
Abstract
Hfq is a global regulator of gene expression in bacteria undergoing adaptation to changing environmental conditions. Its major function is to promote RNA-RNA interactions between regulatory small RNAs (sRNAs) and their target mRNAs. Previously, we demonstrated that Hfq binds many antisense RNAs (asRNAs) in vitro and hypothesized that Hfq may play a role in regulating gene expression via asRNAs. To investigate the E. coli Hfq-binding transcriptome in more detail, we co-immunoprecipitated and deep-sequenced RNAs bound to Hfq in vivo. We detected many new Hfq-binding sRNAs and observed that almost 300 mRNAs bind to Hfq. Among these, several are known to be sRNA targets. We identified 25 novel RNAs, which are transcribed from within protein coding regions and named them intragenic RNAs (intraRNAs). Furthermore, 67 asRNAs were co-immunoprecipitated with Hfq, demonstrating that Hfq binds antisense transcripts in vivo. Northern blot analyses confirmed the deep sequencing results and demonstrated that many of the novel Hfq-binding RNAs identified are regulated by Hfq.
Introduction
Bacteria live in a variety of biological environments prone to sudden changes. In order to adapt, bacteria are capable of rapid metabolic transitions. Gene expression is regulated by proteins, as well as by sRNAs, the latter being particularly important for fast adaptation responses.
The majority of known sRNAs in Escherichia coli act as post-transcriptional regulators through base-pairing with their target mRNAs, resulting in either their up- or downregulation.Citation1 This mode of action allows multiple (functionally related) mRNAs to be regulated by one sRNA, sometimes giving rise to large regulatory networks.Citation2 Regulatory sRNAs are usually transcribed independently of their target mRNAs and act in trans via partial complementarity. While most sRNAs in E. coli were identified as intergenic transcripts, recent work in Salmonella suggested that 3′ UTRs of genes could serve as a reservoir of sRNAs.Citation3
Many of the sRNAs require the global regulator Hfq to overcome the notorious folding and annealing problems of RNA molecules.Citation4,Citation5 Hfq is a protein with RNA chaperone activity, which has evolved to promote not only structural formation, but also intermolecular RNA-RNA interactions. Hfq is a highly conserved small SM-like protein that forms ring-shaped homohexamers, offering at least two surfaces on which RNAs bind and rapidly cycle.Citation6 The sequence specificity of Hfq is not stringent; the distal site binds U-rich RNAs preferentially, while the proximal site prefers AU-rich RNAs. mRNAs that are targeted by sRNAs often bind Hfq via their 5′ UTRs, which is essential for binding to Hfq.Citation7,Citation8 In vitro studies identified an AAYAAYAA motif, that is found predominantly antisense to the protein coding strand, to bind Hfq with high affinity.Citation9 By binding and bringing sRNAs and mRNAs in close proximity, Hfq facilitates base-pairing between the regulatory RNA and its target, becoming an indispensable player in posttranscriptional gene regulation in most bacteria. Notably, most trans-acting sRNAs not only require Hfq for their function, but are also unstable in the absence of Hfq.
Recent developments in high-throughput sequencing have allowed deeper insight into transcriptomes revealing that large, previously neglected parts of genomes are transcribed. As a consequence, antisense transcription was detected in a number of bacterial species including E. coli.Citation10,Citation11 Due to the low abundance and inability to detect antisense RNAs (asRNAs) by traditional biochemical methods, antisense transcription is often considered nonfunctional. Specifically, it was proposed that E. coli asRNAs are the result of transcriptional misfiring because of poor conservation of transcripts and promoters within enteric bacteria.Citation12 However, the recent finding that a large number of antisense RNAs are found in potentially functional double-stranded RNA complexes and are regulated by the dsRNA-specific RNase III indicates a great regulatory potential for this class of RNAs.Citation13
Hfq’s RNA binding capacity has been utilized to identify sRNAs in a variety of bacterial species by employing different techniques, including microarrays and variations of high-throughput sequencing.Citation14-Citation17 In order to further deepen our knowledge of the Hfq-regulated E. coli transcriptome, we identified RNAs bound to Hfq through co-immunoprecipitation of chromosomally tagged Hfq and deep sequencing. By combining state-of-the-art biochemical techniques with bioinformatical and statistical methods, followed by manual data curation, we compiled a comprehensive list and categorization of Hfq-binding RNAs in E. coli. Notably, we identify low abundant, hitherto overlooked, Hfq-binding RNAs, including intragenic and antisense RNAs and distinguish likely functional RNAs from the plethora of pervasive transcripts. Emerging reports of confirmed antisense transcripts and intragenic RNAs indicate that the coding potential of bacterial genomes should be revisited.
Results
Experimental strategy
In order to shed light on the functionality of products of widespread transcription in E. coli, we isolated and deep-sequenced RNA bound to Hfq via co-immunoprecipitation. Two strains were constructed carrying either 3xFlag or HA-Hfq. Extracts of both strains were analyzed by Western blot and showed efficient expression of tagged Hfq (). To ensure the immunoprecipitation of Hfq was efficient and specific, we analyzed the immunoprecipitated samples by Western blot and silver staining (). The immunoprecipitated samples showed enrichment of monomeric, as well as the hexameric Hfq for both epitope-tagged variants ().
Figure 1. Co-immunoprecipitation of chromosomally tagged Hfq and bound RNA. Wild-type and C-terminally tagged-Hfq strains (3xFlag or HA) were grown to exponential phase. Cell lysates were subjected to immunoprecipitation using anti-Flag or anti-HA-coupled beads. Cell lysates (input) and immunoprecipitated fractions (anti-Flag and anti-HA) were separated on 12% SDS-PAA gel. (A) Proteins were transferred onto a nitrocellulose membrane and immuno-blotted with anti-Flag or anti-HA antibody or (B) visualized by Silver staining. Bands corresponding to 3xFlag- (*) and HA-Hfq (#) are indicated. (C) Cells with C-terminally tagged Hfq bearing either 3xFlag or HA, hfq deletion and isogenic wild-type cells were grown to exponential (E) and stationary phase (S). Cell lysates were separated on an 8% SDS-PAA gel, transferred onto a nitrocellulose membrane and immuno-blotted with anti-RpoS antibody. (D) Equal amounts of co-immunoprecipitated RNA were analyzed by chip-based capillary electrophoresis.
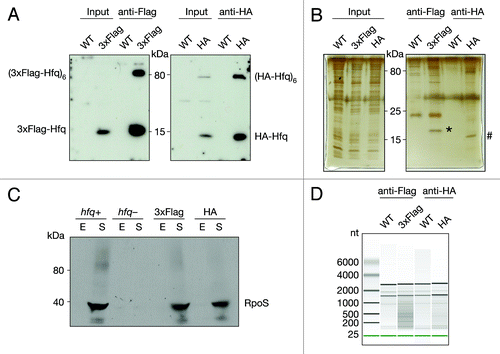
Hfq is a pleiotropic factor found in the center of a large regulatory network. RpoS is known to be upregulated in stationary phase through Hfq-mediated sRNA action.Citation18 To test the functionality of the tagged Hfq variants, we immuno-blotted extracts isolated from cells grown to either exponential or stationary phase and examined RpoS levels (). As expected, levels of RpoS failed to rise in stationary phase when Hfq is absent. In contrast, significant upregulation of RpoS levels, comparable to the wild-type strain, were observed in both Hfq-tagged strains, indicating that both the 3xFlag- and HA-Hfq variants are functional.
Hfq also interacts with proteins, all involved in RNA metabolism: it interacts with RNase E, PNPase, and PAP I; it affects the function of a tRNA-modifying enzyme and binds RNA polymerase through the S1 ribosomal protein.Citation19-Citation23 The proteins listed above or any other potential (direct or indirect) protein partners could affect the output of the co-immunoprecipitation. The purity of the immunoprecipitated fraction was examined by silver staining (). Comparison of 3xFlag and HA-Hfq to the untagged Hfq immunoprecipitations, showed the predominant and specific signal corresponds to Hfq, demonstrating the purity of the immunoprecipitated fraction.
Finally, we purified the co-immunoprecipitated RNA and repeatedly obtained between 2 and 4-fold more RNA from 3xFlag and HA-Hfq compared with the control untagged co-immunoprecipitation. To examine the RNA we performed chip-based capillary electrophoresis. The data showed a significant difference between the RNA profiles from the background control and Hfq co-purified RNA (). The predominate RNAs that co-immunoprecipitated in control experiments corresponded by size to rRNA, therefore the rRNA was considered background. In contrast, RNAs of wide size ranges were found to bind both Hfq-tagged variants.
RNA isolated from the Hfq co-immunoprecipitated fraction was transformed into a cDNA library using a strand-specific, ligation-based protocol and subjected to high-throughput sequencing (Fig. S1A). We intended to use the RNA isolated from control untagged Hfq co-immunoprecipitations as background for assessing the RNA enrichment. However, we repeatedly failed to prepare cDNA libraries from the untagged Hfq strains, likely due to the dramatic difference in quality of co-immunoprecipitated RNA. Therefore, total RNA depleted of rRNA from both 3xFlag and HA-Hfq strains was transformed into cDNA libraries, sequenced and used as background in subsequent analyses.
Deep sequencing and bioinformatical analyses
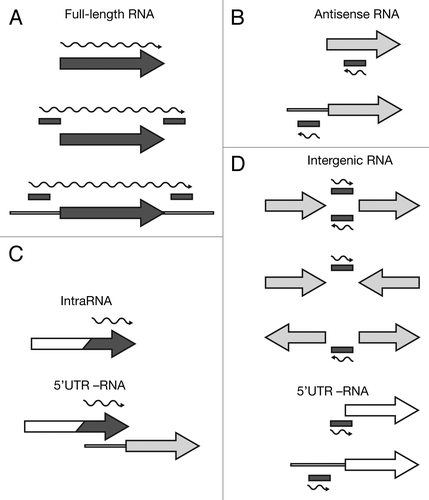
We deep-sequenced cDNA libraries derived from 3xFlag and HA total and Hfq-bound RNA. The reads were mapped to the E. coli MG1655 genome, which resulted in 19–38 Mio reads per data set. From these alignments, we extracted normalized depth-of-coverage (DOC) signals (i.e., the numbers of reads overlapping with a particular genomic position) in a strand-specific manner. In order to extend the analyses beyond the existing annotation, we applied our own peak-finding method to the DOC signals of all four data sets, merged the identified peak-borders and compiled a candidate set of genomic intervals that stem from potentially novel RNAs. We then tested those intervals, as well as all annotated ORFs and RNAs, for significant co-immunoprecipitation with Hfq by applying a protocol for differential expression analyses. Using this method, we identified 309 annotated genes and 786 intervals, which were significantly enriched (adjusted p-value ≤ 0.05). We manually inspected all enriched features and classified them based on their genomic context into four categories of Hfq-binding RNAs: annotated genes (protein and RNA), antisense (as-), intragenic (intra-), and intergenic (igRNAs) (; Tables S1–S4).
Figure 2. Categorization of Hfq-binding RNAs based on deep sequencing. Hfq-binding RNAs were categorized as (A) full-length RNAs, (B) antisense RNAs, (C) intraRNAs, or (D) intergenic RNAs. Annotated coding features (ORFs or RNAs) are represented as arrows and identified intervals are represented as bars. Dark gray elements indicate Hfq-enrichment, while white elements indicate non-enrichment; light gray indicates that an element can be either Hfq-enriched or non-enriched. The wavy line represents the predicted Hfq-binding RNA. For more details see Materials and Methods.
Finally, to check the reliability of our sequencing data and estimated enrichment, we analyzed the co-immunoprecipitated RNA fraction by Northern blot. Northern blot analyses confirmed a significant enrichment of DsrA, a well-known Hfq-binding sRNA, in the co-immunoprecipitated RNA compared to the total RNA (Fig. S1B and C). Accordingly, an RNA that was under-enriched in the deep sequencing data, tmRNA, which is not known to bind Hfq, showed a much lower signal in the co-immunoprecipitated fraction compared with total RNA (Fig. S1D and E).
Validation of the approach
We validated our approach by detecting known Hfq-binding sRNAs and mRNAs. Out of 4,875 annotated features, 297 mRNAs, sRNAs, and tRNAs were recognized as Hfq-enriched (; Table S1). We co-immunoprecipitated a handful of mRNAs that are known sRNA targets (chiP, dppA, fepA, fhlA, flhC, gadX, ompW, and sstT) (Table S1). Furthermore, out of 28 sRNAs reported to bind Hfq, 21 were significantly enriched in our analyses (Table S5). Two sRNAs previously reported to bind Hfq were excluded from our analyses, since the read coverage was too low and insufficient for reliable statistical analyses. Moreover, DicF was enriched with the p-value just above the threshold (padj = 0.052), while 3 other sRNAs were not enriched. SroC, a sRNA shown to bind Hfq in SalmonellaCitation16 is missing from the annotation we used. However, we identified an Hfq-enriched interval overlapping its chromosomal location, substantiating our ability to identify Hfq-binding sRNAs independently of annotation.
The list of detected bacterial sRNAs is steadily increasing due to continuous improvements of high-throughput sequencing assays. Recently, 10 novel sRNAs were identified in E. coli,Citation24 but their function has not been addressed yet. We found Hfq-enriched intervals overlapping three of those sRNAs; here named ig-yhcE-oppA, as-yhcC and 5′ UTR-yejG. The first two were reported to be destabilized in the absence of Hfq.Citation24 Our data provide additional explanation for the observed Hfq stabilization of these sRNAs and extend the list of Hfq-binding sRNAs (Table S6).
We were interested if Hfq-binding RNAs identified in our study showed an enrichment of genes of particular functions. The set of genes tested consisted of annotated coding features that were found to be Hfq-enriched, as well as unique 5′ UTRs identified in both intra- and igRNA categories (Tables S1, S3, and S4; see Discussion). We performed a Gene Ontology (GO) enrichment analysis using FuncAssociate,Citation25 which revealed 15 significantly enriched GO terms (Table S7). The majority of genes enriched by Hfq are involved in nitrate or nitrogen metabolism, anaerobic respiration or electron carrier activity, metal cluster binding and nickel transporting. These results support the current opinion that Hfq is an important factor in stress adaptation.
Antisense RNAs
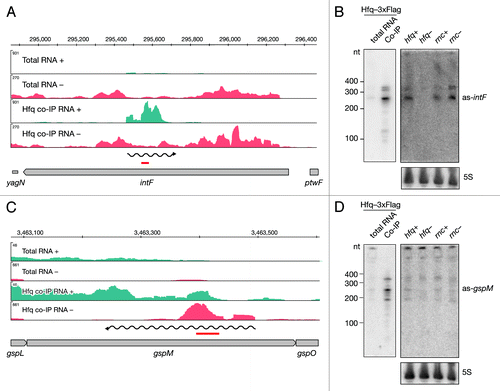
To extend the annotation-based analyses, we used an algorithm that recognizes peaks in read coverage in unannotated (antisense to ORF and intergenic) regions and tested these intervals for significant enrichment in Hfq co-immunoprecipitated libraries. Through manual inspection of the enriched intervals () we identified 67 asRNAs that bind Hfq (Table S2), indicating that these antisense transcripts might be functional.
We identified Hfq-enriched intervals opposite to the intF gene and by Northern blot analyses confirmed binding of a 250 nt long RNA to Hfq (). Additionally two bands of lower intensity were observed around 350 nt. We observed all three species in the total RNA as well. The signal was almost absent in the hfq deletion mutant, suggesting that Hfq stabilizes these RNAs. When we assessed the expression of the as-gspM RNA we observed similar, but low levels in all tested strains (). Again, significant enrichment was confirmed in the Hfq co-immunoprecipitated fraction by Northern blot revealing a predominant band at 230 nt and weaker signals at 180 and 350 nt ().
Figure 3. Identification and verification of antisense RNAs binding to Hfq. Deep sequencing results of as-intF and as-gspM (A and C) represented as averaged coverage maps of 3xFlag- and HA total RNAs and 3xFlag- and HA-co-immunoprecipitated RNAs (Hfq co-IP). The genomic strands are shown in blue (+) and red (-). Note that scales for the + and – strand differ. The genomic location is depicted on top and the genomic context is depicted below. The red bar indicates the position of the oligonucleotide probe that was used in the corresponding Northern. The wavy line represents the predicted position of the novel RNA. Northern blot analyses of as-intF and as-gspM (B and D). Hfq-3xFlag total RNA and RNA co-immunoprecipitated with Hfq-3xFlag (left panel) and total RNA isolated from hfq deletion strain (hfq-), corresponding isogenic wild type (hfq+), RNase III mutant (rnc-) and corresponding isogenic wild-type cells (rnc+) (right panel) were fractionated on a denaturing 8% polyacrylamide gel, electro-blotted onto a nylon membrane and hybridized with radioactively labeled oligonucleotide. Note that different amounts of RNA were loaded; 20 μg total RNA and 1 μg co-IP RNA. Ladder sizes in nt are indicated. 5S RNA was used as a loading control. At least two independent experiments were performed and representative data are shown.
Our data suggest a product of antisense transcription opposite to manX (Fig. S2A). Interestingly, the short Hfq-enriched interval opposite to manX contains an in vitro identified Hfq aptamer.Citation9 We performed Northern blot analyses and detected RNAs of around 220 and 300 nt in the co-immunoprecipitated fraction, confirming our deep sequencing analyses (Fig. S2B). However, the asRNAs were undetectable in the total RNA isolation. Furthermore, we identified an Hfq-enriched interval opposite to the distal 3′ end of yggN (Fig. S2C); a previously reported Hfq aptamerCitation9 maps downstream of the identified interval. Northern blot analyses revealed multiple RNA species binding to Hfq (Fig. S2D). The detected RNAs range from 80 to 400 nt, with the longer RNAs containing the Hfq aptamer. In contrast, only two bands of low intensity corresponding to 250–300 nt were detected in wild-type strains. The signal is almost absent in both mutant strains indicating that Hfq and RNase III stabilize as-yggN.
Intra-RNAs: ORF originating transcripts
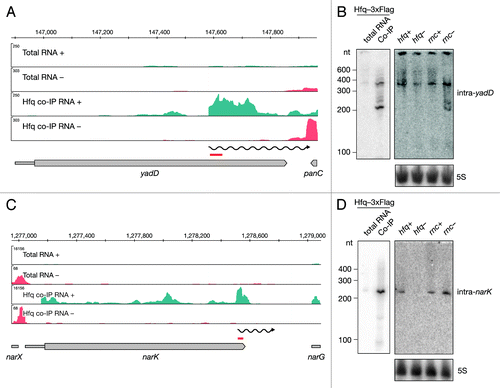
Recently it was proposed that the 3′ UTRs of genes might serve as a reservoir for independent RNAs.Citation3 Upon manual inspection of Hfq-enriched full-length genes, we noticed that 38 of the mRNAs co-immunoprecipitated with Hfq showed a steep increase in coverage within the ORF, often far downstream from the transcriptional/translational start site. We termed these ORF-originating RNAs intragenic RNAs or intraRNAs (; Table S3) and further subcategorized them as 5′ UTRs or true intraRNAs. Thirteen out of 38 Hfq-binding intraRNAs are classified as 5′ UTRs of the downstream genes. For example, our analyses reported nlpD to bind Hfq. However, the observed increase in read coverage overlapped the annotated 5′ UTR of the downstream gene, rpoS. Hfq is known to bind rpoS in its 5′ UTR, which is necessary for sRNA-mediated regulation. The remaining 25 intraRNAs were classified as true intraRNAs (). For example, yadD, the gene convergently transcribed to the downstream gene, showed an increase in read coverage around the last third of the annotated ORF (). In order to independently assess the size of Hfq-bound intra-yadD, we analyzed the co-immunoprecipitated RNA fraction by Northern blot (). We confirmed the binding of two RNAs, approximately 200 and 350 nt long, to Hfq and expression of the 350 nt long RNA in all strains tested. Interestingly, the 200 nt long species was detectable only in the RNase III mutant, suggesting the RNA is degraded by RNase III. In contrast, no such RNase III effect was observed for intra-narK. Northern blot analyses showed a single 230 nt long lowly expressed RNA to be efficiently co-immunoprecipitated with Hfq. The RNA was undetectable in the hfq mutant strain, further confirming its Hfq dependence ().
Figure 4. Identification and verification of novel Hfq-binding intragenic RNAs. Deep sequencing and Northern blot analyses of intraRNAs intra-yadD (A and B) and intra-narK (C and D) as described in and .
Role of Hfq in sRNA and tRNA processing
Hfq is important for the function and stability of many sRNAs and we identified the majority of them in our screen (Table S5). Interestingly, we also identified intervals adjacent to annotated sRNAs (Table S8). We examined the interval identified upstream of spf that overlaps with an Hfq aptamer and determined the sizes of two RNAs in the co-imunoprecipitated fraction (). Two RNAs, 190 and 210 nt long, were efficiently enriched by Hfq and expressed in wild-type strains. Their steady-state levels were significantly decreased in the hfq mutant. In this case, a potential transcriptional start site could not easily be determined based on deep sequencing data. We hypothesized that this transcript may overlap with Spf and probed the Hfq co-immunoprecipitated fraction with a probe specific for the sRNA. As expected, the major band detected and enriched in the Hfq co-immunoprecipitated fraction corresponds to the reported size of Spf, 109 nt. In addition, we detected two RNAs, 190 and 210 nt long, specifically enriched by Hfq. Taken together, the data indicate that the mature Spf is a product of an Hfq-dependent processing event.
Figure 5. Identification and verification of tRNA and sRNA precursors binding to Hfq. Deep sequencing and Northern blot analyses of Spf (A and B) and the metZWV precursor (C and D) as described in and . Violet and red bars indicate the positions of oligonucleotide probes used in the corresponding Northern blots. 6S RNA was used as a loading control. The black bar indicates the position of the Hfq aptamer reported by Lorenz et al.Citation9
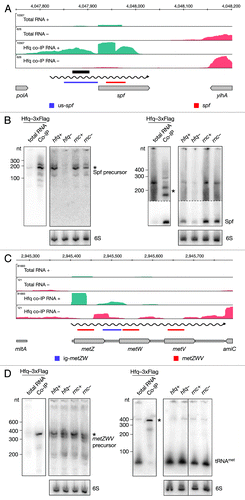
In addition to Hfq enrichment of tRNAmetZ, we identified an Hfq-enriched interval in the metZ and metW intergenic region (; Table S9). A Northern blot performed with a probe specific for the intergenic region, revealed Hfq-binding to a 380 nt long RNA, which is the approximate length of the metZWV operon. Notably, we did not detect a short RNA in the co-immunoprecipitated fraction demonstrating that the spacer region does not bind Hfq independently. To ensure that the long transcript we observed corresponds to the tRNA precursor, we performed a Northern blot using a tRNA-specific probe. The 380 nt long precursor RNA was confirmed to be specifically enriched, while the mature tRNA does not bind Hfq. tRNAmet was detected in all strains tested, but interestingly, an additional precursor RNA was detected in the RNase III mutant strain, indicating a role for RNase III in tRNA maturation. Taken together, our data indicate an Hfq- and RNase III-dependent processing of the metZWV operon.
Intergenic RNAs
After manual categorization of intervals mapping to unannotated regions, we identified 81 putative intergenic RNAs (igRNAs) that bind Hfq (; Table S4). Almost all known sRNAs in E. coli, including those identified in screens of the Hfq-RNA interactome, were found in intergenic regions.Citation15,Citation26,Citation27 Here we report additional 27 intergenic sRNAs. Expression and binding to Hfq was confirmed by Northern blotting for several of these novel sRNAs. As an example we show the RNA encoded between yjgZ and insG (). An RNA, approximately 300 nt long, showed notably lower levels in the hfq deletion mutant strain compared to the isogenic wild type. In contrast, levels of this sRNA were higher in the RNase III mutant strain when compared to the wild type. We also confirmed binding of Hfq to a 320 nt long RNA encoded between zupT and ribB (). However, the expression pattern of this igRNA indicated lower levels in both Hfq and RNase III mutant strains.
Figure 6. Identification and verification of novel Hfq-binding intergenic RNAs. Deep sequencing and Northern blot analyses of intergenic RNAs yjgZ-insG (A and B), zupT-ribB (C and D), 5′ UTR_adhE (E and F) and lrhA-alaA (G and H) as described in and .
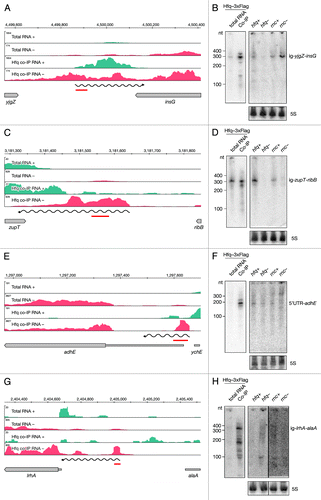
A subcategory of intergenic RNAs (5′ UTR-RNAs) was identified through categorization of Hfq-enriched intervals overlapping the annotated 5′ UTRs of genes (). Importantly, the corresponding downstream genes were not Hfq-enriched. Out of 54 intergenic 5′ UTR-RNAs, 10 are indeed 5′ UTRs of known sRNA targets, while 44 are newly identified components of the Hfq network (Table S4). For example, the coding region of adhE showed no enrichment in the deep sequencing data, but an enriched interval overlaps with the annotated 5′ UTR (). We confirmed binding of a 180 nt long RNA to Hfq by Northern blot and did not detect binding of longer RNAs (). This small RNA showed no apparent difference in expression in the hfq mutant strain, but the signal corresponding to a longer RNA was detectable in the RNase III mutant, suggesting RNase III plays a role in the biogenesis of the short 5′ UTR-adhE.
Our analyses also identified a region upstream of lrhA to be co-immunoprecipitated with Hfq (). The identified interval did not overlap with the annotated 5′ UTR; therefore in our manual categorization we listed this RNA as intergenic ig-lrhA-alaA. Northern blot analyses revealed a corresponding RNA very lowly expressed, virtually undetectable in the total RNA. However, two bands at 150 and 350 nt were observed in the Hfq co-immunoprecipitated fraction (). Although there is a short 5′ UTR annotated upstream of lrhA, in our sequencing data there was no indication of a transcriptional start site in that area. In order to test if the RNA upstream of lrhA is in fact part of the lrhA mRNA, we preformed Northern blot analyses on total RNA separated on an agarose gel (Fig. S3A and B). We detected a long RNA of around 1.5 kb with a probe upstream of lrhA, as well as the signal in the 200–500 nt region. Importantly, the signal was detectable only in the hfq mutant strain, but not in the wild-type strain. Similarly, a shorter product of approximately 1.25 kb was detected in the hfq mutant strain with the probe recognizing the coding region of lrhA, while its levels were significantly lower in the wild type. Taken together, these data indicate that Hfq plays a crucial role in repression of lrhA mRNA.
Discussion
Hfq acts as an RNA chaperone, restructuring both sRNAs and mRNAs into more interaction-favorable conformations.Citation28 In an attempt to better understand the broadness of Hfq action we isolated RNAs that bind Hfq in vivo by co-immunoprecipitation and employed deep sequencing. Our analyses identified a range of Hfq-binding RNAs that can be classified into four major categories: mRNAs (and their 5′UTRs), igRNAs and more surprisingly, intraRNAs, and asRNAs ().
Antisense and ORF-originating intraRNAs and the role of Hfq
High-throughput sequencing has increased detection sensitivity significantly, which enabled the genome-wide detection of antisense transcription across species.Citation10 Reported proportions of genes having antisense counterparts vary significantly between organisms.Citation10 asRNAs are regularly considered low abundant, hence difficult to confirm by independent methods, and are often argued to be products of noise or read-through transcription. By comparing short and long RNA fractions in wild-type and RNase III mutant strains, it was shown that antisense transcripts, although not necessarily regulated themselves, have the potential to regulate their sense counterparts in an RNase III-dependent manner.Citation29 75% of all genes in a number of Gram-positive bacteria were shown to follow this pattern, but the effect was not observed in Salmonella enteritidis, the only Gram-negative species tested. Recently, we identified potentially functional E. coli asRNAs based on their ability to form dsRNA with their sense counterparts in vivo.Citation13 Hfq, as an important player mediating RNA-RNA interactions, is a plausible candidate for mediation of massive dsRNA formation. Whether Hfq is an essential factor for the formation of sense/antisense pairs remains to be tested.
The best-known examples of cis-acting asRNAs, such as components of toxin-antitoxin systems, generally are Hfq-independent. We corroborate this, as we did not co-immunoprecipitate any antitoxin RNAs in our screen. However, it has been shown that Hfq is involved in base-pairing and the following translation inhibition of RNA-IN by a cis-acting RNA-OUT of the Tn10/IS10 system.Citation30,Citation31
We identified 67 asRNAs through Hfq co-immunoprecipitation (Table S2). It is tempting to assume that the function of asRNAs is to regulate their sense counterpart through Hfq-facilitated base pairing. In the well-studied sRNA-regulated mRNAs, both the sRNAs and the mRNAs bind Hfq. Therefore, we hypothesize that both the sense and the antisense RNAs should be immunoprecipitated with Hfq. Alternatively, it has been proposed that pairing between RNAs that display extended perfect complementarity, could be Hfq-independent.Citation10,Citation32
One of the described cis-acting sRNA is GadY; it directs processing of gadXW leading to the accumulation of the two processing products.Citation33 Although it has been shown that GadY binds to and is stabilized by Hfq, it is not clear whether Hfq is required for GadY-mediated gadXW regulation or binding to Hfq is necessary for regulation of possible other trans targets.Citation32 Interestingly, our data identified gadX to be co-immunoprecipitated with Hfq (Table S1), indicating that despite perfect complementarity, base-pairing between two RNAs might require restructuring by the RNA chaperone Hfq.
Among Hfq-binding asRNAs we identified, only nine corresponding sense counterparts bind Hfq (Tables S1, S3, and S4). This might indicate that the majority of asRNAs either do not regulate their cognate sense RNAs in Hfq-dependent manner and/or act in trans. Furthermore, it is possible that Hfq binds only one of the RNAs in an RNA-RNA interaction facilitating a conformational change necessary for pairing. Alternatively, asRNAs could have direct influence on the transcription of its cognate mRNA through transcriptional interference or attenuation.Citation10,Citation34 It remains to be elucidated what would be the role of Hfq in such a mechanism of action.
Recent work suggested that intragenic transcription initiation is massively silenced by histone-like nucleoid structuring protein (H-NS).Citation35 However, there is growing evidence that transcripts, particularly sRNAs, can originate within annotated genes (Lybecker unpublished).3 We term such transcripts intraRNAs and report 25 examples that bind Hfq (Table S3). We identified intra-narK sRNA and confirmed Hfq-dependent expression typical for Hfq-dependent sRNAs (). Most intraRNAs we identified are long enough to code for a short peptide corresponding to the C-terminus end of the mRNA-encoded protein, represented here by intra-yadD (). In other examples, additional alternative putative small ORFs could be predicted in silico, but whether these RNAs are indeed translated or even execute dual function, as a sRNA and peptide coding, remains to be examined.
Some of the newly identified RNAs, in particular asRNAs, were undetectable by Northern blot under the tested growth condition. As most sRNAs are known to be upregulated as a response to stress, analyses of different conditions might be necessary to address expression patterns of asRNAs. Our approach of co-immunoprecipitation with Hfq provided better sensitivity and resolution and allowed distinguishing novel Hfq-binding RNAs from mere noise or background gene expression. Studying particular RNAs individually will be necessary for the elucidation of their biological functions and modes of action. We suggest that the number of transcripts greatly exceeds the existing annotation and that both antisense and intragenic transcription represent a way of extending the coding potential of a relatively small genome.
sRNAs and tRNAs precursors that bind Hfq
In addition to being a major stabilizer and mediator of sRNA function, Hfq has been implicated in maturation of several sRNAs.Citation18,Citation36,Citation37 Our data indicates that Hfq is involved in processing of the Hfq-dependent sRNA Spf, a negative regulator of galK translation.Citation38 We identified an Spf precursor RNA that is stabilized and bound by Hfq (). A functional 53 nt long DicF, a negative regulator of cell division, was shown to be the result of RNase III and RNase E processing of the full-length 190 nt RNA.Citation39,Citation40 Although previously reported to bind Hfq, DicF was just below our significance threshold and did not qualify as Hfq-binding in this study. Interestingly, we found an Hfq-enriched interval upstream of dicF, suggesting Hfq binds the full-length transcript and is involved in its processing. In addition, we identified Hfq-enriched intervals adjacent to 18 other sRNAs (Table S8). It is possible that Hfq-dependent processing of sRNAs is a much wider phenomenon than anticipated.
Our data also identified a set of new igRNAs (Table S4). ig-yjgZ-insG () and ig-ribB-zupT () are destabilized in the absence of Hfq, which is one of the hallmark characteristics of canonical sRNAs. Interestingly, both bona fide sRNAs code for putative small ORFs, 56 and 47 amino acids respectively. Whether these transcripts are indeed trans-acting RNAs and/or code for small peptides, remains to be revealed.
In addition to sRNAs, our attention was drawn to tRNA loci. Due to the repetitive nature of tRNAs, we were not able to assess their enrichment in many cases. However, we identified an Hfq-enriched interval that corresponds to the intergenic region of the metZWV operon and Northern blot analysis revealed that a tRNA precursor was specifically co-immunoprecipitated with Hfq. Our data also indicate that, in addition to many enzymes involved in maturation of tRNAs,Citation41 RNase III, although not indispensible, plays a role. Hfq was shown to bind tRNAs in vitroCitation42 as well as several precursors of proM tRNACitation15 in vivo, which is in agreement with our data (Table S9). We report tRNAvalV and intervals adjacent to several other tRNAs to co-immunoprecipitate with Hfq (Table S9), suggesting Hfq might have a role in tRNA biogenesis.
Hfq binds mRNAs and 5′ UTR-derived RNAs
We identified 274 full-length mRNAs and 58 5′ UTRs (4 intraRNAs and 54 igRNAs), expanding the list of presumable sRNA targets. Although only a handful of mapping experiments have been done to identify the mRNA’s nucleotides directly interacting with Hfq, it is widely accepted that binding occurs via the 5′ UTR and a common (ARN) motif is found in the 5′ UTRs of many known sRNA targets.Citation7,Citation8,Citation43 Hfq binding to 5′ UTRs and subsequent sRNA-mediated regulation (and endoribonucleolitic cleveage) might lead to accumulation of Hfq-bound 5′ UTRs. Accordingly, the 5′ UTR-RNAs we identified (Tables S3 and S4) could be a consequence of sRNA-mediated regulation of corresponding genes.
We found a sRNA derived from the 5′ UTR of full-length adhE to bind Hfq. RNase III might be involved in the biogenesis of the 5’ UTR-adhE, although its stability is not affected by Hfq (). A different effect of Hfq on RNA processing is observed for the lrhA transcript (). In this case, multiple short 5′ UTR derived fragments bind Hfq efficiently. The full-length lrhA transcript and the processed 1.25 kb long RNA are upregulated in absence of Hfq, indicating the entire primary transcript is unstable in the presence of Hfq (; Fig. S3B and C). This processing could be sRNA mediated, but we cannot exclude that Hfq, by binding to the 5′ UTR, facilitates formation of a structure that allows efficient endoribonucleolytic cleavage and contributes to the posttranscriptional regulation independently of sRNAs.
5′ UTRs can contain riboswitches leading to transcriptional termination or anti-termination, translational inhibition or activation.Citation44 To date eight riboswitches are known in E. coli, and nine more have been proposed recently.Citation24 Interestingly, we found three of the newly proposed elements (in this study: ttcC-ynaE, 5′ UTR-ydfK and ybjM) to bind Hfq.Citation24 Further work will be necessary to elucidate the function of these RNAs and determine the role of Hfq. We hypotesize that 5′ UTR RNAs that bind Hfq might act as independent sRNAs, similarly to RNAs with dual roles identified in Listeria monocytogenes.Citation45
Concluding remarks
Hfq binds a variety of RNAs with a broad range of affinities and it has been shown that Hfq-dependence of RNA-RNA pairing can be overcome by increasing RNA concentration.Citation18 Hfq was shown to bind the AAYAAYAA motif, as part of low abundant RNAs, with high affinity.Citation9 This motif is found significantly enriched on noncoding strands, opposite to genes.Citation9 In this study 43 transcripts that contain Hfq aptamers were found to co-immunoprecipitate with Hfq. This suggests that the in vitro-identified motif might contribute to Hfq-RNA interactions in vivo.
Taken together, our data demonstrate that the function of Hfq exceeds the role in sRNA-mediated regulation, and is potentially an important player in mRNA, tRNA and sRNA processing. Also, screening for Hfq-interacting RNAs combined with deep sequencing and stringent statistical analyses, proved to be a valuable tool for the identification of novel functional transcripts, like the intraRNAs, emerging from within ORFs, lowly expressed igRNAs and, in particular, asRNAs.
Materials and Methods
Bacterial strains
The E. coli strains used in this study are listed in Table S10. Cells were grown in LB medium at 37 °C with aeration (200 rpm) to exponential or stationary phase (optical density at 600 nm [OD600] of ~0.5 and ~1.3 respectively). When appropriate, medium was supplemented with kanamycin, tetracycline or ampicillin.
Hfq tagging
The hfq gene was C-terminally tagged with HA or 3xFlag epitopes on the chromosome. The constructs used for epitope introduction were generated as follows. First, the region directly downstream of hfq (DS-hfq) was amplified in a polymerase chain reaction (PCR) from MG1655 strain genomic DNA using primers SalI-DS-Hfq-F and HindIII-DS-Hfq-R, purified and subjected to restriction digest by enzymes SalI and HindIII. pUC19 was used for subcloning; sticky ends were generated through restriction by SalI and HindIII and were subsequently dephosphorylated by CIP (New England Biolabs) and ligated with DS-hfq fragment. Next, kanamycin resistance cassette was amplified from FRT-PGK-gb2-neo-FRT template DNA (Gene Bridges) using primers BamHI-BmtI-Kan-F and SalI-Kan-R, followed by a BamHI and SalI restriction digest, and ligated to pre-digested and dephosphorylated pUC19-DS-hfq plasmid. The epitope-tagged hfq sequence was generated by annealing two overlapping oligonucleotides (Hfq-3xFLAG-Kan-Fwd and Hfq-3xFLAG-Kan-Rev or Hfq-HA-Kan-Fwd and Hfq-HA-Kan-Rev), filling in reaction, A-tailing (GoTaq®, Promega, according to manufacturer’s protocols) and subsequent ligation into pGEM®-T Easy Vector using pGEM®-T Easy Vector System (Promega). The resulting fragments consisted of the 50 distal nucleotides of hfq and 3xFlag or HA sequence. Next, the epitope tagged-hfq fragments were PCR amplified from pGEM®-T Easy-derived plasmid using oligonucleotides XmaI-Hfq-F and BmtI-Flag-R or BmtI-HA-R, subjected to restriction by XmaI and BmtI, and finally ligated to pre-digested and dephosphorylated pUC19-Kan-DS-hfq. All amplifications were performed using Phusion® High-Fidelity DNA Polymerase (New England Biolabs); restriction enzymes (all New England Biolabs) were used per manufacturer’s protocols; ligation reactions of PCR fragments and pUC19 and its derivatives were performed with T4 PNK DNA ligase (New England Biolabs) according to manufacturer’s protocols.
To construct strains used in this study, Quick and Easy E. coli Gene Deletion Kit (Gene Bridges) was used per manufacturer’s protocol. Briefly, MG1655 cells were grown to exponential phase and transformed with pRED/ET (ampicillin) plasmid by electroporation. Cells were then grown to exponential phase in presence of ampicillin; FRT recombinase was induced by addition of L-arabinose (final concentration 0.35%). Linear full-length fragments were amplified with oligonucleotides Hfq-tag-check-Fwd and Hfq-tag-check_Rev by Phusion® High-Fidelity DNA Polymerase (New England Biolabs) from pUC19-derived constructs and electroporated into FRT-expressing cells that were subsequently grown at 37 °C. Correct genomic insertions of both strains were verified by sequencing. All oligonucleotides used are listed in Table S11.
Immunoprecipitation assays
Cell lyses for protein extraction and co-immunoprecipitation were performed as follows. Equivalent number of cells to OD600 = 5 was harvested by centrifugation at 3200 × g at 4 °C for 5 min. Pellets were washed with 4 ml TM buffer (10 mM Tris-HCl pH 7, 10 mM MgCl2) and subsequently frozen at -20 °C to facilitate cell lysis. Cell pellets were then resuspended in 1 ml ice-cold lysis buffer (50 mM Tris-HCl pH 7.5, 5 mM MgCl2, 250 mM NaCl) supplemented with cOmplete mini protease inhibitor (Roche) and 10 U/ml RNasin® (Promega). Cells were lysed by sonication and lysates cleared by centrifugation at 16 100 × g for 30 min at 4 °C. Cell lysates were treated with 10 U Turbo™ DNase I (Roche) at 37 °C for 15 min prior to immunoprecipitation. Fifteen μl of anti-Flag® or anti-HA antibody-conjugated beads (both mouse monoclonal, Sigma) were washed 3 times with TBS (50 mM Tris-HCl pH 7.4, 150 mM NaCl) and added to cell lysates and incubated at 4 °C with rotation overnight. Beads with precipitated RNP-complexes were washed 3 times with 0.5 ml TBS and finally resuspended in 400 μl TBS. RNA was extracted from RNP-complexes by two subsequent phenol/chloroform/isoamyl alcohol (25:24:1) extractions and ethanol-precipitated with NaOAc and glycogen as a carrier. RNA integrity was assessed by analyses of 2 ng of co-immunoprecipitated RNA by a Agilent RNA 6000 Pico Kit.
Protein analyses
For analyses of expression (input) and imunoprecipitation (co-IP) of 3xFlag and HA-Hfq, cell lysate equivalent to OD600 of 0.04 and 1.5 were used for Western blot, while cell lysate equivalent to OD600 of 0.01 and 0.5 were used for Silver stain. For analyses of RpoS expression, 40 μg of protein was used. Protein samples were mixed with loading buffer with no reducing agent and heat-denatured to 95 °C for 5 min, followed by separation on 8 or 12% denaturing Tris-Glycin-SDS-PAGE. Silver staining was performed with Pierce® Silver Stain Kit (Thermo Scientific) following manufacturer’s protocol.
For Western blot, proteins were electro-blotted onto a Hybond ECL membrane (GE Healthcare). Membranes were blocked for 2 h at room temperature in 5% non-fat dried milk in PBS or 5% BSA in TBS with agitation. The membranes were then incubated with primary antibody (anti-Flag® polyclonal rabbit (Sigma), 1:1,000 in 1% BSA in PBS; anti-HA polyclonal rabbit (Sigma), 1:1,000 in PBS-T; anti-RpoS monoclonal mouse (Santa Cruz), 1:1,000 in 5% BSA in TBS) overnight at 4 °C. Subsequently, membranes were incubated with horseradish peroxidase-conjugated secondary antibody (goat anti-rabbit (Sigma), 1:10,000 in PBS-T or goat anti-mouse (Jackson), 1:10,000 in 5% non-fat dried milk in TBS) for 45 min at room temperature. Signal was detected using Amersham™ ECL™ Prime reagent (GE Healthcare).
Total RNA isolation
Total RNA was isolated as described by Lybecker et al.Citation13 Briefly, cells were first mixed with Stop solution (95% ethanol, 5% phenol) in 8:1 ratio to stabilize cellular RNA and harvested by centrifugation at 3000 × g at 4 °C for 5 min. Supernatant was decanted and the pellets were frozen in liquid N2. Pellets were resuspended in lysis buffer (10 mM Tris-HCl pH 8.0, 1 mM EDTA, 0.5 mg/mL lysozyme) and 10% SDS (wt:vol) was added to a final concentration of 0.1%. The lysate was then incubated at 64 °C for 2 min. 1 M NaOAc (pH 5.2) was added to the lysate to a final concentration of 0.1 M. Then an equal volume of water-saturated phenol was added, mixed by inversion and incubated at 64 °C for 6 min with inverting approximately every 40 s. Samples were chilled on ice and centrifuged at 16 100 × g for 10 min at 4 °C. The aqueous layer was transferred to a Phase Lock Gel Heavy tube (5Prime) with an equal volume of phenol/chloroform/isoamyl alcohol (25:24:1), inverted several times and centrifuged for 5 min at 16 100 × g. The aqueous layer was transferred and extraction repeated with chloroform/isoamyl alcohol (24:1). The RNA was then ethanol precipitated from the aqueous layer using NaOAc. Total RNA was treated with Turbo™ DNase I (Roche) following the manufacturer’s protocol. RNA integrity was assessed by agarose gel electrophoresis.
cDNA library preparation
Directional (strand-specific) RNA-seq cDNA libraries were constructed as described by Lybecker et al.Citation13 Briefly, the total and co-immunoprecipitated RNAs were first treated with Turbo™ DNase I (Roche) per manufacturer’s protocol. Total RNA was then depleted of rRNA using the Ribo-Zero™ RNA removal kit for Gram-negative bacteria (Epicenter). 250 ng of RNA was fragmented using the RNA fragmentation reagents (Ambion®) per the manufacturer’s protocol at 70 °C for 5 min. RNA was treated sequentially with tobacco acid pyrophosphatase (Epicenter) and calf intestinal phosphatase (New England Biolabs) per the manufacturer’s protocols to remove 5′ tri- and monophosphates. Finally, RNA was treated with polynucleotide kinase (T4 PNK; New England Biolabs) without ATP to remove 2′-3′ cyclic-phosphates at 37 °C for 4 h in 100 mM Tris-HCl pH 6.5, 100 mM MgAc, 5 mM β-mercaptoethanol. A 3′ RNA adaptor, based on the Illumina multiplexing adaptor sequence (Oligonucleotide sequences © 2007–2014 Illumina, Inc. All rights reserved) blocked at the 3′ end with an inverted dT (5′-GAUCGGAAGA GCACACGUCU [idT]-3′), was phosphorylated at the 5′ end using T4 PNK (New England Biolabs) per the manufacturer’s protocol. The 3′ multiplex RNA adaptor was ligated to the 3′ ends of the total and co-immunoprecipitated RNAs using T4 RNA ligase I (New England Biolabs). RNA was incubated at 20 °C for 6 h in 1X T4 RNA ligase reaction buffer with 1 mM ATP, 20 µM 3′ RNA adaptor, 1 µl DMSO, 5 U of T4 RNA ligase I, and 40 U of RNasin (Promega) in a 10 μl reaction. The excess of oligonucleotide was removed by applying the sample through 3K columns (Pall). The RNAs were phosphorylated at the 5′ ends using T4 PNK (New England Biolabs) per the manufacturer’s protocol to allow for subsequent ligation of the 5′ RNA adaptor. RNA was size-selected (150–300 nt) and purified over a denaturing 8% polyacrylamide/8 M urea/TBE gel. Gel slices were incubated in RNA elution buffer (10 mM Tris-HCl, pH 7.5, 2 mM EDTA, 0.1% SDS, 0.3 M NaOAc) with vigorous shaking at 4 °C overnight. The supernatant was subsequently ethanol precipitated using glycogen as a carrier molecule. The Illumina small RNA 5′ adaptor (5′-GUUCAGAGUU CUACAGUCCG ACGAUC-3′) was ligated to the libraries using T4 RNA ligase I (New England Biolabs) and the excess adaptor was removed as described above. The ligated RNAs were size-selected (200–300 nt) and gel-purified over a denaturing 8% polyacrylamide/8 M urea/TBE gel (as described above). The di-tagged RNA libraries were reverse-transcribed with SuperScript®II reverse transcriptase (Invitrogen) using random nonamers per the manufacturer’s protocol. RNA was removed using RNase H (Promega) per the manufacturer’s protocol and cDNA was amplified in PCR performed using Phusion® High-Fidelity Polymerase (New England Biolabs). cDNA was amplified with Illumina-compatible PCR primers (Table S11) by 15 cycles of PCR. The products were analyzed on an Agilent 2100 Bioanalyzer.
Deep sequencing and bioinformatical analyses
Deep sequencing
Hfq-enriched (co-IP) libraries were sequenced on individual Genome Analyzer IIx lanes (36 bp, single-end); total RNA samples were sequenced multiplexed on one HiSeq 2000 lane (50 bp, single-end) at the CSF NGS unit (http://csf.ac.at/). The reads were mapped with NextGenMap 0.4.10Citation46 against the E. coli genome (strain K12, substrain MG1655), demanding a minimum identity of 90%. Multireads (i.e., reads with mapping quality zero) were pruned and the number of resulting reads were summarized in Table S12.
Coverage signal extraction
Depth-of-coverage signals (i.e., the counts of reads overlapping a particular genomic position) were extracted from the alignments in a strand-specific manner and normalized by the total amount of mapped bases per data set in order to make them comparable among each other. The normalized coverage signals were also converted to the BigWig data format to enable manual data inspection in a genome browser.Citation47
Peak finding
Our own peak-finding method was applied to screen intergenic and antisense regions for coverage peaks stemming from novel (unannotated) RNAs: first, the signal was smoothed using a moving Gaussian kernel and the first derivative at each signal position was approximated by cubic Hermite interpolation. Then potential peaks were identified based on the sign-changes of the derivatives, accepting only peaks that complied with configured minimum/maximum dimensions. In this manner, peaks were called in all four data sets and then merged between data sets if they had an overlap of at least 80% in order to get a final set of candidate intervals based on the peak borders. Next, the set of known ORFs/RNAs was compiled by downloading E. coli MG1655 gene annotations from NCBI (including genes, tRNAs and ncRNAs) and extending this set by missing RefSeq annotations downloaded from UCSC (8 such annotations were added) which resulted in a total of 4,875 annotated genomic features. Intervals that overlapped any annotated feature (ORF or RNA) with more than 80% were discarded and were not considered for further analyses.
Testing for significance
All known annotated ORFs, RNAs and identified intervals that were covered by at least one read in all four data sets were tested for significant “overexpression” in the Hfq co-immunoprecipitated RNA data sets using edgeR.Citation48 In this analysis, 3xFlag and the HA data sets were treated as technical replicates and intervals, ORFs and RNAs with an adjusted p-value ≤ 0.05 were considered significantly enriched.
Interval annotation and categorization
Coding features (ORFs and RNAs) and intervals identified as Hfq-enriched were subjected to manual curation and categorization. Enriched intervals that overlap proximal or distal ends of enriched coding features or annotated 5′ or 3′ UTRs were joined with the ORFs/RNAs to obtain more accurate transcriptional units (TU). If TUs are extended with enriched intervals, the reported 5′ end was approximated based on the steep increase of coverage pattern, and the 3′ end corresponds to the last base of the most distal associated interval. In case no intervals are included in the TU, annotated gene borders are reported. All enriched coding features that display 3′ skewed coverage and a potential 5′ end within the coding region were categorized as intraRNAs. If an intraRNA overlaps an annotated 5′ UTR or the coverage pattern indicates that an intraRNA is part of the downstream transcript, it is subcategorized as a 5′ UTR-RNA. Finally, remaining enriched intervals that display proximal prominent increase in coverage (indicating a 5′ end) or are improbable read-through products (due to the distance to the next annotated gene on the corresponding strand) are categorized as antisense (opposite to coding features and/or 5′ UTR) or intergenic (between coding features). If a putative igRNA overlaps an annotated 5′ UTR or partially maps to the proximal part of a non-enriched coding feature, it is subcategorized as a 5′ UTR-RNA. Reported 5′ ends of intraRNA, igRNAs and asRNAs were manually determined, while the 3′ ends correspond to the last base of the most distal interval.
Gene Ontology enrichment analyses
Gene Ontology (GO) term analysis for 355 genes was performed using FuncAssociate 2.0.Citation25 The gene list was compiled of probable sRNA targets; 297 mRNAs, 4 intra- 5′ UTR-RNAs and 54 ig- 5′ UTR-RNAs. As a background set (gene space) all genes from our annotation set were used. Significantly enriched GO terms were identified by applying an adjusted p-value cutoff of 0.05.
Northern blots
For Northern blot analyses 20 μg of total RNA and 1 μg of co-immunoprecipitated RNA (unless otherwise stated) treated with DNase I (Roche) was separated under denaturing conditions either by a 8% polyacrylamide / 8 M urea / TBE gels in 1X TBE (small transcripts) or a 1% formaldehyde/MOPS agarose gels in 1X MOPS (large lrhA transcript). RNA was denatured in 2X RNA load dye (Fermentas) and heated to 65 °C for 15 min before loading on a gel. RNA was transferred to HybondXL membranes (Ambion) either by electro-blotting at 12 V for 1 h in 0.5X TBE (PAG) or capillary action (formaldehyde-agarose gels). The membranes were UV cross-linked (150 mJ/cm2) and probed with DNA oligonucleotide probes (Table S11) in OligoHyb buffer (Ambion) per the manufacturer’s protocol. DNA oligonucleotide probes were end-labeled with [γ-32P] ATP and T4 PNK (New England Biolabs) per the manufacturer’s protocol.
Data access
Data deposition: Sequences have been deposited at the National Center for Biotechnology Information Sequence Read Archive (Study accession number SRP039345: experiment accession numbers SRX480475, SRX480476, SRX480477, and SRX480478).
| Abbreviations: | ||
| asRNAs | = | antisense RNAs |
| intraRNAs | = | intragenic RNAs |
| sRNAs | = | small RNAs |
| igRNAs | = | intergenic RNAs |
Additional material
Download Zip (2.1 MB)Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank members of the Schroeder laboratory for thoughtful and critical readings of the manuscript, Maarja Lepamets for implementing the peak-calling algorithm, and Bojan Vilagos for assistance with graphical design in the manuscript preparation. We thank members of the Schroeder laboratory for useful discussion and Johanna Stranner for excellent technical assistance. This work was supported by the Austrian Science Fund FWF, grants no. I538 and F4301 to RS and the University of Vienna.
References
- Storz G, Vogel J, Wassarman KM. Regulation by small RNAs in bacteria: expanding frontiers. Mol Cell 2011; 43:880 - 91; http://dx.doi.org/10.1016/j.molcel.2011.08.022; PMID: 21925377
- Papenfort K, Vogel J. Multiple target regulation by small noncoding RNAs rewires gene expression at the post-transcriptional level. Res Microbiol 2009; 160:278 - 87; http://dx.doi.org/10.1016/j.resmic.2009.03.004; PMID: 19366629
- Chao Y, Papenfort K, Reinhardt R, Sharma CM, Vogel J. An atlas of Hfq-bound transcripts reveals 3′ UTRs as a genomic reservoir of regulatory small RNAs. EMBO J 2012; 31:4005 - 19; http://dx.doi.org/10.1038/emboj.2012.229; PMID: 22922465
- Lalaouna D, Simoneau-Roy M, Lafontaine D, Massé E. Regulatory RNAs and target mRNA decay in prokaryotes. Biochim Biophys Acta 2013; 1829:742 - 7; http://dx.doi.org/10.1016/j.bbagrm.2013.02.013; PMID: 23500183
- Schroeder R, Barta A, Semrad K. Strategies for RNA folding and assembly. Nat Rev Mol Cell Biol 2004; 5:908 - 19; http://dx.doi.org/10.1038/nrm1497; PMID: 15520810
- Wagner EGH. Cycling of RNAs on Hfq. RNA Biol 2013; 10:619 - 26; http://dx.doi.org/10.4161/rna.24044; PMID: 23466677
- Soper TJ, Woodson SA. The rpoS mRNA leader recruits Hfq to facilitate annealing with DsrA sRNA. RNA 2008; 14:1907 - 17; http://dx.doi.org/10.1261/rna.1110608; PMID: 18658123
- Salim NN, Feig AL. An upstream Hfq binding site in the fhlA mRNA leader region facilitates the OxyS-fhlA interaction. PLoS One 2010; 5:1 - 11; http://dx.doi.org/10.1371/journal.pone.0013028; PMID: 20927406
- Lorenz C, Gesell T, Zimmermann B, Schoeberl U, Bilusic I, Rajkowitsch L, Waldsich C, von Haeseler A, Schroeder R. Genomic SELEX for Hfq-binding RNAs identifies genomic aptamers predominantly in antisense transcripts. Nucleic Acids Res 2010; 38:3794 - 808; http://dx.doi.org/10.1093/nar/gkq032; PMID: 20348540
- Georg J, Hess WR. cis-antisense RNA, another level of gene regulation in bacteria. Microbiol Mol Biol Rev 2011; 75:286 - 300; http://dx.doi.org/10.1128/MMBR.00032-10; PMID: 21646430
- Dornenburg JE, Devita AM, Palumbo MJ, Wade JT. Widespread antisense transcription in Escherichia coli. MBio 2010; 1:1 - 4; http://dx.doi.org/10.1128/mBio.00024-10; PMID: 20689751
- Raghavan R, Sloan DB, Ochman H. Antisense transcription is pervasive but rarely conserved in enteric bacteria. MBio 2012; 3; http://dx.doi.org/10.1128/mBio.00156-12; PMID: 22872780
- Lybecker M, Zimmermann B, Bilusic I, Tukhtubaeva N, Schroeder R. The double-stranded transcriptome of Escherichia coli. Proc Natl Acad Sci U S A 2014; 111:3134 - 9; http://dx.doi.org/10.1073/pnas.1315974111; PMID: 24453212
- Christiansen JK, Nielsen JS, Ebersbach T, Valentin-Hansen P, Søgaard-Andersen L, Kallipolitis BH. Identification of small Hfq-binding RNAs in Listeria monocytogenes. RNA 2006; 12:1383 - 96; http://dx.doi.org/10.1261/rna.49706; PMID: 16682563
- Zhang A, Wassarman KM, Rosenow C, Tjaden BC, Storz G, Gottesman S. Global analysis of small RNA and mRNA targets of Hfq. Mol Microbiol 2003; 50:1111 - 24; http://dx.doi.org/10.1046/j.1365-2958.2003.03734.x; PMID: 14622403
- Sittka A, Lucchini S, Papenfort K, Sharma CM, Rolle K, Binnewies TT, Hinton JCD, Vogel J. Deep sequencing analysis of small noncoding RNA and mRNA targets of the global post-transcriptional regulator, Hfq. PLoS Genet 2008; 4:e1000163; http://dx.doi.org/10.1371/journal.pgen.1000163; PMID: 18725932
- Dambach M, Irnov I, Winkler WC. Association of RNAs with Bacillus subtilis Hfq. PLoS One 2013; 8:e55156; http://dx.doi.org/10.1371/journal.pone.0055156; PMID: 23457461
- Soper T, Mandin P, Majdalani N, Gottesman S, Woodson SA. Positive regulation by small RNAs and the role of Hfq. Proc Natl Acad Sci U S A 2010; 107:9602 - 7; http://dx.doi.org/10.1073/pnas.1004435107; PMID: 20457943
- Ikeda Y, Yagi M, Morita T, Aiba H. Hfq binding at RhlB-recognition region of RNase E is crucial for the rapid degradation of target mRNAs mediated by sRNAs in Escherichia coli. Mol Microbiol 2011; 79:419 - 32; http://dx.doi.org/10.1111/j.1365-2958.2010.07454.x; PMID: 21219461
- Le Derout J, Folichon M, Briani F, Dehò G, Régnier P, Hajnsdorf E. Hfq affects the length and the frequency of short oligo(A) tails at the 3′ end of Escherichia coli rpsO mRNAs. Nucleic Acids Res 2003; 31:4017 - 23; http://dx.doi.org/10.1093/nar/gkg456; PMID: 12853618
- Mohanty BK, Maples VF, Kushner SR. The Sm-like protein Hfq regulates polyadenylation dependent mRNA decay in Escherichia coli. Mol Microbiol 2004; 54:905 - 20; http://dx.doi.org/10.1111/j.1365-2958.2004.04337.x; PMID: 15522076
- Scheibe M, Bonin S, Hajnsdorf E, Betat H, Mörl M. Hfq stimulates the activity of the CCA-adding enzyme. BMC Mol Biol 2007; 8:92; http://dx.doi.org/10.1186/1471-2199-8-92; PMID: 17949481
- Sukhodolets MV, Garges S. Interaction of Escherichia coli RNA polymerase with the ribosomal protein S1 and the Sm-like ATPase Hfq. Biochemistry 2003; 42:8022 - 34; http://dx.doi.org/10.1021/bi020638i; PMID: 12834354
- Raghavan R, Groisman EA, Ochman H. Genome-wide detection of novel regulatory RNAs in E. coli. Genome Res 2011; 21:1487 - 97; http://dx.doi.org/10.1101/gr.119370.110; PMID: 21665928
- Berriz GF, Beaver JE, Cenik C, Tasan M, Roth FP. Next generation software for functional trend analysis. Bioinformatics 2009; 25:3043 - 4; http://dx.doi.org/10.1093/bioinformatics/btp498; PMID: 19717575
- Wassarman KM, Repoila F, Rosenow C, Storz G, Gottesman S. Identification of novel small RNAs using comparative genomics and microarrays. Genes Dev 2001; 15:1637 - 51; http://dx.doi.org/10.1101/gad.901001; PMID: 11445539
- Vogel J, Bartels V, Tang TH, Churakov G, Slagter-Jäger JG, Hüttenhofer A, Wagner EG. RNomics in Escherichia coli detects new sRNA species and indicates parallel transcriptional output in bacteria. Nucleic Acids Res 2003; 31:6435 - 43; http://dx.doi.org/10.1093/nar/gkg867; PMID: 14602901
- De Lay N, Schu DJ, Gottesman S. Bacterial small RNA-based negative regulation: Hfq and its accomplices. J Biol Chem 2013; 288:7996 - 8003; http://dx.doi.org/10.1074/jbc.R112.441386; PMID: 23362267
- Lasa I, Toledo-Arana A, Dobin A, Villanueva M, de los Mozos IR, Vergara-Irigaray M, Segura V, Fagegaltier D, Penadés JR, Valle J, et al. Genome-wide antisense transcription drives mRNA processing in bacteria. Proc Natl Acad Sci U S A 2011; 108:20172 - 7; http://dx.doi.org/10.1073/pnas.1113521108; PMID: 22123973
- Ross JA, Wardle SJ, Haniford DB. Tn10/IS10 transposition is downregulated at the level of transposase expression by the RNA-binding protein Hfq. Mol Microbiol 2010; 78:607 - 21; http://dx.doi.org/10.1111/j.1365-2958.2010.07359.x; PMID: 20815820
- Ross JA, Ellis MJ, Hossain S, Haniford DB. Hfq restructures RNA-IN and RNA-OUT and facilitates antisense pairing in the Tn10/IS10 system. RNA 2013; 19:670 - 84; http://dx.doi.org/10.1261/rna.037747.112; PMID: 23510801
- Opdyke JA, Kang JG, Storz G. GadY, a small-RNA regulator of acid response genes in Escherichia coli. J Bacteriol 2004; 186:6698 - 705; http://dx.doi.org/10.1128/JB.186.20.6698-6705.2004; PMID: 15466020
- Opdyke JA, Fozo EM, Hemm MR, Storz G. RNase III participates in GadY-dependent cleavage of the gadX-gadW mRNA. J Mol Biol 2011; 406:29 - 43; http://dx.doi.org/10.1016/j.jmb.2010.12.009; PMID: 21147125
- Thomason MK, Storz G. Bacterial antisense RNAs: how many are there, and what are they doing?. Annu Rev Genet 2010; 44:167 - 88; http://dx.doi.org/10.1146/annurev-genet-102209-163523; PMID: 20707673
- Singh SS, Singh N, Bonocora RP, Fitzgerald DM, Wade JT, Grainger DC. Widespread suppression of intragenic transcription initiation by H-NS. Genes Dev 2014; 28:214 - 9; http://dx.doi.org/10.1101/gad.234336.113; PMID: 24449106
- Papenfort K, Said N, Welsink T, Lucchini S, Hinton JCD, Vogel J. Specific and pleiotropic patterns of mRNA regulation by ArcZ, a conserved, Hfq-dependent small RNA. Mol Microbiol 2009; 74:139 - 58; http://dx.doi.org/10.1111/j.1365-2958.2009.06857.x; PMID: 19732340
- Davis BM, Waldor MK. RNase E-dependent processing stabilizes MicX, a Vibrio cholerae sRNA. Mol Microbiol 2007; 65:373 - 85; http://dx.doi.org/10.1111/j.1365-2958.2007.05796.x; PMID: 17590231
- Møller T, Franch T, Højrup P, Keene DR, Bächinger HP, Brennan RG, Valentin-Hansen P. Hfq: a bacterial Sm-like protein that mediates RNA-RNA interaction. Mol Cell 2002; 9:23 - 30; http://dx.doi.org/10.1016/S1097-2765(01)00436-1; PMID: 11804583
- Tétart F, Bouché JP. Regulation of the expression of the cell-cycle gene ftsZ by DicF antisense RNA. Division does not require a fixed number of FtsZ molecules. Mol Microbiol 1992; 6:615 - 20; http://dx.doi.org/10.1111/j.1365-2958.1992.tb01508.x; PMID: 1372677
- Faubladier M, Cam K, Bouché JP. Escherichia coli cell division inhibitor DicF-RNA of the dicB operon. Evidence for its generation in vivo by transcription termination and by RNase III and RNase E-dependent processing. J Mol Biol 1990; 212:461 - 71; http://dx.doi.org/10.1016/0022-2836(90)90325-G; PMID: 1691299
- Arraiano CM, Mauxion F, Viegas SC, Matos RG, Séraphin B. Intracellular ribonucleases involved in transcript processing and decay: precision tools for RNA. Biochim Biophys Acta 2013; 1829:491 - 513; http://dx.doi.org/10.1016/j.bbagrm.2013.03.009; PMID: 23545199
- Lee T, Feig AL. The RNA binding protein Hfq interacts specifically with tRNAs. RNA 2008; 14:514 - 23; http://dx.doi.org/10.1261/rna.531408; PMID: 18230766
- Salim NN, Faner MA, Philip JA, Feig AL. Requirement of upstream Hfq-binding (ARN)x elements in glmS and the Hfq C-terminal region for GlmS upregulation by sRNAs GlmZ and GlmY. Nucleic Acids Res 2012; 40:8021 - 32; http://dx.doi.org/10.1093/nar/gks392; PMID: 22661574
- Serganov A, Nudler E. A decade of riboswitches. Cell 2013; 152:17 - 24; http://dx.doi.org/10.1016/j.cell.2012.12.024; PMID: 23332744
- Loh E, Dussurget O, Gripenland J, Vaitkevicius K, Tiensuu T, Mandin P, Repoila F, Buchrieser C, Cossart P, Johansson J. A trans-acting riboswitch controls expression of the virulence regulator PrfA in Listeria monocytogenes. Cell 2009; 139:770 - 9; http://dx.doi.org/10.1016/j.cell.2009.08.046; PMID: 19914169
- Sedlazeck FJ, Rescheneder P, von Haeseler A. NextGenMap: fast and accurate read mapping in highly polymorphic genomes. Bioinformatics 2013; 29:2790 - 1; http://dx.doi.org/10.1093/bioinformatics/btt468; PMID: 23975764
- Kent WJ, Zweig AS, Barber G, Hinrichs AS, Karolchik D. BigWig and BigBed: enabling browsing of large distributed datasets. Bioinformatics 2010; 26:2204 - 7; http://dx.doi.org/10.1093/bioinformatics/btq351; PMID: 20639541
- Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010; 26:139 - 40; http://dx.doi.org/10.1093/bioinformatics/btp616; PMID: 19910308