
Abstract
The genome of most prokaryotes gives rise to surprisingly complex transcriptomes, comprising not only protein-coding mRNAs, often organized as operons, but also harbors dozens or even hundreds of highly structured small regulatory RNAs and unexpectedly large levels of anti-sense transcripts. Comprehensive surveys of prokaryotic transcriptomes and the need to characterize also their non-coding components is heavily dependent on computational methods and workflows, many of which have been developed or at least adapted specifically for the use with bacterial and archaeal data. This review provides an overview on the state-of-the-art of RNA bioinformatics focusing on applications to prokaryotes.
Introduction
During the last decade, thousands of small RNAs (sRNAs) have been discovered in a widely diverse set of prokaryotes. Beyond the evolutionary ancient “housekeeping” RNA genes encoding tRNAs, rRNAs, RNase P RNA, and SRP RNA (as well as tmRNA and 6S RNA in bacteria), typical genomes harbor dozens or even hundreds of sRNAs with predominantly regulatory roles. Archaea, in addition, have homologs of the small nucleolar RNAs of Eukarya (snoRNAs), directing chemical modifications of rRNAs and other RNA targets. Compared with protein-coding genes, most of the prokaryotic RNAs are still rather poorly characterized in terms of their structure, function, and phylogenetic distribution. In particular, with the advent of high-throughput transcriptomics, large numbers of sRNA candidates have been detected, but so far have not received attention beyond a note of their genomic coordinates.
Computational approaches have been very successful in facilitating, extending, and complementing experimental investigations. In this contribution, we review the state-of-the-art and the limitations of RNA bioinformatics as applied to prokaryotes. Albeit we cover a broad variety of approaches, our presentation emphasizes particular methods and tools that were developed or substantially improved within the Priority Program SPP 1258: Sensory and regulatory RNAs in Prokaryotes funded by the Deutsche Forschungsgemeinschaft from 2007–2013. It is a successful example of a coordinated project in which many new or adapted bioinformatics tools have been developed specifically according to the needs of several experimental groups.
Structure Prediction
The complex three-dimensional structures formed by many functional single-stranded nucleic acids are dominated by base pairing both in terms of the energy of folding and in the sense that much of the shape can be understood in terms of the co-planar arrangement of the bases. At the same time, the status of a nucleotide as either paired or unpaired can be interrogated experimentally by means of chemical or enzymatic probing. This makes secondary structures an important level of description.
The problem of secondary structure prediction is well investigated and described elsewhere.Citation1-Citation3 The most prominent implementations of RNA folding algorithms are mfoldCitation4 and the ViennaRNA Package.Citation5,Citation6 Standard approaches consider only non-crossing structures, a condition that is not always satisfied. Different classes of pseudoknot structures have been definedCitation7 and corresponding prediction algorithms have been implemented, albeit at the expense of higher computational complexity.Citation7-Citation13
The accuracy of secondary structure prediction from single sequences is far from perfect for a wide variety of reasons. Some derive from limitations of the secondary structure model, such as deviations from the additive model, insufficient knowledge of energy parameters, simplified parametrization of multi-loops, and the exclusion of non-standard base pairs. In addition, the precise transcript might be known only partially, or structure motifs are embedded into a larger RNA, which leads to the even harder problem of local structure prediction.Citation14 There are two remedies for these problems: (1) instead of just a single sequence, evolutionary information on patterns of sequence conservation may be taken into account, or (2) experimental evidence such as chemical probing or FRET data may be incorporated into structure prediction.
When accurate sequence alignments can be obtained, these may serve as a basis for computing consensus structures. The simplest approach, implemented e.g., in RNAalifold,Citation15,Citation16 is to extend the RNA folding algorithms to compute a secondary structure that minimizes the average folding energy of the aligned sequences. A more sophisticated phylogenetic model replacing simple averaging is implemented in PETfold.Citation17 At lower levels of sequence conservation, folding and alignment must be computed simultaneously at a much higher computational cost. Several practical approaches exist, from full-fledged implementations of the Sankoff algorithm,Citation18 e.g., in FoldalignCitation19 and Dynalign,Citation20 to computationally much more efficient approximations that restrict themselves to base pairs that are thermodynamically plausible for the individual sequences. Tools of the latter type are LocaRNA and its variants,Citation21-Citation24 and SPARSE.Citation25 A conceptually different approach taken by the RNAshapes packageCitation26 makes use of coarse-grained structures. In all cases, the output consists of a sequence alignment annotated by a consensus structure—exactly the input required later on for homology search.
Experimental data can be integrated into structure prediction either as hard constraints (enforcing or prohibiting certain base pairs) or as soft constraints that distort the ensemble of structure by adding bonus energies or energy penalties to encouraged or discouraged structural elements, resp. Measurement of SHAPE,Citation27 PARS,Citation28 or other chemical or enzymatic probing methods can be converted into pseudo-energies added to paired or unpaired bases, leading to a distortion of the Boltzmann ensemble toward the experimental signal.Citation29,Citation30 Most recently, more sophisticated approaches have appeared toward reconciliating experimental data with the thermodynamic folding approach. RNAassistCitation31 formulates the problem in terms of simultaneously minimizing position-dependent energy penalties and the deviation of observed and predicted probabilities for unpaired nucleotides. SeqFold uses the experimental data to select locally stable secondary structure from the Boltzmann ensemble.Citation32 In ShapeKnots,Citation33 an interative procedure is used to include pseudoknots and SHAPE information. It has been applied to e.g., investigate the structure of a SAM-I riboswitch.
Gene Finding and Transcriptomics
Homology search
The initial gene annotation of a newly sequenced genome is created by comparison with known sequences of related organisms together with the application of de novo prediction methods; in particular, the search of open reading frames of sufficient length. Since non-coding RNAs (ncRNAs) do not offer a similar generic sequence pattern, they are much harder to predict from scratch.Citation34 As a consequence, only a few well-known RNA genes such as tRNAs, RNase P RNA, SPR RNA, and the rRNA subunits, are annotated for most prokaryotic genomes. Both homology search and many of the comparative genomics approaches discussed below are applicable not only to independent sRNAs but also to structured RNA elements, which includes, in particular, riboswitches,Citation35 RNA thermometers,Citation36 and several other cis-acting elements. For brevity, we will simply speak of ncRNAs in the following.
The Rfam database, as the most extensive repository of structured RNAs, lists in its current version 11.0 a total of 605 RNA families with prokaryotic members (527 bacterial and 107 archaeal).Citation37 This number includes, however, a large number of CRISPR RNA repeats, many riboswitches, and other mRNA elements, as well as ubiquitous RNA families such as tRNAs or RNase P. There is, at present, no comprehensive repository of prokaryotic small RNAs. The overwhelming majority of sRNAs discovered after the publication of a reference genome are documented only in the main text of publications or in supplemental material. Despite community efforts and incentives such as free open access publication of RNA family descriptions in this journal,Citation38 only a very moderate number of prokaryotic RNA families have been described in detail and deposited to databases, see e.g. references Citation39–Citation42. As a consequence, the majority of sRNA families remain in practice unavailable for genome annotation pipelines. For the same reason, it is impossible to give an accurate estimate on the total number of bacterial or archaeal sRNA families or to globally assess their phylogenetic distributions with any degree of certainty.
The most widely used tool for homology search is blast. For highly diverged sequences, blast typically reports several small fragments instead of the full-length match to the query sequence. Thus, it is not implicitly the method of choice.Citation43 Specialized ncRNA sequence homology search derivates of blast are available, e.g., blastR.Citation44 Semi-global dynamic programming algorithms such as GotohscanCitation45 are a viable alternative given the small genome size of prokaryotes. This program reports full-length hits, makes subsequent processing of the predicted homologs much easier, and is particularly well-suited for ncRNAs,Citation46 which—in contrast to protein-coding genes—are typically short and evolve rapidly at the sequence level. These properties generally limit the sensitivity of purely sequence-based methods. The information content of the query can be increased by making use of secondary structure conservation as well. Covariance models (CMs), a generalization of HMMs to tree-like structures, provide a convenient technical basis.Citation47 They have to be trained from multiple sequence alignments annotated by a consensus structure. In contrast to blast, which is content with a single query sequence, CMs require a collection of evolutionarily related and alignable homologs as a starting point. With infernal 1.1, a highly efficient implementation of a search tool for CMs has become available that is suitable for large-scale applications.Citation48 Most covariance models, in particular, the models of the Rfam families, are dominated by sequence information.Citation49 At least in this regime, infernal is the most effective tool available. Phylogenetic distance, and hence, decreasing sequence conservation, eventually limits applicability of homology search. It is possible in principle to include thermodynamic stability, either using the idea of thermodynamic matchersCitation50 or employing structural alignments.Citation24 It remains unclear, however, whether such techniques can substantially improve the sensitivity of homology search for distantly related species.
Feature-based gene prediction
sRNApredictCitation51 uses typical features of prokaryotic sRNAs: elevated sequence conservation, putative promoter sequences, and Rho-independent terminator elements. TranstermHP, for instance, is used to predict Rho-independent terminators.Citation52 Its scoring function favors G/C-rich stem loops followed by a poly-T track. It is obviously extremely difficult to detect correct terminator elements in species with a high G/C-content and in those that use structural elements deviating from the canonical terminator structure. In order to increase sensitivity and specificity, sRNApredict focuses on intergenic regions and analyzes the co-occurrence of several of the above-mentioned features. While this strategy works quite well for well-characterized bacterial clades, it is bound to fail in others. Xanthomonas and Helicobacter, for example, lack typical promoter sequences and distinct terminator hairpins.Citation46,Citation53
Transcriptomics
Bacterial (and archaeal) transcriptomics can almost always be performed with a reference genome in place. This simplifies the workflow, which is basically composed of the following steps.
(1) Library preparation: Transcriptome analyses consist of “wet-lab” experiments and “dry-lab” data evaluation. Both components greatly influence the final outcome and it is therefore recommended to design the experimental setup in a cooperative way, such that practical and theoretical issues are discussed at the very beginning. Selection of an appropriate sequencing platform, e.g., 454 or Illumina, and the enrichment or depletion of certain RNA classes, are only two of many design decisions that depend on the research question. The actual experiments are performed and, depending on the sequencing platform and sequencing depth, several gigabytes of RNA transcript data are reported.
(2) Quality check: Sequencing machines typically output FASTQ-formatted files. This extended version of FASTA files is augmented by quality information for each called nucleotide along the sequence. FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc) is commonly used to initially check and visualize the quality of the raw sequencing data. Software suites such as the FASTX-Toolkit (http://hannonlab.cshl.edu/fastx_toolkit) provide several tools to preprocess the raw sequencing reads by e.g., removal of the adaptor and bar code sequences that have been attached during library preparation, or by filtering of low complexity reads. These steps can have a drastic influence on the mapping quality.
(3) Read mapping: A large number of software tools for read mapping has become available that differ widely in their algorithmic basis, memory consumption, speed, and versatility. Mapping strategies furthermore differ in their treatment of reads that map equally good to multiple genomic locations and in their handling of insertions and deletions.Citation54-Citation58 It is therefore important to match the choice of mapping tool to the research question.Citation59 We used segemehlCitation60,Citation61 very successfully in a variety of studies, ranging from dRNA-seq analysis to split read mapping in prokaryotes. In our hands, segemehl has proven to be a flexible and highly accurate framework. This has also been repeatedly shown in benchmarks using real live and simulated data.Citation59,Citation62
Once the mapping step is completed, mapping summary statistics help to verify whether all prior steps have been successful. Transcriptome studies that investigate prokaryotes usually assume that reads map without interruption (“split-free”) and with near perfect sequence identity to the genome. This is, indeed, the case for the overwhelming majority of the reads. There are, however, biological relevant exceptions that usually end up in the “sequencing trash bin.” Examples include transcripts containing self-splicing introns in bacteria, as well as enzymatically spliced and circularized RNAs in archaea. A recent study showed that such “atypical” transcript structures may be much more abundant than expected.Citation63 It remains, however, unclear to what extent rare transcripts of this type are biologically relevant, how many of them are technical artifacts, and to what extent one detects true cellular RNAs that are nevertheless functionally irrelevant. Post-transcriptional modifications may furthermore lead to large local error rates.Citation64
(4) Transcript annotation and classification: The transcripts are then evaluated with respect to the genomic loci they have been mapped to. This covers in general a classification into protein-coding, non-coding, and intergenic regions. For a typical prokaryotic genome, the non-coding portion is mainly comprised of reads that originate from the highly abundant tRNAs and rRNAs and from a few well-characterized house keeping genes such as tmRNA and 6S RNA. In most prokaryotes, only the open reading frames of protein-coding genes are annotated, while regulatory regions of mRNA transcripts, i.e., their UTRs (untranslated regions), are missing and the structure of polycistronic transcripts, i.e., transcripts that contain more than one gene, remains uncertain. Thereby, the number of reads mapping to intergenic regions is overestimated due to this knowledge gap. The detection of polycistronic transcripts can be achieved by using a high-sequencing depth close to saturation. The exact determination of transcriptional units is, however, challenging, as gap-free expression cannot be found even for well-characterized cases such as the cag pathogenicity island of H. pylori.Citation53 Another difficult task is the precise mapping of the genomic positions where transcription is initiated. This challenge has been addressed by specific sequencing library preparation steps; the evaluation of the resulting read patterns is described in more detail in the next subsection on transcription start site (TSS) annotation. The determined TSS maps revealed an unexpected complexity of the transcription unit organization. Transcription is initiated as expected ahead of annotated genes and polycistronic transcripts but also internally and anti-sense to them, and therefore, almost everywhere along the genome. Upstream of the determined TSS, promoter sequence motifs are expected. Textbook knowledge describing two conserved elements, i.e., the -10 and -35 box, has been revised, as these motifs are extremely variable between species. In Xanthomonas and Helicobacter, for instance, only traces of the -10 box are detectable, but no distinct -35 box has been reported.Citation46,Citation53 It seems to be a matter of fact that the current experimental setups enable the detection of TSS with species-specific housekeeping promoters, but alternative binding motifs are still hidden. The sequence between an annotated TSS and the start of a nearby downstream protein-coding gene gives rise to its 5′ UTR. So-called leader-less transcripts that lack 5′ UTRs completely, i.e., translation start and TSS are mapped to (almost) the same position, are abundant in archaea,Citation65,Citation66 but have been thought to be quite rare in bacteria. Surprisingly, dRNA-seq experiments, however, reported a large number of leaderless transcripts and 5′ UTRs lacking Shine-Dalgarno sequence patterns in diverse bacteria.Citation46,Citation53 Besides the possibility to gain new insights into protein-coding genes, most prokaryotic transcriptome studies are set up to detect novel non-coding RNA genes. These are typically identified by the analysis of read accumulations in intergenic regions or anti-sense to annotated genes. The existence of transcription units that might correspond to non-coding genes is verified by independent experiments such as northern blotting, and their exact size is determined by RACE. A single study reveals dozens of novel RNA genes that need to be further characterized. Common tasks are the detection of homologous sequences, structural conservation analysis, evaluation of their coding potential, and target prediction. For a detailed description of these evaluations, we refer to the sections on homology search, comparative genomics, and RNA—RNA interactions, respectively.
TSS annotation
In contrast to translation start sites that can be identified by well-established gene annotation strategies,Citation67,Citation68 surprisingly little is known about transcription start sites (TSS) in most bacteria. Even though a thorough TSS annotation can serve as valuable source of information to (1) understand the architecture of polycistronic transcripts, (2) use it as a paramount hallmark for ncRNA gene annotation, and (3) determine the extent of the 5′ UTR, which often harbors regulatory elements such as riboswitches, RNA thermometer, and sRNA binding sites.
The first successfully applied methods to annotate TSS were primer extensionCitation69 and RACE.Citation70 Both techniques aim to find the 5′ end of partly characterized genes, but suffer from two major drawbacks. First, with these techniques it is not possible to distinguish between 5′ ends of an RNA formed by a transcription initiation event or by an RNA cleavage event, which often occurs in the course of RNA processing. Second, both techniques are difficult to scale up to a genome-wide high-throughput application. Therefore, two RNA-seq-based methods for reliable annotation of TSS in bacterial genomes were developed recently.Citation53,Citation66 Both methods exploit the phosphorylation pattern unique to primary TSS. Mono-nucleotides for transcription are provided to the RNA polymerase in the form of nucleotide triphosphates, which are broken down in the process of transcription elongation and the released energy is used to form a phosphodiester bond between the newly conjoined nucleosides. As a consequence, the first nucleotide still has a triphosphate attached to its 5′ carbon atom. In contrast, if the phosphodiester bond of two consecutive nucleosides is broken by endonucleolytic cleavage, the remaining fragment is a 5′-phosphomonoester.
In the method developed by Wurtzel, et al.,Citation66 the total RNA is treated with tobacco acid pyrophosphatase (TAP), which removes the 5′-triphosphate, and hence, makes the RNA susceptible for the subsequent 5′-sequencing-adaptor ligation. The 3′-adaptor is attached by a random primer. In contrast to a library, which is not TAP-treated, reads associated with primary TSS are enriched in the TAP-treated library.
An alternative methodCitation53 uses the Terminator-5′-phosphate-dependent exonuclease (TEX) to deplete the total RNA of fragments that are not protected from exonuclease degradation by a 5′-triphosphate. As a control, total RNA from the same extraction is processed the same way, but without the TEX treatment. Therefore, in the final analysis step, the differences between the treated (a.k.a. plus) library and the untreated (a.k.a. minus) library have to be screened position-wise for sites with a compelling enrichment of RNA-seq read starts in the plus vs. the minus library. That is why this method was named differential RNA-seq (dRNA-seq).
The first applications of dRNA-seq were manually analyzed by visualizing the reads and assessing the enrichment. Since such a screening is very time-consuming and tedious on genome-scale, and since it involves the subjective assessment of the analyzer, the results suffer from a certain lack of reproducibility and consistency. Therefore, soon after, the first statistical approaches to evaluate dRNA-seq data were proposed. Schmidtke, et al.Citation46 modeled the density of read starts within the genome locally by applying a sliding window approach. Within each window, the distribution of read start counts per position are assumed to follow a Poisson distribution. As a consequence, the differences between the two libraries can be modeled by the Skellam distribution, which allows to calculate the probability to encounter the observed enrichment by chance.
Alternatively, global thresholds are applied to discriminate between significant read enrichment and background noise.Citation74,Citation75 To gain specificity, the TSS calling is split into two steps. First, the relative read coverage increase in the treated library from position i-1 to position i is evaluated. If this increase surpasses a defined threshold, the position is further evaluated whether the ratio of observed transcription initiation between treated and untreated library exceeds a defined threshold. If both tests are passed, the position is annotated as a TSS. The strength of this method, as implemented in the program TTSpredator, lies in the ability to regard dRNA-seq data from different strains and/or growth conditions and dynamically adjust the thresholds if strong signals are observed in one sample. This circumvents a strict a priori threshold definition, which might be difficult to find for a new data set with different sequencing depth, genome size, and TEX treatment efficiency.
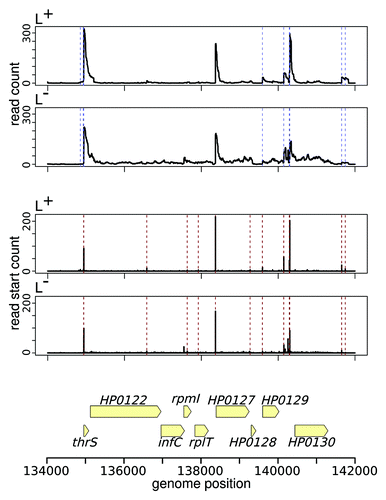
The most recent development in automated TSS annotation from dRNA-seq data, TSSAR,Citation76 picks up the idea from Schmidtke, et al. to model the differences between the treated and untreated library with a Skellam distribution. However, to deduce the parameters from the underlying individual libraries, a zero-inflated Poisson distribution is used instead of a mere Poisson distribution. This allows one to consider the region in focus as a mixture of transcribed and not transcribed segments, where the former are assumed to follow a Poisson distribution and the latter to be zeros with probability 1. The parameters specifying the Skellam distribution are solely deduced from the read density in the transcribed region. The main advantage of TSSAR is the statistical sound analysis resulting in a robust enrichment P value for each genomic position, which in turn, leads to little dependency to a priori defined parameters that can greatly depend on the details of the experimental design and execution. Furthermore, TSSAR is provided as an easy-to-use web service, making its application rather convenient. A comparison of TSSpredator and TSSAR is given in .
Figure 1. Comparison of automated TSS annotation from dRNA-seq data with TTSpredator and TSSAR. The upper plot pair shows the mapped read coverage in the treated (L+) and untreated (L-) library for an exemplary region from H. pylori dRNA-seq data.Citation53 Blue dashed lines indicate TSS annotated by TTS predator (using default parameter). The middle plot pair shows essentially the same data, but only the read start coverage is plotted. This is how TSSAR looks at the data. Dashed red lines indicate TSS annotated by TSSAR (P value cutoff of 10−4). The bottom part shows the positions of the annotated genes in the considered region. The read coverage plots indicate that the data produced by dRNA-seq is more complex than it might appear from the method description; therefore, statistical data analysis is required.
Similar to the eukaryotic research community, the understanding of prokaryotic genomes can benefit from shifting from the established protein-coding gene centered genome annotation to the incorporation of more information on transcripts, with all their diversity in function and architecture. With the recent developments both in wet-lab experiments and computational analysis that allow one to characterize bacterial transcriptomes semi-automated in a high-throughput manner, a comprehensive transcript annotation becomes feasible.
Comparative genomics
Non-coding RNAs are in many cases detectable by comparative genomics alone, i.e., without the benefit of either known homologs or expression data. SIPHTCitation77 makes use of invariant features of many bacterial genes. It identifies candidate loci based on sequence conservation in intergenic regions combined with predicted Rho-independent terminators (downstream) and predicted transcription factor binding sites (upstream). The software also evaluates homology with known sRNAs and cis-regulatory RNA elements. The tool is not directly applicable to some genera such as Helicobacter, which has an A/T-rich genome, and thereby, lacks recognizable terminator hairpins.Citation53
Stabilizing selection acting to preserve secondary structure elements imposes constraints on variations that become fixed in a population, and hence, are observable as differences between orthologous sequences from evolutionarily related organism. In particular, evolutionarily conserved base pairs admit only six of 16 possible nucleotide pairs: GC, CG, AU, UA, GU, and UG. Computer simulations have indicated that RNA sequences still evolve in a drift-like manner even under very strong selection on their secondary structureCitation78,Citation79 so that sequence patterns reflecting the structural constraints rapidly accumulate and become readily detectable already at 10% of sequence divergence.
QrnaCitation80 investigates pair-wise alignments. The algorithm is based on stochastic context-free grammars and estimates the posterior probabilities for an input alignment to be structured RNA, protein-coding, or neither. Its first application to E. coliCitation81 resulted in the prediction of several dozens of novel ncRNAs, many of which have been validated. Multiple sequence alignments convey much more information on substitution patterns than pairwise alignments but are also much harder to simulate as a detailed stochastic model as in EvoFold.Citation82 In RNAz,Citation83 , we have therefore taken a different approach. Two lines of evidence inform about conservation of RNA structures: (1) structural similarity above the level expected from placing the differences at random positions,Citation84 (2) a lower free energy of folding than expected for the same sequence composition. Instead of an explicit stochastic model, RNAz uses machine learning to distinguish between true ncRNAs and decoys with the same dinucleotide content and the same gap pattern as the input alignments. The software is primarily designed for the large genomes of higher eukaryotes but has been employed successfully also for many prokaryotes.Citation85-Citation88 It detects all types of conserved secondary structure elements, including bona fide sRNAs, riboswitches, RNA thermometers, structured cis-acting elements, as well as terminator hairpins. Since its initial publication, several improvements have been introduced. In particular, RNAz 2.0Citation89 makes use of improved consensus structure prediction for assessing structural conservation,Citation16 it explicitly accounts for dinucleotide distribution, and it has been retrained on a much larger training set, including many prokaroytic RNAs. Nevertheless, RNAz still suffers from relatively large false discovery rates (FDR) and a limited accuracy in particular of the boundaries of its predicted structures. Reevaluating the RNAz predictions with structure-based alignment reliability scores computed by LocARNA-PCitation23 not only improves the boundary prediction by more than a factor of three but also halves the FDR.
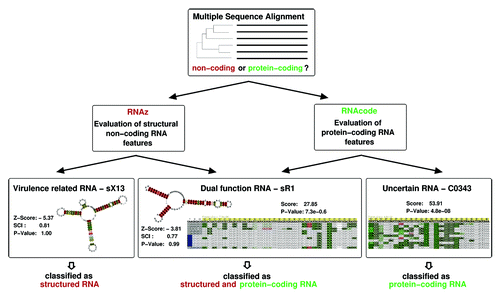
Figure 2. Evolutionary signals are used to classify multiple sequence alignments into non- or protein-coding. RNAz combines structural and thermodynamic descriptors and measures of sequence conservation to detect excess conservation of secondary structure, while RNAcode identifies increased conservation of putative ORFs compared with the observed sequence conservation of the nucleic acid sequences. Well-conserved structured RNAs, such as Xanthomonas sX13, which is involved in virulence-specific gene expression and hfq mRNA regulation, can easily be identifiedCitation71 with RNAz. The E. coli transcript C0343, originally annotated as a small RNA, does not exhibit typical features of a structured RNA. Instead, RNAcode reveals a well-conserved short coding sequence.Citation72 Dual transcripts such as B. subtilis sR1Citation73 are detectable by both RNAz and RNAcode.
A completely different comparative approach is taken by NAPP.Citation90 First, it determines the phylogenetic distribution of conserved sequence elements as well as annotated protein-coding genes. Coherent phylogenetic distribution and co-occurrence in clusters of conserved non-coding elements and coding sequences then indicate that conserved, un-annotated sequences may harbor sRNAs or conserved UTR elements, including riboswitches. An advantage of this approach is that the association with known proteins at least hints at potential functions of the candidate sRNA. A comparison of different computation approaches toward sRNA prediction can be found e.g., in reference Citation90.
Discrimination between coding and non-coding regions poses technical as well as biological challenges not addressed by standard gene finders.Citation91 Ironically, authors working on non-coding RNAs repeatedly had to implement ad hoc solutions to detect coding regions. While longer protein-coding sequences are easily recognized by the absence of stop codons and characteristic, often species-specific patterns of codon usage, it is impossible to reliably detect short peptides of 20 amino acids or less in a single sequence. In complete analogy to RNA secondary structures, however, conservation of peptide sequences constrains the variation of the underlying nucleic acid sequence in characteristic ways. Most obviously, third codon positions are expected to be much more variable. RNAcode,Citation72 , is based on this idea and evaluates for all six possible reading frames whether the amino acids obtained by translating a putative codon is more conserved than expected by the conservation at nucleic acid level. Translated into log odds scores these estimates form the basis of a dynamic programming algorithm that identifies statistically significant conserved peptides in the alignment of nucleic acid sequences. The method was applied e.g., to identify very small peptides as well as annotation errors in H. phylori.Citation53,Citation92
A particular difficulty is posed by transcripts that function both as sRNA by virtue of a conserved secondary structure and at the same time code for a conserved peptide. Well-known examples from the realm of prokaryotes is the Staphylococcus aureus RNAIII, which regulates target genes as sRNA and encodes the 26 amino acid sequence of delta-hemolysin,Citation93 and the Bacillus SR1 RNA involved in the regulation of arginine catabolism.Citation73 The detection of such cases in genome-wide surveys remains difficult, although software for similar tasks has become available. In particular, RNAdecoderCitation94 searches for conserved RNA structure within DNA regions known to be protein-coding; it suffers from very high FDRs, however.Citation95 The intersection of RNAz and RNAcode predictions can provide at least plausible candidates but is certainly not ideal either. To the best of our knowledge, no systematic survey for dual-function RNAs has been conducted in prokaryotes so far.
Estimation of RNA families and classes
The Rfam database divides ncRNAs according to inherent functional, structural, or compositional similarities in more than 2200 different RNA families.Citation37 Rfam’s notion of a clanCitation96 aggregates families that clearly share a common ancestor but are too divergent to be reasonably aligned or groups of families that could be aligned, but have clearly distinct functions. At an even higher level, an RNA class further groups together ncRNA families or clans whose members have no clear homology at the sequence level and presumably do not derive from a common ancestor, but still share common structural properties as a consequence of functional analogy. Prominent examples are microRNAs (miRNAs) and the two distinct classes of snoRNAs (box H/ACA and box C/D).
Current methods for the de novo annotation of ncRNAs rely on unsupervised techniques, such as clustering, to group similar RNAs and subsequent computation of the consensus structure. Using methods implemented in tools like RNAzCitation83 and EvoFold,Citation82 further characteristics that are indicative of functional ncRNA genes are evaluated.
In this framework, the initial clustering phase is a crucial step, and in order to be successful it requires the specification of an appropriate distance or similarity notion that can characterize the functional properties of RNA sequences. The distance measures of course depend on the level of information available and ultimately on the representation used to encode the RNA molecules. These representations can be based on (1) the nucleotide sequence, (2) the connectivity graph of base pairing interactions, or (3) the full three-dimensional conformation. The third option is not yet viable as there is a lack of both experimental techniques to determine 3D conformations of functional RNAs in a large-scale setting (i.e., for machine learning approaches), and of efficient, and sufficiently accurate, modeling techniques to compute these conformations.
Frequently, only sequence information is used since it is directly available from sequencing experiments, of relatively low noise, and it can be manipulated efficiently and with ease by computers.Citation97,Citation98 By construction, any pure sequence-based approach is restricted to RNA families and must fail to detect functional similarity in case of low sequence identity. Indeed, family assignments of structured RNAs obtained from sequence alignments are often wrong when pairwise sequence identities drops below 60%.Citation99 Much lower similarity levels are quite common within a single RNA class. There is therefore a pressing need for similarity and distance notions that efficiently take into account both sequence and structure.
One possible solution is to do structure prediction simultaneous with the construction of alignmentsCitation19,Citation22 as described in the section on structure prediction. This approach was successfully used to classify all known CRISPR repeats.Citation100 However, these alignment-based methods do not scale to efficiently cluster hundred of thousands of candidate ncRNAs predicted by e.g. RNAz screens.
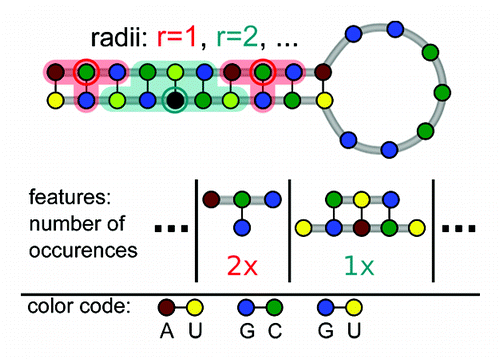
With GraphClust,Citation101 a very different approach has become available. It avoids the alignment phase and the explicit computation of a distance matrix altogether. At the same time it is not restricted to a single structural hypothesis. In order to deal with structural alternatives, abstract shape analysisCitation102 is used to summarize the ensemble of predicted structures. It provides an a priori classification of structures and allows the efficient retrieval of a single representative secondary structure per class, so that each sequence is represented by a small set of sufficiently different secondary structures. Each structure is then interpreted as a labeled graph from which structural features defined as small-localized subgraphs are extracted as outlined in . The resulting sparse feature vectors for each structure amount to a direct generalization of the well-known k-mer similarity from strings to labeled graphs,Citation103 which could be used for clustering.
Figure 3. Features describing a secondary structure graph. Each graph is described by the set of all neighborhood subgraphs (indicated by shaded areas) up to a maximal radius r around a reference nucleotide (marked by a circle).
For large data sets (i.e., > 104 sequences), one cannot afford the quadratic complexity of clustering algorithms that rely on a pairwise distance or similarity information. Instead, GraphClust formulates the clustering problem in terms of approximate nearest neighbor queries, which can be answered with a sub-linear complexity using locality-sensitive hashing.Citation104 The similarity of the k-nearest neighbors can then be used to estimate how compact or dense each neighborhood is within the set of feature vectors so that the most compact non-overlapping neighborhoods can be selected as candidate clusters. Each of these candidate clusters is then refined using alignment techniques designed to discard incompatible RNA sequences. A corresponding covariance model is employed to scan the original data set for similar sequences that were missed by graph-based pre-clustering. The entire procedure is then iterated on the remaining instances producing in each round a user-defined number of clusters that can later be merged to decrease the final cluster fragmentation.
GraphClust was successfully applied to cluster bacterial ncRNAs. Using a benchmark set of 363 ncRNAs, GraphClust detected 43 high-quality clusters representing 38 families.Citation101 In this benchmark, additional genomic context was added to simulate the application scenario of unknown precise transcript boundaries. The quality of clustering (measured with the F-measure or with the Rand index) was higher than the state-of-the-art clustering using LocARNA. Thus, GraphClust can successfully determine RNA classes for bacterial ncRNAs, even when the precise transcript boundaries are unknown.
RNA-RNA Interactions
Models for predicting sRNA–mRNA interactions
The rise of high-throughput methods, first tiling arrays and now RNA-seq, to characterize transcriptomes, had led to an explosion in the number of identified sRNAs in prokaryotes; more than 100 sRNAs have been reported in most species (e.g., refs. Citation105–Citation108). Most sRNAs studied to date form base pair interactions with mRNAs to post-transcriptionally regulate their targets’ translation and stability.Citation109 The functional characterization of novel sRNAs thus involves identification of their interaction partners together with the precise interaction sites. A promising strategy to cope with the steadily increasing number of discovered but uncharacterized sRNAs is computational prediction of candidate sRNA targets, followed by experimental verification using transcriptomics and proteomics approaches.
Computational methods for predicting RNA–RNA interactions fall into four main classes. The following section gives an overview of the available methods and tools with an emphasis on sRNA–mRNA interaction prediction (previously also reviewed in refs. Citation110 and Citation111). summarizes web-based applications designed for genome-wide sRNA target predictions.
Table 1. Web server for genome-scale prediction of sRNA target genes
The first class of methods evaluates the stability of the duplex formed between two RNA molecules aiming to find the loci in both partners that yield the energetically most favorable hybridization. Only base pairs between the two RNAs are evaluated, while their intramolecular structure is ignored. The most popular tools of this type are RNAhybrid,Citation119 RNAduplex and RNAplex,Citation120 DINAMelt,Citation121,Citation122 and RIsearch.Citation123 Methods of this class are primarily tailored for predicting potential binding sites of short RNAs (like eukaryotic miRNAs) in large target RNAs as they tend to maximize the hybridization length. The prediction is based on a modified version of the secondary structure prediction algorithm of reference Citation124 that omits multi-loops. A simplified loop energy model was introduced by RNAplex. This tool also allows one to favor shorter interactions by per-nucleotide penalties. RIsearch further simplifies the nearest-neighbor energy model by a local alignment-like algorithmCitation125 that uses dinucleotide scoring. Its main application is the efficient pre-filtering of interaction candidates in genome-wide screens; the resulting putative interactions can later be evaluated with more complex interaction prediction approaches. The web server TargetRNACitation126,Citation127 was specifically designed for the prediction of bacterial sRNA targets; it provides two scoring schemes: (1) scoring of individual base pairs by a local alignment-like algorithm or (2) duplex minimum free energy (mfe) similar to RNAhybrid. Recently, its successor TargetRNA2 was released (unpublished).
Methods of the second class determine a joint secondary structure of two RNAs, i.e., a common structure including both intra- and intermolecular base pairs. The two input RNA sequences are concatenated and then folded by an RNA folding algorithm such as Zuker’s algorithm,Citation124 which is extended to handle the loop containing the concatenation point energetically as an external loop. Tools implementing this idea are, for example, PairFoldCitation128 and RNAcofold.Citation129 The sRNATarget web serverCitation117,Citation130 computes the mfe structure of the concatenated sequence to derive interaction features, such as length-normalized free energy, seed match length, and A/U-content in single-stranded regions. A naïve Bayes classifier based on these features is then applied to discriminate sRNA–mRNA interactions from non-interacting sRNAs and mRNAs. The main disadvantage of all concatenation-based approaches is their restriction on the allowed interaction types. The underlying RNA folding algorithm can only predict pseudoknot-free secondary structures, although many interaction sites are actually located in loop regions.Citation131 Interactions between two stem loops (loop–loop interactions) represent a pseudoknot in the context of the concatenated sequences, and therefore, cannot be predicted by these approaches.
The third class comprises interaction prediction methods that model the competition between formation of duplex and intramolecular base pairs by the structural accessibility of the interaction sites. This strategy is supported by two systematic studies, which showed that functional interaction sites are typically well-accessible in both sRNAs and their target mRNAs.Citation132,Citation133 The tools IntaRNACitation114 and RNAupCitation134,Citation135 calculate the thermodynamics of RNA–RNA interactions as sum of two energy contributions: (1) the energy required to make the sRNA and target interaction sites accessible, which is calculated from the ensemble of all secondary structures, and (2) the hybridization energy of the two interacting subsequences. IntaRNA additionally incorporates seed regions, i.e., regions of (nearly) perfect sequence complementarity that are thought to initiate interaction formation. The IntaRNA web serverCitation113 allows for genome-scale sRNA target predictions followed by functional enrichment analysis of top target predictions and visualization of putative interaction regions. RNAplex optionally approximates interaction site accessibility by position-specific per-nucleotide penalties.Citation116 An sRNA target prediction web server on top of RNAplex is implemented by the software RNApredator.Citation115 The web server sTarPicker combines ideas from accessibility-based and concatenation-based approaches.Citation118 Putative seed interactions are extended by computing a joint secondary structure of sRNA and mRNA. The predictions are then classified into true and false interaction predictions based on the interaction features A/U-content, hybridization energy, accessibility, and seed length. All methods represented by this class can predict complex interactions like loop–loop interactions, but interactions are restricted to one locus. For RNA–RNA interactions involving two or more interaction sites as, e.g., OxyS–fhlACitation136 and RNAIII–rot,Citation93 only one of the interaction sites can be predicted. Whether formation of interactions at multiple loci is a common principle and frequently required for regulation by sRNAs in vivo is still an open question. The sRNA RNAIII, for example, binds its target coa in Staphylococcus aureus both via an imperfect duplex and a loop–loop interaction, but the former interaction alone is sufficient for in vivo repression.Citation137
Several tools of the third class have been successfully applied to identify sRNA targets in various prokaryotic species. IntaRNA, for example, aided in finding that the cyanobacterial sRNA Yfr1 inhibits translation of two outer membrane proteinsCitation138 and that the sRNA PhrS stimulates translation of the quorum-sensing regulator pqsR in Pseudomonas.Citation139 But sRNA–mRNA interactions are not restricted to the bacterial domain of life. Jäger, et al.,Citation140 for example, showed by a combination of computational and experimental approaches that the archaeal sRNA162 targets both a cis- and a trans-encoded mRNA via two distinct domains.
Methods of the final class can predict more complex joint secondary structures and also allow for multiple interaction sites. The IRIS toolCitation141 introduced a model that maximizes the number of base pairs. Alkan et al.Citation142 then presented a more realistic energy model. The type of joint structures considered in this study were the basis for several subsequent approaches to predict mfe structures,Citation143-Citation145 to compute the partition function of joint secondary structures,Citation146,Citation147 and to sample joint secondary structures.Citation148 All these algorithms have a high time and space complexity, in practice precluding genome-wide application. Except for IRIS, all methods of this class are also not able to handle pseudoknotted structures or crossing interactions. Consequently, they still cannot predict instances like the two loop–loop interactions between RNAIII and rot in Staphylococcus aureus as these constitute a crossing interaction.Citation93
Comparative sRNA target prediction
Genome-scale prediction of sRNA target genes is a computationally challenging task and all methods presented above suffer from a high false positive rate. Starting from the observation that the target binding site in the sRNA is marked by high-sequence conservation across related species,Citation132,Citation133 comparative target prediction for conserved sRNAs appears to be a promising strategy to reduce the number of false positive predictions.
PETcofold was the first comparative method for the prediction of RNA–RNA interactions and joint secondary structures.Citation149-Citation151 Using two multiple alignments of RNA sequences as input, PETcofold predicts conserved RNA–RNA interactions and RNA structures taking into account covariance information arising from compensatory base pair exchanges. Such an alignment-based strategy will predominantly report duplexes in which the interaction base pairing is conserved across species. Its applicability is, therefore, limited to a subclass of interactions that exhibit broad evolutionary conservation. The same constraint applies to other comparative joint secondary structures prediction approaches such as ripalign.Citation152
Interactions with conserved base pairing pattern cover only a subset of all observed interactions; conservation of target complementarity can range from marginal to full conservation even for different targets of the same sRNA.Citation133 This observation is particularly challenging for alignment-based approaches as it is not known a priori whether the interaction between a specific sRNA and mRNA is well conserved or not. CopraRNA introduced a very promising alternative strategy overcoming fixed input sequence alignments.Citation112
As for other comparative approaches, CopraRNA’s main idea is to combine the target prediction in several species. But in contrast to the above-mentioned approaches, CopraRNA does neither enforce conservation of the interaction site nor of the interaction pattern. Rather, it performs target prediction in each organism independently and then combines the evidence for all these predictions (see ). The basic assumption is that only the target regulation by the sRNA is required to be conserved, but the specific base-pairing pattern can be variable and the interaction site might have even been shifted, especially in the mRNA. For a functional interaction, it is often sufficient to have a binding in proximity to the ribosomal binding site without the necessity of a fixed position.
Figure 4. Comparative prediction of sRNA targets as implemented in the CopraRNA pipeline. For a given pair of sRNA and mRNA sequences, the associated homologs are selected. In the next step, the best interaction in each species is determined and scored by its P value. Finally, all species-specific P values are combined into a single joint P value while taking the evolutionary distances into account.

In order to combine the single evidences of an interaction from each organism, one could naïvely use the average of all calculated scores. This approach has, however, two caveats: (1) the scores are not normalized and depend, e.g., on the G/C-content of the organism, and (2) closely related species are likely to have similar scores due to their similarity in sequence composition. Concerning the first point, a way to normalize the score is to use P values instead of raw scores. Since each sRNA has typically only few functional interactions (for example, a total of 21 direct targets has previously been reported for the well-characterized sRNA GcvBCitation153), one can use the score distribution of all genome-wide predicted interactions for a given sRNA in one organism as background to calculate the P values. For the second point, one first has to determine how P values from different organism can be combined. Even though intuitively a good solution, the product of P values does not constitute a P value anymore as it is not uniform across the background. For that purpose, one has to use a transformation. In CopraRNA, the inverse normal method of HartungCitation154 was used since it additionally allows to weight the P- values, thus correcting for the evolutionary distance of the species.
Open Questions
Many questions and computational problems remain open. Although experimental and computational methods are now in place to identify transcription start sites, the corresponding termination sites still cannot be determined reliably, in particular, when they are not associated with Rho-independent terminator structures. Even less is known about other forms of RNA processing, such as cleavage and editing: Where does it occur? How do processing patterns look like in RNA-seq data?
Although it has become clear that sRNAs are abundant in most prokaryotes, we still lack a clear picture of their phylogenetic distribution. In particular, distant homologies have remained largely unexplored. The abundance of pseudoknots and complex interaction structures is still unknown, at least in part due to the high-computational cost but also the limited reliability of prediction algorithms in particular when applied to single sequences. The RNA chaperone Hfq facilitates pairing of sRNA and target mRNA in diverse bacterial lineages.Citation155 The still unknown rules governing the binding of Hfq to specific sRNAs in what appears to be a highly dynamic molecular mechanismCitation156 are likely to provide a dramatic improvement for predicting functional sRNA–mRNA interactions, and thus, for the functional annotation of sRNAs. Eventually, the goal would be to complete the whole bacterial gene regulatory network. Due to their influence on RNA–RNA interactions, this must also include the determination of protein–RNA interactions. Furthermore, not only the sRNA targets, but also the transcriptional regulation of the sRNA itself has to be understood. This would allow one to apply the systems biology toolbox to explore the dynamics of the full gene regulatory network, which is most likely to be altered by the introduction of sRNAs into the network.
The recent time has seen the development of a plethora of high-throughput approaches like CLIP-seq to further investigate the gene regulatory network. It can also be seen that these new experimental techniques require a constant development of appropriate bioinformatic tools. The constant mutual development of experimental techniques and associated bioinformatic methods was well established in the Priority Program SPP 1258, which thus can serve as a blueprint for similar collaborative projects.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
This work was supported by the DFG under the auspices of Priority Program SPP 1258: Sensory and regulatory RNAs in Prokaryotes, grant nos. BA 2168/2 and STA 850/7.
References
- Ding Y. Statistical and Bayesian approaches to RNA secondary structure prediction. RNA 2006; 12:323 - 31; http://dx.doi.org/10.1261/rna.2274106; PMID: 16495231
- Bompfünewerer AF, Backofen R, Bernhart SH, Hertel J, Hofacker IL, Stadler PF, Will S. Variations on RNA folding and alignment: lessons from Benasque. J Math Biol 2008; 56:129 - 44; http://dx.doi.org/10.1007/s00285-007-0107-5; PMID: 17611759
- Seetin MG, Mathews DH. RNA structure prediction: an overview of methods. Methods Mol Biol 2012; 905:99 - 122; PMID: 22736001
- Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res 2003; 31:3406 - 15; http://dx.doi.org/10.1093/nar/gkg595; PMID: 12824337
- Hofacker IL, Fontana W, Stadler PF. Bonhoe_er LS, Tacker M, and Schuster P. Fast folding and comparison of RNA secondary structures. Monatsh Chem 1994; 125:167 - 88; http://dx.doi.org/10.1007/BF00818163
- Lorenz R, Bernhart SH, Höner Zu Siederdissen C, Tafer H, Flamm C, Stadler PF, Hofacker IL. ViennaRNA Package 2.0. Algorithms Mol Biol 2011; 6:26; http://dx.doi.org/10.1186/1748-7188-6-26; PMID: 22115189
- Liu B, Mathews DH, Turner DH. RNA pseudoknots: folding and finding. F1000 Biol Rep 2010; 2:8; PMID: 20495679
- Möhl M, Will S, Backofen R. Fixed parameter tractable alignment of RNA structures including arbitrary pseudoknots. In Proceedings of the 19th Annual Symposium on Combinatorial Pattern Matching (CPM 2008), LNCS, pages 69–81. Springer-Verlag, 2008.
- Bindewald E, Kluth T, Shapiro BA. CyloFold: secondary structure prediction including pseudoknots. Nucleic Acids Res 2010; 38:W368-72; http://dx.doi.org/10.1093/nar/gkq432; PMID: 20501603
- Möhl M, Will S, Backofen R. Lifting prediction to alignment of RNA pseudoknots. J Comput Biol 2010; 17:429 - 42; http://dx.doi.org/10.1089/cmb.2009.0168; PMID: 20377455
- Bon M, Orland H. TT2NE: a novel algorithm to predict RNA secondary structures with pseudoknots. Nucleic Acids Res 2011; 39:e93; http://dx.doi.org/10.1093/nar/gkr240; PMID: 21593129
- Reidys CM, Huang FWD, Andersen JE, Penner RC, Stadler PF, Nebel ME. Topology and prediction of RNA pseudoknots. Bioinformatics, 2011, 27:1076–1085. Addendum in. Bioinformatics 2012; 28:300; http://dx.doi.org/10.1093/bioinformatics/btr643; PMID: 22106334
- Möhl M, Salari R, Will S, Backofen R, Sahinalp SC. Sparsification of RNA structure prediction including pseudoknots. Algorithms Mol Biol 2010; 5:39; http://dx.doi.org/10.1186/1748-7188-5-39; PMID: 21194463
- Lange SJ, Maticzka D, Möhl M, Gagnon JN, Brown CM, Backofen R. Global or local? Predicting secondary structure and accessibility in mRNAs. Nucleic Acids Res 2012; 40:5215 - 26; http://dx.doi.org/10.1093/nar/gks181; PMID: 22373926
- Hofacker IL, Fekete M, Stadler PF. Secondary structure prediction for aligned RNA sequences. J Mol Biol 2002; 319:1059 - 66; http://dx.doi.org/10.1016/S0022-2836(02)00308-X; PMID: 12079347
- Bernhart SH, Hofacker IL, Will S, Gruber AR, Stadler PF. RNAalifold: improved consensus structure prediction for RNA alignments. BMC Bioinformatics 2008; 9:474; http://dx.doi.org/10.1186/1471-2105-9-474; PMID: 19014431
- Seemann SE, Gorodkin J, Backofen R. Unifying evolutionary and thermodynamic information for RNA folding of multiple alignments. Nucleic Acids Res 2008; 36:6355 - 62; http://dx.doi.org/10.1093/nar/gkn544; PMID: 18836192
- Sankoff D. Simultaneous solution of the RNA folding, alignment, and proto-sequence problems. SIAM J Appl Math 1985; 45:810 - 25; http://dx.doi.org/10.1137/0145048
- Torarinsson E, Havgaard JH, Gorodkin J. Multiple structural alignment and clustering of RNA sequences. Bioinformatics 2007; 23:926 - 32; http://dx.doi.org/10.1093/bioinformatics/btm049; PMID: 17324941
- Harmanci AO, Sharma G, Mathews DH. Efficient pairwise RNA structure prediction using probabilistic alignment constraints in Dynalign. BMC Bioinformatics 2007; 8:130; http://dx.doi.org/10.1186/1471-2105-8-130; PMID: 17445273
- Backofen R, Will S. Local sequence-structure motifs in RNA. J Bioinform Comput Biol 2004; 2:681 - 98; http://dx.doi.org/10.1142/S0219720004000818; PMID: 15617161
- Will S, Reiche K, Hofacker IL, Stadler PF, Backofen R. Inferring noncoding RNA families and classes by means of genome-scale structure-based clustering. PLoS Comput Biol 2007; 3:e65; http://dx.doi.org/10.1371/journal.pcbi.0030065; PMID: 17432929
- Will S, Joshi T, Hofacker IL, Stadler PF, Backofen R. LocARNA-P: Accurate boundary prediction and improved detection of structured RNAs for genome-wide screens. RNA 2012; 18:900 - 14; http://dx.doi.org/10.1261/rna.029041.111; PMID: 22450757
- Will S, Siebauer MF, Heyne S, Engelhardt J, Stadler PF, Reiche K, Backofen R. LocARNAscan: Incorporating thermodynamic stability in sequence and structure-based RNA homology search. Algorithms Mol Biol 2013; 8:14; http://dx.doi.org/10.1186/1748-7188-8-14; PMID: 23601347
- Will S, Schmiedl C, Miladi M, Möhl M, Backofen R. SPARSE: Quadratic time simultaneous alignment and folding of RNAs without sequence-based heuristics. In Deng M, Jiang R, Sun F, and Zhang X, eds., Proceedings of the 17th International Conference on Research in Computational Molecular Biology (RECOMB 2013), volume 7821 of Lect. Notes Comp. Sci., pages 289–290, Berlin, Heidelberg, 2013. Springer.
- Reeder J, Giegerich R. RNA secondary structure analysis using the RNAshapes package. Curr Protoc Bioinformatics 2009; Chapter 12:Unit12.8; http://dx.doi.org/10.1002/0471250953.bi1208s26; PMID: 19496058
- Low JT, Weeks KM. SHAPE-directed RNA secondary structure prediction. Methods 2010; 52:150 - 8; http://dx.doi.org/10.1016/j.ymeth.2010.06.007; PMID: 20554050
- Kertesz M, Wan Y, Mazor E, Rinn JL, Nutter RC, Chang HY, Segal E. Genome-wide measurement of RNA secondary structure in yeast. Nature 2010; 467:103 - 7; http://dx.doi.org/10.1038/nature09322; PMID: 20811459
- Deigan KE, Li TW, Mathews DH, Weeks KM. Accurate SHAPE-directed RNA structure determination. Proc Natl Acad Sci U S A 2009; 106:97 - 102; http://dx.doi.org/10.1073/pnas.0806929106; PMID: 19109441
- Zarringhalam K, Meyer MM, Dotu I, Chuang JH, Clote P. Integrating chemical footprinting data into RNA secondary structure prediction. PLoS One 2012; 7:e45160; http://dx.doi.org/10.1371/journal.pone.0045160; PMID: 23091593
- Washietl S, Hofacker IL, Stadler PF, Kellis M. RNA folding with soft constraints: reconciliation of probing data and thermodynamic secondary structure prediction. Nucleic Acids Res 2012; 40:4261 - 72; http://dx.doi.org/10.1093/nar/gks009; PMID: 22287623
- Ouyang Z, Snyder MP, Chang HY. SeqFold: genome-scale reconstruction of RNA secondary structure integrating high-throughput sequencing data. Genome Res 2013; 23:377 - 87; http://dx.doi.org/10.1101/gr.138545.112; PMID: 23064747
- Hajdin CE, Bellaousov S, Huggins W, Leonard CW, Mathews DH, Weeks KM. Accurate SHAPE-directed RNA secondary structure modeling, including pseudoknots. Proc Natl Acad Sci U S A 2013; 110:5498 - 503; http://dx.doi.org/10.1073/pnas.1219988110; PMID: 23503844
- Backofen R, Bernhart SH, Flamm C, Fried C, Fritzsch G, Hackermüller J, Hertel J, Hofacker IL, Missal K, Mosig A, et al, Athanasius F Bompfünewerer Consortium. RNAs everywhere: genome-wide annotation of structured RNAs. J Exp Zool B Mol Dev Evol 2007; 308:1 - 25; PMID: 17171697
- Breaker RR. Prospects for riboswitch discovery and analysis. Mol Cell 2011; 43:867 - 79; http://dx.doi.org/10.1016/j.molcel.2011.08.024; PMID: 21925376
- Kortmann J, Narberhaus F. Bacterial RNA thermometers: molecular zippers and switches. Nat Rev Microbiol 2012; 10:255 - 65; http://dx.doi.org/10.1038/nrmicro2730; PMID: 22421878
- Burge SW, Daub J, Eberhardt R, Tate J, Barquist L, Nawrocki EP, Eddy SR, Gardner PP, Bateman A. Rfam 11.0: 10 years of RNA families. Nucleic Acids Res 2013; 41:D226 - 32; http://dx.doi.org/10.1093/nar/gks1005; PMID: 23125362
- Gardner PP, Gardner AG. A home for RNA families at RNA Biology. RNA Biol 2009; 6:2 - 4; http://dx.doi.org/10.4161/rna.6.1.7635
- Gierga G, Voss B, Hess WR. The Yfr2 ncRNA family, a group of abundant RNA molecules widely conserved in cyanobacteria. RNA Biol 2009; 6:222 - 7; http://dx.doi.org/10.4161/rna.6.3.8921; PMID: 19502815
- Findeiss S, Schmidtke C, Stadler PF, Bonas U. A novel family of plasmid-transferred anti-sense ncRNAs. RNA Biol 2010; 7:120 - 4; http://dx.doi.org/10.4161/rna.7.2.11184; PMID: 20220307
- del Val C, Romero-Zaliz R, Torres-Quesada O, Peregrina A, Toro N, Jiménez-Zurdo JI. A survey of sRNA families in α-proteobacteria. RNA Biol 2012; 9:119 - 29; http://dx.doi.org/10.4161/rna.18643; PMID: 22418845
- Steif A, Meyer IM. The hok mRNA family. RNA Biol 2012; 9:1399 - 404; http://dx.doi.org/10.4161/rna.22746; PMID: 23324554
- Freyhult EK, Bollback JP, Gardner PP. Exploring genomic dark matter: a critical assessment of the performance of homology search methods on noncoding RNA. Genome Res 2007; 17:117 - 25; http://dx.doi.org/10.1101/gr.5890907; PMID: 17151342
- Bussotti G, Raineri E, Erb I, Zytnicki M, Wilm A, Beaudoing E, Bucher P, Notredame C. BlastR--fast and accurate database searches for non-coding RNAs. Nucleic Acids Res 2011; 39:6886 - 95; http://dx.doi.org/10.1093/nar/gkr335; PMID: 21624887
- Hertel J, de Jong D, Marz M, Rose D, Tafer H, Tanzer A, Schierwater B, Stadler PF. Non-coding RNA annotation of the genome of Trichoplax adhaerens. Nucleic Acids Res 2009; 37:1602 - 15; http://dx.doi.org/10.1093/nar/gkn1084; PMID: 19151082
- Schmidtke C, Findeiss S, Sharma CM, Kuhfuss J, Hoffmann S, Vogel J, Stadler PF, Bonas U. Genome-wide transcriptome analysis of the plant pathogen Xanthomonas identifies sRNAs with putative virulence functions. Nucleic Acids Res 2012; 40:2020 - 31; http://dx.doi.org/10.1093/nar/gkr904; PMID: 22080557
- Eddy SR, Durbin R. RNA sequence analysis using covariance models. Nucleic Acids Res 1994; 22:2079 - 88; http://dx.doi.org/10.1093/nar/22.11.2079; PMID: 8029015
- Nawrocki EP, Eddy SR. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 2013; 29:2933 - 5; http://dx.doi.org/10.1093/bioinformatics/btt509; PMID: 24008419
- Nawrocki EP. Structural RNA homology search and alignment using covariance models. PhD thesis, Washington University, Saint Louis, 2009. Figure 1.9.
- Höchsmann T, Höchsmann M, Giegerich R. Thermodynamic matchers: strengthening the significance of RNA folding energies. Comput Syst Bioinformatics Conf 2006; 111 - 21; PMID: 17369630
- Livny J, Fogel MA, Davis BM, Waldor MK. sRNAPredict: an integrative computational approach to identify sRNAs in bacterial genomes. Nucleic Acids Res 2005; 33:4096 - 105; http://dx.doi.org/10.1093/nar/gki715; PMID: 16049021
- Kingsford CL, Ayanbule K, Salzberg SL. Rapid, accurate, computational discovery of Rho-independent transcription terminators illuminates their relationship to DNA uptake. Genome Biol 2007; 8:R22; http://dx.doi.org/10.1186/gb-2007-8-2-r22; PMID: 17313685
- Sharma CM, Hoffmann S, Darfeuille F, Reignier J, Findeiss S, Sittka A, Chabas S, Reiche K, Hackermüller J, Reinhardt R, et al. The primary transcriptome of the major human pathogen Helicobacter pylori. Nature 2010; 464:250 - 5; http://dx.doi.org/10.1038/nature08756; PMID: 20164839
- Trapnell C, Salzberg SL. How to map billions of short reads onto genomes. Nat Biotechnol 2009; 27:455 - 7; http://dx.doi.org/10.1038/nbt0509-455; PMID: 19430453
- Ruffalo M, LaFramboise T, Koyutürk M. Comparative analysis of algorithms for next-generation sequencing read alignment. Bioinformatics 2011; 27:2790 - 6; http://dx.doi.org/10.1093/bioinformatics/btr477; PMID: 21856737
- Schbath S, Martin V, Zytnicki M, Fayolle J, Loux V, Gibrat JF. Mapping reads on a genomic sequence: an algorithmic overview and a practical comparative analysis. J Comput Biol 2012; 19:796 - 813; http://dx.doi.org/10.1089/cmb.2012.0022; PMID: 22506536
- Hatem A, Bozdağ D, Toland AE, Çatalyürek ÜV. Benchmarking short sequence mapping tools. BMC Bioinformatics 2013; 14:184; http://dx.doi.org/10.1186/1471-2105-14-184; PMID: 23758764
- Engström PG, Steijger T, Sipos B, Grant GR, Kahles A, Alioto T, Behr J, Bertone P, Bohnert R, Campagna D, et al, RGASP Consortium. Systematic evaluation of spliced alignment programs for RNA-seq data. Nat Methods 2013; 10:1185 - 91; http://dx.doi.org/10.1038/nmeth.2722; PMID: 24185836
- Caboche S, Audebert C, Lemoine Y, Hot D.. Comparison of mapping algorithms used in high-throughput sequencing: application to Ion Torrent data. BMC Genomics 2014; In press
- Hoffmann S, Otto C, Kurtz S, Sharma CM, Khaitovich P, Vogel J, Stadler PF, Hackermüller J. Fast mapping of short sequences with mismatches, insertions and deletions using index structures. PLoS Comput Biol 2009; 5:e1000502; http://dx.doi.org/10.1371/journal.pcbi.1000502; PMID: 19750212
- Hoffmann S, Otto C, Doose G, Tanzer A, Langenberger D, Christ S, Kunz M, Holdt L, Teupser D, Hackermüeller J, et al. A multi-split mapping algorithm for circular RNA, splicing, trans-splicing, and fusion detection. Genome Biol 2014; 15:R34; http://dx.doi.org/10.1186/gb-2014-15-2-r34; PMID: 24512684
- Otto C, Stadler PF, Hoffmann S. Lacking alignments? The next generation sequencing mapper segemehl revisited. Bioinformatics 2014; accepted http://dx.doi.org/10.1093/bioinformatics/btu146; PMID: 24626854
- Doose G, Alexis M, Kirsch R, Findeiß S, Langenberger D, Machné R, Mörl M, Hoffmann S, Stadler PF. Mapping the RNA-Seq trash bin: unusual transcripts in prokaryotic transcriptome sequencing data. RNA Biol 2013; 10:1204 - 10; http://dx.doi.org/10.4161/rna.24972; PMID: 23702463
- Findeiss S, Langenberger D, Stadler PF, Hoffmann S. Traces of post-transcriptional RNA modifications in deep sequencing data. Biol Chem 2011; 392:305 13; http://dx.doi.org/10.1515/BC.2011.043; PMID: 21345160
- Brenneis M, Hering O, Lange C, Soppa J. Experimental characterization of Cis-acting elements important for translation and transcription in halophilic archaea. PLoS Genet 2007; 3:e229; http://dx.doi.org/10.1371/journal.pgen.0030229; PMID: 18159946
- Wurtzel O, Sapra R, Chen F, Zhu Y, Simmons BA, Sorek R. A single-base resolution map of an archaeal transcriptome. Genome Res 2010; 20:133 - 41; http://dx.doi.org/10.1101/gr.100396.109; PMID: 19884261
- Meyer F, Goesmann A, McHardy AC, Bartels D, Bekel T, Clausen J, Kalinowski J, Linke B, Rupp O, Giegerich R, et al. GenDB--an open source genome annotation system for prokaryote genomes. Nucleic Acids Res 2003; 31:2187 - 95; http://dx.doi.org/10.1093/nar/gkg312; PMID: 12682369
- Vallenet D, Labarre L, Rouy Z, Barbe V, Bocs S, Cruveiller S, Lajus A, Pascal G, Scarpelli C, Médigue C. MaGe: a microbial genome annotation system supported by synteny results. Nucleic Acids Res 2006; 34:53 - 65; http://dx.doi.org/10.1093/nar/gkj406; PMID: 16407324
- Duchêne M, Schweizer A, Lottspeich F, Krauss G, Marget M, Vogel K, von Specht BU, Domdey H. Sequence and transcriptional start site of the Pseudomonas aeruginosa outer membrane porin protein F gene. J Bacteriol 1988; 170:155 - 62; PMID: 2447060
- Frohman MA, Dush MK, Martin GR. Rapid production of full-length cDNAs from rare transcripts: amplification using a single gene-specific oligonucleotide primer. Proc Natl Acad Sci U S A 1988; 85:8998 - 9002; http://dx.doi.org/10.1073/pnas.85.23.8998; PMID: 2461560
- Schmidtke C, Abendroth U, Brock J, Serrania J, Becker A, Bonas U. Small RNA sX13: a multifaceted regulator of virulence in the plant pathogen Xanthomonas. PLoS Pathog 2013; 9:e1003626; http://dx.doi.org/10.1371/journal.ppat.1003626; PMID: 24068933
- Washietl S, Findeiß S, Müller S, Kalkhof S, von Bergen M, Hofacker IL, Stadler PF, Goldman N. RNAcode: robust prediction of protein coding regions in comparative genomics data. RNA 2011; 17:578 - 94; http://dx.doi.org/10.1261/rna.2536111; PMID: 21357752
- Gimpel M, Preis H, Barth E, Gramzow L, Brantl S. SR1--a small RNA with two remarkably conserved functions. Nucleic Acids Res 2012; 40:11659 - 72; http://dx.doi.org/10.1093/nar/gks895; PMID: 23034808
- Herbig A, Sharma C, Nieselt K.. Automated transcription start site prediction for comparative transcriptomics using the SuperGenome. EMBnet. journal 2013; 19; http://dx.doi.org/10.14806/ej.19.A.617
- Dugar G, Herbig A, Förstner KU, Heidrich N, Reinhardt R, Nieselt K, Sharma CM. High-resolution transcriptome maps reveal strain-specific regulatory features of multiple Campylobacter jejuni isolates. PLoS Genet 2013; 9:e1003495; http://dx.doi.org/10.1371/journal.pgen.1003495; PMID: 23696746
- Amman F, Wolfinger MT, Lorenz R, Hofacker IL, Stadler PF, Findeiss S. TSSAR: TSS annotation regime for dRNA-seq data. BMC Bioinformatics 2014; In press
- Livny J, Teonadi H, Livny M, Waldor MK. High-throughput, kingdom-wide prediction and annotation of bacterial non-coding RNAs. PLoS One 2008; 3:e3197; http://dx.doi.org/10.1371/journal.pone.0003197; PMID: 18787707
- Schuster P, Fontana W, Stadler PF, Hofacker IL. From sequences to shapes and back: a case study in RNA secondary structures. Proc Biol Sci 1994; 255:279 - 84; http://dx.doi.org/10.1098/rspb.1994.0040; PMID: 7517565
- Huynen MA, Stadler PF, Fontana W. Smoothness within ruggedness: the role of neutrality in adaptation. Proc Natl Acad Sci U S A 1996; 93:397 - 401; http://dx.doi.org/10.1073/pnas.93.1.397; PMID: 8552647
- Rivas E, Eddy SR. Noncoding RNA gene detection using comparative sequence analysis. BMC Bioinformatics 2001; 2:8; http://dx.doi.org/10.1186/1471-2105-2-8; PMID: 11801179
- Rivas E, Klein RJ, Jones TA, Eddy SR. Computational identification of noncoding RNAs in E. coli by comparative genomics. Curr Biol 2001; 11:1369 - 73; http://dx.doi.org/10.1016/S0960-9822(01)00401-8; PMID: 11553332
- Pedersen JS, Bejerano G, Siepel A, Rosenbloom K, Lindblad-Toh K, Lander ES, Kent J, Miller W, Haussler D. Identification and classification of conserved RNA secondary structures in the human genome. PLoS Comput Biol 2006; 2:e33; http://dx.doi.org/10.1371/journal.pcbi.0020033; PMID: 16628248
- Washietl S, Hofacker IL, Stadler PF. Fast and reliable prediction of noncoding RNAs. Proc Natl Acad Sci U S A 2005; 102:2454 - 9; http://dx.doi.org/10.1073/pnas.0409169102; PMID: 15665081
- Gruber AR, Bernhart SH, Hofacker IL, Washietl S. Strategies for measuring evolutionary conservation of RNA secondary structures. BMC Bioinformatics 2008; 9:122; http://dx.doi.org/10.1186/1471-2105-9-122; PMID: 18302738
- Sonnleitner E, Sorger-Domenigg T, Madej MJ, Findeiss S, Hackermüller J, Hüttenhofer A, Stadler PF, Bläsi U, Moll I. Detection of small RNAs in Pseudomonas aeruginosa by RNomics and structure-based bioinformatic tools. Microbiology 2008; 154:3175 - 87; http://dx.doi.org/10.1099/mic.0.2008/019703-0; PMID: 18832323
- Schilling D, Findeiss S, Richter AS, Taylor JA, Gerischer U. The small RNA Aar in Acinetobacter baylyi: a putative regulator of amino acid metabolism. Arch Microbiol 2010; 192:691 - 702; http://dx.doi.org/10.1007/s00203-010-0592-6; PMID: 20559624
- del Val C, Rivas E, Torres-Quesada O, Toro N, Jiménez-Zurdo JI. Identification of differentially expressed small non-coding RNAs in the legume endosymbiont Sinorhizobium meliloti by comparative genomics. Mol Microbiol 2007; 66:1080 - 91; http://dx.doi.org/10.1111/j.1365-2958.2007.05978.x; PMID: 17971083
- Hot D, Slupek S, Wulbrecht B, D’Hondt A, Hubans C, Antoine R, Locht C, Lemoine Y. Detection of small RNAs in Bordetella pertussis and identification of a novel repeated genetic element. BMC Genomics 2011; 12:207; http://dx.doi.org/10.1186/1471-2164-12-207; PMID: 21524285
- Gruber AR, Findeiß S, Washietl S, Hofacker IL, Stadler PF. RNAz 2.0: improved noncoding RNA detection. Pac Symp Biocomput 2010; 15:69 - 79; PMID: 19908359
- Ott A, Idali A, Marchais A, Gautheret D. NAPP: the nucleic acid phylogenetic profile database. Nucleic Acids Res 2012; 40:D205 - 9; http://dx.doi.org/10.1093/nar/gkr807; PMID: 21984475
- Dinger ME, Pang KC, Mercer TR, Mattick JS. Differentiating protein-coding and noncoding RNA: challenges and ambiguities. PLoS Comput Biol 2008; 4:e1000176; http://dx.doi.org/10.1371/journal.pcbi.1000176; PMID: 19043537
- Müller SA, Findeiß S, Pernitzsch SR, Stadler PF, Hofacker IL, Sharma CM, von Bergen M, Kalkhof S. Proteogenomic analysis of the Helicobacter pylori strain 26695 genome. J Proteomics 2013; 86:27 - 42; PMID: 23665149
- Boisset S, Geissmann T, Huntzinger E, Fechter P, Bendridi N, Possedko M, Chevalier C, Helfer AC, Benito Y, Jacquier A, et al. Staphylococcus aureus RNAIII coordinately represses the synthesis of virulence factors and the transcription regulator Rot by an antisense mechanism. Genes Dev 2007; 21:1353 - 66; http://dx.doi.org/10.1101/gad.423507; PMID: 17545468
- Pedersen JS, Meyer IM, Forsberg R, Simmonds P, Hein J. A comparative method for finding and folding RNA secondary structures within protein-coding regions. Nucleic Acids Res 2004; 32:4925 - 36; http://dx.doi.org/10.1093/nar/gkh839; PMID: 15448187
- Findeiss S, Engelhardt J, Prohaska SJ, Stadler PF. Protein-coding structured RNAs: A computational survey of conserved RNA secondary structures overlapping coding regions in drosophilids. Biochimie 2011; 93:2019 - 23; http://dx.doi.org/10.1016/j.biochi.2011.07.023; PMID: 21835221
- Gardner PP, Daub J, Tate J, Moore BL, Osuch IH, Griffiths-Jones S, Finn RD, Nawrocki EP, Kolbe DL, Eddy SR, et al. Rfam: Wikipedia, clans and the “decimal” release. Nucleic Acids Res 2011; 39:suppl 1 D141 - 5; http://dx.doi.org/10.1093/nar/gkq1129; PMID: 21062808
- Shi Y, Tyson GW, DeLong EF. Metatranscriptomics reveals unique microbial small RNAs in the ocean’s water column. Nature 2009; 459:266 - 9; http://dx.doi.org/10.1038/nature08055; PMID: 19444216
- Kunin V, Sorek R, Hugenholtz P. Evolutionary conservation of sequence and secondary structures in CRISPR repeats. Genome Biol 2007; 8:R61; http://dx.doi.org/10.1186/gb-2007-8-4-r61; PMID: 17442114
- Gardner PP, Wilm A, Washietl S. A benchmark of multiple sequence alignment programs upon structural RNAs. Nucleic Acids Res 2005; 33:2433 - 9; http://dx.doi.org/10.1093/nar/gki541; PMID: 15860779
- Lange SJ, Alkhnbashi OS, Rose D, Will S, Backofen R. CRISPRmap: an automated classification of repeat conservation in prokaryotic adaptive immune systems. Nucleic Acids Res 2013; 41:8034 - 44; http://dx.doi.org/10.1093/nar/gkt606; PMID: 23863837
- Heyne S, Costa F, Rose D, Backofen R. GraphClust: alignment-free structural clustering of local RNA secondary structures. Bioinformatics 2012; 28:i224 - 32; http://dx.doi.org/10.1093/bioinformatics/bts224; PMID: 22689765
- Giegerich R, Voss B, Rehmsmeier M. Abstract shapes of RNA. Nucleic Acids Res 2004; 32:4843 - 51; http://dx.doi.org/10.1093/nar/gkh779; PMID: 15371549
- Costa F, Grave KD. Fast neighborhood subgraph pairwise distance kernel. In Proceedings of the 26 th International Conference on Machine Learning, pages 255–262. Omnipress, 2010.
- Indyk P, Motwani R. Approximate nearest neighbors: towards removing the curse of dimensionality. In Proceedings of the thirtieth annual ACM symposium on Theory of computing, pages 604–613. ACM, 1998.
- Kröger C, Dillon SC, Cameron ADS, Papenfort K, Sivasankaran SK, Hokamp K, Chao Y, Sittka A, Hébrard M, Händler K, et al. The transcriptional landscape and small RNAs of Salmonella enterica serovar Typhimurium. Proc Natl Acad Sci U S A 2012; 109:E1277 - 86; http://dx.doi.org/10.1073/pnas.1201061109; PMID: 22538806
- Raghavan R, Groisman EA, Ochman H. Genome-wide detection of novel regulatory RNAs in E. coli. Genome Res 2011; 21:1487 - 97; http://dx.doi.org/10.1101/gr.119370.110; PMID: 21665928
- Mitschke J, Georg J, Scholz I, Sharma CM, Dienst D, Bantscheff J, Voss B, Steglich C, Wilde A, Vogel J, et al. An experimentally anchored map of transcriptional start sites in the model cyanobacterium Synechocystis sp. PCC6803. Proc Natl Acad Sci U S A 2011; 108:2124 - 9; http://dx.doi.org/10.1073/pnas.1015154108; PMID: 21245330
- Jäger D, Sharma CM, Thomsen J, Ehlers C, Vogel J, Schmitz RA. Deep sequencing analysis of the Methanosarcina mazei Gö1 transcriptome in response to nitrogen availability. Proc Natl Acad Sci U S A 2009; 106:21878 - 82; http://dx.doi.org/10.1073/pnas.0909051106; PMID: 19996181
- Storz G, Vogel J, Wassarman KM. Regulation by small RNAs in bacteria: expanding frontiers. Mol Cell 2011; 43:880 - 91; http://dx.doi.org/10.1016/j.molcel.2011.08.022; PMID: 21925377
- Backofen R, Hess WR. Computational prediction of sRNAs and their targets in bacteria. RNA Biol 2010; 7:33 - 42; http://dx.doi.org/10.4161/rna.7.1.10655; PMID: 20061798
- Tjaden B. Biocomputational identification of bacterial small rnas and their target binding sites. In Mallick B and Ghosh Z, eds., Regulatory RNAs, pages 273–293. Springer Berlin Heidelberg, 2012.
- Wright PR, Richter AS, Papenfort K, Mann M, Vogel J, Hess WR, Backofen R, Georg J. Comparative genomics boosts target prediction for bacterial small RNAs. Proc Natl Acad Sci U S A 2013; 110:E3487 - 96; http://dx.doi.org/10.1073/pnas.1303248110; PMID: 23980183
- Smith C, Heyne S, Richter AS, Will S, Backofen R. Freiburg RNA Tools: a web server integrating INTARNA, EXPARNA and LOCARNA. Nucleic Acids Res 2010; 38:Suppl W373 7; http://dx.doi.org/10.1093/nar/gkq316; PMID: 20444875
- Busch A, Richter AS, Backofen R. IntaRNA: efficient prediction of bacterial sRNA targets incorporating target site accessibility and seed regions. Bioinformatics 2008; 24:2849 - 56; http://dx.doi.org/10.1093/bioinformatics/btn544; PMID: 18940824
- Eggenhofer F, Tafer H, Stadler PF, Hofacker IL. RNApredator: fast accessibility-based prediction of sRNA targets. Nucleic Acids Res 2011; 39:W149 54; http://dx.doi.org/10.1093/nar/gkr467; PMID: 21672960
- Tafer H, Amman F, Eggenhofer F, Stadler PF, Hofacker IL. Fast accessibility-based prediction of RNA-RNA interactions. Bioinformatics 2011; 27:1934 - 40; http://dx.doi.org/10.1093/bioinformatics/btr281; PMID: 21593134
- Cao Y, Zhao Y, Cha L, Ying X, Wang L, Shao N, Li W. sRNATarget: a web server for prediction of bacterial sRNA targets. Bioinformation 2009; 3:364 - 6; http://dx.doi.org/10.6026/97320630003364; PMID: 19707302
- Ying X, Cao Y, Wu J, Liu Q, Cha L, Li W. sTarPicker: a method for efficient prediction of bacterial sRNA targets based on a two-step model for hybridization. PLoS One 2011; 6:e22705; http://dx.doi.org/10.1371/journal.pone.0022705; PMID: 21799937
- Rehmsmeier M, Steffen P, Höchsmann M, Giegerich R. Fast and effective prediction of microRNA/target duplexes. RNA 2004; 10:1507 - 17; http://dx.doi.org/10.1261/rna.5248604; PMID: 15383676
- Tafer H, Hofacker IL. RNAplex: a fast tool for RNA-RNA interaction search. Bioinformatics 2008; 24:2657 - 63; http://dx.doi.org/10.1093/bioinformatics/btn193; PMID: 18434344
- Dimitrov RA, Zuker M. Prediction of hybridization and melting for double-stranded nucleic acids. Biophys J 2004; 87:215 - 26; http://dx.doi.org/10.1529/biophysj.103.020743; PMID: 15240459
- Markham NR, Zuker M. DINAMelt web server for nucleic acid melting prediction. Nucleic Acids Res 2005; 33:W577 81; http://dx.doi.org/10.1093/nar/gki591; PMID: 15980540
- Wenzel A, Akbasli E, Gorodkin J. RIsearch: fast RNA-RNA interaction search using a simplified nearest-neighbor energy model. Bioinformatics 2012; 28:2738 - 46; http://dx.doi.org/10.1093/bioinformatics/bts519; PMID: 22923300
- Zuker M, Stiegler P. Optimal computer folding of large RNA sequences using thermodynamics and auxiliary information. Nucleic Acids Res 1981; 9:133 - 48; http://dx.doi.org/10.1093/nar/9.1.133; PMID: 6163133
- Smith TF, Waterman MS. Identification of common molecular subsequences. J Mol Biol 1981; 147:195 - 7; http://dx.doi.org/10.1016/0022-2836(81)90087-5; PMID: 7265238
- Tjaden B, Goodwin SS, Opdyke JA, Guillier M, Fu DX, Gottesman S, Storz G. Target prediction for small, noncoding RNAs in bacteria. Nucleic Acids Res 2006; 34:2791 - 802; http://dx.doi.org/10.1093/nar/gkl356; PMID: 16717284
- Tjaden B. TargetRNA: a tool for predicting targets of small RNA action in bacteria. Nucleic Acids Res 2008; 36:W109 13; http://dx.doi.org/10.1093/nar/gkn264; PMID: 18477632
- Andronescu M, Zhang ZC, Condon A. Secondary structure prediction of interacting RNA molecules. J Mol Biol 2005; 345:987 - 1001; http://dx.doi.org/10.1016/j.jmb.2004.10.082; PMID: 15644199
- Bernhart SH, Tafer H, Mückstein U, Flamm C, Stadler PF, Hofacker IL. Partition function and base pairing probabilities of RNA heterodimers. Algorithms Mol Biol 2006; 1:3; http://dx.doi.org/10.1186/1748-7188-1-3; PMID: 16722605
- Zhao Y, Li H, Hou Y, Cha L, Cao Y, Wang L, Ying X, Li W. Construction of two mathematical models for prediction of bacterial sRNA targets. Biochem Biophys Res Commun 2008; 372:346 - 50; http://dx.doi.org/10.1016/j.bbrc.2008.05.046; PMID: 18501192
- Brunel C, Marquet R, Romby P, Ehresmann C. RNA loop-loop interactions as dynamic functional motifs. Biochimie 2002; 84:925 - 44; http://dx.doi.org/10.1016/S0300-9084(02)01401-3; PMID: 12458085
- Peer A, Margalit H. Accessibility and evolutionary conservation mark bacterial small-rna target-binding regions. J Bacteriol 2011; 193:1690 - 701; http://dx.doi.org/10.1128/JB.01419-10; PMID: 21278294
- Richter AS, Backofen R. Accessibility and conservation: general features of bacterial small RNA-mRNA interactions?. RNA Biol 2012; 9:954 - 65; http://dx.doi.org/10.4161/rna.20294; PMID: 22767260
- Mückstein U, Tafer H, Hackermüller J, Bernhart SH, Stadler PF, Hofacker IL. Thermodynamics of RNA-RNA binding. Bioinformatics 2006; 22:1177 - 82; http://dx.doi.org/10.1093/bioinformatics/btl024; PMID: 16446276
- Mückstein U, Tafer H, Bernhart SH, Hernandez-Rosales M, Vogel J, Stadler PF, Hofacker IL. Translational control by RNA-RNA interaction: Improved computation of RNA-RNA binding thermodynamics. In Elloumi M, Küng J, Linial M, Murphy R, Schneider K, and Toma C, eds., Bioinformatics Research and Development, volume 13 of Communications in Computer and Information Science, pages 114–127. Springer-Verlag Berlin Heidelberg, 2008.
- Argaman L, Altuvia S. fhlA repression by OxyS RNA: kissing complex formation at two sites results in a stable antisense-target RNA complex. J Mol Biol 2000; 300:1101 - 12; http://dx.doi.org/10.1006/jmbi.2000.3942; PMID: 10903857
- Chevalier C, Boisset S, Romilly C, Masquida B, Fechter P, Geissmann T, Vandenesch F, Romby P. Staphylococcus aureus RNAIII binds to two distant regions of coa mRNA to arrest translation and promote mRNA degradation. PLoS Pathog 2010; 6:e1000809; http://dx.doi.org/10.1371/journal.ppat.1000809; PMID: 20300607
- Richter AS, Schleberger C, Backofen R, Steglich C. Seed-based INTARNA prediction combined with GFP-reporter system identifies mRNA targets of the small RNA Yfr1. Bioinformatics 2010; 26:1 - 5; http://dx.doi.org/10.1093/bioinformatics/btp609; PMID: 19850757
- Sonnleitner E, Gonzalez N, Sorger-Domenigg T, Heeb S, Richter AS, Backofen R, Williams P, Hüttenhofer A, Haas D, Bläsi U. The small RNA PhrS stimulates synthesis of the Pseudomonas aeruginosa quinolone signal. Mol Microbiol 2011; 80:868 - 85; http://dx.doi.org/10.1111/j.1365-2958.2011.07620.x; PMID: 21375594
- Jäger D, Pernitzsch SR, Richter AS, Backofen R, Sharma CM, Schmitz RA. An archaeal sRNA targeting cis- and trans-encoded mRNAs via two distinct domains. Nucleic Acids Res 2012; 40:10964 - 79; http://dx.doi.org/10.1093/nar/gks847; PMID: 22965121
- Pervouchine DD. IRIS: intermolecular RNA interaction search. Genome Inform 2004; 15:92 - 101; PMID: 15706495
- Alkan C, Karakoç E, Nadeau JH, Sahinalp SC, Zhang K. RNA-RNA interaction prediction and antisense RNA target search. J Comput Biol 2006; 13:267 - 82; http://dx.doi.org/10.1089/cmb.2006.13.267; PMID: 16597239
- Chitsaz H, Backofen R, Sahinalp SC. biRNA: Fast RNARNA binding sites prediction. In Salzberg S and Warnow T, eds., Proc. of the 9th Workshop on Algorithms in Bioinformatics (WABI), volume 5724 of Lecture Notes in Computer Science, pages 25–36. Springer Berlin / Heidelberg, 2009.
- Salari R, Backofen R, Sahinalp SC. Fast prediction of RNA-RNA interaction. Algorithms Mol Biol 2010; 5:5; http://dx.doi.org/10.1186/1748-7188-5-5; PMID: 20047661
- Salari R, Möhl M, Will S, Sahinalp SC, Backofen R. Time and space efficient RNA-RNA interaction prediction via sparse folding. In Berger B, ed., Proc. of RECOMB 2010, volume 6044 of Lecture Notes in Computer Science, pages 473–490. Springer-Verlag Berlin Heidelberg, 2010.
- Chitsaz H, Salari R, Sahinalp SC, Backofen R. A partition function algorithm for interacting nucleic acid strands. Bioinformatics 2009; 25:i365 - 73; http://dx.doi.org/10.1093/bioinformatics/btp212; PMID: 19478011
- Huang FWD, Qin J, Reidys CM, Stadler PF. Partition function and base pairing probabilities for RNA-RNA interaction prediction. Bioinformatics 2009; 25:2646 - 54; http://dx.doi.org/10.1093/bioinformatics/btp481; PMID: 19671692
- Huang FWD, Qin J, Reidys CM, Stadler PF. Target prediction and a statistical sampling algorithm for RNA-RNA interaction. Bioinformatics 2010; 26:175 - 81; http://dx.doi.org/10.1093/bioinformatics/btp635; PMID: 19910305
- Seemann SE, Richter AS, Gesell T, Backofen R, Gorodkin J. PETcofold: predicting conserved interactions and structures of two multiple alignments of RNA sequences. Bioinformatics 2011; 27:211 - 9; http://dx.doi.org/10.1093/bioinformatics/btq634; PMID: 21088024
- Seemann SE, Richter AS, Gorodkin J, Backofen R. Hierarchical folding of multiple sequence alignments for the prediction of structures and RNA-RNA interactions. Algorithms Mol Biol 2010; 5:22; http://dx.doi.org/10.1186/1748-7188-5-22; PMID: 20492641
- Seemann SE, Menzel P, Backofen R, Gorodkin J. The PETfold and PETcofold web servers for intra- and intermolecular structures of multiple RNA sequences. Nucleic Acids Res 2011; 39:W107 11; http://dx.doi.org/10.1093/nar/gkr248; PMID: 21609960
- Li AX, Marz M, Qin J, Reidys CM. RNA-RNA interaction prediction based on multiple sequence alignments. Bioinformatics 2011; 27:456 - 63; http://dx.doi.org/10.1093/bioinformatics/btq659; PMID: 21134894
- Sharma CM, Papenfort K, Pernitzsch SR, Mollenkopf HJ, Hinton JCD, Vogel J. Pervasive post-transcriptional control of genes involved in amino acid metabolism by the Hfq-dependent GcvB small RNA. Mol Microbiol 2011; 81:1144 - 65; http://dx.doi.org/10.1111/j.1365-2958.2011.07751.x; PMID: 21696468
- Hartung J. A note on combining dependent tests of significance. Biom J 1999; 41:849 - 55; http://dx.doi.org/10.1002/(SICI)1521-4036(199911)41:7<849::AID-BIMJ849>3.0.CO;2-T
- Vogel J, Luisi BF. Hfq and its constellation of RNA. Nat Rev Microbiol 2011; 9:578 - 89; http://dx.doi.org/10.1038/nrmicro2615; PMID: 21760622
- Fender A, Elf J, Hampel K, Zimmermann B, Wagner EGH. RNAs actively cycle on the Sm-like protein Hfq. Genes Dev 2010; 24:2621 - 6; http://dx.doi.org/10.1101/gad.591310; PMID: 21123649