
Abstract
The symbiotic α-rhizobia Sinorhizobium meliloti, Bradyrhizobium japonicum, Rhizobium etli and the related plant pathogen Agrobacterium tumefaciens are important model organisms for studying plant-microbe interactions. These metabolically versatile soil bacteria are characterized by complex lifestyles and large genomes. Here we summarize the recent knowledge on their small non-coding RNAs (sRNAs) including conservation, function, and interaction of the sRNAs with the RNA chaperone Hfq. In each of these organisms, an inventory of hundreds of cis- and trans-encoded sRNAs with regulatory potential was uncovered by high-throughput approaches and used for the construction of 39 sRNA family models. Genome-wide analyses of hfq mutants and co-immunoprecipitation with tagged Hfq revealed a major impact of the RNA chaperone on the physiology of plant-associated α-proteobacteria including symbiosis and virulence. Highly conserved members of the SmelC411 family are the AbcR sRNAs, which predominantly regulate ABC transport systems. AbcR1 of A. tumefaciens controls the uptake of the plant-generated signaling molecule GABA and is a central regulator of nutrient uptake systems. It has similar functions in S. meliloti and the human pathogen Brucella abortus. As RNA degradation is an important process in RNA-based gene regulation, a short overview on ribonucleases in plant-associated α-proteobacteria concludes this review.
Introduction
Soil bacteria interacting with plants are of high agricultural importance. They include several research model organisms which belong to the Rhizobiales order within the much larger α-proteobacterial phylum. Among them are the so-called α-rhizobia which are capable of entering a symbiosis with leguminous plants, such as Bradyrhizobium japonicum, Rhizobium etli and Sinorhizobium meliloti, the nitrogen fixing endosymbionts of soybean, common bean and alfalfa, respectively.Citation1-Citation3 This order also includes plant pathogens belonging to the Agrobacterium/Rhizobium lineage. A well-studied representative is A. tumefaciens, also used for genetic engineering of plants.Citation4 A. tumefaciens, R. etli and S. meliloti belong to the family of Rhizobiaceae while B. japonicum is a member of the distinct family of Bradyrhizobiaceae in the Rhizobiales. Recent high-throughput analyses revealed that these α-proteobacterial species harbor hundreds of RNAs with regulatory potential.Citation5-Citation12 It is the challenge now to understand their physiological roles and the underlying molecular mechanisms. A systematic comparison of the sRNA repertoires of selected model organisms may help to understand the commonalities and differences between these phylogenetically and ecologically related bacteria.
The α-rhizobia and A. tumefaciens are metabolically versatile soil bacteria specifically interacting with host plants. B. japonicum, R. etli and S. meliloti infect a quite limited spectrum of leguminous plants leading to the formation of specialized root organs named nodules, in which the differentiated bacteria (bacteroids) live as endosymbionts, obtain nutrients from the plant and convert atmospheric nitrogen into ammonia to the benefit of the host.Citation2,Citation3,Citation13 In contrast, A. tumefaciens is a broad-host-range phytopathogen that transfers bacterial T-DNA into plant cells and leads to the formation of tumors (crown galls) on most dicotyledonous and some monocotyledonous plants.Citation4 Interestingly, there are fundamental similarities in the mechanisms of microbe-plant interactions and the interactions between pathogenic bacteria and mammalian cells.Citation14 Beside their agricultural importance, this is one of the reasons for the intense investigation of the three α-rhizobial species and A. tumefaciens as model organisms.
All four plant-associated species persist in the soil under a variety of environmental conditions, compete with other bacteria in the rhizosphere, interact with the plant and, in the case of rhizobia, differentiate into bacteroids. These complex lifestyles are reflected by large genomes (5.7 Mb in A. tumefaciens, 6.5 Mb in R. etli, 6.7 Mb in S. meliloti and 9 kb in B. japonicum) coding for a plethora of protein transcription factors.Citation15-Citation19 Comparatively little is known about the regulation of gene expression at the posttranscriptional level in these bacteria. Small non-coding RNAs (sRNAs) are major players in RNA-based gene regulation together with the RNA chaperone Hfq and ribonucleases (RNases) or RNase-containing complexes.Citation20,Citation21 They participate in the response and adaptation to diverse stress conditions, and are important for the regulation of quorum sensing and virulence.Citation22-Citation25 The capability of A. tumefaciens, B. japonicum, R. etli and S. meliloti to occupy diverse ecological niches is mediated by complex regulatory networks which include riboregulation as well. This view is supported by the large amount of cis- and trans-encoded sRNAs detected by genome-wide analyses in these plant-associated bacteria.Citation5-Citation12
Since conservation of sRNAs is mostly limited to closely related bacteria, the phylogenetic relationships between α-rhizobia and Agrobacterium species allow the functional comparison of sRNAs with different degrees of conservation in function and regulation. Here, we summarize the current knowledge on the non-coding RNome as inferred from genome-wide surveys and discuss the incipient insights into biological functions and mechanisms of riboregulation in these organisms.
Transcriptome landscape: inventory of non-coding RNAs in α-rhizobia and the plant-pathogen A. tumefaciens
Genome-wide identification of small non-coding RNAs in the Rhizobiales has been pioneered by computational predictions in S. meliloti and R. etli.Citation5-Citation7,Citation9 These were based on comparative genomics combining features characteristic of trans-encoded sRNAs (trans-sRNAs): transcription from genomic regions separating protein-encoding genes, association with promoter and/or Rho-independent transcriptional terminator sequence motifs, sequence conservation between closely related species, and conservation and thermodynamic stability of the predicted RNA secondary structure. However, complementation of these in silico approaches by microarray- and RNA-seq based surveys of the non-coding transcriptome of the model α-rhizobial species S. meliloti, R. etli and B. japonicum and the phytopathogen A. tumefaciens represented the major breakthrough in this field. Here we give an overview of inventories of the non-coding transcriptomes of these α-proteobacteria, which provides a firm foundation for comparative and functional studies of regulatory RNAs.
The non-coding transcriptome of S. meliloti
S. meliloti is well-studied for its engagement in a nitrogen fixing symbiosis with its plant hosts Medicago, Melilotus, and Trigonella. Strain Rm1021 possesses a large, tripartite genome composed of a chromosome (3.54 Mb), and the megaplasmids pSymA (1.35 Mb) and pSymB (1.68 Mb).Citation15 Several RNA-seq based approaches were undertaken in this S. meliloti strain and its close relative Rm2011. These screens aimed at profiling the transcriptome landscape and identifying sRNAs. Differential RNA-seq (dRNA-seq) of size-fractionated RNA using the Genome Sequencer FLX Titanium System (Roche Diagnostics) was applied to discover short RNAs of a size range from 50 to 350 nucleotides in RNA populations derived from exponential and stationary growing batch cultures, and from six stress conditions.Citation8 dRNA-seq involves enrichment of RNAs with primary 5′-ends by terminal exonuclease (TEX) treatment of one of the samples and its comparison to a non-treated sample still containing the original amount of processed 5′-monophosphorylated transcripts.Citation26 This approach revealed a total of 1,125 sRNA candidates of which 173 were grouped to trans-sRNAs, 117 to cis-encoded antisense sRNAs (asRNAs), 379 to mRNA leader sequences, and 456 to sense sRNAs overlapping coding regions. Short RNAs derived from 5′-UTRs of mRNAs may include metabolite-controlled riboswitches and RNA thermometers. This approach identified a surprisingly low number of antisense transcripts compared with RNA-seq studies of other prokaryotes.
An RNA-seq strategy designed for genome-wide identification of transcriptional start sites (TSS) of S. meliloti from 16 different growth and stress conditions revealed 3,720 asRNAs that were assigned to approximately 35% of the protein-coding genes.Citation27 Thus, the lower number of growth conditions and the size fractionation of RNA populations analyzed in the first approach was likely responsible for the small number of antisense transcripts identified.Citation8 Genome-wide TSS mapping not only increased the number of known asRNAs, but also identified new members of the classes of trans-sRNAs and sense RNAs, and identified TSS of mRNAs.Citation27 A total of 17,001 TSS falling into six categories were reported in this study: mRNA (4,430 TSS assigned to 2,657 protein-coding genes), leaderless mRNAs (171), putative mRNAs (425), internal sense transcripts (7,650), asRNA (3,720), and trans-sRNAs (605 TSS associated with 440 trans-sRNAs). This raised the number of trans-sRNAs to 550 in S. meliloti. Trans-sRNAs were found on all three replicons of the multipartite genome of S. meliloti. Taking into account the individual sizes of the three replicons these sRNAs were overrepresented on pSymA.Citation27
Both RNA-seq approaches that aimed at identifying primary 5′-ends revealed a strikingly high number of sense transcripts overlapping coding regions, but not involving an open reading frame (ORF).Citation8,Citation27 Such sense RNAs have also been identified in similar studies of other prokaryotic transcriptomes. It is likely that a high proportion of these sense RNAs were degradation products derived from the corresponding mRNAs that survived the enrichment procedure. However, upstream promoter motifs were predicted for 632 sense RNAs.Citation8,Citation27 Whether these transcripts have a functional role remains to be investigated.
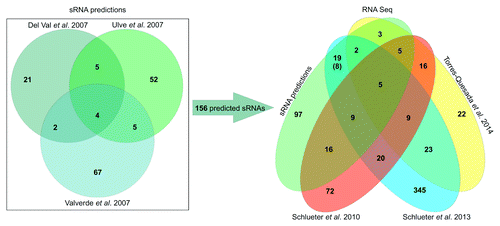
The precise determination of TSS by RNA-seq involving enrichment of primary transcripts allowed for identification of promoter motifs and improved consensus sequences.Citation8,Citation27 The RNA-seq approaches clearly outperformed previous microarray-based strategies.Citation7,Citation8 Comparison of RNA-seq based identification to computational predictions of trans-sRNAs demonstrated the power of deep sequencing strategies that rediscovered 59 out of 156 predicted candidates and identified numerous novel trans-sRNAs ().Citation8,Citation27,Citation28
Figure 1. Overview of computational predicted and experimentally identified sRNAs in S. meliloti. On the left, Venn-diagram illustrating the number of potential sRNA encoding regions predicted by del Val et al.,Citation5 Ulve et al.,Citation6 and Valverde et al.Citation7 On the right, Venn-diagram representing sRNA transcripts identified via RNA-seq studies by Schlüter et al.,Citation8 Schlüter et al.,Citation27 and Torres-Quesada et al.Citation28 and a comparison to the 156 predicted candidates. Number in brackets: clustered sRNA genes (at least two experimentally identified neighboring sRNA genes) which were spuriously predicted and summarized as a single sRNA gene region were counted as individual transcription units.
The non-coding transcriptome of R. etli
In silico searches were combined with transcriptome profiles of R. etli CFN42 at various time points during free-living growth and during symbiosis with Phaseolus vulgaris.Citation9 This study aimed at the identification of transcriptionally active intergenic regions (IGR) applying a whole-genome-high-resolution tiling oligonucleotide array. The expression profiles suggested 89 non-coding RNAs mapping to all replicons of the R. etli CFN42 genome, the 4.38 Mb chromosome and the six megaplasmids encompassing additional 2.15 Mb.Citation19 66 of these candidates were classified as novel non-coding RNAs since they did not group to well-characterized house-keeping RNAs and had not been reported before in R. etli or related α-proteobacteria. These candidates, comprising 17 trans-sRNAs and 49 putative asRNAs, overlapped with the group of computationally predicted non-coding RNAs.Citation9 Several of these non-coding RNAs were differentially expressed in free-living conditions and during symbiosis. Clustering identified three classes characterized by dominant expression during exponential growth, in the stationary phase, or in planta implying a role in adaptation to different environmental factors.Citation9
The non-coding transcriptome of A. tumefaciens
Two deep sequencing strategies uncovered the transcriptome of the plant pathogen A. tumefaciens under non-virulent and virulence induced conditions.Citation11,Citation12 Wilms et al.Citation11 used the Roche 454 platform and libraries from cells grown in minimal medium with or without the phenolic compound and virulence gene inducer Acetosyringone (AS). A total number of 348,998 cDNAs reads were mapped to the genome. The 228 identified sRNA candidates (152 trans-sRNAs, 76 cis-asRNAs) are distributed on all replicons: 129 on the circular chromosome (trans-sRNA: 96, asRNA: 33), 59 on the linear chromosome (trans-sRNA: 41, asRNA: 18), 20 on the At plasmid (trans-sRNA: 8, asRNA: 12) and 20 on the Ti plasmid (trans-sRNA: 7, asRNA: 13). Among them were the known sRNAs RepE, AbcR1, and AbcR2.Citation29 22 of these identified sRNAs were independently confirmed by northern blot analysis. The observation that several sRNAs were differentially expressed in response to growth media, growth phase, temperature, or pH, and that one sRNA from the Ti-plasmid was massively induced in the presence of AS suggests that sRNAs play a role in various cellular processes including virulence.
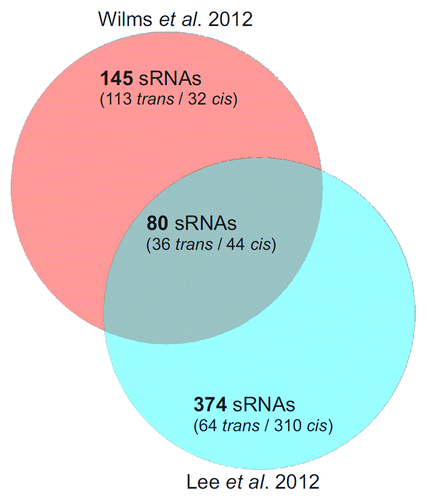
The virulence-induced sRNA Ti2 is encoded downstream of a promoter strictly dependent on the VirA/G two-component system.Citation11 Its location in the 5′-UTR of the atu6155virK operon suggests that it may be a processed or prematurely terminated product of the UTR as proposed by Lee et al.Citation12 The latter study made use of an Illumina GAII platform to sequence cDNA libraries from cells grown in nutrient-rich medium at mid-log phase, nutrient-rich medium at late stationary phase, and minimal medium with or without AS. Mapping of a total number of 490,552 million reads identified 475 highly expressed non-coding RNAs distributed across all four replicons (101 trans-RNAs, 354 antisense RNAs and 20 5′-UTRs), including RepE, AbcR1 and AbcR2. Disregarding the 5′-UTRs, 210 were on the circular chromosome (trans-sRNA: 56, asRNA: 154), 158 on the linear chromosome (trans-sRNA: 33, asRNA: 125), 41 on the At plasmid (trans-sRNA: 8, asRNA: 33) and 44 on the Ti plasmid (trans-sRNA: 4, asRNA: 42). Fifteen sRNAs were induced and 7 were suppressed by the virulence inducer AS. A total number of 374 novel sRNA were found in this study (). Interestingly, this study found fewer trans-sRNAs but many more antisense RNAs than Wilms et al.Citation11 demonstrating the value of several independent approaches in sRNA discovery (see also ).
Figure 2. Comparison of two A. tumefaciens dRNA-seq studies. Venn diagram comparing the sRNAs identified by Wilms et al.Citation11 and Lee et al.Citation12
Both studies reported the presence of antisense sRNAs transcribed from the reverse strand of virulence genes, for example of virC2 (required for excision of the T-DNA) and virB9 (component of the type IV secretion system) suggesting that they might play regulatory roles in the plant infection process. Two candidates antisense to atsD (might be important for bacterial attachment on plants) and antisense to virB10 (Atu6176, essential component of type IV secretion system) were tested for their role in A. tumefaciens virulence. However, their depletion by anti-antisense technology did not significantly affect tumor genesis in three different virulence assays suggesting that the real targets of these cis-antisense sRNAs might not be the sense-strand genes.Citation12 Likewise, deletion of the massively AS-induced sRNA Ti2 did not significantly affect tumor genesis of Kalanchoe daigremontiana leaves and potatoe tumor discs (unpublished data).
The non-coding transcriptome of B. japonicum USDA 110
The initial genome-wide search for sRNAs in B. japonicum USDA 110 relied on computational predictions based on comparison of genomes of Bradyrhizobium sp.BTAi1 and B. japonicum USDA 110 to the genome of the free-living purple bacterium Rhodopseudomonas palustris BisB5a.Citation10 Approximately 100 so-called “clusters” representing potential trans-sRNAs were predicted, and seven candidate sRNAs (BjrC2a, BjrC2b, BjrC2c, BjrC68, BjrC80, BjrC174 and BjrC1505) were verified by northern blot hybridization in addition to conserved sRNAs with housekeeping function like SRP RNA, RNase P RNA, tmRNA and 6S rRNA. The detected sRNA candidates have a different degree of conservation. Three of these sRNAs (BjrC2a, b and c) belong to the previously described RNA family RF00519.Citation30 Multiple imperfect copies of RF00519 sRNAs are present in many species in the Rhizobiales including S. meliloti and A. tumefaciens. Another well conserved sRNA is BjrC1505, homologs of which are present in Bradyrhizobiaceae and some genera of Rhizobiaceae like Brucella and Mesorhizobium but not in Sinorhizobium and Agrobacterium/Rhizobium. The other sRNAs are limited to Bradyrhizobiaceae. The least conserved BjrC68 corresponds to the 3′-UTR of blr0613 and its homologs are found only in the closely related genera Bradyrhizobium and Rhodopseudomonas.Citation10
As in most similar studies, the expression of the verified sRNAs was monitored by northern blot hybridization under diverse stresses and at different growth phases. The results were directly compared with data from microarray and dRNA-seq analyses and displayed similar tendencies - increases or decreases in the amounts of the particular sRNA in bacteroids were supported by at least two of the methods.Citation10 Specific accumulation of full-length forms or processing/degradation products of sRNAs in nodules suggested functional relevance in symbiosis for BjrC174, BjrC68 and BjrC80. Interestingly, although the amount of 6S RNA was not increased in the stationary phase in liquid cultures, it was increased in nodules suggesting a similar role for 6S RNA in the non-dividing bacteroids of B. japonicum USDA 110 as in stationary cultures of E. coli.Citation10,Citation31,Citation32 Using dRNA-seq, comparable levels of “product RNAs” (pRNAs) of 10 to 17 nt in length were detected in free-living bacteria and in bacteroids.Citation10 The pRNAs are short primary transcripts synthesized by the RNA polymerase using 6S RNA as template during the process of release, and their detection suggests turnover of 6S RNA of B. japonicum USDA 110 under both free-living and symbiotic conditions.Citation33
α-proteobacterial sRNA families
Constructing sRNA family models (RFMs) from transcripts observed in an RNA-seq study serves two purposes. For a transcript with a known or suspected regulatory function, this information can be transferred to related bacteria that host a family member in their genome, and experimental evidence available from different organisms may be interpreted in combination. For an sRNA transcript without associated functional evidence, successful construction of an RFM provides evidence for conservation in sequence and/or structure, and indirectly hints at the existence of a conserved function. Furthermore, the specific pattern of conservation observed may point to functions related to the lifestyle of a closely related group.
Starting from the sRNAs identified by Schlüter et al.,Citation8 a set of 52 trans-sRNAs was chosen, some of which apparently were multiple homologs of the same sRNA on different replicons of S. meliloti. From these, 39 family models were constructed employing two different bioinformatics approaches.Citation34
• Covariance models (CMs) are stochastic models, capturing sequence and structure conservation in an alignment of family members. CMs can be automatically constructed by INFERNAL, given such an alignment.Citation35
• Thermodynamic matchers (TDMs) are RNA folding programs, based on the established thermodynamic model, but tailored to a specific structural motif. Production of such matchers is supported by the graphical editor LOCOMOTIF and by the Bellman's GAP system.Citation36,Citation37
CMs are the more commonly used approach, which is employed e.g., with the Rfam database. The method is applicable when sequence conservation is high enough to obtain a reliable multiple alignment from candidate sequences. 37 out of the 39 family models were constructed in this fashion. In two cases where structure was conserved, but sequence was highly diverged (except for short loop motifs), the TDM approach was applied. TDMs focus on structure and folding energy; they can ignore sequence conservation in some parts, e.g., in helices, and yet insist on conserved sequence motifs elsewhere, e.g., in loops. Building such a matcher requires human design decisions and some experimentation, and hence, it is more laborious than the standard route to family model construction. The precise workflows employed were described by Reinkensmeier et al.Citation34
del Val et al.Citation38 applied a similar approach to analyze the distribution of six sRNA candidates (SmrC7/SmelC023, SmrC9/SmelC289, SmrC45/SmelC706, SmrC15/SmelC411, SmrC14/SmelC397, SmrB35/SmelB053) identified in S. meliloti 1021.Citation5 Conservation and occurrence of sRNAs in the Rhizobiales derived from these studies is summarized in . Three of these sRNAs, SmelB053, SmelC023, and SmelC289, were investigated in both comparative studies that suggested consistent phylogenetic distributions. Microsynteny of the genomic regions containing sRNA genes further supported 27 of the 39 RFMs.Citation34
Figure 3. Distribution of trans-sRNAs in the Rhizobiales. The circular plot is based on a simplified phylogenetic tree adopted from Reinkensmeier et al. and del Val et al.Citation34,Citation38 Sequenced strains which harbor at least one sRNA of a defined RNA family model (RFM) are illustrated. Each circular row incorporates all sRNA members of an RFM and shows their assignment to members of the Rhizobiales. The two RFMs separated by the green circle were defined exclusively by del Val et al.Citation38 The RFM nomenclature in brackets indicates the availability of alternative RFMs calculated by del Val et al.Citation38 (not shown here). Grey, chromosomal sRNAs; light blue, sRNAs encoded on pSymB-like replicons; dark-blue, sRNAs encoded on pSymA-like replicons. Multi-colored rows indicate RFMs with several RNA gene homologs per strain/genome: Yellow, single gene; orange, two homologs; pale violet red, three homologs; green, four or more homologs. Species glossary: R. l., Rhizobium leguminosarum; R. e., Rhizobium etli; A. r., Agrobacterium radiobacter; A. t., Agrobacterium tumefaciens; A. v., Agrobacterium vitis; A. sp., Agrobacterium species, S. m., Sinorhizobium meliloti; S. e., Sinorhizobium meliloti; S. f., Sinorhizobium fredii; B. a., Brucella abortus; B. m., Brucella melitensis; B. s., Brucella suis; B. mi., Brucella microti; B. o., Brucella ovis; B. c., Brucella canis; O. a., Ochrobactrum antrophi; M. l., Mesorhizobium loti; M. c., Mesorhizobium cicero; M. sp., Mesorhizobium species; P. l., Parvibaculum lavamentivorans; M. e., Methylobacterium extorquens; M. c., Methylobacterium chloromethanicum; X. a., Xanthobacter autotrophicus; St. n., Starkeya novella; Az. c., Azorhizobium caulinodans; Rh. p., Rhodopseudomonas palustris; N. w., Nitrobacter winogradskyi; Be. i., Beijerinckia indica; Met. s., Methylocella silvestris; Rm. v., Rhodomicrobium vannielii.
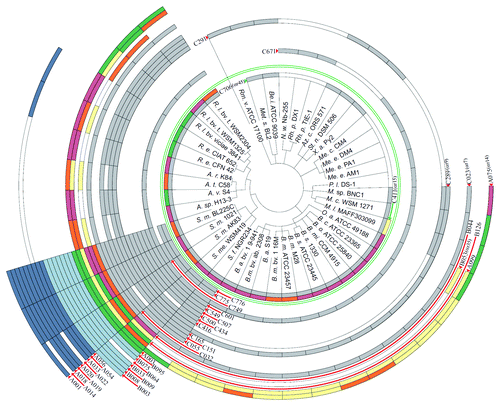
RNA families founded by S. meliloti sRNA candidates encoded by the megaplasmids pSymA or pSymB were restricted to Sinorhizobium species (). The only exception to this observation was SmelA033 that was also found in R. etli strains. This is in agreement with the assumption that the large rhizobial plasmids mostly contain accessory genetic information.Citation39 Most sRNA families were restricted to the Rhizobiaceae and only eight families were present in all species of the Rhizobiaceae investigated in these comparative studies, including five Rhizobium, five Sinorhizobium, and four Agrobacterium strains ().Citation34,Citation38 Among these was the family SmelC411 (SmrC15) including the AbcR sRNAs that have been first described and functionally studied in A. tumefaciens and S. meliloti ().Citation29,Citation40 The AbcR family contains two highly conserved modules resembling the anti-Shine–Dalgarno sequence, one with a 5′-CCUCCC-3′ sequence (M1) within loop 1 and a second 5′-GUUCCCCUC-3′ sequence (M2) between the first and the second loop (). Homologs of AbcR1 and AbcR2 (SmrC15/SmrC16, Smr15c1/Smr15c2, SmelC411/SmelC412, Sra41, and Sm3/Sm3′, ReD01, ReC58/ReC59) have been reported in the closely related plant symbionts S. meliloti, S. fredii, R. etli, Rhizobium gallicum, Rhizobium giardinii, Rhizobium leguminosarum, and in the human pathogen Brucella abortus.Citation5-Citation7,Citation31,Citation41 In several Sinorhizobium species, comparative sequence analyses predicted a third member of this family on megaplasmid pSymA in addition to the chromosomal members AbcR1 and AbcR2.
Figure 4. Structural Alignment of AbcR1/2 (SmelC411), SmelA075, and SmelA099. Consensus secondary structures are colored according to the Vienna RNA conservation coloring scheme.Citation110 A. Structural alignment of RFM αr15 (AbcR1/2) defined by del Val et al.Citation38 In yellow, anti-Shine-Dalgarno (aSD) sequences (modules M1 and M2) in single-stranded regions and loop structures. Pale red, Hfq binding site. B. Structural alignment of RFM SmelA075 and SmelA099 defined by Reinkensmeier et al.Citation34
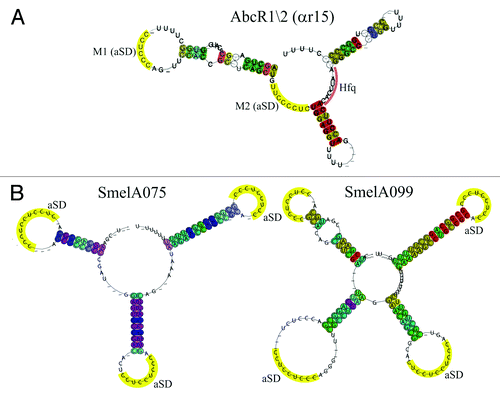
Only ten sRNA families showed broad conservation in the Rhizobiales, but neither occurred in all subtaxa of this order. These RFMs mostly showed only conservation of short sequence motifs combined with structural similarities. Among these are the RFMs founded by SmelA075 and SmelA099 (). Like the AbcR1 family, both contain highly conserved 5′-CCUCCUCCC-3′ loop motifs resembling the anti-Shine–Dalgarno (aSD) sequence. sRNAs composed of two to four stem loops containing this conserved loop motif were also found in closely related as well as more distant α-proteobacteria.Citation34,Citation42 The Rhodobacter spaeroides sRNA family RSs0680 showing similar features has been implicated in the oxidative stress response.Citation42 Comprehensive sequence and structural conservation facilitates identification of sRNA homologs in close relatives and also suggests functional conservation. Though, prediction of homologs in more distantly related bacteria is complicated by poor conservation. This may result in spurious and misleading assignments of RNAs to RFMs. Nevertheless, the conservation patterns of trans-sRNAs and the frequent occurrence of paralogs in the Rhizobiales point to the evolutionary history of this class of riboregulators and imply that duplication events followed by divergent evolution probably provide an adaptive advantage.
RNA-binding proteins in α-rhizobia and A. tumefaciens
sRNA activity in many bacterial species involves RNA-binding proteins. One of the best studied examples are CsrA/RsmA family RNA-binding proteins acting as regulators of translation.Citation43 Csr/Rsm mimic sRNAs compete with the target mRNA for binding of these proteins thereby releasing the translational block. With exception of a CsrA/RsmA-like protein encoded by the cryptic plasmid pMBA19a in S. meliloti MBA19, α-proteobacterial genomes lack genes homologous to csrA/rsmA.Citation44
In contrast, hfq encoding an RNA-binding protein predicted to be present in at least 50% of all bacterial species was identified in all α-proteobacterial genomes sequenced so far.Citation21 Hfq proteins found in this group of bacteria share strong sequence similarities with enterobacterial Hfq proteins suggesting a similar quaternary organization into a hexameric toroid with at least two different RNA binding surfaces.Citation45 This is further supported by successful complementation of an E. coli hfq mutant by the S. meliloti hfq gene and vice versa.Citation46,Citation47 Hfq assists RNA-mediated gene regulation.Citation21,Citation48 Its functional role exceeds the interaction with trans-sRNAs and their mRNA targets to binding of asRNAs and tRNAs.Citation49-Citation53 Translational control or regulation of mRNA decay mediated by binding of Hfq to mRNAs in a trans-sRNA independent manner has been reported.Citation54,Citation55 Like in other groups of bacteria, α-proteobacterial hfq genes appear to be strongly expressed and autoregulated at posttranscriptional level.Citation46,Citation47,Citation56
In most α-proteobacteria, hfq mutants were affected in a broad range of phenotypes, including growth behavior, adaptation to stress factors, motility, cell morphology, host colonization, or virulence. The A. tumefaciens hfq mutant was delayed in growth, impaired in motility, showed altered cell morphology and was attenuated in tumor formation.Citation57 A. caulinodans and Rhizobium leguminosarum bv. viciae hfq mutants were deficient in symbiotic nitrogen fixation.Citation46,Citation58,Citation59 A number of different studies characterized phenotypes caused by hfq knockouts and their impact on the S. meliloti transcriptome and proteome. S. meliloti hfq mutants were delayed in growth, impaired in motility and more sensitive to heat, oxidative, or pH stresses. They showed alterations in the utilization of nitrogen sources, in profiles of quorum sensing signals, and symbiotic traits.Citation45,Citation56,Citation60-Citation63 Competitiveness for infection, nodule development, intracellular survival of bacteroids and nitrogen fixation was negatively affected in S. meliloti hfq mutants. These pleiotropic phenotypes can largely be traced back to global changes in gene expression patterns at RNA and protein levels. Although the various profiling studies involved different S. meliloti strains, growth conditions, and host plants some common observations were made. Massive differential expression of genes mediating tolerance to a variety of stress factors and to genes involved in transport of small molecules relevant to carbon and nitrogen metabolism suggests a major role of Hfq in physiology of S. meliloti.Citation56,Citation60-Citation62 These findings were further supported by an A. tumefaciens hfq mutant showing overproduction of several ABC transporter components when compared with the wild type.Citation57
A positive contribution of Hfq to the post-transcriptional regulation of nifA, encoding the major transcriptional regulator of nitrogen fixation, has been observed in several free-living and symbiotic α-proteobacterial diazotrophs.Citation46,Citation59,Citation64 Furthermore, R. leguminosarum Hfq negatively regulates the broad range amino acid transport systems Aap and Bra which have a key role in nitrogen exchange between the bacteroid and the infected plant cell and suppressor mutations in hfq rescued R. leguminosarum GOGAT mutants that were deficient in nitrogen fixation.Citation65 α-proteobacterial hfq mutants impaired in virulence and symbiosis, as observed for several symbiotic α-rhizobia, phytopathogenic A. tumefaciens, and animal-pathogenic Brucella species, imply that Hfq is not only important for free-living states but also for establishment and maintenance of chronic microbial infections of eukaryotic hosts.Citation66,Citation67
Co-immunoprecipitation (CoIP) of an epitope-tagged Hfq and its associated RNA in S. meliloti followed by PCR identified a few selected trans-sRNAs.Citation40,Citation62 Recently, Hfq-CoIP combined with deep sequencing of the recovered RNA generated a genome-wide atlas of S. meliloti Hfq-bound transcripts in five growth/stress conditions.Citation28 This study suggested 1,315 Hfq-bound transcripts derived from non-coding RNAs, both trans-encoded (6.4%) and antisense (asRNAs) (6.3%), and mRNAs (86%) with the largest proportion of RNAs enriched from stress conditions. However, only 14% of trans-sRNAs and 2% of asRNAs were identified in previous studies. Hfq was associated with 18% of the predicted S. meliloti mRNAs encoding proteins related to transport and metabolism, σE2-dependent stress responses, quorum sensing, flagella biosynthesis, ribosome and membrane assembly, or symbiotic nitrogen fixation. This suggests that in S. meliloti stress responses and symbiotic gene expression are substantially affected by Hfq-mediated post-transcriptional control.
Another candidate for an RNA binding protein that may be related to riboregulation is the S. meliloti SMc01113 protein, a homolog to E. coli YbeY. This protein is broadly conserved in bacteria and comprises an RNA-binding region reminiscent of the MID domain of eukaryotic Argonaute proteins involved in RNA silencing.Citation68 Phenotypes of a SMc01113 mutant are pleiotropic and resemble those of hfq mutants.Citation68 In E. coli, YbeY modulates Hfq-dependent and Hfq-independent sRNA-mRNA interactions.Citation69 Furthermore, YbeY is related to rRNA processing and transcription termination and recently, it was found that YbeY is an RNase playing a role in rRNA maturation and quality control.Citation70-Citation72 The functional role of YbeY-like proteins in α-proteobacteria remains to be investigated.
Regulatory functions of non-coding RNAs: AbcR1 and AbcR2
The function of most sRNAs in the Rhizobiales is unexplored. Apart from housekeeping rRNAs and tRNAs, the regulatory 6S RNA, and the housekeeping 4.5S and RNase P RNAs are ubiquitous in bacteria and their genes have also been identified in α-proteobacterial genomes.Citation73-Citation76 Random mutagenesis led to the identification of the B. japonicum tmRNA encoded by sra (for symbiotic ribonucleic acid). A B. japonicum sra mutant was deficient in nodulation and nitrogen fixation.Citation77 In silico searches confirmed the prevalence of the tmRNA also in α-proteobacteria.Citation76, Citation78, Citation79 In S. meliloti this RNA was identified in genome-wide profiling and Northern analysis. It accumulated under stress conditions.Citation6 Two stable 214 nt and 82 nt RNAs derived from an unstable precursor were reported.Citation8 These likely correspond to the mRNA and tRNA domains. The plasmid-encoded incA antisense RNA mediates incompatibility within the large repABC family of α-proteobacterial plasmids.Citation80 A vast number of α-rhizobia carries symbiotic plasmids of this family.
Apart from RepE, which regulates Ti plasmid replication, AbcR1 (ABC transporter regulator 1) was the first functionally characterized sRNA in A. tumefaciens.Citation29,Citation81 At least three target mRNAs of AbcR1, all encoding proteins related to ABC transport systems, were identified by one dimensional protein gels and computer predictions.Citation29 Expression of these three targets is downregulated by AbcR1. Among them is atu2422 encoding the binding protein for GABA (γ-amino butyric acid), a plant-derived defense molecule that interferes with quorum sensing in Agrobacterium.Citation82,Citation83 AbcR1 inhibits initiation of atu2422 translation through direct RNA-RNA interaction masking the ribosome binding site and is the first described bacterial sRNA that controls uptake of a plant-generated signaling molecule.Citation29 Most recent data suggests that AbcR1 is a central regulator of nutrient uptake systems in A. tumefaciens.Citation84 The sRNA has numerous targets, most of them coding for ABC transporter components. Two single-stranded regions of AbcR1 expose conserved single-stranded anti-SD sequences (module M1 and M2, ) and are able to basepair with target mRNAs either in the translation initiation region or up to 500 nucleotides downstream into the coding region.Citation84
AbcR1 is encoded in an intergenic region in tandem with the related sRNA AbcR2.Citation29 Their transcripts are maximally expressed in the late exponential phase. To date, there is no evidence for AbcR2-mediated gene regulation in A. tumefaciens. Homologs of AbcR1 and AbcR2 have been also reported in the closely related plant symbiont S. meliloti, in other α-rhizobia and in more distantly related α-proteobacteria.Citation5-Citation7,Citation31,Citation41 In contrast to A. tumefaciens, AbcR1 and AbcR2 are differently expressed in S. meliloti, suggesting that they operate at diverse conditions.Citation5,Citation31 One protein of an ABC transporter, the amino acid binding protein LivK, was over-represented in the periplasmic proteome of an S. meliloti AbcR1 mutant. As in A. tumefaciens, AbcR2 does not play a role in livK regulation and the role of this sRNA remains to be elucidated.Citation40 This is remarkably different in B. abortus, where both AbcR1 and AbcR2 seem to have a regulatory and somewhat redundant function.Citation41 Quantitative proteomics and microarray analysis revealed elevated levels of multiple proteins and transcripts related to ABC transport systems in the double mutant. At least three of these transcripts were shown to be controlled by AbcR1 or AbcR2 alone.
The rhizobial AbcR1 was shown to belong to the large group of Hfq-associated sRNAs and a putative binding sequence for Hfq was predicted ().Citation28,Citation31,Citation41,Citation57,Citation62 Both AbcR1 and AbcR2 were enriched in Hfq-CoIP-RNA libraries.Citation28 Moreover, the livK mRNA (SMc01946) was also among the Hfq-bound transcripts identified in this study. A double-plasmid reporter system assay showed downregulation of SMa0495 and prbA (SMc01642) by both AbcR1 and AbcR2.Citation28,Citation40 SMa0495 and pbrA encode periplasmic solute binding proteins, the latter probably mediating uptake of proline betaine both at high and low osmolarities. Putative involvement of Hfq in this regulation was further supported by enrichment of the SMa0495 and prbA mRNAs in the Hfq-CoIP-RNA libraries derived from exponentially growing and salt-shocked cultures.Citation28
Quantitative proteomics of the Hfq-regulon in A. tumefaciens and S. meliloti or Hfq-coimmunoprecipitations in R. sphaeroides supported the notion that transport processes (like ABC transport) in α-proteobacteria are regulated by AbcR1 and other sRNAs as has been reported for many other bacteria.Citation42,Citation57,Citation63
Tools for in vivo analysis of sRNA-mRNA interactions in Rhizobiaceae
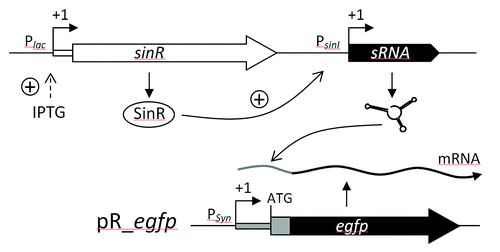
Computational and experimental screens have uncovered a staggering number of trans-sRNAs in plant-associated α-proteobacteria that demand functional characterization. Identification of the mRNA targets is crucial to fully understand the biological function of a given sRNA. Biocomputational algorithms can help to in silico predict conserved sRNA-target recognition at the genomic scale, but sRNA-mRNAs interactions require in vivo validation.Citation85 Similar to the reporter system developed for enterobacteria, a double-plasmid reporter assay has been implemented in S. meliloti.Citation40,Citation86 Basically, the sRNA and the translational fusion of a candidate target mRNA to a reporter gene are co-expressed from constitutive promoters to primarily assay post-transcriptional regulation. This system has confirmed the regulation of several targets genes encoding ABC transporters by the AbcR1/2 sRNAs.Citation28,Citation40 S. meliloti cells were co-transformed with a low-copy reporter plasmid (pR_egfp) and a mid-copy plasmid constitutively expressing the sRNA from a modified Plac promoter (pSRK_C). The translational fusion of the mRNA region of interest to egfp was expressed under the control of a constitutive promoter to specifically detect posttranscriptional effects rather than transcriptional regulation.
As shown by Torres-Quesada et al.Citation40 sRNA expression from the modified Plac promoter allowed for target identification. However, constitutive overexpression of some sRNAs may have indirect pleiotropic, toxic or lethal effects. IPTG-inducible sRNA overexpression driven by the native Plac promoter would partially overcome these limitations. However, it does not ensure sRNA yields that are high enough to test weak sRNA-mRNA regulatory pairs in most α-proteobacteria since regulatory effects can be masked by the contribution of native transcription from the target gene. Therefore, we have modified an IPTG-inducible system that relies on the S. meliloti sinR-sinI quorum sensing genes (M. McIntosh, personal communication) to drive strong pulse overexpression of a desired sRNA. The sinI gene encoding an N-acyl homoserine lactone synthase is the target gene of the LuxR-type transcriptional regulators SinR and ExpR.Citation87 This inducible expression construct for sRNA genes takes advantage of the strong activation of the PsinI promoter upon moderate induction of sinR expression even in the absence of ExpR ().Citation86,Citation88 S. meliloti sinR- sinI- expR- cells harboring the target reporter fusion of interest were transformed with plasmids carrying this construct for pulse overexpression of S. meliloti sRNA genes. GFP fluorescence of the resulting transformants confirmed the expected effects of the sRNAs on the expression of their predicted mRNA targets in S. meliloti. Furthermore, this system was also proven to be suitable for induced sRNA overexpression in other members of the Rhizobiaceae, such as S. medicae, S. fredii, R. tropicii, R. radiobacter, R. etli, and A. tumefaciens (M. McIntosh, M. Robledo, personal commmunication).
Figure 5. sRNA pulse overexpression construct based on IPTG inducible expression of sinR which activates PsinI-driven sRNA gene expression. The effect of sRNA overproduction is monitored applying a reporter construct based on plasmid pR_egfp.Citation40 PSyn is a constitutive promoter derived from Plac.
RNA degradation
The understanding of the mechanisms for processing and degradation of RNA (ribolysis) is a prerequisite for the understanding of RNA-based gene regulation for the following reasons: 1) Abundance and availability of an sRNA is important for its impact in the cell.Citation89,Citation90 2) The abundance (steady-state amount) of an sRNA is determined by the rate of decay in addition to the rate of transcription, and the rate of decay is a subject of regulation.Citation31,Citation91,Citation92 3) Some sRNAs are generated by processing of primary transcripts.Citation93,Citation94 4) Often the interaction between sRNAs and their mRNA targets regulates the stability of the target and in many cases both molecules are degraded simultaneously.Citation20,Citation95,Citation96 Generally two different mechanisms participate in this degradation. On the one hand, binding of an sRNA to the target RNA may result in a cleavage site specifically recognized by the double-strand specific ribonuclease (RNase) III.Citation97,Citation98 On the other hand, the imperfect basepairing of sRNAs and mRNAs is often mediated by Hfq, which directly binds to RNase E, a single strand specific RNase with major importance for decay of RNA in many bacteria. In this way RNase E is guided by an sRNA and Hfq to its cleavage site on an mRNA.Citation20 The guiding sRNA carries a monophosphate at the 5′-end and can be rapidly degraded by RNase E and other RNases which prefer processed RNAs.Citation99-Citation101 On the other hand, Hfq stabilizes the sRNAs which do not interact with the target protecting them from RNase E cleavage.Citation31,Citation102
The impact of RNA processing and degradation on sRNA-mediated gene regulation is best understood in Enterobacteriaceae.Citation20,Citation95,Citation99,Citation103 For most bacteria with genome-wide mapped sRNAs this information is still missing. In the case of plant-associated α-proteobacteria, some progress was made for the sRNA AbcR1 (originally named SmrC16), the levels of which are decreased in the stationary phase and in the absence of Hfq in S. meliloti.Citation5,Citation31,Citation40 In both cases increased degradation contributes to the decrease in the amount of the sRNA. In the wild type S. meliloti, the half-life of AbcR1 was 2.5-fold shorter during transition to the stationary phase when compared with the exponential growth phase, showing regulation of the AbcR1 expression at the posttranscriptional level.Citation31 In a Δhfq mutant, the half-life was decreased 2-fold during logarithmic growth and 6.5-fold during transition to the stationary phase when compared with the wild type.Citation31 This suggests a similar role for S. meliloti Hfq in riboregulation like in E. coli.Citation21 Most probably RNase E is responsible for the posttranscriptional regulation of AbcR1 expression, since the amounts and the stability of AbcR1 are strongly increased in an RNase E mutant of S. meliloti (E. Evguenieva-Hackenberg and A. Becker, unpublished). Probably RNase E and Hfq physically interact in α-proteobacteria like in E. coli. Their association was documented in Rhizobium leguminosarum, where Hfq is necessary for the RNase E-dependent activation of the translation of NifA.Citation59
The set of predicted degradative RNases in plant-associated α-proteobacteria includes the above mentioned endoribonucleases RNase E and RNase III, the 3′-5′ exoribonucleases RNase R and polynucleotide phosphorylase (PNPase), and RNase J, which is not present in E. coli but is an endo- and 5′-3′ exoribonuclease in B. subtilis.Citation14-Citation19,Citation104,Citation105 The analysis of mini-Tn5 mutants with insertions in the genes encoding RNase E and RNase J enabled first insights into mechanisms of RNA processing and degradation in S. meliloti. Both mutants showed specific and overlapping effects in a microarray analysis which suggested a role for both RNases in quorum sensing (E. Evguenieva-Hackenberg and A. Becker, unpublished).Citation106 RNase E was found to be necessary for the 5′-degradation of sinI mRNA encoding an autoinducer synthase. It specifically cleaves in the 5′-UTR of this mRNA between the Shine-Dalgarno sequence and the start codon in an Hfq-independent manner.Citation107 The role of RNase J in riboregulation is still not clear but it is responsible for the maturation of 5′-ends of rRNA in S. meliloti.Citation108
Conclusions
It is now well established that sRNAs are very abundant in plant-associated bacteria. Antisense transcripts and trans-sRNAs constitute the largest class of riboregulators in these bacteria. The functional role of only a small number of these riboregulators has been deciphered. Nonetheless, it is already emerging that some of them are global regulators with an important impact on bacterial physiology. Identifying the target genes of other sRNAs by bioinformatic and experimental methods and revealing the mechanistic principles of RNA-mediated regulation in these bacteria is going to be an interesting task for future research. Experimental tools tailored to plant-associated α-proteobacteria are being developed and will significantly promote functional analysis of sRNAs in these bacteria. Studies of the role of the ubiquitous RNA chaperone Hfq in several Rhizobiaceae suggested a broad impact of this protein on riboregulation, but also provided evidence for trans-RNAs that may not require Hfq for their function. This implies Hfq-independent regulatory mechanisms or the presence of additional rhizobial RNA chaperones involved in riboregulation that still await to be studied. A particularly fascinating endeavor will be to study the role of sRNAs in plant-microbe interaction, not only from the bacterial site but also from the plant perspective. It is conceivable that the plant response to contact with a bacterial symbiont or pathogen involves microRNAs or other regulatory RNAs in the plant. A promising technique to look into this on a global scale would be dual RNA-seq of microbe and host.Citation109
| Abbreviations: | ||
| AS | = | acetosyringone |
| asRNA | = | cis-acting antisense RNA |
| CM | = | covariance model |
| dRNA-seq | = | differential RNA-seq |
| GABA | = | γ-amino butyric acid |
| IGR | = | intergenic region |
| sRNA | = | small non-coding RNA |
| ORF | = | open reading frame |
| RFM | = | RNA family model |
| RNase | = | ribonuclease |
| TDM | = | thermodynamic matcher |
| trans-sRNA | = | trans-encoded sRNA |
| TSS | = | transcriptional start site |
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
This work was supported by the following grants from the Deutsche Forschungsgemeinschaft (DFG) in the framework of the priority program SPP 1258 “Sensory and Regulatory RNAs in Prokaryotes”: BE 2121/3–1, BE 2121/4–1, BE 2121/5–2, FN 240/8–1, GI 178/4–1, GI 178/4–2, Ev42/4–1, and Ev42/4–2.
References
- Kuykendall LD, Elkan GH. Rhizobium japonicum derivatives differing in nitrogen-fixing efficiency and carbohydrate utilization. Appl Environ Microbiol 1976; 32:511 - 9; PMID: 988784
- Segovia L, Young JP, Martínez-Romero E. Reclassification of American Rhizobium leguminosarum biovar phaseoli type I strains as Rhizobium etli sp. nov. Int J Syst Bacteriol 1993; 43:374 - 7; http://dx.doi.org/10.1099/00207713-43-2-374; PMID: 8494746
- Jones KM, Kobayashi H, Davies BW, Taga ME, Walker GC. How rhizobial symbionts invade plants: the Sinorhizobium-Medicago model. Nat Rev Microbiol 2007; 5:619 - 33; http://dx.doi.org/10.1038/nrmicro1705; PMID: 17632573
- Gelvin SB. Agrobacterium-mediated plant transformation: the biology behind the “gene-jockeying” tool. Microbiol Mol Biol Rev 2003; 67:16 - 37; http://dx.doi.org/10.1128/MMBR.67.1.16-37.2003; PMID: 12626681
- del Val C, Rivas E, Torres-Quesada O, Toro N, Jiménez-Zurdo JI. Identification of differentially expressed small non-coding RNAs in the legume endosymbiont Sinorhizobium meliloti by comparative genomics. Mol Microbiol 2007; 66:1080 - 91; http://dx.doi.org/10.1111/j.1365-2958.2007.05978.x; PMID: 17971083
- Ulvé VM, Sevin EW, Chéron A, Barloy-Hubler F. Identification of chromosomal alpha-proteobacterial small RNAs by comparative genome analysis and detection in Sinorhizobium meliloti strain 1021. BMC Genomics 2007; 8:467; http://dx.doi.org/10.1186/1471-2164-8-467; PMID: 18093320
- Valverde C, Livny J, Schlüter JP, Reinkensmeier J, Becker A, Parisi G. Prediction of Sinorhizobium meliloti sRNA genes and experimental detection in strain 2011. BMC Genomics 2008; 9:416; http://dx.doi.org/10.1186/1471-2164-9-416; PMID: 18793445
- Schlüter JP, Reinkensmeier J, Daschkey S, Evguenieva-Hackenberg E, Janssen S, Jänicke S, Becker JD, Giegerich R, Becker A. A genome-wide survey of sRNAs in the symbiotic nitrogen-fixing alpha-proteobacterium Sinorhizobium meliloti.. BMC Genomics 2010; 11:245; http://dx.doi.org/10.1186/1471-2164-11-245; PMID: 20398411
- Vercruysse M, Fauvart M, Cloots L, Engelen K, Thijs IM, Marchal K, Michiels J. Genome-wide detection of predicted non-coding RNAs in Rhizobium etli expressed during free-living and host-associated growth using a high-resolution tiling array. BMC Genomics 2010; 11:53; http://dx.doi.org/10.1186/1471-2164-11-53; PMID: 20089193
- Madhugiri R, Pessi G, Voss B, Hahn J, Sharma CM, Reinhardt R, Vogel J, Hess WR, Fischer HM, Evguenieva-Hackenberg E. Small RNAs of the Bradyrhizobium/Rhodopseudomonas lineage and their analysis. RNA Biol 2012; 9:47 - 58; http://dx.doi.org/10.4161/rna.9.1.18008; PMID: 22258152
- Wilms I, Overlöper A, Nowrousian M, Sharma CM, Narberhaus F. Deep sequencing uncovers numerous small RNAs on all four replicons of the plant pathogen Agrobacterium tumefaciens.. RNA Biol 2012; 9:446 - 57; http://dx.doi.org/10.4161/rna.17212; PMID: 22336765
- Lee K, Huang X, Yang C, Lee D, Ho V, Nobuta K, Fan JB, Wang K. A genome-wide survey of highly expressed non-coding RNAs and biological validation of selected candidates in Agrobacterium tumefaciens.. PLoS One 2013; 8:e70720; http://dx.doi.org/10.1371/journal.pone.0070720; PMID: 23950988
- Fischer HM. Genetic regulation of nitrogen fixation in rhizobia. Microbiol Rev 1994; 58:352 - 86; PMID: 7968919
- Paulsen IT, Seshadri R, Nelson KE, Eisen JA, Heidelberg JF, Read TD, Dodson RJ, Umayam L, Brinkac LM, Beanan MJ, et al. The Brucella suis genome reveals fundamental similarities between animal and plant pathogens and symbionts. Proc Natl Acad Sci U S A 2002; 99:13148 - 53; http://dx.doi.org/10.1073/pnas.192319099; PMID: 12271122
- Galibert F, Finan TM, Long SR, Puhler A, Abola P, Ampe F, Barloy-Hubler F, Barnett MJ, Becker A, Boistard P, et al. The composite genome of the legume symbiont Sinorhizobium meliloti.. Science 2001; 293:668 - 72; http://dx.doi.org/10.1126/science.1060966; PMID: 11474104
- Goodner B, Hinkle G, Gattung S, Miller N, Blanchard M, Qurollo B, Goldman BS, Cao Y, Askenazi M, Halling C, et al. Genome sequence of the plant pathogen and biotechnology agent Agrobacterium tumefaciens C58. Science 2001; 294:2323 - 8; http://dx.doi.org/10.1126/science.1066803; PMID: 11743194
- Wood DW, Setubal JC, Kaul R, Monks DE, Kitajima JP, Okura VK, Zhou Y, Chen L, Wood GE, Almeida NF Jr., et al. The genome of the natural genetic engineer Agrobacterium tumefaciens C58.. Science 2001; 294:2317 - 23; http://dx.doi.org/10.1126/science.1066804; PMID: 11743193
- Kaneko T, Nakamura Y, Sato S, Minamisawa K, Uchiumi T, Sasamoto S, Watanabe A, Idesawa K, Iriguchi M, Kawashima K, et al. Complete genomic sequence of nitrogen-fixing symbiotic bacterium Bradyrhizobium japonicum USDA110.. DNA Res 2002; 9:189 - 97; http://dx.doi.org/10.1093/dnares/9.6.189; PMID: 12597275
- González V, Santamaría RI, Bustos P, Hernández-González I, Medrano-Soto A, Moreno-Hagelsieb G, Janga SC, Ramírez MA, Jiménez-Jacinto V, Collado-Vides J, et al. The partitioned Rhizobium etli genome: genetic and metabolic redundancy in seven interacting replicons. Proc Natl Acad Sci U S A 2006; 103:3834 - 9; http://dx.doi.org/10.1073/pnas.0508502103; PMID: 16505379
- Morita T, Maki K, Aiba H. RNase E-based ribonucleoprotein complexes: mechanical basis of mRNA destabilization mediated by bacterial noncoding RNAs. Genes Dev 2005; 19:2176 - 86; http://dx.doi.org/10.1101/gad.1330405; PMID: 16166379
- Vogel J, Luisi BF. Hfq and its constellation of RNA. Nat Rev Microbiol 2011; 9:578 - 89; http://dx.doi.org/10.1038/nrmicro2615; PMID: 21760622
- Papenfort K, Pfeiffer V, Mika F, Lucchini S, Hinton JC, Vogel J. SigmaE-dependent small RNAs of Salmonella respond to membrane stress by accelerating global omp mRNA decay. Mol Microbiol 2006; 62:1674 - 88; http://dx.doi.org/10.1111/j.1365-2958.2006.05524.x; PMID: 17427289
- Lenz DH, Mok KC, Lilley BN, Kulkarni RV, Wingreen NS, Bassler BL. The small RNA chaperone Hfq and multiple small RNAs control quorum sensing in Vibrio harveyi and Vibrio cholerae.. Cell 2004; 118:69 - 82; http://dx.doi.org/10.1016/j.cell.2004.06.009; PMID: 15242645
- Boisset S, Geissmann T, Huntzinger E, Fechter P, Bendridi N, Possedko M, Chevalier C, Helfer AC, Benito Y, Jacquier A, et al. Staphylococcus aureus RNAIII coordinately represses the synthesis of virulence factors and the transcription regulator Rot by an antisense mechanism. Genes Dev 2007; 21:1353 - 66; http://dx.doi.org/10.1101/gad.423507; PMID: 17545468
- Heroven AK, Böhme K, Rohde M, Dersch P. A Csr-type regulatory system, including small non-coding RNAs, regulates the global virulence regulator RovA of Yersinia pseudotuberculosis through RovM. Mol Microbiol 2008; 68:1179 - 95; http://dx.doi.org/10.1111/j.1365-2958.2008.06218.x; PMID: 18430141
- Sharma CM, Hoffmann S, Darfeuille F, Reignier J, Findeiss S, Sittka A, Chabas S, Reiche K, Hackermüller J, Reinhardt R, et al. The primary transcriptome of the major human pathogen Helicobacter pylori.. Nature 2010; 464:250 - 5; http://dx.doi.org/10.1038/nature08756; PMID: 20164839
- Schlüter JP, Reinkensmeier J, Barnett MJ, Lang C, Krol E, Giegerich R, Long SR, Becker A. Global mapping of transcription start sites and promoter motifs in the symbiotic α-proteobacterium Sinorhizobium meliloti 1021. BMC Genomics 2013; 14:156; http://dx.doi.org/10.1186/1471-2164-14-156; PMID: 23497287
- Torres-Quesada O, Reinkensmeier J, Schlüter J-P, Robledo M, Peregrina A, Giegerich R, Toro N, Becker A, Jiménez-Zurdo JI. Genome-wide profiling of Hfq-binding RNAs uncovers extensive post-transcriptional rewiring of major stress response and symbiotic regulons in Sinorhizobium meliloti.. RNA Biol 2014; 11; In press http://dx.doi.org/10.4161/rna.28239; PMID: 24786641
- Wilms I, Voss B, Hess WR, Leichert LI, Narberhaus F. Small RNA-mediated control of the Agrobacterium tumefaciens GABA binding protein. Mol Microbiol 2011; 80:492 - 506; http://dx.doi.org/10.1111/j.1365-2958.2011.07589.x; PMID: 21320185
- Corbino KA, Barrick JE, Lim J, Welz R, Tucker BJ, Puskarz I, Mandal M, Rudnick ND, Breaker RR. Evidence for a second class of S-adenosylmethionine riboswitches and other regulatory RNA motifs in alpha-proteobacteria. Genome Biol 2005; 6:R70; http://dx.doi.org/10.1186/gb-2005-6-8-r70; PMID: 16086852
- Voss B, Hölscher M, Baumgarth B, Kalbfleisch A, Kaya C, Hess WR, Becker A, Evguenieva-Hackenberg E. Expression of small RNAs in Rhizobiales and protection of a small RNA and its degradation products by Hfq in Sinorhizobium meliloti.. Biochem Biophys Res Commun 2009; 390:331 - 6; http://dx.doi.org/10.1016/j.bbrc.2009.09.125; PMID: 19800865
- Wassarman KM, Storz G. 6S RNA regulates E. coli RNA polymerase activity. Cell 2000; 101:613 - 23; http://dx.doi.org/10.1016/S0092-8674(00)80873-9; PMID: 10892648
- Wassarman KM, Saecker RM. Synthesis-mediated release of a small RNA inhibitor of RNA polymerase. Science 2006; 314:1601 - 3; http://dx.doi.org/10.1126/science.1134830; PMID: 17158328
- Reinkensmeier J, Schlüter JP, Giegerich R, Becker A. Conservation and occurrence of trans-encoded sRNAs in the Rhizobiales. Genes (Basel) 2011; 2:925 - 56; http://dx.doi.org/10.3390/genes2040925; PMID: 24710299
- Nawrocki EP, Kolbe DL, Eddy SR. Infernal 1.0: inference of RNA alignments. Bioinformatics 2009; 25:1335 - 7; http://dx.doi.org/10.1093/bioinformatics/btp157; PMID: 19307242
- Reeder J, Reeder J, Giegerich R. Locomotif: from graphical motif description to RNA motif search. Bioinformatics 2007; 23:i392 - 400; http://dx.doi.org/10.1093/bioinformatics/btm179; PMID: 17646322
- Sauthoff G, Möhl M, Janssen S, Giegerich R. Bellman’s GAP--a language and compiler for dynamic programming in sequence analysis. Bioinformatics 2013; 29:551 - 60; http://dx.doi.org/10.1093/bioinformatics/btt022; PMID: 23355290
- del Val C, Romero-Zaliz R, Torres-Quesada O, Peregrina A, Toro N, Jiménez-Zurdo JI. A survey of sRNA families in α-proteobacteria. RNA Biol 2012; 9:119 - 29; http://dx.doi.org/10.4161/rna.18643; PMID: 22418845
- Young JP, Crossman LC, Johnston AW, Thomson NR, Ghazoui ZF, Hull KH, Wexler M, Curson AR, Todd JD, Poole PS, et al. The genome of Rhizobium leguminosarum has recognizable core and accessory components. Genome Biol 2006; 7:R34; http://dx.doi.org/10.1186/gb-2006-7-4-r34; PMID: 16640791
- Torres-Quesada O, Millán V, Nisa-Martínez R, Bardou F, Crespi M, Toro N, Jiménez-Zurdo JI. Independent activity of the homologous small regulatory RNAs AbcR1 and AbcR2 in the legume symbiont Sinorhizobium meliloti.. PLoS One 2013; 8:e68147; http://dx.doi.org/10.1371/journal.pone.0068147; PMID: 23869210
- Caswell CC, Gaines JM, Ciborowski P, Smith D, Borchers CH, Roux CM, Sayood K, Dunman PM, Roop Ii RM. Identification of two small regulatory RNAs linked to virulence in Brucella abortus 2308. Mol Microbiol 2012; 85:345 - 60; http://dx.doi.org/10.1111/j.1365-2958.2012.08117.x; PMID: 22690807
- Berghoff BA, Glaeser J, Sharma CM, Zobawa M, Lottspeich F, Vogel J, Klug G. Contribution of Hfq to photooxidative stress resistance and global regulation in Rhodobacter sphaeroides.. Mol Microbiol 2011; 80:1479 - 95; http://dx.doi.org/10.1111/j.1365-2958.2011.07658.x; PMID: 21535243
- Babitzke P, Romeo T. CsrB sRNA family: sequestration of RNA-binding regulatory proteins. Curr Opin Microbiol 2007; 10:156 - 63; http://dx.doi.org/10.1016/j.mib.2007.03.007; PMID: 17383221
- Watson RJ, Heys R. Replication regions of Sinorhizobium meliloti plasmids. Plasmid 2006; 55:87 - 98; http://dx.doi.org/10.1016/j.plasmid.2005.08.003; PMID: 16202450
- Sobrero P, Valverde C. The bacterial protein Hfq: much more than a mere RNA-binding factor. Crit Rev Microbiol 2012; 38:276 - 99; http://dx.doi.org/10.3109/1040841X.2012.664540; PMID: 22435753
- Kaminski PA, Desnoues N, Elmerich C. The expression of nifA in Azorhizobium caulinodans requires a gene product homologous to Escherichia coli HF-I, an RNA-binding protein involved in the replication of phage Q beta RNA. Proc Natl Acad Sci U S A 1994; 91:4663 - 7; http://dx.doi.org/10.1073/pnas.91.11.4663; PMID: 8197116
- Sobrero P, Valverde C. Evidences of autoregulation of hfq expression in Sinorhizobium meliloti strain 2011. Arch Microbiol 2011; 193:629 - 39; http://dx.doi.org/10.1007/s00203-011-0701-1; PMID: 21484295
- Storz G, Vogel J, Wassarman KM. Regulation by small RNAs in bacteria: expanding frontiers. Mol Cell 2011; 43:880 - 91; http://dx.doi.org/10.1016/j.molcel.2011.08.022; PMID: 21925377
- Sittka A, Lucchini S, Papenfort K, Sharma CM, Rolle K, Binnewies TT, Hinton JC, Vogel J. Deep sequencing analysis of small noncoding RNA and mRNA targets of the global post-transcriptional regulator, Hfq. PLoS Genet 2008; 4:e1000163; http://dx.doi.org/10.1371/journal.pgen.1000163; PMID: 18725932
- Sittka A, Sharma CM, Rolle K, Vogel J. Deep sequencing of Salmonella RNA associated with heterologous Hfq proteins in vivo reveals small RNAs as a major target class and identifies RNA processing phenotypes. RNA Biol 2009; 6:266 - 75; http://dx.doi.org/10.4161/rna.6.3.8332; PMID: 19333007
- Lorenz C, Gesell T, Zimmermann B, Schoeberl U, Bilusic I, Rajkowitsch L, Waldsich C, von Haeseler A, Schroeder R. Genomic SELEX for Hfq-binding RNAs identifies genomic aptamers predominantly in antisense transcripts. Nucleic Acids Res 2010; 38:3794 - 808; http://dx.doi.org/10.1093/nar/gkq032; PMID: 20348540
- Chao Y, Papenfort K, Reinhardt R, Sharma CM, Vogel J. An atlas of Hfq-bound transcripts reveals 3′ UTRs as a genomic reservoir of regulatory small RNAs. EMBO J 2012; 31:4005 - 19; http://dx.doi.org/10.1038/emboj.2012.229; PMID: 22922465
- Dambach M, Irnov I, Winkler WC. Association of RNAs with Bacillus subtilis Hfq. PLoS One 2013; 8:e55156; http://dx.doi.org/10.1371/journal.pone.0055156; PMID: 23457461
- Vytvytska O, Moll I, Kaberdin VR, von Gabain A, Bläsi U. Hfq (HF1) stimulates ompA mRNA decay by interfering with ribosome binding. Genes Dev 2000; 14:1109 - 18; PMID: 10809669
- Mohanty BK, Maples VF, Kushner SR. The Sm-like protein Hfq regulates polyadenylation dependent mRNA decay in Escherichia coli.. Mol Microbiol 2004; 54:905 - 20; http://dx.doi.org/10.1111/j.1365-2958.2004.04337.x; PMID: 15522076
- Barra-Bily L, Pandey SP, Trautwetter A, Blanco C, Walker GC. The Sinorhizobium meliloti RNA chaperone Hfq mediates symbiosis of S. meliloti and alfalfa. J Bacteriol 2010; 192:1710 - 8; http://dx.doi.org/10.1128/JB.01427-09; PMID: 20081033
- Wilms I, Möller P, Stock AM, Gurski R, Lai EM, Narberhaus F. Hfq influences multiple transport systems and virulence in the plant pathogen Agrobacterium tumefaciens.. J Bacteriol 2012; 194:5209 - 17; http://dx.doi.org/10.1128/JB.00510-12; PMID: 22821981
- Kaminski PA, Elmerich C. The control of Azorhizobium caulinodans nifA expression by oxygen, ammonia and by the HF-I-like protein, NrfA. Mol Microbiol 1998; 28:603 - 13; http://dx.doi.org/10.1046/j.1365-2958.1998.00823.x; PMID: 9632262
- Zhang Y, Hong G. Post-transcriptional regulation of NifA expression by Hfq and RNase E complex in Rhizobium leguminosarum bv. viciae.. Acta Biochim Biophys Sin (Shanghai) 2009; 41:719 - 30; http://dx.doi.org/10.1093/abbs/gmp060; PMID: 19727520
- Barra-Bily L, Fontenelle C, Jan G, Flechard M, Trautwetter A, Pandey SP, Walker GC, Blanco C. Proteomic alterations explain phenotypic changes in Sinorhizobium meliloti lacking the RNA chaperone Hfq. J Bacteriol 2010; 192:1719 - 29; http://dx.doi.org/10.1128/JB.01429-09; PMID: 20081032
- Gao M, Barnett MJ, Long SR, Teplitski M. Role of the Sinorhizobium meliloti global regulator Hfq in gene regulation and symbiosis. Mol Plant Microbe Interact 2010; 23:355 - 65; http://dx.doi.org/10.1094/MPMI-23-4-0355; PMID: 20192823
- Torres-Quesada O, Oruezábal RI, Peregrina A, Jofré E, Lloret J, Rivilla R, Toro N, Jiménez-Zurdo JI. The Sinorhizobium meliloti RNA chaperone Hfq influences central carbon metabolism and the symbiotic interaction with alfalfa. BMC Microbiol 2010; 10:71; http://dx.doi.org/10.1186/1471-2180-10-71; PMID: 20205931
- Sobrero P, Schlüter JP, Lanner U, Schlosser A, Becker A, Valverde C. Quantitative proteomic analysis of the Hfq-regulon in Sinorhizobium meliloti 2011. PLoS One 2012; 7:e48494; http://dx.doi.org/10.1371/journal.pone.0048494; PMID: 23119037
- Drepper T, Raabe K, Giaourakis D, Gendrullis M, Masepohl B, Klipp W. The Hfq-like protein NrfA of the phototrophic purple bacterium Rhodobacter capsulatus controls nitrogen fixation via regulation of nifA and anfA expression. FEMS Microbiol Lett 2002; 215:221 - 7; http://dx.doi.org/10.1111/j.1574-6968.2002.tb11394.x; PMID: 12399038
- Mulley G, White JP, Karunakaran R, Prell J, Bourdes A, Bunnewell S, Hill L, Poole PS. Mutation of GOGAT prevents pea bacteroid formation and N2 fixation by globally downregulating transport of organic nitrogen sources. Mol Microbiol 2011; 80:149 - 67; http://dx.doi.org/10.1111/j.1365-2958.2011.07565.x; PMID: 21276099
- Robertson GT, Roop RM Jr.. The Brucella abortus host factor I (HF-I) protein contributes to stress resistance during stationary phase and is a major determinant of virulence in mice. Mol Microbiol 1999; 34:690 - 700; http://dx.doi.org/10.1046/j.1365-2958.1999.01629.x; PMID: 10564509
- Roop RM 2nd, Robertson GT, Ferguson GP, Milford LE, Winkler ME, Walker GC. Seeking a niche: putative contributions of the hfq and bacA gene products to the successful adaptation of the brucellae to their intracellular home. Vet Microbiol 2002; 90:349 - 63; http://dx.doi.org/10.1016/S0378-1135(02)00220-1; PMID: 12414155
- Pandey SP, Minesinger BK, Kumar J, Walker GC. A highly conserved protein of unknown function in Sinorhizobium meliloti affects sRNA regulation similar to Hfq. Nucleic Acids Res 2011; 39:4691 - 708; http://dx.doi.org/10.1093/nar/gkr060; PMID: 21325267
- Pandey SP, Winkler JA, Li H, Camacho DM, Collins JJ, Walker GC. Central role for RNase YbeY in Hfq-dependent and Hfq-independent small-RNA regulation in bacteria. BMC Genomics 2014; 15:121; http://dx.doi.org/10.1186/1471-2164-15-121; PMID: 24511998
- Davies BW, Köhrer C, Jacob AI, Simmons LA, Zhu J, Aleman LM, Rajbhandary UL, Walker GC. Role of Escherichia coli YbeY, a highly conserved protein, in rRNA processing. Mol Microbiol 2010; 78:506 - 18; http://dx.doi.org/10.1111/j.1365-2958.2010.07351.x; PMID: 20807199
- Grinwald M, Ron EZ. The Escherichia coli translation-associated heat shock protein YbeY is involved in rRNA transcription antitermination. PLoS One 2013; 8:e62297; http://dx.doi.org/10.1371/journal.pone.0062297; PMID: 23638028
- Jacob AI, Köhrer C, Davies BW, RajBhandary UL, Walker GC. Conserved bacterial RNase YbeY plays key roles in 70S ribosome quality control and 16S rRNA maturation. Mol Cell 2013; 49:427 - 38; http://dx.doi.org/10.1016/j.molcel.2012.11.025; PMID: 23273979
- Wassarman KM. 6S RNA: a regulator of transcription. Mol Microbiol 2007; 65:1425 - 31; http://dx.doi.org/10.1111/j.1365-2958.2007.05894.x; PMID: 17714443
- Egea PF, Stroud RM, Walter P. Targeting proteins to membranes: structure of the signal recognition particle. Curr Opin Struct Biol 2005; 15:213 - 20; http://dx.doi.org/10.1016/j.sbi.2005.03.007; PMID: 15837181
- Altman S. A view of RNase P. Mol Biosyst 2007; 3:604 - 7; http://dx.doi.org/10.1039/b707850c; PMID: 17700860
- http://rfam.sanger.ac.uk/
- Ebeling S, Kündig C, Hennecke H. Discovery of a rhizobial RNA that is essential for symbiotic root nodule development. J Bacteriol 1991; 173:6373 - 82; PMID: 1717438
- Keiler KC, Shapiro L, Williams KP. tmRNAs that encode proteolysis-inducing tags are found in all known bacterial genomes: A two-piece tmRNA functions in Caulobacter.. Proc Natl Acad Sci U S A 2000; 97:7778 - 83; http://dx.doi.org/10.1073/pnas.97.14.7778; PMID: 10884408
- Williams KP. The tmRNA Website: invasion by an intron. Nucleic Acids Res 2002; 30:179 - 82; http://dx.doi.org/10.1093/nar/30.1.179; PMID: 11752287
- MacLellan SR, Smallbone LA, Sibley CD, Finan TM. The expression of a novel antisense gene mediates incompatibility within the large repABC family of α-proteobacterial plasmids. Mol Microbiol 2005; 55:611 - 23; http://dx.doi.org/10.1111/j.1365-2958.2004.04412.x; PMID: 15659174
- Chai Y, Winans SC. A small antisense RNA downregulates expression of an essential replicase protein of an Agrobacterium tumefaciens Ti plasmid. Mol Microbiol 2005; 56:1574 - 85; http://dx.doi.org/10.1111/j.1365-2958.2005.04636.x; PMID: 15916607
- Chevrot R, Rosen R, Haudecoeur E, Cirou A, Shelp BJ, Ron E, Faure D. GABA controls the level of quorum-sensing signal in Agrobacterium tumefaciens.. Proc Natl Acad Sci U S A 2006; 103:7460 - 4; http://dx.doi.org/10.1073/pnas.0600313103; PMID: 16645034
- Planamente S, Moréra S, Faure D. In planta fitness-cost of the Atu4232-regulon encoding for a selective GABA-binding sensor in Agrobacterium. Commun Integr Biol 2013; 6:e23692; http://dx.doi.org/10.4161/cib.23692; PMID: 23710277
- Overlöper A, Kraus A, Gurski R, Wright PR, Georg J, Hess WR, Narberhaus F. Two separate modules of the conserved regulatory RNA AbcR1 address multiple target mRNAs in and outside of the translation initiation region. RNA Biol 2014; 11; http://dx.doi.org/10.4161/rna.29145; PMID: 24921646
- Wright PR, Richter AS, Papenfort K, Mann M, Vogel J, Hess WR, Backofen R, Georg J. Comparative genomics boosts target prediction for bacterial small RNAs. Proc Natl Acad Sci U S A 2013; 110:E3487 - 96; http://dx.doi.org/10.1073/pnas.1303248110; PMID: 23980183
- Urban JH, Vogel J. Translational control and target recognition by Escherichia coli small RNAs in vivo.. Nucleic Acids Res 2007; 35:1018 - 37; http://dx.doi.org/10.1093/nar/gkl1040; PMID: 17264113
- McIntosh M, Meyer S, Becker A. Novel Sinorhizobium meliloti quorum sensing positive and negative regulatory feedback mechanisms respond to phosphate availability. Mol Microbiol 2009; 74:1238 - 56; http://dx.doi.org/10.1111/j.1365-2958.2009.06930.x; PMID: 19889097
- McIntosh M, Czuppon P, Best K, Becker A, Pfaffelhuber P. Modeling quorum sensing in Sinorhizobium meliloti.. International Journal of Biomathematics and Biostatistics 2013; 2:59 - 74
- Chen S, Zhang A, Blyn LB, Storz G. MicC, a second small-RNA regulator of Omp protein expression in Escherichia coli.. J Bacteriol 2004; 186:6689 - 97; http://dx.doi.org/10.1128/JB.186.20.6689-6697.2004; PMID: 15466019
- Dühring U, Axmann IM, Hess WR, Wilde A. An internal antisense RNA regulates expression of the photosynthesis gene isiA.. Proc Natl Acad Sci U S A 2006; 103:7054 - 8; http://dx.doi.org/10.1073/pnas.0600927103; PMID: 16636284
- Basineni SR, Madhugiri R, Kolmsee T, Hengge R, Klug G. The influence of Hfq and ribonucleases on the stability of the small non-coding RNA OxyS and its target rpoS in E. coli is growth phase dependent. RNA Biol 2009; 6:584 - 94; http://dx.doi.org/10.4161/rna.6.5.10082; PMID: 20016254
- Madhugiri R, Basineni SR, Klug G. Turn-over of the small non-coding RNA RprA in E. coli is influenced by osmolarity. Mol Genet Genomics 2010; 284:307 - 18; http://dx.doi.org/10.1007/s00438-010-0568-x; PMID: 20717695
- Kim KS, Lee Y. Regulation of 6S RNA biogenesis by switching utilization of both sigma factors and endoribonucleases. Nucleic Acids Res 2004; 32:6057 - 68; http://dx.doi.org/10.1093/nar/gkh939; PMID: 15550566
- Papenfort K, Said N, Welsink T, Lucchini S, Hinton JC, Vogel J. Specific and pleiotropic patterns of mRNA regulation by ArcZ, a conserved, Hfq-dependent small RNA. Mol Microbiol 2009; 74:139 - 58; http://dx.doi.org/10.1111/j.1365-2958.2009.06857.x; PMID: 19732340
- Massé E, Escorcia FE, Gottesman S. Coupled degradation of a small regulatory RNA and its mRNA targets in Escherichia coli.. Genes Dev 2003; 17:2374 - 83; http://dx.doi.org/10.1101/gad.1127103; PMID: 12975324
- Rasmussen AA, Eriksen M, Gilany K, Udesen C, Franch T, Petersen C, Valentin-Hansen P. Regulation of ompA mRNA stability: the role of a small regulatory RNA in growth phase-dependent control. Mol Microbiol 2005; 58:1421 - 9; http://dx.doi.org/10.1111/j.1365-2958.2005.04911.x; PMID: 16313626
- Afonyushkin T, Vecerek B, Moll I, Bläsi U, Kaberdin VR. Both RNase E and RNase III control the stability of sodB mRNA upon translational inhibition by the small regulatory RNA RyhB. Nucleic Acids Res 2005; 33:1678 - 89; http://dx.doi.org/10.1093/nar/gki313; PMID: 15781494
- Viegas SC, Silva IJ, Saramago M, Domingues S, Arraiano CM. Regulation of the small regulatory RNA MicA by ribonuclease III: a target-dependent pathway. Nucleic Acids Res 2011; 39:2918 - 30; http://dx.doi.org/10.1093/nar/gkq1239; PMID: 21138960
- Bandyra KJ, Said N, Pfeiffer V, Górna MW, Vogel J, Luisi BF. The seed region of a small RNA drives the controlled destruction of the target mRNA by the endoribonuclease RNase E. Mol Cell 2012; 47:943 - 53; http://dx.doi.org/10.1016/j.molcel.2012.07.015; PMID: 22902561
- Mackie GA. Ribonuclease E is a 5′-end-dependent endonuclease. Nature 1998; 395:720 - 3; http://dx.doi.org/10.1038/27246; PMID: 9790196
- Evguenieva-Hackenberg E, Klug G. New aspects of RNA processing in prokaryotes. Curr Opin Microbiol 2011; 14:587 - 92; http://dx.doi.org/10.1016/j.mib.2011.07.025; PMID: 21945217
- Moll I, Afonyushkin T, Vytvytska O, Kaberdin VR, Bläsi U. Coincident Hfq binding and RNase E cleavage sites on mRNA and small regulatory RNAs. RNA 2003; 9:1308 - 14; http://dx.doi.org/10.1261/rna.5850703; PMID: 14561880
- Viegas SC, Pfeiffer V, Sittka A, Silva IJ, Vogel J, Arraiano CM. Characterization of the role of ribonucleases in Salmonella small RNA decay. Nucleic Acids Res 2007; 35:7651 - 64; http://dx.doi.org/10.1093/nar/gkm916; PMID: 17982174
- Arraiano CM, Andrade JM, Domingues S, Guinote IB, Malecki M, Matos RG, Moreira RN, Pobre V, Reis FP, Saramago M, et al. The critical role of RNA processing and degradation in the control of gene expression. FEMS Microbiol Rev 2010; 34:883 - 923; PMID: 20659169
- Mathy N, Bénard L, Pellegrini O, Daou R, Wen T, Condon C. 5′-to-3′ exoribonuclease activity in bacteria: role of RNase J1 in rRNA maturation and 5′ stability of mRNA. Cell 2007; 129:681 - 92; http://dx.doi.org/10.1016/j.cell.2007.02.051; PMID: 17512403
- Pobigaylo N, Wetter D, Szymczak S, Schiller U, Kurtz S, Meyer F, Nattkemper TW, Becker A. Construction of a large signature-tagged mini-Tn5 transposon library and its application to mutagenesis of Sinorhizobium meliloti.. Appl Environ Microbiol 2006; 72:4329 - 37; http://dx.doi.org/10.1128/AEM.03072-05; PMID: 16751548
- Baumgardt K, Charoenpanich P, McIntosh M, Schikora A, Stein E, Thalmann S, Kogel KH, Klug G, Becker A, Evguenieva-Hackenberg E. RNase E affects the expression of the acyl-homoserine lactone synthase gene sinI in Sinorhizobium meliloti.. J Bacteriol 2014; 196:1435 - 47; http://dx.doi.org/10.1128/JB.01471-13; PMID: 24488310
- Madhugiri R, Evguenieva-Hackenberg E. RNase J is involved in the 5′-end maturation of 16S rRNA and 23S rRNA in Sinorhizobium meliloti.. FEBS Lett 2009; 583:2339 - 42; http://dx.doi.org/10.1016/j.febslet.2009.06.026; PMID: 19540834
- Westermann AJ, Gorski SA, Vogel J. Dual RNA-seq of pathogen and host. Nat Rev Microbiol 2012; 10:618 - 30; http://dx.doi.org/10.1038/nrmicro2852; PMID: 22890146
- Gruber AR, Lorenz R, Bernhart SH, Neuböck R, Hofacker IL. The Vienna RNA websuite. Nucleic Acids Res 2008; 36:W70-4; http://dx.doi.org/10.1093/nar/gkn188; PMID: 18424795