
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
We report here an in-depth characterization of the aptamer domain of the transcriptional adenine-sensing riboswitch (pbuE) by NMR and fluorescence spectroscopy. By NMR studies, the structure of two aptamer sequences with different lengths of the helix P1, the central element involved in riboswitch conformational switching, was characterized. Hydrogen-bond interactions could be mapped at nucleotide resolution providing information about secondary and tertiary structure, structure homogeneity and dynamics. Our study reveals that the elongation of helix P1 has pronounced effects not only on the local but on the global structure of the apo aptamer domain. The structural differences induced by stabilizing helix P1 were found to be linked to changes of the ligand binding affinity as revealed from analysis of kinetic and thermodynamic data obtained from stopped-flow fluorescence studies. The results provide new insight into the sequence-dependent fine tuning of the structure and function of purine-sensing riboswitches.
Introduction
Riboswitches are regulatory RNA elements found in all three domains of life but predominantly in bacteria.Citation1,Citation2 Riboswitches are mostly located within the 5′-untranslated region (5′UTR) of mRNAs. Up to now, more than 25 riboswitch classes binding to different metabolites (coenzymes, nucleotide derivates, amino acids, and ions) have been identified.Citation3 They regulate either transcription or translation by stabilizing alternative mRNA structure in a ligand-dependent manner. In addition, further types of riboswitches have recently been identified that act as ribozymes or trans-acting riboswitches.Citation4,Citation5 Reorganization on the secondary and/or tertiary structure level upon ligand binding transmits an ON- or an OFF-signal for further gene transcription or translation. The so-called aptamer domain of a riboswitch contains the ligand binding pocket and often adopts a sophisticated three-dimensional structure able to bind ligands with high specificity and affinity. The mode of RNA binding to ligands is well understood from crystal structures of numerous RNA-ligand complexes.Citation6,Citation7 Further details of the binding mode have been revealed in structure-activity-relationship analyses utilizing chemical derivatives of cognate ligands together with RNA mutational studies.Citation8 In addition, several NMR-spectroscopic investigations have added to the current understanding of how the ligand is binding to and recognized by its respective riboswitch.Citation9-Citation14 Recent biophysical studies have also addressed the structural features of ligand-free aptamer domains from different riboswitch classes to contribute to our understanding of their complex folding pathways.Citation11,Citation12,Citation15,Citation16 Central structural rearrangements involve the switching sequence that is either part of the aptamer domain or the expression platform (). Both conformations of the switching sequence are mutually exclusive, and this mutual exclusion provides the basis of the switch mechanism.
Figure 1. Sequence and secondary structure of purine-sensing riboswitches. (A) Secondary structure of the full-length pbuE adenine-sensing riboswitch without adenine (OFF-state) and of the aptamer domain with bound adenine (ON-state). The switching sequence in the aptamer domain is shaded in gray. Nucleotides in red are conserved in greater than 90% of the known examples for this riboswitch class. (B) P1long aptamer domain with 4 additional GC base pairs within helix P1. (C) Surface presentation of the X-ray structure of the aptamer domain of the add adenine riboswitch.Citation21 Helices P1, P2, P3 and the binding pocket are distinguished by different colors using the same coloring schema as in D. (D) Alignment of five representative sequences of mRNA domains that conform with the purine riboswitch consensus sequence. Organism’s abbreviations: Bacillus subtilis (BS), Clostridium perfringens (CP), Vibrio vulnificus (VV). Specificity for binding of either adenine or guanine is established by WC base pairing to the riboswitch nucleotide at position 74 (U for adenine, C for guanine). Regions with colored background represent helices P1, P2, and P3. Junctions connecting helices are notated J1–2, J2–3, and J3–1. Nucleotides in red are conserved in greater than 90% of the known examples for this riboswitch class.
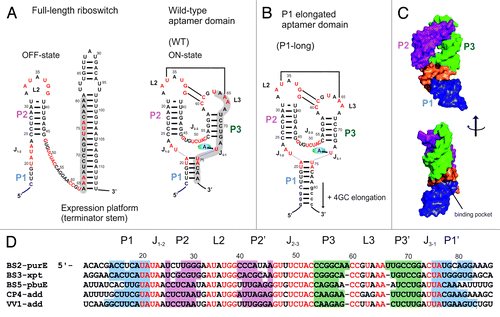
Inspection of the X-ray structures of purine-binding riboswitch aptamer domains shows that these riboswitches bury on average 90% of the accessible surface of their effector moleculesCitation7 (e.g., adenine binding aptamer domain, ). X-ray structure determination of ligand-free aptamer domains has been successful only for a few riboswitches. Yet, crystal packing apparently stabilizes only one conformation from the ensemble of conformers accessible to the ligand-free aptamer in solution.Citation17,Citation18 Consequently, in these cases crystallization of the ligand-bound aptamer structure utilizing soaking approaches has been hampered by the near inaccessibility of the binding pocket in the bound states.Citation17,Citation18
From NMR-spectroscopic investigations, analysis by small angle X-ray scattering and in-line probing studies, it is shown that riboswitches can be subdivided into two groups: One group of riboswitches experience only local structural changes while the second group undergoes global structural rearrangement upon ligand binding.Citation6,Citation19,Citation20
The purine-sensing riboswitches have been particularly well studied in the past due to their relatively small size. Their aptamer domains adopt a complex three-dimensional structure and exhibit high diversity in function and ligand specificity.Citation21 They comprise both off- as well as on-switches. For example, the add adenine riboswitchCitation22 containing a translation repressor stem in the free structure has been reported to regulate translation while the pbuE adenine riboswitch investigated here regulates transcription and forms a transcription terminator in the absence of ligand. The secondary structure of the conserved aptamer domain of the purine-sensing riboswitches is characterized by a three-way-junction (). In the apo state, helices P2 and P3 are fully or partially preformed while the binding pocket is rather flexible.Citation10,Citation12,Citation13,Citation23 Only when ligand is bound, nucleotides of the binding pocket adopt a persistent structure characterized by an intricate network of hydrogen bonds and stacking interactions.Citation14,Citation21 Long-range interactions between loops L2 and L3 stabilize a homogenous global fold. In several studies it has been shown that the ability of these loops to interact is a prerequisite for ligand binding because tertiary interactions are essential for structural pre-organization of the binding competent aptamer domain.Citation10,Citation24 In addition, recent studies performed in our lab revealed that increased flexibility of the apo-state conformational ensemble compared with holo state can be partially compensated by excess of Mg2+.Citation10,Citation24
Reports regarding the structure of helix P1 in the apo state have been controversial as outlined in the review article by Garst et al., 2010.Citation25 Analyses of different guanine- and adenine-sensing riboswitches by SHAPE (selective 2′-hydroxyl acylation analyzed by primer extension), smFRET (single molecule FRET) and NMR spectroscopy confirm that the helix P1 is stable in the apo state and becomes further stabilized by ligand binding and/or low temperature.Citation12,Citation13,Citation23,Citation24,Citation26 By contrast, results from single molecule force microscopy by Greenleaf et al., 2008 suggest that in the apo state of pbuE adenine riboswitch helix P1 is not stable but becomes persistently formed only after ligand binding.Citation27 These results seem surprising since helix P1 is part of the regulatory switching sequence and participates in shaping of the binding pocket. Indeed, part of the reason for these different results is the fact that different riboswitch sequences have been investigated. Though adenine and guanine-sensing riboswitches share the same global fold, the sequence of helix P1 naturally differs substantially in length and/or base pair composition leading to predicted differences in base pairing patterns. Especially, differences in length of the helix P1 arise due to either different riboswitches regulating different genes in different organisms () or from additional base pairs that have been introduced for the purpose of efficient in vitro synthesis.
In this contribution, we therefore investigate in detail the effect of the length of helix P1 on the free aptamer structure and the ligand-induced RNA folding pathway. The apo and holo structures of two constructs of the pbuE adenine-sensing riboswitch with different length of helix P1 (; wild-type and P1-elongated aptamer domains) have been investigated by NMR spectroscopy. In addition, ligand binding kinetics and affinity have been studied by stopped-flow fluorescence spectroscopy. Results show that differences exist in terms of structure in the apo state as well as in terms of binding affinity whereas the structure in the holo state is retained.
Results
NMR resonance assignment and structural model
Formation of canonical and non-canonical base pairs has major impact on secondary and tertiary structure of RNA. Imino protons act as hydrogen-bond donors and become protected from bulk solvent exchange if they are involved in stable base pair formation. Only in this case, their NMR signals appear in spectra with characteristic proton chemical shifts of 10–15 ppm. Moreover, imino protons of adjacent base pairs give rise to NOE cross-peaks in 2D-1H,1H-NOESY spectra that can be used for assignment and analysis of long-range interactions. Direct monitoring of imino protons in persistent hydrogen-bonding interactions in RNA is a particular strength of NMR spectroscopy providing information about base pairing conformers, structure homogeneity and dynamics with nucleotide specific resolution.
P1-long aptamer domain with elongated helix P1: + Mg2+ + adenine
We first discuss the NMR characterization of an aptamer domain of the pbuE adenine riboswitch that has been stabilized by four additional GC base pairs within helix P1 (P1-long, )—a situation that closely resembles natively occurring riboswitches with extended P1 stems such as the add riboswitch from Vibrio vulnificus with a seven base pair P1 (). The solution structure of this adenine-sensing aptamer domain with stabilized helix P1 has been characterized previously in our group.Citation26 The imino region of the 1H,15N-HSQC of the aptamer domain shows well-resolved and sharp signals in the presence of 1.5 eq adenine and 5 mM Mg2+ (). NMR resonance assignment unambiguously shows formation of a homogeneous and compact structure, which is in agreement with the X-ray structure of the homologous add adenine riboswitch with the same architecture (for sequence see ).Citation21 Helices P1, P2, and P3 are completely formed, while only terminal residues G13 and G14 are not visible, likely due to the reduced stability and the resulting fast exchange of imino protons with the bulk water solvent. Imino signals within the binding pocket (G46, U47, U49, U51, U74) can be detected. Surprisingly, the imino signal of residue U22 that is involved in formation of the binding pocket is only visible in the ligand-bound state when Mg2+ is not present. This observation could be in principle due to loss of stable hydrogen bonding but more likely due to chemical exchange e. g. caused by transient Mg2+-binding that renders the NMR signal undetectable. As expected from the X-ray structure of a homologous adenine riboswitch, loop-loop interactions are formed as evidenced by distinct chemical shifts of imino signals for U34, G37, and G38 involved in base quartet formation with C64 and C65 of loop L3. There are only two weak unassigned signals that do not show any NOE cross peaks in the 1H,1H-NOESY-spectrum.
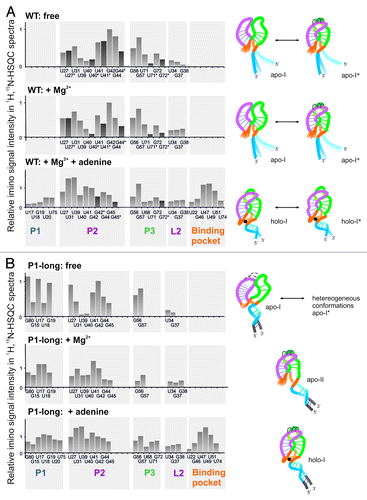
Figure 2. Mg2+ and adenine binding to the P1-long aptamer domain ([RNA] = 0.5 mM). Imino regions of the 2D- 1H,15N-HSQC spectra (left) and 2D-1H,1H-NOESY spectra with 200 ms mixing time (right) measured at 900 MHz and 283 K. Color code for assignments is adapted to the presentation of the secondary structures in D. (A) Overlay of the imino regions with Mg2+ (5 mM) and/or adenine (1.5 eq). Inset shows the 1D projection of the imino signal of the residue U22. (B) RNA with Mg2+ (5 mM). (C) Free RNA. (D) Model for secondary structures of the aptamer domain according to assignments in A, B, and C. Base pairs with assigned imino proton are indicated by a solid line. Dashed lines in apo-I state indicate less table base pairs according to the lower signal intensities of loop residues. Adenine is depicted with a red circle.
![Figure 2. Mg2+ and adenine binding to the P1-long aptamer domain ([RNA] = 0.5 mM). Imino regions of the 2D- 1H,15N-HSQC spectra (left) and 2D-1H,1H-NOESY spectra with 200 ms mixing time (right) measured at 900 MHz and 283 K. Color code for assignments is adapted to the presentation of the secondary structures in D. (A) Overlay of the imino regions with Mg2+ (5 mM) and/or adenine (1.5 eq). Inset shows the 1D projection of the imino signal of the residue U22. (B) RNA with Mg2+ (5 mM). (C) Free RNA. (D) Model for secondary structures of the aptamer domain according to assignments in A, B, and C. Base pairs with assigned imino proton are indicated by a solid line. Dashed lines in apo-I state indicate less table base pairs according to the lower signal intensities of loop residues. Adenine is depicted with a red circle.](/cms/asset/051c3188-135d-4895-9c1e-5c2f79099b57/krnb_a_10929439_f0002.gif)
P1-long aptamer domain with elongated helix P1: + Mg2+
The 1H,15N-HSQC spectrum of the ligand-free P1-long aptamer domain in excess of Mg2+ shows a considerably reduced number of imino signals (). The signals are broad and vary significantly in their intensities. Such line shapes indicate that the structure is more flexible adopting different conformations, which interconvert with exchange rates of the order of µs to ms and thus give rise to an averaged signal on the NMR time scale. A second set of NMR signals that would indicate a second persistent conformation cannot be detected. The signals for U20, U22, G45, G46, U47, U49, U51, U71, G72, U74, U75 located in the ligand-binding core and at the ends of the helices facing the core region are completely absent. Interestingly, only two base pairs are visible in helix P3 (G56, G57). In contrast, the isolated helix P3 displays four imino signals (G57, G56, U71, and G72) (Fig. S1) in the same experiment and under the same conditions. The disappearance of imino signals of helix P3 in the P1-long aptamer domain might be due to the destabilization of base pairs or due to induced conformational flexibility of the helix that reduces the intensity of NMR signals below detectability. Three imino signals of U34, G37, and G38 indicate the presence of stable loop-loop interactions. There are three remaining signals that cannot be assigned due to missing NOE cross-peaks.
P1-long aptamer domain with elongated P1-helix: free
As discussed in our previous work, absence of both ligand and Mg2+ leads to destabilization of the global fold that results in the formation of alternative conformers.Citation26 In the absence of both ligand and Mg2+, the 1H, 15N-HSQC-spectrum shows several new peaks that differ in intensity and line shape. The RNA structure is heterogeneous and only a single conformation or only structural elements common to all members of the ensemble can be assigned unambiguously. This main conformation (apo-I) is characterized by the formation of the predicted helices P1, P2, and P3. Helix P1 is formed with five base pairs whose imino proton signals show clear NOE-based connectivities (G19, U18, U17, G80, G15). Helix P2 is less stable lacking a stable U31-U39 terminal base pair. In particular, the peak intensity of U39 is significantly reduced and the signal can hardly be detected. Signals for U40 and U41 are present in the 1H,15N-HSQC-spectrum but lack cross peaks in a 1H,1H-NOESY-spectrum supporting the notion that helix P2 is destabilized in the apical region in absence of Mg2+. By contrast, signals for G42, U27, G44, and G45 can be clearly assigned in NOESY-spectra. For helix P3, still only two base pairs can be detected (G56-C69, G57-C70). A large number of imino signals remain unassigned revealing prominent heterogeneity of the RNA structure.
Mg2+-induced changes to the ligand-free P1-long RNA
Analysis of the chemical shift perturbation upon Mg2+-titration reveals distinct responses of the various conformers present in the absence of Mg2+ (). Imino signals of the main conformation in the apo state (apo-I) keep their positions but become more intense and narrow. Addition of Mg2+ stabilizes tertiary contacts between helix P2 and P3 by stabilizing the loop-loop interactions between loops L2 and L3. Due to Mg2+-induced stabilization, the imino signal of residue G37 becomes more intense and continuously shifts toward the Mg2+-bound conformer (apo-II). This is indicative for fast chemical exchange at time scales faster than µs. In contrast, other unassigned peaks (apo-I*) mainly disappear upon Mg2+-titration without strong chemical shift changes. This behavior can be associated with slow chemical exchange or an intermediate state without hydrogen bonds.
Figure 3. Mg2+-induced changes of the P1-long aptamer domain ([RNA] = 0.5 mM): Overlay of imino regions of 2D- 1H,15N-HSQC spectra with different Mg2+-concentrations (0 to 5 mM). Insets show expansions of different imino signals. Signals labeled with a star (*) belong to heterogeneous conformations that are not assigned (apo-I*) and disappear upon addition of Mg2+ without chemical shift changes. Assigned signals (e.g., G37, U31) from the apo-I conformation shift to the position of signals corresponding to the apo-II conformation as indicated by an arrow.
![Figure 3. Mg2+-induced changes of the P1-long aptamer domain ([RNA] = 0.5 mM): Overlay of imino regions of 2D- 1H,15N-HSQC spectra with different Mg2+-concentrations (0 to 5 mM). Insets show expansions of different imino signals. Signals labeled with a star (*) belong to heterogeneous conformations that are not assigned (apo-I*) and disappear upon addition of Mg2+ without chemical shift changes. Assigned signals (e.g., G37, U31) from the apo-I conformation shift to the position of signals corresponding to the apo-II conformation as indicated by an arrow.](/cms/asset/bbc5c550-71b7-4988-a301-e815b235db5a/krnb_a_10929439_f0003.gif)
Wild-type (aptamer domain with native P1 helix): + Mg2+ + adenine
We now discuss the NMR characterization of the wild-type aptamer domain of the pbuE adenine riboswitch with the native helix P1 (WT, ), which contains five predicted base pairs. The 1H,15N-HSQC spectrum in the presence of Mg2+ and adenine reveals that the aptamer domain is completely folded including the binding pocket (U22, G46, U47, U49, U51, U74) and stable loop-loop interactions (U34, G37, G38). In contrast to the P1-long discussed above (), the imino signal of residue U22 is visible in the presence of both adenine and Mg2+ and shows strong NOE cross-peaks to residue U47. Helix P2 and P3 are completely formed, while only part of helix P1 can be detected by three unambiguously assigned imino protons (G19, U20, U75). The assignment of the imino signals of U18 and U17 is based on chemical shift comparison with the 1H, 15N-spectrum of a P1-helix long but here both signals lack NOE cross-peaks. Moreover, we detect a second minor conformation visible for residues G42*, G45*, and G72*. From the significant chemical shift difference for the two signals corresponding to residue G72 the second conformation (G72*) likely represents a conformation involved in different stacking interactions between G72 and A24 that is involved in formation of the binding pocket. Such structural heterogeneity of the ligand-bound complex observed here by NMR spectroscopy is in line with results obtained by measuring 2-AP fluorescence life times in a previous study.Citation28 The data in this study indicate that in a second conformer of the ligand-bound aptamer domain 2-AP experiences less stacking interactions within the binding pocket and therefore the fluorescence signal is changed.
Figure 4. Mg2+ and adenine binding to the wild-type aptamer domain ([RNA] = 0.5 mM). Imino regions of the 2D- 1H,15N-HSQC spectra (left) and 2D-1H,1H-NOESY spectra with 200 ms mixing time (right) measured at 900 MHz and 283 K. NOE connectivities highlighted by solid lines indicate the major conformation, dashed lines (and *) indicate the minor conformation. The color code used for visualizing the assignments is also adopted in the presentation of the secondary structure in D. (A) RNA with Mg2+ (5 mM) and adenine (1.5 eq). (B) RNA with Mg2+ (5 mM). (C) Free RNA. (D) Model for secondary structures of the aptamer domain according to assignments in A, B and C. Base pairs with assigned imino protons are indicated by solid line. Adenine is depicted with a red circle.
![Figure 4. Mg2+ and adenine binding to the wild-type aptamer domain ([RNA] = 0.5 mM). Imino regions of the 2D- 1H,15N-HSQC spectra (left) and 2D-1H,1H-NOESY spectra with 200 ms mixing time (right) measured at 900 MHz and 283 K. NOE connectivities highlighted by solid lines indicate the major conformation, dashed lines (and *) indicate the minor conformation. The color code used for visualizing the assignments is also adopted in the presentation of the secondary structure in D. (A) RNA with Mg2+ (5 mM) and adenine (1.5 eq). (B) RNA with Mg2+ (5 mM). (C) Free RNA. (D) Model for secondary structures of the aptamer domain according to assignments in A, B and C. Base pairs with assigned imino protons are indicated by solid line. Adenine is depicted with a red circle.](/cms/asset/22c54e66-4bf7-4771-85d8-048ea0c8182e/krnb_a_10929439_f0004.gif)
Wild type: + Mg2+
The 1H,15N-HSQC spectrum of ligand-free wild-type aptamer domain with Mg2+ shows broad signals and many signals that appear to be double peaks (). Indeed, careful assignment reveals that the apo structure of the wild-type construct adopts at least two conformations. Since separate signal sets can be detected for the two major conformations, they interconvert with rates of the order of ms to s, i.e., they are in slow exchange. Temperature-dependent changes of the equilibrium indicate slow exchange (Fig. S2). In the major conformer (apo-I), helix P2 is stably formed and can be detected by NMR through imino signals of U27, U39, U40, U41, G42, G44, and G45. The second conformation (apo-I*) for helix P2 shows the same number of base pairs but imino signals appear less intense. The largest difference in intensity is detected for the U40 signal: in the minor conformation, the imino proton of U40 shows no NOE cross-peaks to the imino proton signals of the U31-U39 base pair. The less intense U40 signal and the loss of the NOE cross-peaks to the loop closing base pair U31-U39 indicate a weakening of the loop-loop interactions between L2 and L3. Assignment of the imino signal of residue U40* is based on comparison of its NOE cross-peaks to other protons (2–9 ppm) with NOE cross-peaks of the residue U40 in the major conformation (data not shown).
For helix P3, imino signals for four base pairs (G56, G57, U71, G72) can be assigned. Interestingly, this observation implies that the P3-helix in the wild-type aptamer domain is more stable than in the P1-long (). Residues G72 and U71 show two well-resolved peaks (; G72/G72*, U71/U71*) while G56 and G57 give rise to broadened single peaks. No signals from helix P1 can be assigned based on NOE cross-peaks.
Wild-type: free
In the absence of Mg2+, NMR spectra show peaks at the same position compared with the spectra in the presence of 5 mM Mg2+, but the intensities of different peaks are modulated. In the absence of Mg2+, helix P2 in the major conformation is less stabilized and the imino signals of the non-canonical U31-U39 base pair are less intense. Obviously, Mg2+ stabilizes the compact structure where loop-loop interactions are established but it has no effect on formation of helix P1.
Our data show that the wild-type aptamer domain undergoes defined dynamics driven by the slow process of opening and closing of the loop-loop interactions. The equilibrium between the closed (apo-I) and open (apo-I*) conformation is Mg2+ and temperature dependent. At 20 °C, the equilibrium is shifted to the conformation without tertiary contacts as indicated by changes in signal intensities (Fig. S2).
Influence of the length of helix P1 on the aptamer structure
The assignment of 1H,15N-HSQC-spectra of the two constructs allows us to identify secondary and tertiary structural elements of the aptamer domain. shows the intensities of individual imino proton signals in 1H,15N-HSQC-spectra grouped into structural regions (helices P1, P2, and P3, binding pocket and loop-loop interactions [L2]).
Figure 5. Intensity of imino protons of wild-type (A) and P1-long (B) RNA in 1H, 15N-HSQC spectra ( and ) in the free state (top), with Mg2+ (middle), with Mg2+ and/or adenine (bottom). Assignments with and without star (*) indicate two different conformations of the same residue; the intensity of the second peak is indicated by dark gray bar. Signal intensities are normalized to the intensity of the residue G42 in each spectrum. Imino signals are grouped into structural elements (helices P1, P2, P3, binding pocket, loop L2) and translated into structural models on the right side. The color code of structural elements is the same as in .
For illustrative purposes we translate our data in a structural cartoon long that utilizes the X-ray structure of the add riboswitch ().
From the comparison of both constructs in the apo form, it can be concluded that the length and stability of helix P1 has a previously unobserved effect on the global apo structure of the aptamer domain and not only on structural elements in the immediate vicinity of P1. In the P1-long, elongation of helix P1 destabilizes helices P2, P3, as well as the loop-loop interactions. Apparently, formation of helix P1 globally destabilizes the entire structure which is counterintuitive at the first view. However, inspection of the X-ray structure and information from molecular dynamics simulation of the add riboswitch provide some support for this observation.Citation29,Citation30 MD simulations have been conducted starting from the X-ray structure with adenine and after removal of adenine. During the course of the MD simulation after removal of the ligand, the binding pocket and helices P1 and P3 become destabilized; in particular, P1 unwinds. Our NMR data confirm this destabilization and show that for the pbuE wild-type aptamer domain removal of adenine leads to complete melting of helix P1. This result agrees with the conclusion from single molecule force microscopy.Citation27
Influence of the length of helix P1 on thermal stability and ligand binding affinity
The thermal stabilities of wild-type and P1-long aptamer domains have been investigated with CD spectroscopy measuring temperature-dependent unfolding and folding curves under different conditions (free, with adenine and/or Mg2+). Extracted melting temperatures are summarized in and show that the P1-long is more stable than the wild-type aptamer domain under all conditions as expected due to the addition of four GC base pairs. The maximal difference in stability of 9 °C is found in the ligand-bound state in the presence of Mg2+, and the overall thermal stability is strongly influenced by the presence of Mg2+. Addition of adenine in the absence of Mg2+ does not change the melting temperatures for both constructs. In contrast, the cooperativity of the melting process is only changed for the P1-long aptamer domain (Fig. S3); we conclude that ligand release takes place prior to global thermal melting induced by local structural changes.
Figure 6. Thermal stability and binding kinetics of the wild-type and P1-long aptamer domains. (A) Melting point (Tm) derived from the first derivative of the melting curve measured with CD spectroscopy at different conditions. (B) Ligand dissociation constants, association and dissociation rate constants at different temperatures obtained from fluorescence spectroscopy with 2-AP (50 nM) in the presence of 4mM Mg2+.
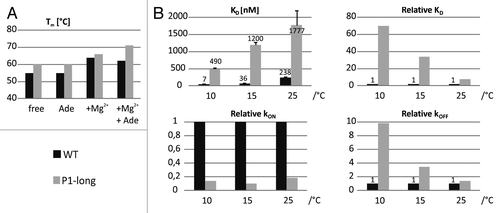
Our NMR analysis reveals unexpected differences between the wild-type and the P1-long aptamer domains, raising the question whether such differences have an effect on ligand binding behavior. In order to determine the ligand binding affinity, we utilized fluorescence spectroscopy and 2-AP as a fluorescent adenine analog in the presence of Mg2+ following previous work by Wickiser et al. in 2005.Citation31 Association and dissociations rate constants (kON, kOFF) have been determined at different RNA concentrations in stopped-flow fluorescence-detected experiments allowing calculation of the ligand dissociation constant (KD) and differences in the free energy of the ligand binding reaction (). Stopped-flow experiments have been measured first at the same temperature (10 °C) as NMR experiments have been conducted and second at two higher temperatures (15 °C, 25 °C). Data are summarized in and .
Table 1. Kinetic and thermodynamic data for 2-AP binding to the adenine-sensing aptamer domain obtained by fluorescence spectroscopy
As it can be seen from KD values there is the expected trend that ligand affinity is decreased at higher temperatures for both constructs mainly because the dissociation rate constants are markedly increased at higher temperatures (). Much more unexpected differences are revealed by the comparison between the wild-type and P1-long aptamer domains. A very strong effect of the length of helix P1 on complex stability is observed. The binding affinity of the P1-long aptamer domain is weaker for all three temperatures while the destabilizing effect of the elongated helix P1 is most pronounced at low temperature where the KD differs by a factor of 70. Changes of both association and dissociation rates due to the stabilization of helix P1 contribute to the lower affinity of the P1-long RNA. Interestingly, relative differences in dissociation rates are much more temperature-dependent than association rates. In particular, at lower temperature the dissociation rate of the P1-long aptamer domain is 10-fold faster than for the wild-type.
Discussion
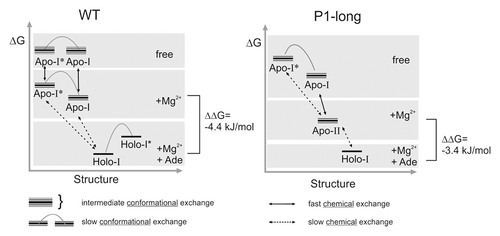
In the present study, we used NMR and fluorescence spectroscopies to investigate structure, the conformational dynamics and the binding affinities of the aptamer domain of the pbuE adenine-sensing riboswitch with different lengths of the helix P1. In transcriptional riboswitches, the genetic decision occurs right after the binding-competent aptamer domain has been transcribed. In the absence of ligand, the full-length riboswitch is trapped in the off-state that is not able to bind adenine (). It is therefore reasonable to consider only the aptamer domain for the investigation of binding affinities. In the wild-type construct, a sequence with the native base pairing pattern of the aptamer domain was investigated. For this construct, in the ligand-free state at 10 °C, the aptamer domain is formed with helices P2 and P3 adopting two conformations interconverting in the slow time regime (ms-s) ( and ). The two conformations differ in the extent of pre-formation of the loop-loop interactions but both lack detectable signals for helix P1. Addition of Mg2+ shifts the equilibrium toward a structure with loop-loop interactions but does not stabilize helix P1. By contrast, at higher temperature of 20 °C when the loop-loop interactions are weakened it is possible to assign imino signals for G19, U18, and U17 based on NOE cross-peaks (Fig. S2). This observation gives further evidence for the competition between loop-loop interactions, helices P2 and P3 on the one side and helix P1 on the other.
In contrast, in the P1-long aptamer domain the formation of helix P1 is enforced by adding four GC base pairs. Such elongation of helix P1 mimics the naturally occurring diversity within different riboswitches (). For instance, the add adenine riboswitch comprises a helix P1 with seven base pairs while the helix P1 of the pbuE adenine riboswitch forms only five base pairs (). Indeed, the free state of the P1-long is characterized by formation of a three-way-junction including helices P1, P2, and P3 ( and ). However, in this case, helices P2, P3, and native loop-loop interactions are destabilized compared with the wild-type construct especially in absence of Mg2+. Several imino signals corresponding to alternative base pairs of heterogeneous conformers are present but cannot be clearly assigned. However, these signals disappear when Mg2+ is added while the folded structure with stable helices P2 and native loop-loop interactions is strongly stabilized leading to narrower imino proton signals. Addition of Mg2+ recovers native loop-loop interactions but still helix P3 remains destabilized with only two detectable base pairs.
Comparison of ligand-bound states in the presence of Mg2+ reveals a very good agreement between imino spectra of wild-type and P1-long aptamer domains. Nevertheless, a small difference is observed for residue U22 of the binding pocket. In the P1-long aptamer domain, the imino signal of U22 is not visible in presence of Mg2+. Such difference might be important because residue U22 forms part of the binding pocket and is involved in the formation of a base triple with A52 and A73 that is stacked on the bound ligand. This observation is indicating that global stabilization of the structure by divalent cations has some effect on the local geometry of the binding pocket only when helix P1 is elongated.
From stopped-flow fluorescence experiments we can conclude that observed structural differences between the apo and holo states of wild-type and P1-long RNAs lead to considerable differences in the binding affinity for 2-AP. On the one hand, the thermal stability is increased by elongation of helix P1 but on the other hand the thermodynamic stability of the RNA-ligand complex is in fact reduced. Dissection of kinetic data at different temperatures reveals that both reduced association and increased dissociation rates contribute to the reduced binding affinity of the P1-long aptamer domain. One possible explanation for the reduced association rate is the steric effect occurring when helix P1 with additional GC base pairs is formed in the free state and therefore hinders the entry of the ligand to the binding pocket. Moreover, in the wild-type aptamer domain helix P2 and P3 are more stable which might be beneficial for better pre-organization of the binding pocket. The fact that elongation of helix P1 has a negative effect on complex stability and thus increases the disassociation rate constants is rather unexpected. However, especially at low temperature (10 °C) large differences in dissociation rates clearly indicate that also subtle structural differences observed by NMR analysis correspond to pronounced effects on complex stability. One possible explanation might be that the interhelical twist angle between P2 and P3 in the free state is altered by the presence of a stable P1 stem induced by the additional GC base pairs. This subtle conformational change might then be transmitted to the binding pocket leading to geometry changes. Such small structural changes nevertheless might be sufficient to affect binding affinity. Removal of Mg2+, in contrast, weakens helix stability and thereby reduces shearing forces which seem to arise from the stabilized helix P1.
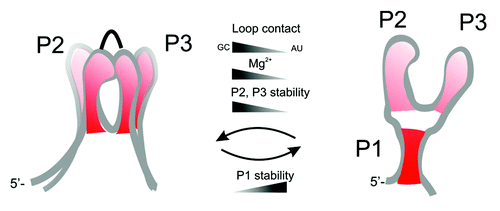
Here, for the first time it is shown, that in addition to nucleotide mutations in the loop regions and changes in the Mg2+ concentration, also changes in stability of helix P1 can have a similar effect on ligand binding affinity. This points toward very balanced and long-range effects of local structural changes in the aptamer domain of purine-sensing riboswitches. These observed coupled effects, Mg2+ concentration, loop-loop interactions and stability of helix P1, induce different packing of the three-way-junction which seems to be crucial for appropriate organization of the binding pocket (). In the biological context, where loop-loop interactions are maintained by conserved nucleotides and Mg2+ concentration is not coupled to the regulation requirements, altering the stability of helices might be the only way how different thermodynamic and kinetic features can be realized in order to accomplish different requirements for efficient kinetic control of transcription and for discrimination between chemically similar ligands. Local changes in the helix-helix interactions influence the packing of the aptamer domain in a long-range manner.
Figure 7. Global packing of the helices and organization of the aptamer domain. Helix-helix interactions are determined by different factors that influence stability of helices P1, P2, and P3.
The comparison of both constructs provides furthermore new insight into the folding pathways of the aptamer domain during transcription. It is safe to assume that during the vectorial process of transcription helices P2 and P3 are formed first, which can align side-by-side via stable loop-loop interactions. In our previous work it has been shown that loop-loop interactions are even stable in shortened constructs that only comprise helices P2 and P3.Citation13 As transcription proceeds, helix P1 can be formed depending on the sequence of the riboswitch and stabilities of helices P1, P2, and P3. In the case of the pbuE adenine switch, helix P1 is short and remains open in the ligand-free state and can only be formed when adenine is added. This agrees with a finding by single molecule force microscopy by Greenleaf et al., 2008.Citation27 For purine-sensing riboswitches that have a longer or more stable helix P1 (P1-long, xpt guanine riboswitch), the folding pathway is likely different. In this case, after transcription of the full aptamer domain, helix P1 is formed while at the same time helices P2, P3 and loop-loop interactions are destabilized. This has also been observed for the 2’deoxyguanosine riboswitch that has a stable helix P1.Citation9 By contrast, for the xpt guanine riboswitch with stable helix P1 but also GC-rich helices P2 and P3 () it has been shown that helices P2, P3 and loop-loop interactions are stable in the free state as well.Citation10,Citation12 We conclude that strong unwinding of helices P2 and P3 induced by the tightening of helix P1 can be compensated by more stable base pairing in P2 and P3 of the xpt-riboswitch.
In summary, we show that competition between different structural elements within the aptamer domain of the adenine riboswitch does take place. Mutual interference of structural elements is dependent on the stability of individual helices, which is determined by sequence. Our first conclusion is that the stability of individual helices might adjust the balance of three-way-junction packing, enable different degrees of pre-organization, and fine-tune kinetic and thermodynamic features (), which are necessary for gene regulatory mechanisms. Second, we propose from our findings that competition and rearrangement between structural elements in the free state is responsible to prevent the aptamer domain from being trapped into bound-state like structures that are not accessible to the ligand. These findings will be of significant importance for the development of riboswitch aptamer modules as regulation units in synthetic biology applications.
Figure 8. Schematic energy-structure diagram for Mg2+ and adenine dependent equilibria of the wild-type (left) and P1-long (right) aptamer domains.
Materials and Methods
Synthesis of labeled adenine
The 15N-labeled adenine was synthesized as described previously.Citation14 The concentration of the labeled adenine was calculated using UV absorbance at 261 nm and an extinction coefficient of 13.400 mol−1cm−1.
RNA preparation
RNAs were prepared by in vitro transcription as described before.Citation14 15N labeled rNTPs were purchased from Silantes GmbH. NMR samples were prepared in H2O/D2O (9:1) using the following NMR buffer conditions: 25 mM potassium phosphate, pH ~6.2, 50 mM potassium chloride.
Nuclear magnetic resonance (NMR) spectroscopy
The NMR experiments were recorded on a Bruker NMR spectrometer (900 MHz) with a 5 mm z-axis gradient TXI-HCN cryogenic probe using standard 1H, 15N-HSQC and 1H,1H-NOESY pulse sequences. Data were processed and analyzed using the program TOPSPIN 2.1 (Bruker Biospin).
Stopped-flow fluorescence spectroscopy
2-aminopurine (2-AP) was obtained from Sigma-Aldrich and dissolved in buffer (2 mM Mg2+, 25 mM potassium phosphate, 50 mM potassium chloride, pH 6.2). The concentration of 2-AP was calculated using UV absorbance at 305 nm and an extinction coefficient of 13.400 mol−1cm−1. The RNA was dissolved in buffer containing 2 mM Mg2+, 25 mM potassium phosphate, 50 mM potassium chloride at pH 6.2.
For the binding experiments, we used a stopped-flow apparatus (Applied Photophysics π*-180 with a dead time of 1–2 ms) with a Xe-light source (wavelength of 305 nm) while detection was performed using a 320 nm cut-off filter. The rate constants were determined under pseudo first order conditions. We used equal volumes of solutions containing RNA and 2-AP for mixing. The RNA concentration was varied from 100 nM - 1 µM in excess over 2-AP (50 nM constant). Rate constants were determined at 10 °C, 15 °C, and 25 °C. The fluorescence decay was fitted using a three-parameter single exponential function. The resulting apparent rate constants used for analysis are averaged values of at least three independent measurements.
Association (kon) and dissociation (koff) constants were determined by plotting the reciprocal apparent rate constant (τ) vs. the RNA concentration using the following equation:
Free activation energies (ΔGa) for the reaction of ligand binding and complex dissociation were determined by using following equation:
Circular dichroism (CD) spectroscopy
CD melting profiles were recorded on a Jasco J-810 instrument within the temperature range of 4–94 °C using a quartz cuvette of 1mm path length. RNA samples were measured in NMR-buffer with final concentrations for RNA of 5–10 µM, for Mg2+ of 4 mM, and adenine of 56 µM. The melting profiles were followed at a wavelength of 264 nm and data were collected with heating rates of 1 °C/min. The temperatures of unfolding transitions (Tm) were extracted from the first derivative of the fitted CD profile. The heating and cooling profiles could be superimposed indicating absence of hysteresis effect in thermal melting.
Additional material
Download Zip (253.1 KB)Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
Work in the groups of H.S. and J.W. is supported by the DFG and the state of Hessen (BMRZ). H.S. is a member of the DFG-funded cluster of excellence: macromolecular complexes. A.R. was supported by SPP 1258: Sensory and regulatory RNAs in Prokayrotes, J.W. and H.S. are supported by SFB902: Molecular Principles of RNA-based regulation.
Acknowledgments
We thank Dr. Boris Fürtig and Anna Wacker for helpful discussion and Elke Stirnal and Dr. Christian Richter for technical support.
References
- Winkler WC, Breaker RR. Genetic control by metabolite-binding riboswitches. Chembiochem 2003; 4:1024 - 32; http://dx.doi.org/10.1002/cbic.200300685; PMID: 14523920
- Mandal M, Breaker RR. Gene regulation by riboswitches. Nat Rev Mol Cell Biol 2004; 5:451 - 63; http://dx.doi.org/10.1038/nrm1403; PMID: 15173824
- Breaker RR. Riboswitches and the RNA World. Cold Spring Harb Perspect Biol 2012; 4:a003566; http://dx.doi.org/10.1101/cshperspect.a003566; PMID: 21106649
- Winkler WC, Nahvi A, Roth A, Collins JA, Breaker RR. Control of gene expression by a natural metabolite-responsive ribozyme. Nature 2004; 428:281 - 6; http://dx.doi.org/10.1038/nature02362; PMID: 15029187
- Loh E, Dussurget O, Gripenland J, Vaitkevicius K, Tiensuu T, Mandin P, Repoila F, Buchrieser C, Cossart P, Johansson J. A trans-acting riboswitch controls expression of the virulence regulator PrfA in Listeria monocytogenes. Cell 2009; 139:770 - 9; http://dx.doi.org/10.1016/j.cell.2009.08.046; PMID: 19914169
- Schwalbe H, Buck J, Fürtig B, Noeske J, Wöhnert J. Structures of RNA switches: insight into molecular recognition and tertiary structure. Angew Chem Int Ed Engl 2007; 46:1212 - 9; http://dx.doi.org/10.1002/anie.200604163; PMID: 17226886
- Edwards TE, Klein DJ, Ferré-D’Amaré AR. Riboswitches: small-molecule recognition by gene regulatory RNAs. Curr Opin Struct Biol 2007; 17:273 - 9; http://dx.doi.org/10.1016/j.sbi.2007.05.004; PMID: 17574837
- Mandal M, Breaker RR. Adenine riboswitches and gene activation by disruption of a transcription terminator. Nat Struct Mol Biol 2004; 11:29 - 35; http://dx.doi.org/10.1038/nsmb710; PMID: 14718920
- Wacker A, Buck J, Mathieu D, Richter C, Wöhnert J, Schwalbe H. Structure and dynamics of the deoxyguanosine-sensing riboswitch studied by NMR-spectroscopy. Nucleic Acids Res 2011; 39:6802 - 12; http://dx.doi.org/10.1093/nar/gkr238; PMID: 21576236
- Buck J, Noeske J, Wöhnert J, Schwalbe H. Dissecting the influence of Mg2+ on 3D architecture and ligand-binding of the guanine-sensing riboswitch aptamer domain. Nucleic Acids Res 2010; 38:4143 - 53; http://dx.doi.org/10.1093/nar/gkq138; PMID: 20200045
- Fürtig B, Buck J, Manoharan V, Bermel W, Jäschke A, Wenter P, Pitsch S, Schwalbe H. Time-resolved NMR studies of RNA folding. Biopolymers 2007; 86:360 - 83; http://dx.doi.org/10.1002/bip.20761; PMID: 17595685
- Buck J, Fürtig B, Noeske J, Wöhnert J, Schwalbe H. Time-resolved NMR methods resolving ligand-induced RNA folding at atomic resolution. Proc Natl Acad Sci U S A 2007; 104:15699 - 704; http://dx.doi.org/10.1073/pnas.0703182104; PMID: 17895388
- Noeske J, Buck J, Fürtig B, Nasiri HR, Schwalbe H, Wöhnert J. Interplay of ‘induced fit’ and preorganization in the ligand induced folding of the aptamer domain of the guanine binding riboswitch. Nucleic Acids Res 2007; 35:572 - 83; http://dx.doi.org/10.1093/nar/gkl1094; PMID: 17175531
- Noeske J, Richter C, Grundl MA, Nasiri HR, Schwalbe H, Wöhnert J. An intermolecular base triple as the basis of ligand specificity and affinity in the guanine- and adenine-sensing riboswitch RNAs. Proc Natl Acad Sci U S A 2005; 102:1372 - 7; http://dx.doi.org/10.1073/pnas.0406347102; PMID: 15665103
- Buck J, Fürtig B, Noeske J, Wöhnert J, Schwalbe H. Time-resolved NMR spectroscopy: ligand-induced refolding of riboswitches. Methods Mol Biol 2009; 540:161 - 71; http://dx.doi.org/10.1007/978-1-59745-558-9_12; PMID: 19381559
- Rieder R, Lang K, Graber D, Micura R. Ligand-induced folding of the adenosine deaminase A-riboswitch and implications on riboswitch translational control. Chembiochem 2007; 8:896 - 902; http://dx.doi.org/10.1002/cbic.200700057; PMID: 17440909
- Serganov A, Huang L, Patel DJ. Structural insights into amino acid binding and gene control by a lysine riboswitch. Nature 2008; 455:1263 - 7; http://dx.doi.org/10.1038/nature07326; PMID: 18784651
- Stoddard CD, Montange RK, Hennelly SP, Rambo RP, Sanbonmatsu KY, Batey RT. Free state conformational sampling of the SAM-I riboswitch aptamer domain. Structure 2010; 18:787 - 97; http://dx.doi.org/10.1016/j.str.2010.04.006; PMID: 20637415
- Montange RK, Batey RT. Riboswitches: emerging themes in RNA structure and function. Annu Rev Biophys 2008; 37:117 - 33; http://dx.doi.org/10.1146/annurev.biophys.37.032807.130000; PMID: 18573075
- Baird NJ, Ferré-D’Amaré AR. Idiosyncratically tuned switching behavior of riboswitch aptamer domains revealed by comparative small-angle X-ray scattering analysis. RNA 2010; 16:598 - 609; http://dx.doi.org/10.1261/rna.1852310; PMID: 20106958
- Serganov A, Yuan Y-R, Pikovskaya O, Polonskaia A, Malinina L, Phan AT, Hobartner C, Micura R, Breaker RR, Patel DJ. Structural basis for discriminative regulation of gene expression by adenine- and guanine-sensing mRNAs. Chem Biol 2004; 11:1729 - 41; http://dx.doi.org/10.1016/j.chembiol.2004.11.018; PMID: 15610857
- Reining A, Nozinovic S, Schlepckow K, Buhr F, Fürtig B, Schwalbe H. Three-state mechanism couples ligand and temperature sensing in riboswitches. Nature 2013; 499:355 - 9; http://dx.doi.org/10.1038/nature12378; PMID: 23842498
- Stoddard CD, Gilbert SD, Batey RT. Ligand-dependent folding of the three-way junction in the purine riboswitch. RNA 2008; 14:675 - 84; http://dx.doi.org/10.1261/rna.736908; PMID: 18268025
- Lemay J-F, Penedo JC, Tremblay R, Lilley DMJ, Lafontaine DA. Folding of the adenine riboswitch. Chem Biol 2006; 13:857 - 68; http://dx.doi.org/10.1016/j.chembiol.2006.06.010; PMID: 16931335
- Garst AD, Edwards AL, Batey RT. Riboswitches: Structures and Mechanisms. Cold Spring Harb Perspect Biol 2011; 3:a003533; http://dx.doi.org/10.1101/cshperspect.a003533; PMID: 20943759
- Noeske J, Schwalbe H, Wöhnert J. Metal-ion binding and metal-ion induced folding of the adenine-sensing riboswitch aptamer domain. Nucleic Acids Res 2007; 35:5262 - 73; http://dx.doi.org/10.1093/nar/gkm565; PMID: 17686787
- Greenleaf WJ, Frieda KL, Foster DAN, Woodside MT, Block SM. Direct observation of hierarchical folding in single riboswitch aptamers. Science 2008; 319:630 - 3; http://dx.doi.org/10.1126/science.1151298; PMID: 18174398
- Eskandari S, Prychyna O, Leung J, Avdic D, O’Neill MA. Ligand-directed dynamics of adenine riboswitch conformers. J Am Chem Soc 2007; 129:11308 - 9; http://dx.doi.org/10.1021/ja073159l; PMID: 17713907
- Sharma M, Bulusu G, Mitra A. MD simulations of ligand-bound and ligand-free aptamer: molecular level insights into the binding and switching mechanism of the add A-riboswitch. RNA 2009; 15:1673 - 92; http://dx.doi.org/10.1261/rna.1675809; PMID: 19625387
- Villa A, Wöhnert J, Stock G. Molecular dynamics simulation study of the binding of purine bases to the aptamer domain of the guanine sensing riboswitch. Nucleic Acids Res 2009; 37:4774 - 86; http://dx.doi.org/10.1093/nar/gkp486; PMID: 19515936
- Wickiser JK, Cheah MT, Breaker RR, Crothers DM. The kinetics of ligand binding by an adenine-sensing riboswitch. Biochemistry 2005; 44:13404 - 14; http://dx.doi.org/10.1021/bi051008u; PMID: 16201765