27,Citation29 Despite of the presumed null or very low expression of the nitrogen fixation genes in aerobically growing free-living rhizobia, we found that the nifA mRNA as well as the FixK-dependent mRNA cluster coding for components of the electron transport chain associated to the nitrogenase complex (i.e., fixNOQGHIS) were enriched in CoIP–RNA (). Interestingly, the mRNA encoding RpoN (SMc01139), the σ54 factor of the RNA polymerase holoenzyme that drives transcription of the nifHDKE (nitrogenase components) and fixABCX (proteins involved in electron transport) operons, was also cataloged as Hfq-bound.
Finally, another large set of mRNAs found to be highly enriched in CoIP–RNA (50-to 100-fold in some libraries) encodes proteins involved in translation, particularly elongation factors (six mRNAs) and the large and small ribosomal subunits (40 mRNAs) (Table S3).
Enrichment patterns of mRNAs in CoIP–RNA
Unlike the identified Hfq sRNAs, the enrichment patterns of mRNAs in CoIP RNA were more complex (). In a subset of cases (including most of the genes mentioned above), cDNA reads mapped over the entire length of monocistronic or polycistronic mRNAs, covering also virtual or experimentally determined 5′/3′-UTRs (). However, many other Hfq mRNAs were not evenly enriched in the CoIP–RNA libraries, but rather most cDNA reads clustered over defined stretches of the transcripts. The ABC transporter mRNAs tauA (SMb21526), SMa1646, and potG (SMc00771), all enriched in the Hfq-salt library, exemplify the three different clustering patterns of cDNA reads over particular mRNA portions (). The tauA gene is the first in the operon encoding a putative taurine ABC transporter and specifies the periplasmic component of the complex. cDNAs from tauA mainly clustered in the 5′ region of this mRNA ranging from the transcription start site (TSS) into the N-terminal coding sequence. Both SMa1646 and potG mRNAs code for the ATP-binding protein of ABC systems for the uptake of nitrogen compounds (amino acids and polyamines). SMa1646 corresponds to the last cistron of a likely polycistronic transcript, whose cDNA sequences almost exclusively mapped to an 83-nt region immediately downstream of the SMa1646 stop codon. Finally, cDNAs from potG were almost exclusively derived from a 159-nt stretch internal to the coding sequence. This type of distribution of cDNA reads over the Hfq-bound mRNAs has rarely been found in our study, being the sinI mRNA another example (). This variety of enrichment patterns in CoIP–RNA could be related to the primary binding sites of Hfq as well as to the Hfq-dependent mechanisms of post-transcriptional regulation of each particular mRNA.
Validation of predicted Hfq-dependent sRNA–mRNA regulatory pairs
Enrichment by Hfq CoIP combined with misregulation in the hfq mutant may indicate post-transcriptional control of certain mRNAs by Hfq-dependent sRNAs. In S. meliloti targeting of the livK mRNA by the so-called AbcR1 sRNA is the only sRNA–mRNA pair experimentally validated to date.Citation42 AbcR1 and its homologous AbcR2 sRNA are tandemly encoded in a S. meliloti chromosomal IGR and both have been reported to be Hfq-dependent.Citation29,Citation41,Citation42 Accordingly, these sRNAs were enriched in our Hfq CoIP–RNA libraries (Fig. S4A). Similarly, the livK mRNA (SMc01946), which codes for the periplasmic component of an ABC transporter involved in branched-chain amino acid uptake, has been reported to be negatively regulated by Hfq and cataloged as Hfq-bound in this study (Fig. S4B). A number of additional target mRNAs of AbcR1 and/or AbcR2, mostly coding for ABC transport systems, have been computationally predicted.Citation31,Citation42 We selected three of these mRNAs, namely SMa0495, prbA (SMc01642), and oppA (SMb21196), which as livK code for amino acid binding proteins, are Hfq-bound and upregulated in an hfq mutant (Table S3), to further assess their regulation by AbcR1/2 in vivo using a double-plasmid reporter system.Citation42 This assay failed to demonstrate targeting of oppA (not shown) but revealed downregulation of SMa0495 and prbA by both AbcR1 and AbcR2 ().
Figure 6. Targeting of the SMa0495 (A) and prbA (B) mRNAs by the AbcR1 and AbcR2 sRNAs. IntaRNA predicted duplexes are shown, with the RBS and AUG start codons of the SMa0495 and prbA mRNAs underlined. In these diagrams, numberings denote positions relative to the AUG start codon of the mRNA and the TSS of AbcR1 and AbcR2. The predicted minimum hybridization energy (E) is indicated in each case. The IGB diagrams show the fold enrichment (vertical axis) of these mRNAs in the indicated Hfq CoIP–RNA library. Schematics of the reporter fusions cloned in plasmid pR-EGFP are shown below the genomic information of each mRNA. The histograms show the fluorescence of the reporter S. meliloti double deletion mutant (∆R1/2) co-transformed with the target fusions and plasmids pSRK (empty control vector), pSRK-R1 (R1+), or pSRK-R2 (R2+). Values reported are means and standard deviation of 48 fluorescence measurements, i.e., four determinations in three independent exponential cultures of four double transconjugants representing each plasmid combination. Fluorescence values were normalized to culture OD600 (F/OD). Background fluorescence from strains harboring pSRK and the empty pR-EGFP plasmid instead of the target fusions was subtracted from the fluorescence of target fusions. Relevant subsections of 2D-PAGE gels revealing downregulation of the periplasmic protein PrbA (double arrowhead) by both AbcR1 and AbcR2 sRNAs are also shown. Periplasmic protein extracts were obtained from exponential cultures of S. meliloti single mutants ∆R1 and ∆R2 carrying pSRK or either pSRK-R1 (R1+) or pSRK-R2 (R2+) as indicated. Expression/absence (+/−) of AbcR1 and AbcR2 (R1/2) in each strain is indicated in brackets. Note that AbcR1 is highly expressed from the chromosome whereas AbcR2 is almost absence in exponentially growing bacteria.Citation42
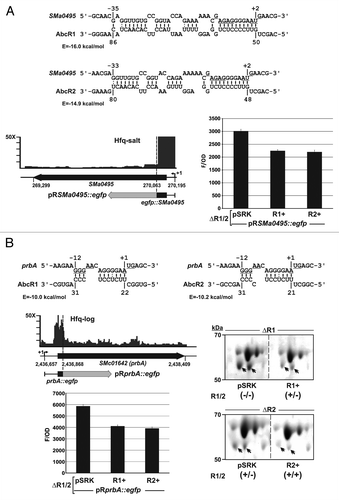
As for livK,Citation42 AbcR1 and AbcR2 are predicted to target specifically the ribosome binding site (RBS) and flanking nucleotides in the SMa0495 and prbA mRNAs through anti Shine-Dalgarno motifs (positions 21–31 or 50–59 within the sRNAs) (). SMa0495 and prbA were enriched in the Hfq-log and Hfq-salt libraries, respectively, with cDNA reads mostly clustering over their 5′-regions, including the respective 5′-UTR (). Therefore, two genomic DNA fragments spanning from the native TSS to the 16th and 18th codons of SMa0495 and prbA mRNAs, respectively, were translationally fused to the N terminus of EGFP in plasmids pRSMa0495::egfp () and pRprbA::egfp (). These plasmids were used as reporters of AbcR1 and AbcR2 activity in a S. meliloti 1021 AbcR1/2 double deletion mutant (ΔR1/2). Fluorescence of exponential cultures of the reporter strains co-expressing pRSMa0495::egfp and AbcR1 or AbcR2 from plasmids pSRK-R1 (R1+) or pSRK-R2 (R2+) was 25% less than that of control strains co-transformed with the same target fusion and the empty vector pSRK (). Similarly, expression of both AbcR1 and AbcR2 resulted in reduction of the fluorescence activity of plasmid pRprbA::egfp by 33% ().
A previous analysis of the S. meliloti 1021 AbcR1- and AbcR2-dependent periplasmic proteome from exponentially growing TY bacterial cultures unequivocally demonstrated downregulation of LivK by AbcR1.Citation42 However, a deeper examination of the same Coomassie-stained 2D gels revealed a few more subtle changes in protein abundance that, nonetheless, had not been further confirmed by other approaches. Interestingly, mass spectrometry (MALDI-TOF) identified PrbA (MW 58.4 kDa and pI 5.84) as one of these differentially accumulated polypeptides (, right panel). Gels resolved two forms of PrbA (probably because of a post-translational modification) whose accumulation decreased in S. meliloti 1021 single deletion mutants ΔR1 and ΔR2 transformed with plasmids pSRK-R1 and pSRK-R2, respectively, when compared with that in control strains carrying pSRK (, right panel). This finding correlates with the AbcR1/2-dependent fluorescence of pRprbA::egfp, further supporting direct downregulation of prbA by both sRNAs.
Altogether, these data validate pbrA and SMa0495 as common targets of AbcR1 and AbcR2 sRNAs, thus rendering our data set as a valuable tool to search for mRNAs targeted by other S. meliloti trans-acting sRNAs in an Hfq-dependent manner.
Discussion
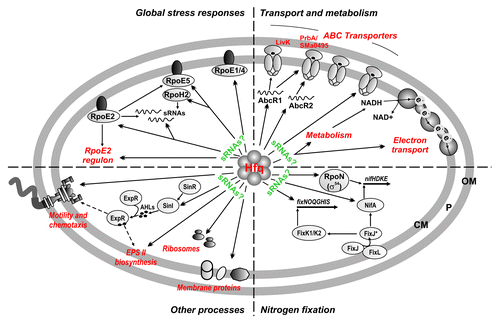
The Sm-like protein Hfq is increasingly recognized as a major hub in RNA transactions underlying virtually any adaptive reprogramming of gene expression in bacteria. The model legume symbiont S. meliloti encodes a functional Hfq homolog, which, as its characterized counterparts in related α-proteobacteria, has been shown to influence a diversity of free-living traits and the symbiotic interaction.Citation27-Citation32 Here we have explored the RNA-binding features of S. meliloti Hfq by deep sequencing characterization of the population of transcripts specifically associated in vivo to a FLAG-tagged variant of the protein. To our knowledge, such a study has no precedent in the group of α-proteobacteria interacting with eukaryotes. Our findings thus provide the first global insights into the Hfq-mediated post-transcriptional control of relevant S. meliloti gene networks that to date have been almost exclusively viewed from the perspective of transcriptional regulation ().
Figure 7. Summary of the Hfq–RNA interactions in S. meliloti. Double arrowheads stand for direct targeting by Hfq of sRNAs and mRNAs encoding transport and metabolic enzymes, proteins of the ExpR/Sin and global stress regulons, regulators of nitrogen fixation, and ribosome and membrane components. Single arrowheads denote reported transcriptional regulation. Hfq-bound sRNAs could be on the basis of the post-transcriptional regulation of some mRNA genes. This work extended the list of validated AbcR1/2 targets to the prbA and SMa0495 ABC transporter mRNAs. OM, outer membrane; P, periplasmic space; CM; cytoplasmic membrane.
Originally described in Salmonella, the methodology used here is emerging as the generic approach of choice for successful genome-wide identification of Hfq RNA substrates in bacteria.Citation7,Citation9,Citation11-Citation13 To target a broad fraction of the transcriptome, we analyzed CoIP–RNA representing several rather than a single growth/stress condition. This strategy delivered a large catalog of RNA species, almost exclusively derived from putative regulatory sRNAs and mRNAs. Hfq-binding RNAs were mostly recovered from bacteria grown under stress, further highlighting the major impact of this protein into bacterial adaptive traits.
Limited but functionally significant influence of Hfq in S. meliloti sRNA pathways
Trans-acting sRNAs are likely the predominant class of Hfq partner transcripts in certain bacterial species. Consequently, pull-down with Hfq has been an ideal strategy to successfully recover most of the expressed trans-sRNAs in these bacteria (e.g., Salmonella).Citation7,Citation9,Citation12 However, our screen failed to identify as Hfq-bound a large fraction (~86%) of the ~600 annotated S. meliloti trans-sRNAs,Citation23,Citation39 confirming preliminary observations on a very small subset of these transcripts.Citation29 Furthermore, the number of newly identified transcripts represents a minor addition (< 3%) to this catalog. This finding could be partially explained by the reduced number of biological conditions tested compared with previous RNAseq surveys (5 vs 16 growth/stress conditions)Citation23,Citation39 and the stringent threshold (≥ 5-fold enrichment with respect to the controls) imposed to score the RNA species as Hfq-bound. However, current data more likely anticipate that in S. meliloti the impact of Hfq in trans-sRNA activity could be more limited than expected from the paradigms learned from model enterobacteria, as suggested also for some Gram-positive bacteria.Citation13,Citation50 A plausible candidate to complement or even fulfill the role of Hfq in riboregulation is the widely conserved Argonaute-like protein YbeY (SMc01113), whose loss-of-function has been shown to produce phenotypes and to compromise trans-sRNA stability similar to Hfq in S. meliloti.Citation51 In contrast to trans-sRNAs, asRNAs seem to be a minor fraction of the Hfq substrates in bacteria.Citation7,Citation9,Citation11-Citation13 Nonetheless, Hfq has been shown to negatively regulate transposition in E. coli by promoting pairing of an asRNA to the IS10 transposase mRNA.Citation52,Citation53 RNAseq-based transcriptomic profiling of S. meliloti has evidenced pervasive antisense transcription involving approximately 35% of the annotated protein-coding genes.Citation23 CoIP recovered only 2% of the annotated S. meliloti asRNAs, none of which transcribed opposite to genes encoding transposable elements, further supporting the presumably discrete influence of Hfq in asRNA-mediated regulation of gene expression. Considering the sizes of the three S. meliloti replicons, both Hfq-bound trans-sRNAs and asRNAs were overrepresented on megaplasmid pSymA. This replicon bias had been observed for the whole set of S. meliloti trans-sRNAs but not for the asRNAs.Citation23 Rhizobial species exhibit a multi-replicon pangenomic structure, being pSymA a major source of intraspecific genetic and phenotypic variability.Citation54 Hfq has been shown to assist the activity of horizontally acquired genes in Salmonella.Citation7,Citation9,Citation12 Therefore, it is reasonable to speculate that this protein may also aid to integrate acquired sRNAs into core S. meliloti pathways.
A major role of Hfq in riboregulation is promoting sRNA stability, more likely by protecting non-coding transcripts from ribonucleolytic degradation.Citation55,Citation56 However, binding alone does not necessarily imply requirement of Hfq for effective target regulation.Citation50 Previous studies have revealed that Hfq also influences the stability of trans-sRNAs in S. meliloti.Citation41,Citation42,Citation51 Our results show that Hfq selectively stabilizes CoIP-enriched trans-sRNAs and asRNAs, suggesting that binding to Hfq is required for the activity of these transcripts in vivo.
The differential expression of the bacterial riboregulators is usually a predictor of their physiological function. Approximately 94% of the Hfq-bound sRNAs (both trans-sRNAs and asRNAs) identified in this study were retrieved from bacteria grown under abiotic stress conditions and most under salt stress. The S. meliloti σ factors RpoH1/2 and RpoE2 have been shown to govern large and scarcely overlapping arrays of genes activated in response to a variety of stress conditions including heat shock, oxidative stress, pH oscillations, hyperosmolarity, and entry into stationary phase upon carbon or nitrogen starvation, which are often faced by rhizobia both in soil and in planta.Citation19-Citation23,Citation57 The available genome-wide map of S. meliloti TSS and promoter motifs along with predictions on the newly identified sRNA loci assigned 26 Hfq-bound sRNAs to either the RpoH1/2 or RpoE2 regulons,Citation23 which further explains the dynamics of these sRNAs as inferred from their RPKM-normalized recovery rates.
Several factors such as differences in specificity of binding to Hfq and yields of reverse transcription over particular secondary structures can bias the recovery of some sRNAs by pull-down with Hfq. However, expression profiling of a subset of Hfq-bound sRNAs by northern blot probing, either reported previously (e.g., SmelC289, SmelA075, SmelA060, AbcR1, and AbcR2)Citation36,Citation39,Citation42 or presented here (SmelB126, SmelB075, SMc_Hfq_ncRNA_6, and SMc_Hfq_asRNA_18), suggest accumulation profiles of these transcripts that, overall, paralleled their relative recovery rates in CoIP–RNA. Therefore, our data set provides a reliable reference for the identification of sRNAs potentially involved in major S. meliloti pathways underlying general stress adaptation and the hyperosmotic stress response.
Hfq mostly targets mRNAs of major stress response and symbiotic regulons
The largest fraction of transcripts directly targeted by Hfq derived from mRNAs corresponding to approximately 18% of the predicted S. meliloti protein-coding genes. This avidity for mRNA binding is similar to that reported in Salmonella and Bacillus subtillis,Citation7,Citation9,Citation12,Citation13 but markedly higher than that of the α-proteobacterium Rhodobacter sphaeroides Hfq, for which only 47 mRNA ligands have been identified.Citation11 Gram-positive bacteria and α-proteobacteria (including S. meliloti and R. sphaeroides) encode Hfq homologs that lack the C-terminal region of their enterobacterial counterparts, which has been largely assumed to be required for mRNA binding and regulation.Citation58 In the divergent bacterial species E. coli and S. meliloti, Hfq has been shown to regulate its own translation likely by direct binding to the hfq mRNA leader.Citation30,Citation59 Further supporting its ability for effective autoregulation, the hfq mRNA was found within the pool of RNA substrates of the naturally truncated S. meliloti and R. sphaeroides Hfq homologs in this and previous studies, respectively.Citation11 Therefore, this data adds uncertainty to the role of the C-terminal domain of the bacterial Hfq proteins in mRNA regulation.Citation58,Citation60
Our study revealed that S. meliloti Hfq associates to mRNAs encoding functionally diverse proteins involved in cellular processes relevant to both the free-living and symbiotic rhizobial lifestyles namely, transport and metabolism, flagella biosynthesis, QS, ribosome and membrane assembly, global stress responses mediated by alternative σ factors, and symbiotic nitrogen fixation (). Many of these processes are similarly impacted by Hfq in a diversity of phylogenetically distant bacteria, further pointing to the existence of broadly conserved Hfq-dependent pathways fundamental for maintaining cell homeostasis.Citation7,Citation11,Citation13,Citation61
A significant subset of mRNAs previously found to be downregulated in the absence of Hfq were unambiguously scored as Hfq-bound in our survey, thus providing further understanding to the phenotypic pleiotropy associated to the deletion of hfq in S. meliloti.Citation27-Citation32 The reported overall sensibility to abiotic stress (e.g., heat shock, pH alterations, or nutrient limitation) and reduced infection rates of the hfq mutants can be partly explained by the requirement of Hfq for the expression of large groups of genes encoding certain metabolic enzymes, QS and motility and chemotaxis determinants, almost half of the S. meliloti alternative σ factors, and key components of the RpoE2 regulon. The latter include enzymes involved in detoxification of reactive oxygen species (ROS) produced during aerobic growth and legume infection. Similarly, direct positive Hfq-mediated regulation of structural and regulatory genes (fix and nif) specifying nitrogen fixation traits provides additional explanations to the symbiotic defects of the mutants.Citation62
Inferring Hfq-dependent regulatory mechanisms
cDNA clusters from most of the mRNAs positively regulated by Hfq were found to be evenly distributed over the entire sequence of the corresponding genes. This enrichment pattern hints at Hfq-mediated stabilization of these transcripts either in an sRNA-independent manner involving active competition with RNase E for binding to a large number of sites in the mRNAsCitation55 or through the activity of Hfq-dependent trans-sRNAs.Citation63,Citation64 Activation of the expression of these genes could also involve promotion of translation by a sRNACitation65 or directly by Hfq. The latter scenario has been suggested for the Rhizobium leguminosarum bv. viciae nifA mRNA.Citation26 The proposed mechanism involves Hfq-mediated recruitment of RNase E for specific cleavage within the 5′-UTR of the mRNA, leading to disruption of inhibitory structures over the RBS.
In certain cases, specific regions of the mRNAs, rather than the full-length transcripts, appeared enriched in the CoIP–RNA libraries. These fragments, mostly derived from UTRs, could correspond to novel Hfq-dependent trans-sRNAs resulting from parallel transcriptional output or post-transcriptional processing of certain mRNAs.Citation12,Citation66-Citation68 Alternatively, these enrichment patterns likely reveal primary high-affinity binding sites for Hfq that remain protected during exoribonucleolytic degradation of the message upon base pairing with a trans-acting sRNA. Indeed, these cDNA clusters preferentially mapped to the 5′ regions of the S. meliloti mRNAs, which are the most common binding sites of this type of riboregulators in bacteria.Citation2
The classical model of RNA cycling on Hfq predicts that the hexameric Hfq ring uses its two faces to simultaneously bind the sRNA–mRNA regulatory pairs for antisense interaction.Citation69 In line with this notion, many targets of well-characterized Salmonella sRNAs have been identified within the pool of Hfq-associated transcripts.Citation7 Similarly, among the identified Hfq-bound mRNAs, those encoding the ABC transporters PrbA and SMa0495 were validated here as new negatively regulated targets of the S. meliloti Hfq-dependent AbcR1 and AbcR2 sRNAs. The reported upregulation of prbA and SMa0495 in the absence of Hfq and the overrepresentation of their 5′ regions in CoIP–RNA are consistent with a canonical mechanism for AbcR1 and AbcR2 activity involving antisense interaction with complementary sequences within or in the vicinity of the RBS of their targets, leading to blocking of translation and subsequent mRNA decay. Our findings further support the hypothesis that the AbcR family of α-proteobacterial sRNAs is involved in the fine-tuning of nutrient uptake by selective repression of multiple ABC transporters of nitrogen compounds, similar to GcvB sRNA in enterobacteria.Citation42,Citation70-Citation74
In conclusion, the comprehensive catalog of primary RNA targets of the global regulator Hfq in S. meliloti provides a valuable resource to dissect unprecedented stress response and symbiotic post-transcriptional regulons as well as the regulatory mechanisms underlying Hfq and sRNA activity.
Materials and Methods
Bacterial strains, plasmids, media, and growth conditions for CoIP experiments
All the bacterial strains and plasmids used in this work with their relevant characteristics are listed in Table S4. Rhizobial strains were routinely grown in complex tryptone–yeast TY mediumCitation75 at 30 °C. E. coli strains were grown in Luria–Bertani (LB) medium at 37 °C. Media were supplemented with antibiotics if required at the following final concentrations (µg/ml): streptomycin (Sm) 250, ampicillin (Ap) 200, tetracycline (Tc) 10, erythromycin (Er) 100, gentamicin (Gm) 45, and kanamycin (Km) 50 for E. coli and 180 for rhizobia.
The S. meliloti strains used in CoIP experiments were Sm2B3001 as the wild-type strain,Citation76 SmpWsinIFLAG (expressing a C-terminal 3xFLAG-tagged SinI protein), and SmhfqFLAG (expressing a C-terminal 3xFLAG-tagged Hfq protein). For construction of strain SmpWsinIFLAG, the SinI coding sequence devoid of its TGA stop codon was PCR amplified using Sm2B3001 genomic DNA as template and primers sinI-NdeI-fwd and sinI-FX-rev (Table S5), the latter carrying the 3xFLAG sequence followed by a TGA stop codon. The PCR product was restricted with NdeI and XbaI and inserted into the corresponding sites of the pSRKGm vector,Citation77 which carries an inducible Lac promoter closely upstream the NdeI restriction site. The resulting plasmid was mobilized to Sm2B3001 by conjugation. Strain SmhfqFLAG was obtained by allelic replacement of the chromosomal Sm2B3001 hfq gene using the pK18mobsacB-derived suicide plasmid pK18Hfq3xFlag.Citation29
For CoIP, strain SmhfqFLAG was cultured in 200 ml broth under five different growth/stress conditions. Exponential and stationary cultures were obtained in TY medium by growing bacteria to OD600 0.6 and 2.8, respectively. Temperature shifts for cold (20 °C) and heat (40 °C) shocks were applied for 30 min to exponentially growing bacteria in TY. Salt stress was imposed by addition of 0.4 M NaCl for 1 h to exponential cultures in minimal GMS medium.Citation78 Strains Sm2B3001 and SmpWsinIFLAG, both expressing untagged Hfq, were used as controls in the experiments. Sm2B3001 was cultured in TY to exponential and stationary phases whereas SmpWsinIFLAG was subjected to cold, heat, and salt shocks as described in the presence of 1 µM IPTG to induce sinIFLAG expression. Two independent cultures were obtained for each strain and growth condition.
CoIP–RNA preparation and RNAseq
CoIP was performed in rhizobial lysates using the ANTI-FLAG M2 affinity gel (Sigma #A220) according to a published protocol.Citation29 The process was monitored by western blot analysis with an ANTI-FLAG monoclonal antibody (Sigma #F7425) as reported.Citation29 CoIP–RNA was further purified from the isolated RNA–protein complexes by phenol/chloroform/isoamyl alcohol extraction and ethanol precipitation. Equivalent CoIP–RNA quantities from the two independent pull-down experiments with SmhfqFLAG in each of the five growth conditions were finally pooled. Similarly, two control samples were obtained by pooling the CoIP–RNAs from strains Sm2B3001 and SmpWsinIFLAG.
CoIP–RNA pools were further processed by Vertis Biotechnologie AG to generate a total of seven strand-specific cDNA libraries, namely Hfq-log, Hfq-stat, Hfq-cold, Hfq-heat, Hfq-salt (from strain SmhfqFLAG), C-wt (from Sm2B3001), and C-SinI (from SmpWsinIFLAG). For this, CoIP–RNA samples were poly(A)-tailed using poly(A) polymerase. Then, the 5′PPP structures were removed using tobacco acid pyrophosphatase (TAP). Afterwards, an RNA adaptor was ligated to the 5′-phosphate of the RNA. First-strand cDNA synthesis was performed using an oligo(dT)-adaptor primer and the M-MLV reverse transcriptase. The resulting cDNAs were PCR-amplified to about 20–30 ng/μl using a high fidelity DNA polymerase. During PCR cycles, barcode sequences were attached to the 5′-ends of the cDNAs. The cDNAs were purified using the Agencourt AMPure XP kit (Beckman Coulter Genomics) and analyzed by capillary electrophoresis.
The primers used for PCR amplification were designed for TruSeq sequencing according to the instructions of Illumina. Adaptor sequences flanking the cDNA inserts were harbored by primers TrueSeq_Sense_primer and TrueSeq Antisense NNNNNN primer Barcode (Table S5). The combined length of the flanking sequences is 146 bases.
The libraries were sequenced on an Illumina GAIIx sequencer with 100 cycles. After sequencing, reads were demultiplexed based on their barcodes and adapters were removed.
Identification of Hfq-dependent transcripts/features
Genome sequences and annotations of S. meliloti reference strain 1021 (accessions NC_003037, NC_003047 and NC_003078) were downloaded from the Rhizogate Portal.Citation79 Next to 6296 protein-coding genes, the annotation of S. meliloti 1021 includes 545 trans-sRNAs, 3028 asRNAs, and the positions of TSS defining the 5′-UTR of 2832 mRNAs, all discovered in previous studies.Citation23,Citation39
Sequencing reads from the five Hfq and the two control (C) CoIP cDNA libraries were preprocessed and individually mapped to the S. meliloti 1021 genome using Segemehl with standard parameters.Citation80 Reads that did not map to the reference genome were trimmed to remove alien nucleotide sequences (e.g., polyA tails or adaptor sequences) and then subjected to a second mapping round. Reads mapping to a unique genomic location were kept for further analysis.
For detection of Hfq-dependent features, changes of relative transcript abundance between Hfq- and C-CoIP libraries were assessed. The quantification of transcript abundances was done for each sample and started with the aggregation of reads over all annotated S. meliloti 1021 genomic features. In case of protein-coding genes, the genomic coordinates of the UTRs, if available, were included in the analysis. Read counts per feature were obtained in a three-step procedure. Initially, reads overlapping single features were assigned to these features. To determine the origin of a read which overlapped multiple features, the definition of the intersection-nonempty counting mode of the HTSeq framework was applied.Citation81 Reads that could be disambiguated were assigned to their originating features. In an intermediate step, for each feature, an RPKM value was calculated from the read counts. Finally, the remaining ambiguous reads were allocated to the corresponding features in the ratio of their RPKM values.Citation82
After the calculation of feature read counts for the individual libraries, each of the five Hfq- and their corresponding C-CoIP library pairs were normalized, using the normalization strategy of the DESeq software.Citation83 Starting with feature read counts as input, DESeq estimated global size factors to normalize each library. The resulting normalized feature read counts (i.e., the relative transcript abundances) were the basis for the fold change calculation.
Features were defined to be bound by Hfq if (1) in at least one Hfq–CoIP library increased transcript abundance, relative to the control, by a factor higher than 5-fold was estimated, and (2) they were represented by at least 30 reads in the respective Hfq–CoIP library.
Identification of novel sRNAs
In addition to the discovery of Hfq-dependent features, a global screen to identify new unannotated trans-sRNAs and asRNAs was performed. First, reads from all five Hfq–CoIP libraries were pooled and clustered to transcripts. Transcripts with a minimal length of 50 nt and a continuous coverage of at least 10 reads were selected if they also met the Hfq dependency criteria. Transcripts that passed the filter step were classified based on the genomic position relative to annotated features. As missing UTR information can lead to misclassified transcripts, we applied the minimal transcriptional unit (MTU) model to the S. meliloti annotation.Citation23 The MTU model extends the annotation of a protein coding gene, which has incomplete UTR annotations, by virtual 5′- and 3′-UTRs of length 54 nt and 20 nt, respectively.
Transcripts represent trans-encoded sRNAs if they do not overlap any features, while transcripts overlapping a gene in antisense orientation are classified as cis-encoded antisense sRNA. The resulting novel trans-sRNAs and asRNAs were named according to the following conventions, SMx_Hfq_ncRNA_n and SMx_Hfq_asRNA_n, respectively, where “x” stands for the S. meliloti genomic replicon (i.e., c, chromosome; a, plasmid pSymA; b, plasmid pSymB).
Novel sRNA promoter profile search
Based on established promoter models,Citation23 profile searches for putative promoters specific for RpoD, RpoE2, RpoH1, RpoH2, RpoH1/2, and RpoN were performed upstream of the novel sRNAs using PoSSuMsearch.Citation84
RNA isolation and northern analysis
Total RNA from strain Sm2B3001 grown under the conditions of CoIP experiments was isolated by acid phenol/chloroform extraction as reported previously.Citation36 To assess Hfq-dependent sRNA decay, Sm2B3001 and its derivative SmΔhfq, obtained by deletion of the chromosomal Sm2B3001 hfq gene using the suicide plasmid pK18Δhfq,Citation29 were grown in 150 ml of TY broth until exponential phase, and transcription was terminated by rifampicin addition at final concentration of 800 μg/ml. Aliquots (10 ml) of the cultures were withdrawn immediately before rifampicin addition and at different time-points after the arrest of transcription for RNA extraction.
For northern analysis RNA samples (10 μg) were separated on 6% polyacrylamide/7M urea gels, blotted onto nylon membranes, and probed with 5′-end radiolabeled 25-mer oligonucleotides specific for selected sRNAs (Table S5) as described.Citation36 Hybridization signal intensities were quantified with the Quantity One software package (Bio-Rad).
Validation of predicted AbcR1 and AbcR2 mRNA targets
Antisense interactions between AbcR1/2 and their putative target mRNAs were searched for with IntaRNA.Citation85 Reporter plasmids, pRSMa0495::egfp and pRprbA::egfp, constitutively expressing translational fusions of the 5′ regions of SMa0495 and prbA (containing the predicted AbcR1/2 interaction site) to the N terminus of EGFP were then constructed by insertion of DNA fragments amplified from the Sm2B3001 genome with the primer pairs SMa0495_F/SMa0495_R and SMc01642_F/SMc01642_R, respectively (Table S5), into the low-copy pR-EGFP vector.
Reporter plasmids were individually transferred by conjugation to a S. meliloti 1021 AbcR1/2 double deletion mutant (ΔR1/2) harboring plasmids pSRK-R1, pSRK-R2 (constitutively expressing AbcR1 and AbcR2 sRNAs, respectively), or the empty vector pSRK.Citation42 Four double transconjugants for each pSRK-target fusion combination were grown in TY medium to exponential phase and fluorescence of 100 µl of bacterial cell cultures was measured on Tecan Infinite M200 reader (Tecan Trading AG) using in 96-well microtiter plates (Greiner). Background fluorescence was determined from strains harboring pSRK and the empty pR-EGFP plasmid instead of the target fusions and subtracted from the fluorescence of target fusions.
The AbcR1/2-dependent periplasmic proteome was previously determined.Citation42 New spots corresponding to differentially accumulated polypeptides were excised from gels and further processed for protein identification as reported.Citation42
| Abbreviations: | ||
| sRNA | = | small non-coding RNA |
| asRNA | = | antisense sRNA |
| CoIP | = | co-immunoprecipitation |
| ECF | = | extracytoplasmic function σ factor |
| UTR(s) | = | untranslated region(s) |
| IGR(s) | = | intergenic region(s) |
| IGB | = | integrated genome browser |
| MTU | = | minimal transcription unit |
| COG | = | clusters of orthologous groups |
| QS | = | quorum sensing |
| ORF(s) | = | open reading frame(s) |
| GMS | = | glucose mannitol salt medium |
| AHL(s) | = | acyl-homoserine lactone(s) |
| TSS | = | transcription start site(s) |
| RBS | = | ribosome binding site |
| ROS | = | reactive oxygen species |
| TAP | = | tobacco acid pyrophosphatase |
| bp | = | base pair |
| nt | = | nucleotide(s) |
Additional material
Download Zip (1.3 MB)Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
This work has been funded by ERDF-cofinanced grants AGL2009-07925 and CSD2009-00006 (Consolider-Ingenio Program) from the Spanish Ministerio de Economía y Competitividad (formerly Ministerio de Ciencia e Innovación) to Jiménez-Zurdo JI and Toro N, respectively. Torres-Quesada O was recipient of a FPI fellowship from the same Ministry and Peregrina A is recipient of a CSIC fellowship in the frame of the JAEDoc Program. Vicenta Millán for technical assistance and the core facilities of EEZ-CSIC for routine sequencing of plasmid constructs are acknowledged. Becker A acknowledges support from the LOEWE program of the State of Hesse, Germany (in the framework of the Center for Synthetic Microbiology, SYNMIKRO, Marburg) and from the German Research Foundation (SPP 1258, grant Be 2121/5-2). Work of Robledo M at SYNMIKRO was funded by a postdoctoral fellowship from the Alexander von Humboldt Foundation. We thank Konrad Förster (Institute of Molecular Infection Biology, University of Würzburg) and Richard Reinhart (Max Planck Genome Centre Cologne) for sequencing of the cDNA libraries.
References
- Sorek R, Cossart P. Prokaryotic transcriptomics: a new view on regulation, physiology and pathogenicity. Nat Rev Genet 2010; 11:9 - 16; http://dx.doi.org/10.1038/nrg2695; PMID: 19935729
- Storz G, Vogel J, Wassarman KM. Regulation by small RNAs in bacteria: expanding frontiers. Mol Cell 2011; 43:880 - 91; http://dx.doi.org/10.1016/j.molcel.2011.08.022; PMID: 21925377
- Waters LS, Storz G. Regulatory RNAs in bacteria. Cell 2009; 136:615 - 28; http://dx.doi.org/10.1016/j.cell.2009.01.043; PMID: 19239884
- Vogel J, Luisi BF. Hfq and its constellation of RNA. Nat Rev Microbiol 2011; 9:578 - 89; http://dx.doi.org/10.1038/nrmicro2615; PMID: 21760622
- Sobrero P, Valverde C. The bacterial protein Hfq: much more than a mere RNA-binding factor. Crit Rev Microbiol 2012; 38:276 - 99; http://dx.doi.org/10.3109/1040841X.2012.664540; PMID: 22435753
- Christiansen JK, Nielsen JS, Ebersbach T, Valentin-Hansen P, Søgaard-Andersen L, Kallipolitis BH. Identification of small Hfq-binding RNAs in Listeria monocytogenes.. RNA 2006; 12:1383 - 96; http://dx.doi.org/10.1261/rna.49706; PMID: 16682563
- Sittka A, Lucchini S, Papenfort K, Sharma CM, Rolle K, Binnewies TT, Hinton JC, Vogel J. Deep sequencing analysis of small noncoding RNA and mRNA targets of the global post-transcriptional regulator, Hfq. PLoS Genet 2008; 4:e1000163; http://dx.doi.org/10.1371/journal.pgen.1000163; PMID: 18725932
- Sonnleitner E, Sorger-Domenigg T, Madej MJ, Findeiss S, Hackermüller J, Hüttenhofer A, Stadler PF, Bläsi U, Moll I. Detection of small RNAs in Pseudomonas aeruginosa by RNomics and structure-based bioinformatic tools. Microbiology 2008; 154:3175 - 87; http://dx.doi.org/10.1099/mic.0.2008/019703-0; PMID: 18832323
- Sittka A, Sharma CM, Rolle K, Vogel J. Deep sequencing of Salmonella RNA associated with heterologous Hfq proteins in vivo reveals small RNAs as a major target class and identifies RNA processing phenotypes. RNA Biol 2009; 6:266 - 75; http://dx.doi.org/10.4161/rna.6.3.8332; PMID: 19333007
- Lorenz C, Gesell T, Zimmermann B, Schoeberl U, Bilusic I, Rajkowitsch L, Waldsich C, von Haeseler A, Schroeder R. Genomic SELEX for Hfq-binding RNAs identifies genomic aptamers predominantly in antisense transcripts. Nucleic Acids Res 2010; 38:3794 - 808; http://dx.doi.org/10.1093/nar/gkq032; PMID: 20348540
- Berghoff BA, Glaeser J, Sharma CM, Zobawa M, Lottspeich F, Vogel J, Klug G. Contribution of Hfq to photooxidative stress resistance and global regulation in Rhodobacter sphaeroides.. Mol Microbiol 2011; 80:1479 - 95; http://dx.doi.org/10.1111/j.1365-2958.2011.07658.x; PMID: 21535243
- Chao Y, Papenfort K, Reinhardt R, Sharma CM, Vogel J. An atlas of Hfq-bound transcripts reveals 3′ UTRs as a genomic reservoir of regulatory small RNAs. EMBO J 2012; 31:4005 - 19; http://dx.doi.org/10.1038/emboj.2012.229; PMID: 22922465
- Dambach M, Irnov I, Winkler WC. Association of RNAs with Bacillus subtilis Hfq. PLoS One 2013; 8:e55156; http://dx.doi.org/10.1371/journal.pone.0055156; PMID: 23457461
- Vytvytska O, Moll I, Kaberdin VR, von Gabain A, Bläsi U. Hfq (HF1) stimulates ompA mRNA decay by interfering with ribosome binding. Genes Dev 2000; 14:1109 - 18; PMID: 10809669
- Mohanty BK, Maples VF, Kushner SR. The Sm-like protein Hfq regulates polyadenylation dependent mRNA decay in Escherichia coli.. Mol Microbiol 2004; 54:905 - 20; http://dx.doi.org/10.1111/j.1365-2958.2004.04337.x; PMID: 15522076
- Jones KM, Kobayashi H, Davies BW, Taga ME, Walker GC. How rhizobial symbionts invade plants: the Sinorhizobium-Medicago model. Nat Rev Microbiol 2007; 5:619 - 33; http://dx.doi.org/10.1038/nrmicro1705; PMID: 17632573
- Galibert F, Finan TM, Long SR, Pühler A, Abola P, Ampe F, Barloy-Hubler F, Barnett MJ, Becker A, Boistard P, et al. The composite genome of the legume symbiont Sinorhizobium meliloti.. Science 2001; 293:668 - 72; http://dx.doi.org/10.1126/science.1060966; PMID: 11474104
- Ono Y, Mitsui H, Sato T, Minamisawa K. Two RpoH homologs responsible for the expression of heat shock protein genes in Sinorhizobium meliloti.. Mol Gen Genet 2001; 264:902 - 12; http://dx.doi.org/10.1007/s004380000380; PMID: 11254138
- Oke V, Rushing BG, Fisher EJ, Moghadam-Tabrizi M, Long SR. Identification of the heat-shock sigma factor RpoH and a second RpoH-like protein in Sinorhizobium meliloti.. Microbiology 2001; 147:2399 - 408; PMID: 11535780
- Sauviac L, Philippe H, Phok K, Bruand C. An extracytoplasmic function sigma factor acts as a general stress response regulator in Sinorhizobium meliloti.. J Bacteriol 2007; 189:4204 - 16; http://dx.doi.org/10.1128/JB.00175-07; PMID: 17400745
- Flechard M, Fontenelle C, Blanco C, Goude R, Ermel G, Trautwetter A. RpoE2 of Sinorhizobium meliloti is necessary for trehalose synthesis and growth in hyperosmotic media. Microbiology 2010; 156:1708 - 18; http://dx.doi.org/10.1099/mic.0.034850-0; PMID: 20223801
- Barnett MJ, Bittner AN, Toman CJ, Oke V, Long SR. Dual RpoH sigma factors and transcriptional plasticity in a symbiotic bacterium. J Bacteriol 2012; 194:4983 - 94; http://dx.doi.org/10.1128/JB.00449-12; PMID: 22773790
- Schlüter JP, Reinkensmeier J, Barnett MJ, Lang C, Krol E, Giegerich R, Long SR, Becker A. Global mapping of transcription start sites and promoter motifs in the symbiotic α-proteobacterium Sinorhizobium meliloti 1021. BMC Genomics 2013; 14:156; http://dx.doi.org/10.1186/1471-2164-14-156; PMID: 23497287
- Kaminski PA, Desnoues N, Elmerich C. The expression of nifA in Azorhizobium caulinodans requires a gene product homologous to Escherichia coli HF-I, an RNA-binding protein involved in the replication of phage Q beta RNA. Proc Natl Acad Sci U S A 1994; 91:4663 - 7; http://dx.doi.org/10.1073/pnas.91.11.4663; PMID: 8197116
- Drepper T, Raabe K, Giaourakis D, Gendrullis M, Masepohl B, Klipp W. The Hfq-like protein NrfA of the phototrophic purple bacterium Rhodobacter capsulatus controls nitrogen fixation via regulation of nifA and anfA expression. FEMS Microbiol Lett 2002; 215:221 - 7; http://dx.doi.org/10.1111/j.1574-6968.2002.tb11394.x; PMID: 12399038
- Zhang Y, Hong G. Post-transcriptional regulation of NifA expression by Hfq and RNase E complex in Rhizobium leguminosarum bv. viciae. Acta Biochim Biophys Sin (Shanghai) 2009; 41:719 - 30; http://dx.doi.org/10.1093/abbs/gmp060; PMID: 19727520
- Barra-Bily L, Pandey SP, Trautwetter A, Blanco C, Walker GC. The Sinorhizobium meliloti RNA chaperone Hfq mediates symbiosis of S. meliloti and alfalfa. J Bacteriol 2010; 192:1710 - 8; http://dx.doi.org/10.1128/JB.01427-09; PMID: 20081033
- Gao M, Barnett MJ, Long SR, Teplitski M. Role of the Sinorhizobium meliloti global regulator Hfq in gene regulation and symbiosis. Mol Plant Microbe Interact 2010; 23:355 - 65; http://dx.doi.org/10.1094/MPMI-23-4-0355; PMID: 20192823
- Torres-Quesada O, Oruezabal RI, Peregrina A, Jofré E, Lloret J, Rivilla R, Toro N, Jiménez-Zurdo JI. The Sinorhizobium meliloti RNA chaperone Hfq influences central carbon metabolism and the symbiotic interaction with alfalfa. BMC Microbiol 2010; 10:71; http://dx.doi.org/10.1186/1471-2180-10-71; PMID: 20205931
- Sobrero P, Valverde C. Evidences of autoregulation of hfq expression in Sinorhizobium meliloti strain 2011. Arch Microbiol 2011; 193:629 - 39; http://dx.doi.org/10.1007/s00203-011-0701-1; PMID: 21484295
- Sobrero P, Schlüter JP, Lanner U, Schlosser A, Becker A, Valverde C. Quantitative proteomic analysis of the Hfq-regulon in Sinorhizobium meliloti 2011. PLoS One 2012; 7:e48494; http://dx.doi.org/10.1371/journal.pone.0048494; PMID: 23119037
- Barra-Bily L, Fontenelle C, Jan G, Flechard M, Trautwetter A, Pandey SP, Walker GC, Blanco C. Proteomic alterations explain phenotypic changes in Sinorhizobium meliloti lacking the RNA chaperone Hfq. J Bacteriol 2010; 192:1719 - 29; http://dx.doi.org/10.1128/JB.01429-09; PMID: 20081032
- Robertson GT, Roop RM Jr.. The Brucella abortus host factor I (HF-I) protein contributes to stress resistance during stationary phase and is a major determinant of virulence in mice. Mol Microbiol 1999; 34:690 - 700; http://dx.doi.org/10.1046/j.1365-2958.1999.01629.x; PMID: 10564509
- Roop RM 2nd, Robertson GT, Ferguson GP, Milford LE, Winkler ME, Walker GC. Seeking a niche: putative contributions of the hfq and bacA gene products to the successful adaptation of the brucellae to their intracellular home. Vet Microbiol 2002; 90:349 - 63; http://dx.doi.org/10.1016/S0378-1135(02)00220-1; PMID: 12414155
- Wilms I, Möller P, Stock AM, Gurski R, Lai EM, Narberhaus F. Hfq influences multiple transport systems and virulence in the plant pathogen Agrobacterium tumefaciens.. J Bacteriol 2012; 194:5209 - 17; http://dx.doi.org/10.1128/JB.00510-12; PMID: 22821981
- del Val C, Rivas E, Torres-Quesada O, Toro N, Jiménez-Zurdo JI. Identification of differentially expressed small non-coding RNAs in the legume endosymbiont Sinorhizobium meliloti by comparative genomics. Mol Microbiol 2007; 66:1080 - 91; http://dx.doi.org/10.1111/j.1365-2958.2007.05978.x; PMID: 17971083
- Ulvé VM, Sevin EW, Chéron A, Barloy-Hubler F. Identification of chromosomal alpha-proteobacterial small RNAs by comparative genome analysis and detection in Sinorhizobium meliloti strain 1021. BMC Genomics 2007; 8:467; http://dx.doi.org/10.1186/1471-2164-8-467; PMID: 18093320
- Valverde C, Livny J, Schlüter JP, Reinkensmeier J, Becker A, Parisi G. Prediction of Sinorhizobium meliloti sRNA genes and experimental detection in strain 2011. BMC Genomics 2008; 9:416; http://dx.doi.org/10.1186/1471-2164-9-416; PMID: 18793445
- Schlüter JP, Reinkensmeier J, Daschkey S, Evguenieva-Hackenberg E, Janssen S, Jänicke S, Becker JD, Giegerich R, Becker A. A genome-wide survey of sRNAs in the symbiotic nitrogen-fixing alpha-proteobacterium Sinorhizobium meliloti.. BMC Genomics 2010; 11:245; http://dx.doi.org/10.1186/1471-2164-11-245; PMID: 20398411
- Jiménez-Zurdo JI, Valverde C, Becker A. Insights into the noncoding RNome of nitrogen-fixing endosymbiotic α-proteobacteria. Mol Plant Microbe Interact 2013; 26:160 - 7; http://dx.doi.org/10.1094/MPMI-07-12-0186-CR; PMID: 22991999
- Voss B, Hölscher M, Baumgarth B, Kalbfleisch A, Kaya C, Hess WR, Becker A, Evguenieva-Hackenberg E. Expression of small RNAs in Rhizobiales and protection of a small RNA and its degradation products by Hfq in Sinorhizobium meliloti.. Biochem Biophys Res Commun 2009; 390:331 - 6; http://dx.doi.org/10.1016/j.bbrc.2009.09.125; PMID: 19800865
- Torres-Quesada O, Millán V, Nisa-Martínez R, Bardou F, Crespi M, Toro N, Jiménez-Zurdo JI. Independent activity of the homologous small regulatory RNAs AbcR1 and AbcR2 in the legume symbiont Sinorhizobium meliloti.. PLoS One 2013; 8:e68147; http://dx.doi.org/10.1371/journal.pone.0068147; PMID: 23869210
- Reinkensmeier J, Schlüter J-P, Giegerich R, Becker A. Conservation and occurrence of trans-encoded sRNAs in the Rhizobiales. Genes 2011; 2:925 - 56; http://dx.doi.org/10.3390/genes2040925
- del Val C, Romero-Zaliz R, Torres-Quesada O, Peregrina A, Toro N, Jiménez-Zurdo JI. A survey of sRNA families in α-proteobacteria. RNA Biol 2012; 9:119 - 29; http://dx.doi.org/10.4161/rna.18643; PMID: 22418845
- Ulvé VM, Chéron A, Trautwetter A, Fontenelle C, Barloy-Hubler F. Characterization and expression patterns of Sinorhizobium meliloti tmRNA (ssrA). FEMS Microbiol Lett 2007; 269:117 - 23; http://dx.doi.org/10.1111/j.1574-6968.2006.00616.x; PMID: 17241239
- Hoang HH, Becker A, González JE. The LuxR homolog ExpR, in combination with the Sin quorum sensing system, plays a central role in Sinorhizobium meliloti gene expression. J Bacteriol 2004; 186:5460 - 72; http://dx.doi.org/10.1128/JB.186.16.5460-5472.2004; PMID: 15292148
- Gao M, Chen H, Eberhard A, Gronquist MR, Robinson JB, Rolfe BG, Bauer WD. sinI- and expR-dependent quorum sensing in Sinorhizobium meliloti.. J Bacteriol 2005; 187:7931 - 44; http://dx.doi.org/10.1128/JB.187.23.7931-7944.2005; PMID: 16291666
- Gurich N, González JE. Role of quorum sensing in Sinorhizobium meliloti-Alfalfa symbiosis. J Bacteriol 2009; 191:4372 - 82; http://dx.doi.org/10.1128/JB.00376-09; PMID: 19395488
- Charoenpanich P, Meyer S, Becker A, McIntosh M. Temporal expression program of quorum sensing-based transcription regulation in Sinorhizobium meliloti.. J Bacteriol 2013; 195:3224 - 36; http://dx.doi.org/10.1128/JB.00234-13; PMID: 23687265
- Romby P, Charpentier E. An overview of RNAs with regulatory functions in gram-positive bacteria. Cell Mol Life Sci 2010; 67:217 - 37; http://dx.doi.org/10.1007/s00018-009-0162-8; PMID: 19859665
- Pandey SP, Minesinger BK, Kumar J, Walker GC. A highly conserved protein of unknown function in Sinorhizobium meliloti affects sRNA regulation similar to Hfq. Nucleic Acids Res 2011; 39:4691 - 708; http://dx.doi.org/10.1093/nar/gkr060; PMID: 21325267
- Ross JA, Wardle SJ, Haniford DB. Tn10/IS10 transposition is downregulated at the level of transposase expression by the RNA-binding protein Hfq. Mol Microbiol 2010; 78:607 - 21; http://dx.doi.org/10.1111/j.1365-2958.2010.07359.x; PMID: 20815820
- Ross JA, Ellis MJ, Hossain S, Haniford DB. Hfq restructures RNA-IN and RNA-OUT and facilitates antisense pairing in the Tn10/IS10 system. RNA 2013; 19:670 - 84; http://dx.doi.org/10.1261/rna.037747.112; PMID: 23510801
- Galardini M, Mengoni A, Brilli M, Pini F, Fioravanti A, Lucas S, Lapidus A, Cheng JF, Goodwin L, Pitluck S, et al. Exploring the symbiotic pangenome of the nitrogen-fixing bacterium Sinorhizobium meliloti.. BMC Genomics 2011; 12:235; http://dx.doi.org/10.1186/1471-2164-12-235; PMID: 21569405
- Folichon M, Arluison V, Pellegrini O, Huntzinger E, Régnier P, Hajnsdorf E. The poly(A) binding protein Hfq protects RNA from RNase E and exoribonucleolytic degradation. Nucleic Acids Res 2003; 31:7302 - 10; http://dx.doi.org/10.1093/nar/gkg915; PMID: 14654705
- Moon K, Gottesman S. Competition among Hfq-binding small RNAs in Escherichia coli.. Mol Microbiol 2011; 82:1545 - 62; http://dx.doi.org/10.1111/j.1365-2958.2011.07907.x; PMID: 22040174
- Mitsui H, Sato T, Sato Y, Ito N, Minamisawa K. Sinorhizobium meliloti RpoH1 is required for effective nitrogen-fixing symbiosis with alfalfa. Mol Genet Genomics 2004; 271:416 - 25; http://dx.doi.org/10.1007/s00438-004-0992-x; PMID: 15007732
- Vecerek B, Rajkowitsch L, Sonnleitner E, Schroeder R, Bläsi U. The C-terminal domain of Escherichia coli Hfq is required for regulation. Nucleic Acids Res 2008; 36:133 - 43; http://dx.doi.org/10.1093/nar/gkm985; PMID: 18000007
- Vecerek B, Moll I, Bläsi U. Translational autocontrol of the Escherichia coli hfq RNA chaperone gene. RNA 2005; 11:976 - 84; http://dx.doi.org/10.1261/rna.2360205; PMID: 15872186
- Olsen AS, Møller-Jensen J, Brennan RG, Valentin-Hansen P. C-terminally truncated derivatives of Escherichia coli Hfq are proficient in riboregulation. J Mol Biol 2010; 404:173 - 82; http://dx.doi.org/10.1016/j.jmb.2010.09.038; PMID: 20888338
- Guisbert E, Rhodius VA, Ahuja N, Witkin E, Gross CA. Hfq modulates the sigmaE-mediated envelope stress response and the sigma32-mediated cytoplasmic stress response in Escherichia coli.. J Bacteriol 2007; 189:1963 - 73; http://dx.doi.org/10.1128/JB.01243-06; PMID: 17158661
- Gong Z, Zhu J, Yu G, Zou H. Disruption of nifA gene influences multiple cellular processes in Sinorhizobium meliloti.. J Genet Genomics 2007; 34:783 - 9; http://dx.doi.org/10.1016/S1673-8527(07)60089-7; PMID: 17884688
- Fröhlich KS, Papenfort K, Fekete A, Vogel J. A small RNA activates CFA synthase by isoform-specific mRNA stabilization. EMBO J 2013; 32:2963 - 79; http://dx.doi.org/10.1038/emboj.2013.222; PMID: 24141880
- Papenfort K, Sun Y, Miyakoshi M, Vanderpool CK, Vogel J. Small RNA-mediated activation of sugar phosphatase mRNA regulates glucose homeostasis. Cell 2013; 153:426 - 37; http://dx.doi.org/10.1016/j.cell.2013.03.003; PMID: 23582330
- Salvail H, Caron MP, Bélanger J, Massé E. Antagonistic functions between the RNA chaperone Hfq and an sRNA regulate sensitivity to the antibiotic colicin. EMBO J 2013; 32:2764 - 78; http://dx.doi.org/10.1038/emboj.2013.205; PMID: 24065131
- Vogel J, Bartels V, Tang TH, Churakov G, Slagter-Jäger JG, Hüttenhofer A, Wagner EG. RNomics in Escherichia coli detects new sRNA species and indicates parallel transcriptional output in bacteria. Nucleic Acids Res 2003; 31:6435 - 43; http://dx.doi.org/10.1093/nar/gkg867; PMID: 14602901
- Kawano M, Reynolds AA, Miranda-Rios J, Storz G. Detection of 5′- and 3′-UTR-derived small RNAs and cis-encoded antisense RNAs in Escherichia coli.. Nucleic Acids Res 2005; 33:1040 - 50; http://dx.doi.org/10.1093/nar/gki256; PMID: 15718303
- Loh E, Dussurget O, Gripenland J, Vaitkevicius K, Tiensuu T, Mandin P, Repoila F, Buchrieser C, Cossart P, Johansson J. A trans-acting riboswitch controls expression of the virulence regulator PrfA in Listeria monocytogenes.. Cell 2009; 139:770 - 9; http://dx.doi.org/10.1016/j.cell.2009.08.046; PMID: 19914169
- Wagner EGH. Cycling of RNAs on Hfq. RNA Biol 2013; 10:619 - 26; http://dx.doi.org/10.4161/rna.24044; PMID: 23466677
- Sharma CM, Darfeuille F, Plantinga TH, Vogel J. A small RNA regulates multiple ABC transporter mRNAs by targeting C/A-rich elements inside and upstream of ribosome-binding sites. Genes Dev 2007; 21:2804 - 17; http://dx.doi.org/10.1101/gad.447207; PMID: 17974919
- Pulvermacher SC, Stauffer LT, Stauffer GV. Role of the Escherichia coli Hfq protein in GcvB regulation of oppA and dppA mRNAs. Microbiology 2009; 155:115 - 23; http://dx.doi.org/10.1099/mic.0.023432-0; PMID: 19118352
- Sharma CM, Papenfort K, Pernitzsch SR, Mollenkopf H-J, Hinton JCD, Vogel J. Pervasive post-transcriptional control of genes involved in amino acid metabolism by the Hfq-dependent GcvB small RNA. Mol Microbiol 2011; 81:1144 - 65; http://dx.doi.org/10.1111/j.1365-2958.2011.07751.x; PMID: 21696468
- Wilms I, Voss B, Hess WR, Leichert LI, Narberhaus F. Small RNA-mediated control of the Agrobacterium tumefaciens GABA binding protein. Mol Microbiol 2011; 80:492 - 506; http://dx.doi.org/10.1111/j.1365-2958.2011.07589.x; PMID: 21320185
- Caswell CC, Gaines JM, Ciborowski P, Smith D, Borchers CH, Roux CM, Sayood K, Dunman PM, Roop Ii RM. Identification of two small regulatory RNAs linked to virulence in Brucella abortus 2308. Mol Microbiol 2012; 85:345 - 60; http://dx.doi.org/10.1111/j.1365-2958.2012.08117.x; PMID: 22690807
- Beringer JE. R factor transfer in Rhizobium leguminosarum.. J Gen Microbiol 1974; 84:188 - 98; http://dx.doi.org/10.1099/00221287-84-1-188; PMID: 4612098
- Bahlawane C, McIntosh M, Krol E, Becker A. Sinorhizobium meliloti regulator MucR couples exopolysaccharide synthesis and motility. Mol Plant Microbe Interact 2008; 21:1498 - 509; http://dx.doi.org/10.1094/MPMI-21-11-1498; PMID: 18842098
- Khan SR, Gaines J, Roop RM 2nd, Farrand SK. Broad-host-range expression vectors with tightly regulated promoters and their use to examine the influence of TraR and TraM expression on Ti plasmid quorum sensing. Appl Environ Microbiol 2008; 74:5053 - 62; http://dx.doi.org/10.1128/AEM.01098-08; PMID: 18606801
- Zevenhuizen LPTM, van Neerven ARW. Gel-forming capsular polysaccharide of Rhizobium leguminosarum and Rhizobium trifolii.. Carbohydr Res 1983; 124:166 - 71; http://dx.doi.org/10.1016/0008-6215(83)88366-9
- Becker A, Barnett MJ, Capela D, Dondrup M, Kamp PB, Krol E, Linke B, Rüberg S, Runte K, Schroeder BK, et al. A portal for rhizobial genomes: RhizoGATE integrates a Sinorhizobium meliloti genome annotation update with postgenome data. J Biotechnol 2009; 140:45 - 50; http://dx.doi.org/10.1016/j.jbiotec.2008.11.006; PMID: 19103235
- Hoffmann S, Otto C, Kurtz S, Sharma CM, Khaitovich P, Vogel J, Stadler PF, Hackermüller J. Fast mapping of short sequences with mismatches, insertions and deletions using index structures. PLoS Comput Biol 2009; 5:e1000502; http://dx.doi.org/10.1371/journal.pcbi.1000502; PMID: 19750212
- Anders S. HTSeq: Analysing high-throughput sequencing data with Python. URL http://www-huber embl de/users/anders/HTSeq/doc/overview html 2010.
- Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods 2008; 5:621 - 8; http://dx.doi.org/10.1038/nmeth.1226; PMID: 18516045
- Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol 2010; 11:R106; http://dx.doi.org/10.1186/gb-2010-11-10-r106; PMID: 20979621
- Beckstette M, Homann R, Giegerich R, Kurtz S. Significant speedup of database searches with HMMs by search space reduction with PSSM family models. Bioinformatics 2009; 25:3251 - 8; http://dx.doi.org/10.1093/bioinformatics/btp593; PMID: 19828575
- Smith C, Heyne S, Richter AS, Will S, Backofen R. Freiburg RNA Tools: a web server integrating INTARNA, EXPARNA and LOCARNA. Nucleic Acids Res 2010; 38:Suppl W373 - 7; http://dx.doi.org/10.1093/nar/gkq316; PMID: 20444875