Abstract
Dengue virus (DENV) is a rapidly re-emerging flavivirus that causes dengue fever (DF), dengue hemorrhagic fever (DHF) and dengue shock syndrome (DSS), diseases for which there are no available therapies or vaccines. The DENV-2 positive-strand RNA genome contains 5’ and 3’ untranslated regions (UTRs) that have been shown to form secondary structures required for virus replication and interaction with host cell proteins. In order to comprehensively identify host cell factors that bind the DENV-2 UTRs, we performed RNA chromatography, using the DENV-2 5’ and 3’ UTRs as “bait”, combined with quantitative mass spectrometry. We identified several proteins, including DDX6, G3BP1, G3BP2, Caprin1, and USP10, implicated in P body (PB) and stress granule (SG) function, and not previously known to bind DENV RNAs. Indirect immunofluorescence microscopy showed these proteins to colocalize with the DENV replication complex. Moreover, DDX6 knockdown resulted in reduced amounts of infectious particles and viral RNA in tissue culture supernatants following DENV infection. DDX6 interacted with DENV RNA in vivo during infection and in vitro this interaction was mediated by the DB1 and DB2 structures in the 3’ UTR, possibly by formation of a pseudoknot structure. Additional experiments demonstrate that, in contrast to DDX6, the SG proteins G3BP1, G3BP2, Caprin1 and USP10 bind to the variable region (VR) in the 3’ UTR. These results suggest that the DENV-2 3’ UTR is a site for assembly of PB and SG proteins and, for DDX6, assembly on the 3’ UTR is required for DENV replication.
Introduction
The four Dengue viruses (DENV 1–4) are enveloped, positive strand RNA viruses of the family Flaviviridae, which also includes the yellow fever, West Nile and Hepatitis C viruses. The DENV positive strand RNA has several important roles in viral replication, it acts as: mRNA for translation of viral proteins, template for the synthesis of the negative strand in viral transcription, and genome packaged into progeny virions. The DENV RNA is translated into a single polyprotein that is subsequently cleaved into the structural and non-structural proteins by a combination of cellular and viral proteases. The structural proteins, capsid (C), pre-membrane/membrane (prM/M) and envelope (E), are assembled into the infectious virions along with DENV RNA genome. The non-structural proteins, NS1, NS2A, NS2B, NS3, NS4A, NS4B and NS5, perform a variety of functions in the host cell during infection. NS3 and NS5 are directly implicated in viral RNA replication, with NS3 acting as a RNA helicase and NS5 acting as the RNA-dependent RNA polymerase. To initiate viral RNA replication, the positive strand viral RNA is transcribed into a complementary negative RNA strand that acts as the template for synthesis of new positive strands. This newly synthesized positive strand DENV RNA can be translated into additional viral proteins or packaged into progeny virions for subsequent rounds of infection.Citation1
DENV RNA contains secondary structures in the 5′ and 3′ UTRs that mediate translation and viral RNA transcription.Citation2–Citation6 The 5′ stem-loop A (SLA) is required for DENV RNA replication in vitro and in the DENV replicon system, and interacts directly with DENV NS5.Citation7 The cHP element is located adjacent to the start codon for the virus polyprotein and has been implicated in virus translation and replication in DENV and replicon systems.Citation8,Citation9 Using luciferase reporter constructs containing the DENV 3′ UTR and 3′ SLA mutants, Holden and Harris demonstrated that the 3′ SLA enhances translation.Citation10 The A2 and A3 structures (also called DB2 and DB1, respectively) in the 3′ UTR are required for RNA replication but not translation of a DENV replicon.Citation11 Interestingly, a 30 nucleotide deletion corresponding to the DB1 structure attenuates all four DENV serotypes and is currently being tested as a vaccine candidate.Citation12
Long-range RNA-RNA interactions mediate circularization of the DENV genome via complementary sequences in the 5′ and 3′ UTR. The cyclization sequence (CS) mediates base-pairing via complementary sequences in the 5′ and 3′ UTR and is required for virus replication but not for translation.Citation13 Additional complementary sequences contained in the 5′ and 3′ termini have also been shown to be required for DENV replication, notably the upstream of AUG region (UAR) and downstream of AUG region (DAR).Citation14,Citation15 Studies using either mutagenesis to disrupt sequence complementarity or morpholino oligonucleotides that target the 5′ or 3′ stem-loops, CS or UAR, demonstrate that both local RNA secondary structures and long-range interactions are critical for DENV virus replication.Citation11,Citation14,Citation16
As obligate parasites, viruses rely on host cell proteins to establish successful infection and accomplish viral replication. In both human and mosquito cells, Sjogren syndrome antigen B protein (SSB), formerly known as La autoantigen protein, has been shown to bind to the 5′ and 3′ UTRs of DENV RNA and in human cells forms a complex with NS3 and NS5.Citation17,Citation18 Additional experiments demonstrated that SSB inhibits DENV RNA replication in vitro.Citation18 Another study demonstrated that the Y-box binding protein (YBX1), as well as hnRNP A1, A2/B1 and Q, bind to the DENV 3′ UTR and that YBX1 has a negative effect on DENV translation.Citation19 The stress granule protein TIA1 cytotoxic granule-associated RNA binding protein-like 1 (TIAL1) has been shown to colocalize with sites of DENV replication.Citation20 While the role of TIAL1 in DENV replication remains unclear, studies using West Nile virus (WNV), a related flavivirus, show that the accumulation of TIAL1 in replication complexes may facilitate viral transcription of the positive strand RNA from the negative strand template.Citation21 More recently, the polypyrimidine tract-binding protein (PTB) was shown to interact with DENV RNA and to be required for RNA accumulation, but not for translation.Citation22 Finally, genome-wide screens utilizing RNA interference have identified hundreds of host cell proteins required for WNV and DENV replication, highlighting that DENV virus relies heavily on the host cell to accomplish virus replication.Citation23,Citation24
In a screen for host cell proteins that physically interact with the DENV-2 5′ and 3′ UTRs, we identified several proteins associated with P-bodies (PBs) and stress granules (SG). One of these was DDX6 (also RCK/p54), a DEAD box RNA helicase that was previously implicated in translational and miRNA-mediated silencing.Citation25–Citation27 In Xenopus oocytes, the DDX6 homolog, Xp54, has been shown to be a core component of mRNP particles that store and silence maternal mRNAs during early development.Citation28 In mammalian cells, DDX6 has been shown to localize to and be required for the formation of processing (P) bodies, which are sites of translational silencing and mRNA storage in somatic cells.Citation29
In addition to maternal and somatic translation regulation, DDX6 has been implicated in the replication of several viruses.Citation30 The Brome mosaic virus (BMV)-Saccharomyces cerevisiae system expresses positive-strand RNAs from a plant alphavirus in yeast cells and has been used in genetic screens for cellular factors required for BMV replication. A screen for factors required for BMV gene expression identified the yeast homolog of DDX6, Dhh1p, which was also shown to be required for recruitment of BMV RNA from sites of translation to sites of virus replication.Citation30,Citation31 Hepatitis C (HCV), a member of the Flaviviridae family, has also been shown to require DDX6 for viral transcription in the HCV replicon system.Citation32,Citation33 In addition, DDX6, HCV core protein and HCV RNA were shown to form a complex in vivo.Citation32
These studies underscore the importance of viral RNA secondary structures and their interactions with both viral and host cell proteins. Using RNA affinity chromatography methodology coupled to stable isotopic labeling in cell culture (SILAC) and quantitative mass spectrometry, we identified 61 host cell proteins that specifically interact with the DENV RNA 5′ and 3′ UTRs. Several of the proteins identified have previously been associated with PB and SG, including DDX6, G3BP1, G3BP2, Caprin1 and USP10. Indirect immunofluorescence microscopy demonstrates that DDX6, Caprin1, G3BP1, G3BP2 and USP10 colocalize with sites of DENV-2 replication, suggesting that the interactions identified by RNA chromatography combined with MS can be recapitulated in DENV-infected cells. DDX6 knockdown leads to a significant reduction of infectious DENV-2 particles in tissue culture media of infected cells and reduced amounts of DENV-2 RNA. DDX6 interacts with DENV-2 RNA in vivo and, using the RNA affinity chromatography methodology, binds to the DB2 and DB1 structures in the DENV-2 3′ UTR. The interaction between DDX6 and the DB structures required formation of a potential pseudoknot structure, suggesting that the complex tertiary structures in the DENV 3′ UTR would be required for DDX6 function. Furthermore, DDX6, Caprin1, G3BP1, G3BP2 and USP10 show different binding preferences for specific structures in the DENV-2 3′ UTR. These results suggest that the DENV UTRs act as assembly sites for DDX6 and SG proteins and that proper assembly of these RBPs have functional consequences for DENV replication.
Results
Purification and identification of host cell proteins that bind to the DENV-2 5′ and 3′ UTRs.
We identified host cell proteins that associate with an RNA containing sequences from the 5′ and 3′ UTR of DENV-2 NGC and a tobramycin RNA aptamer (). Sequences from the 5′ UTR (nts 1–172) and 3′ UTR (nts 10,242–10,724) were amplified from DENV-2 NGC. An oligonucleotide containing streptavidin and tobramycin aptamer sequences and overlapping sequence from the 5′ and 3′ UTR was used to fuse the 5′ UTR, aptamer sequences and 3′ UTR, respectively, using splice overlap extension (SOE) PCR. In addition, the oligonucleotide used to amplify the 5′ UTR incorporated a T7 promoter for in vitro transcription. As a control, similar length sequences from the DENV-2 NGC NS2A coding region were cloned using similar methods as those used to generate the 5′ and 3′ UTR construct. A 14 nt complementary sequence was engineered into the control construct in order to induce formation of a double-stranded RNA region. PCR-amplified templates were used for in vitro transcription reactions in the presence of 2′-Fluoro-UTP and 2′-Fluoro-CTP to render the bait RNA more resistant to RNases. To generate beads for RNA chromatography, tobramycin was covalently coupled to NHS-Sepharose beads as described in Hartmuth et al. 2004. 30 µg of purified DENV-2 control or 5′ and 3′ UTR RNA was resuspended in RNA binding buffer (see Materials and Methods), heated to 95°C for 5 minutes, cooled to room temperature and combined with 15 µL of tobramycin beads. Following incubation at 4°C, the tobramycin-RNA beads were washed to remove unbound RNA and incubated with cell lysates as described below.
In order to facilitate identification of proteins that associate with the DENV-2 5′ and 3′ UTRs, we used stable isotopic labeling of amino acids in cell culture (SILAC) and quantitative mass spectrometry.Citation35,Citation36 This methodology is advantageous because it allows both identification and quantification of proteins in a complex mixture, and allows direct comparison of recovered protein from the DENV-2 control and 5′ and 3′ UTR RNA samples. HeLa cells were used in these experiments because they had previously been optimized for use in the SILAC labeling and quantitative mass spectrometry in the Blackstock laboratory. In the experiments described here we utilized uninfected cell lysates for RNA chromatography. For SILAC-based RNA pull-down experiments HeLa cells were grown in media containing normal isotopes of L-lysine-(12C614N2) (K0) and L-arginine-(12C614N4) (R0) for ‘light’ sample and L-lysine-(13C615N2) (K8) and L-arginine-(13C615N4) (R10) for ‘heavy’ sample. After six cell divisions the cultures were uniformly labeled with heavy isotopes, as confirmed by quantitative mass spectrometric analysis (Sup. Fig. 1). Labeled cells were washed twice with cold PBS, harvested by scraping and lysed in RNA affinity chromatography buffer (see Materials and Methods). Protein concentration in the lysates was determined using the BioRad Protein Assay calibrated using a BSA standard curve. In two experiments (forward experiments), the DENV-2 control RNA was incubated with light cell lysate and the DENV-2 5′ and 3′ UTR RNA was incubated with heavy cell lysate. In a third experiment (reverse experiment), the DENV-2 control RNA was incubated with heavy cell lysate and the DENV-2 5′ and 3′ UTR RNA was incubated with light cell lysate. For all experiments, following incubation with cell lysate the DENV-2 control and 5′ and 3′ UTR RNA beads were combined and washed. Control and 5′ and 3′ UTR RNA and associated factors were eluted from the beads and eluates were subjected to LC-MS analysis as described in Materials and Methods.
MS data were analyzed by the MaxQuant program.Citation37 The integrated intensity of the peptide peak, as determined by MaxQuant, reflects the peptide abundance and the fold-change represents the ratio of peptides quantified in the experimental sample labeled with heavy isotopes (K8R10) versus the control sample with light (K0R0) for forward experiment. Most proteins identified were present in a 1:1 ratio and corresponded to the normal distribution of non-specific binding proteins. For statistical purposes, the greater the deviation in the peptide ratio from 1:1, the greater the specificity of the selective RNA binding of the corresponding protein. Using the high mass accuracy of the peptide precursor ion measurement, identification of SILAC pairs, and the statistical significance calculated by MaxQuant, it was possible to confidently differentiate the specific interacting partners from the population of nonspecific binding proteins. The specific binding partners were further verified by a reverse experiment (the experimental sample labeled with light isotopes and the control sample with heavy). Proteins that preferentially associate with the DENV-2 5′ and 3′ UTR RNA compared to the DENV-2 control RNA generated a ratio >1.0 in the forward experiment and a ratio <1.0 in the reverse experiment. Only ratios with the product of significance in the forward and reverse experiments (pforward x preverse) < 0.0001 were considered as specific binding partners of DENV-2 5′ and 3′ UTR RNA (Table S1). To select the interacting partners with the highest confidence we used pforward x preverse < 1e-10 and minimum log2 SILAC ratio of 2 for the forward exeriment and maximum-2 for the reverse experiment for this analysis (red dots in upper left quadrant in ).
All of the proteins identified in the screen with a pforward x preverse < 1e-10 have documented functions in various aspects of RNA metabolism (). Interestingly, many of these have previously been implicated in formation or function of processing bodies (PBs) and stress granules (SGs).Citation38 PBs and SGs are cytoplasmic RNA granules that, following polysome disassembly, act as sites for storage and/or degradation of mRNAs. Many proteins localize to both P bodies and SGs, but each class of RNA granules appears to have unique functions. The Argonaute proteins localize to P bodies where they function in the process of miRNA silencing and mRNA decay. SGs contain components of the ribosome and function as sites of translational silencing in response to cellular stress. Among the proteins with pforward x preverse < 1e-10, Caprin1, DDX6, USP10, G3BP1, G3BP2, EDC3 and FMR1 have all been shown to localize or interact with proteins in P bodies, SGs or both RNA granules.Citation38 Our results suggested that the DENV RNA interacts with P body and SG components in infected cells.
In addition to P body and SG proteins, several other DENV-2 5′ and 3′ UTR interacting factors have documented roles in RNA metabolism. HNRNPA2B1 and HNRNPA1, which were about 5-fold enriched with a pforward x preverse < 1e-9 for binding to the DENV 5′ and 3′ UTR relative to the control RNA, were reviously shown to interact with DENV RNA and have been implicated in pre-mRNA splicing.Citation19 Interestingly, though DENV RNA does not undergo splicing, several other splicing factors were identified that specifically interact with the DENV 5′ and 3′ UTR, including U2AF1, U2AF2, PABPN1 and several other HNRNPs.Citation39 In order to better understand whether the proteins identified in this study have any previously described interactions, we performed an analysis of protein interaction networks using the String 8.3 database.Citation40 Based on this analysis, we found that there were significant interactions between many of the proteins identified (Fig. S2). There was a core interaction network involving the splicing factors discussed above, with other factors forming additional interaction networks with weak links to the core network. Notably, among the proteins in , DDX6, EDC3 and LSM12 form an interaction network with connections to the core network, while Caprin1, G3BP1 and USP10 form a separate interaction network also linked to the core network. While additional work is necessary to validate these interaction networks, the String 8.3 analysis suggests that multiple complexes of RNA binding proteins associated with the DENV 5′ and 3′ UTRs.
PB and SG proteins colocalize with the DENV-2 replication complex in infected cells.
To determine the subcellular localization of the PB and SG proteins during DENV-2 infection, we performed indirect immunofluorescence microscopy using antibodies specific to DDX6, Caprin1, G3BP1, G3BP2 and USP10. To detect DENV RNA, we used an antibody specific to dsRNA that has been used previously as a marker for sites of DENV replication.Citation41 HuH-7 cells were plated on glass coverslips and infected with DENV-2 NGC at an MOI of 1.0. After 24 hours, the cells were fixed, permeabilized and stained with the indicated antibody against cellular proteins and a monoclonal antibody specific to dsRNA. For comparison, uninfected cells were plated on glass coverslips, incubated 24 hours, fixed and stained with the same antibodies. During the final wash, DAPI was added to the wash buffer and the coverslips were sealed and analyzed using by epifluorescence microscopy.
As a negative control for colocalization with the DENV replication complex, we probed uninfected and infected cells with antibody specific for stem-loop binding protein (SLBP), an RNA binding protein that associates with a stem-loop in the 3′ UTR of histone mRNAs.Citation42 While this protein binds to RNA secondary structures, it was not expected to colocalize with the DENV replication complex since it was not identified in the original screen for DENV-2 5′ and 3′ UTR-binding proteins. In both uninfected and infected cells, SLBP localized either to the nucleus or throughout the cell ( and B). In infected cells, SLBP and dsRNA did not have overlapping patterns of localization (). In uninfected cells, DDX6 localized strongly with P body-like structures (). In infected cells, DDX6 localized to P body-like structures as well as sites of DENV replication indicated by dsRNA signal (). The SG component Caprin1 localized to the perinuclear space in uninfected and infected cells and also sites of DENV replication in infected cells ( and F). G3BP1 localized to the cytoplasm in uninfected and infected cells, with slightly stronger staining in the perinuclear space in DENV-infected cells, overlapping with sites of replication ( and H). G3BP2 localized to the cytoplasm and appeared to relocalize to perinuclear sites of DENV replication 24 hours post-infection ( and J). Finally, USP10 localized throughout uninfected cells and localized to the perinuclear space corresponding to sites of DENV replication in infected cells ( and L). These results suggest that DDX6, Caprin1, G3BP1, G3BP2 and USP10, but not SLBP, colocalized with DENV replication sites. The localization of the proteins identified by the RNA affinity chromatography to the DENV replication complex provided an in vivo validation for our methodology.
DDX6 is required for efficient assembly or release of infectious particles.
We wanted to test whether proteins identified in the screen had a role in DENV-2 replication. DDX6 has previously been implicated in HCV replication and translational silencing.Citation26,Citation32 Given its previously defined role in Flaviviridae replication, we focused on the potential function of DDX6 in DENV-2 replication. We performed siRNA-mediated knockdown of DDX6 expression followed by DENV infection and analyzed levels of infectious viral particles and viral RNA in the media of infected cells. We performed knockdowns in HuH-7 cells using either a control siRNA complementary to GFP RNA (siGFP) or three different siRNAs targeting DDX6 RNA (siDDX6_1, siDDX6_2 or siDDX6_3), followed by infection with DENV-2 NGC at an MOI of 0.5. We analyzed DDX6 protein expression in infected cells at 24 hours post-infection. The siRNA targeting GFP had no significant effect on DDX6 expression. DDX6 levels were significantly decreased in cells transfected with siRNAs targeting DDX6 at 24 hours post-infection (). DDX6 levels, which were quantified and normalized relative to cellular GAPDH levels using the Odyssey Infrared Imaging system (LiCor), were found to be reduced to 50% or less compared with DDX6 levels in cells treated with siGFP (one-way ANOVA, p < 0.0001, Dunnett's Multiple Comparison Test, p < 0.01) ().
To investigate if DDX6 knockdown had an effect on the formation or release of infectious DENV particles, we assayed the tissue culture media for the ability to form virus foci in BHK21 cells. Diluted tissue culture media were used to infect BHK21 cell monolayers. After four days, cells were fixed and probed for viral foci using the DENV E protein-specific 3H5 monoclonal antibody and an anti-mouse secondary antibody. Viral foci were visualized using the Odyssey system and counted. We found that at 24 hours post-infection, there was, on average, a 2- to 3-fold reduction of virus focus forming units (ffu) in supernatants from DDX6 knockdown compared to the siGFP treatment (one-way ANOVA, p = 0.0022, Dunnett's Multiple Comparison Test, p < 0.01) ().
To determine if DDX6 knockdown had an effect on packaging or release of DENV RNA, we performed real-time PCR on cDNAs from viral RNA isolated from the media at 24 hours post-infection. Using the same samples from , total RNA was harvested, cDNA was synthesized using random hexamer primers and utilized as a template for DENV-specific real-time PCR. As a control for RNA recovery from the tissue culture media, 25 ng of an in vitro transcript of sequence from the D. melanogaster Boule mRNA was added to each sample prior to Trizol extraction and used as a normalization control. Viral RNA levels in supernatants from DDX6 knockdown were significantly reduced, on average, to 2- to 3-fold of the siGFP-treated viral RNA level 24 hours post-infection (one-way ANOVA, p = 0.0024, Dunnett's Multiple Comparison Test, p < 0.01) (). These results suggest that DDX6 knockdown significantly reduces the production and/or release of infectious particles from DENV-infected cells. Furthermore, these results validate using the RNA chromatography method for discovery of functionally relevant host cell proteins.
DDX6 associates with DENV-2 RNA during infection.
DDX6 knockdowns above suggested a host factor function for DDX6 in the production and/or release of DENV infectious particles. To extend this observation, we wanted to confirm the interaction we observed between DDX6 and the DENV UTRs by testing whether DDX6 interacted with DENV RNA during bona fide virus infection. We investigated whether an interaction between DDX6 and DENV-2 RNA could be detected by RNA immunoprecipitation in cells infected with DENV-2 NGC. HuH-7 cells were infected at an MOI of 0.5 and incubated for 24 hours. Infected cells were washed with cold PBS, harvested by scraping and lysed in RNA affinity chromatography buffer (see Materials and Methods). Cell lysates were used for immunoprecipitation using non-specific IgG or a polyclonal antibody specific to DDX6 bound to protein A beads. After extensive washing, total RNA was harvested from the beads using Trizol reagent and analyzed by RT-PCR for β-actin, c-myc and DENV-2 RNA. As expected, we detected β-actin and c-myc RNAs in both uninfected and infected lysates, but DENV-2 RNA only in infected cell lysates ( and lanes 1 to 4). As a negative control for immunoprecipitation, we performed RT-PCR using oligonucleotides specific for β-actin RNA, which is not expected to interact with DDX6 in vivo, and indeed DDX6-specific antibodies did not immunoprecipitate β-actin RNA ( and lanes 5 to 12). As a positive control, we performed RT-PCR using oligonucleotides specific for c-myc RNA, which has previously been shown to interact with DDX6.Citation43 The DDX6-specific antibody, but not non-specific IgG, immunoprecipitated c-myc RNA in uninfected and infected cells, though the signal (input and immunoprecipitated) was reduced in infected cells ( and lanes 5 to 12). Finally, we performed RT-PCR using oligonucleotides specific for DENV-2 RNA and found that the DDX6-specific antibody, but not non-specific IgG, efficiently immunoprecipitated DENV-2 RNA from infected cell lysates ( and lanes 9 to 12). These results demonstrate that DDX6 specifically associated with DENV-2 RNAs during infection.
In order to quantify the amount of DENV-2 RNA associated with DDX6 during infection, we repeated the RNA immunoprecipitation described above in triplicate followed by analysis by real-time PCR. Following immunoprecipitation, an in vitro transcript corresponding to a short sequence from the D. melanogaster Boule mRNA was added to each sample prior to Trizol extraction to act as a recovery and normalization control. RNA samples were treated similarly as described above for preparation of cDNA, but the samples were then subjected to real-time PCR for Boule and DENV-2 RNA. Samples were quantified based on a standard curve of DENV-2 RNA and converted to molecules of DENV-2 RNA. We found that DDX6 IP enriched for DENV-2 RNA approximately 1,000-fold compared to the control IP from infected cell lysates (one-way ANOVA, p = 0.0045, Dunnett's Multiple Comparison Test, p < 0.01). These results, which suggest an interaction in vivo, provide additional validation of the RNA chromatography.
DDX6 binds to the DB2 and DB1 structures in the DENV-2 3′ UTR.
Since DDX6 was demonstrated to function in viral replication and interact with DENV RNA in vivo, we wanted to determine what sequences were required for the interaction between DDX6 and the DENV UTRs. We used RNA affinity chromatography to identify the DDX6 binding site in the DENV-2 UTRs using a series of RNAs containing deletions in their secondary structures. We performed RNA affinity chromatography similarly to that described in except that we analyzed binding by western blotting. As expected from our MS results, DDX6 preferentially bound to the DENV-2 5′ and 3′ UTR over the control RNA ( and lanes 2 and 5). We tested whether DDX6 preferentially bound to the DENV-2 5′ or 3′ UTR. As negative controls for DDX6 binding, we tested constructs containing the full-length control, the 5′ control sequence alone and the 3′ control sequence alone, and found that DDX6 did not bind to any of these RNAs ( and lanes 2, 3 and 4). An additional negative control for binding to the UTR constructs, TIAL1, is presented below. We tested constructs containing the DENV-2 5′ and 3′ UTRs, the 5′ UTR alone or the 3′ UTR alone and found that DDX6 preferentially bound to the 3′ UTR ( and lanes 3–5).
In order to narrow down the minimal sequence required for DDX6 binding, we tested a series of deletion mutants in the variable region (VR) and found that VR Stem-loops I, II, III and IV were not required for DDX6 binding ( and lanes 4–8). As expected, DDX6 bound to the construct containing both the 5′ and 3′ UTR or the 3′ UTR alone, but not the control construct ( and lanes 2–4). These results demonstrate that stem-loops I-IV in the VR were not required for DDX6 binding to the DENV-2 3′ UTR.
We tested an additional deletion that removed the entire VR sequence and found that it was not required for DDX6 binding ( and lane 6). As expected, DDX6 bound to the construct containing both the 5′ and 3′ UTR or the 3′ UTR alone, but not the control RNA ( and lanes 2 to 4). A construct with deletions of the SLA, SLB, CS and DB1 bound DDX6 ( and lane 5). We tested constructs that contained deletions in the VR and additional structures and determined that, while a construct containing the SLA, SLB, CS and DB1 bound DDX6, a construct containing the SLA, SLB and CS alone did not ( and lanes 7 and 8). This result was puzzling since both the construct containing deletions in the SLA, SLB, CS and DB1 and the construct containing deletions in the VR and DB2 bound DDX6 ( and lanes 5 and 7). If there was a single binding site for DDX6, then progressive deletions from the 5′ and 3′ end would be expected to identify a single structure or sequence sufficient for binding. This result suggests that there are multiple structures or sequences in the 3′ UTR that bind DDX6. However, it has been noted before that DB2 and DB1 are both structurally similar and highly conserved, suggesting that these homologous structures could act as redundant DDX6 binding sites.Citation44
To test whether DB2 and DB1 are the DDX6 binding sites, additional deletion mutants were generated that removed the SLA, SLB and CS sequences or the VR and DB sequences from the 3′ UTR. As expected, DDX6 bound to the construct containing both the 5′ and 3′ UTR or the 3′ UTR alone, but not the control RNA ( and lanes 2–4). A construct containing the VR, DB2 and DB1 bound DDX6 ( and lane 5), however, an RNA containing only the SLA, SLB and CS did not ( and lane 6). We had previously established that the VR was not required for DDX6 binding ( and lane 6). Since the DB2 and DB1 are very similar structures, we tested these sequences together and separately for their ability to bind DDX6. RNAs containing both DB2 and DB1 or each DB in isolation bound DDX6 ( and lanes 7–9). These results demonstrate that DDX6 interacts with the DENV-2 3′ UTR via the DB2 and DB1 structures. We suggest that DDX6 functions in DENV replication via its interaction with the DB structures.
DDX6 binds to predicted pseudoknot structures formed by the DB2 and DB1 sequences in the DENV-2 3′ UTR.
In order to map the sequences required for the DDX6 interaction, we employed the RNA affinity chromatography method using a series of DENV-2 3′ UTR RNAs containing deletions in their secondary structures. As demonstrated in , DDX6 bound to an RNA containing only the DB1 and DB2 structures, with each of the individual dumbbell structures sufficient for interaction with DDX6 ( and lanes 31 and 32). DB2 and DB1 are predicted to form pseudoknots by basepairing between the terminal stem-loop (pk2′ and pk1′, respectively) and the downstream sequences (pk2 and pk1, respectively) ().Citation44 More recently, Manzano et al. demonstrated that the terminal loop sequences in the DB2 and DB1 (referred to as TL1 and TL2, respectively) predicted to form the pseudoknot structures contribute to translation and RNA amplification using a DENV-2 replicon system.Citation45 We wanted to determine whether base-pairing between the pk2′ and pk2 sequences, or pk1′ and pk1 sequences, was required for DDX6 binding to DB2 and DB1. We generated point mutants that would disrupt basepairing between pk2′ and pk2 ( and pk2′ mut), pk1′ and pk1 ( and pk1′ mut) or both ( and pk2′/pk1′ mut and pk2/pk1 mut). In addition, we generated compensatory mutants that would restore basepairing between pk2′ and pk2 as well as pk1′ and pk1 ( and pk2′/pk1′/pk2/pk1 mut). To preserve as much of the adjacent RNA structures as possible, these mutations were made in the full-length DENV-2 3′ UTR. The nucleotide changes used to generate the pk mutants are listed in Supplemental Table 2.
We found that DDX6 binding to the 3′ UTR was reduced in the presence of point mutations in pk2′ and pk1′ ( and lanes 3–5). When point mutations were introduced into both pk2′ and pk1′, DDX6 binding was further reduced ( and lane 6). Binding was also reduced when point mutations were introduced into pk2 and pk1, the predicted sites for basepairing adjacent to the DB2 and DB1 loop structures ( and lane 7). Compensatory mutations, predicted to restore base-pairing between the loop and downstream RNA sequences did not restore DDX6 binding ( and lane 8). These results indicate that binding of DDX6 to the DENV-2 3′ UTR RNA required the same nucleotides predicted to form the pseudoknots in the DB2 and DB1 structures. DDX6 binding to the DENV-2 3′ UTR, however, required additional sequence information, since the compensatory mutations, which are predicted to restore pseudoknot structures, did not restore binding.
Other PB and SG proteins interact with different structures in the DENV-2 3′ UTR.
In order to confirm and extend our analysis of the interaction of host cell proteins with the DENV 5′ and 3′ UTRs, we performed RNA affinity chromatography using different sequences from the DENV 5′ and 3′ UTRs. We expanded our target proteins from DDX6 to include other SG proteins identified by MS, including Caprin1, G3BP1, G3BP2 and USP10. Since DDX6 was shown to function in DENV replication () and was shown to bind to the DB structures in the 3′ UTR ( and ), we wanted to determine if other SG proteins bound to the same sequences in the DENV-2 3′ UTR. We performed RNA affinity chromatography similarly to that described in . TIAL1, which has previously shown to bind the negative strand 3′ UTR of WNV,Citation46 did not bind any of the positive strand DENV RNAs used in this assay ( and lanes 2–10). As expected DDX6, Caprin1, G3BP1, G3BP2 and USP10 preferentially bound the DENV-2 3′ and 5′ UTR RNA over the control RNA ( and lanes 2 and 3). DDX6, Caprin1, G3BP1, G3BP2 and USP10 did not bind the 5′ UTR but showed strong binding to the 3′ UTR ( and lanes 4 and 5). 3′ UTR sequences upstream of the CS were necessary and sufficient to bind these five proteins (lanes 7 and 8). DDX6 bound the DB2 and DB1 sequences in the 3′ UTR ( and mutation series in left part and lanes 6–10) whereas Caprin1, G3BP1, G3BP2 and USP10 bound sequences in the VR ( and mutation series in left part and lanes 6, 9 & 10). These results confirm that several of the proteins identified by RNA affinity chromatography and SILAC-MS bind to the DENV-2 5′ and 3′ UTR RNA. Further, these results demonstrate that the DENV-2 3′ UTR acts as an assembly site for PB and SG proteins. Finally, since they demonstrate differential binding preference for the DB or VR structures in the 3′ UTR, DDX6 and the other four proteins analyzed may play different functional roles in DENV replication.
Discussion
Using a novel RNA affinity chromatography strategy coupled with SILAC-MS technology, we identified 61 RNA-binding proteins that specifically interacted with the DENV-2 5′ and 3′ UTRs. Among the top eight hits with pforward x preverse < 1e-10, seven have previously described functions in PBs and SGs. PB and SG proteins have been implicated in pro- and anti-viral functions for other viruses,Citation30 but this study represents the most extensive characterization of host cell RNA binding proteins that interact with the DENV-2 UTRs and is the first study to implicate multiple PB and SG proteins in DENV replication. The colocalization of each of these proteins with sites of DENV replication further implicated these proteins in either a pro- or anti-viral role in the host cell.
The connection between PBs, SGs and DENV replication is not completely clear. DENV replication complexes have been shown to colocalize with, and assemble virion on, ER membranes, though the precise dynamics of this process is not well understood.Citation41 In addition, egress of assembled DENV particles has been shown to require several membrane-associated and endolysosomal proteins.Citation47 A recent study using HCV, another member of Flaviviridae family, has demonstrated that DDX6 colocalizes with lipid droplets (LDs), which are ER-derived membranes, during infection.Citation48 LDs were previously demonstrated to be required for HCV replication and also for production of infectious DENV virus.Citation49,Citation50 One intriguing possibility is that LDs are sites of packaging and that the process of egress and release of fully assembled infectious virus begins at this site. Recent work by Gibbings et al. has demonstrated that PB and SG components copurify with multi-vesicular bodies and Tahbaz et al. have demonstrated that Dicer and the RISC effector protein Ago2, which also colocalizes with PBs, are present in membrane fractions of cells.Citation51–Citation53 Our results demonstrate that several PB and SG proteins colocalize with DENV replication sites and bind to different sites on DENV RNA. This work suggests an intimate connection between PBs, SGs and endolysosomal membranes, which could play a pro-viral role in DENV replication. The assembly of PB and SG proteins on the viral RNA could coordinate the various steps of virion assembly and regulate its interaction with the viral replication complexes and cellular membranes.
For DDX6, the data clearly demonstrate that it has a pro-viral role in the assembly or release of infectious particles. We determined, using two independent methodologies, that DDX6 and DENV RNA interact both in vitro and in vivo. Furthermore, the binding site for DDX6 was mapped to the DB structures in the DENV-2 3′ UTR, whereas Caprin1, G3BP1, G3BP2 and USP10 preferentially bound to the VR region of the DENV-2 3′ UTR, suggesting that different structures within the DENV 3′ UTR could act as assembly sites for protein complexes. Since Caprin1, G3BP1, G3BP2 and USP10 have different sites of interaction with the viral RNA, it is likely that these have different roles in viral replication. SGs are assembled in response to cell stress, which can include virus infection.Citation54 G3BP1, as well as TIAL1, have been implicated in assembly of SGs, and assembly of SGs is inhibited during WNV infection.Citation20,Citation55 While TIAL1 was shown to have a function in viral transcription, the disruption of SGs by WNV could also be a way to subvert the host cell response to infection. It seems possible that the various mRNP complexes that bind to the DENV 3′ UTR are competing and the viral RNA would act as a platform for recruitment of protein complexes that function in various steps of the virus life cycle.
The DENV RNA must act as the protein-coding mRNA, the template for replication, and the viral genome. However, the sorting of viral RNAs from sites of translation to replication and subsequent packaging, is a poorly understood process. It is possible that the assembly of PB and SG proteins on the 3′ UTR could regulate whether the viral RNA undergoes translation, replication or packaging. DDX6 has been shown to have a role in the recruitment of viral RNAs from sites of translation to sites of replication in the BMV-Saccharomyces cerevisiae system.Citation56 Additionally, DDX6 has a well-established role in translational silencing.Citation25–Citation27 DDX6 is thought to play a role in mRNP remodeling in SGs and this determines whether mRNAs within SGs will be translated, stored or degraded.Citation27 Though the sites of DENV replication and translation have yet to be separated, it is possible that DDX6 is acting to sort viral RNAs throughout the entire process, from translation and replication to the sites of viral packaging and egress.
Materials and Methods
Cell culture and DENV-2 infection.
HeLa and HuH-7 cells were maintained in DMEM (Invitrogen) supplemented with 10% FBS (HyClone), 10 U/mL penicillin and 10 µg/mL streptomycin (Pen/Strep, Gibco). For SILAC labeling, HeLa cells were maintained in SILAC-DMEM (Thermo) supplemented with 10% dialyzed FBS (Thermo) and Pen/Strep for at least six cell divisions in order to achieve uniform incorporation. Light media was supplemented with 0.8 mM L-lysine:HCl and 0.4 mM L-arginine:HCl (Sigma). Light media was supplemented with 0.8 mM L-lysine:HCl and 0.4 mM L-arginine:HCl (Sigma). Heavy media was supplemented with 0.8 mM L-lysine:2HCl (U-13C6, U-15N2) and 0.4 mM L-arginine:HCl (U-13C6, U-15N4) (Cambridge Isotope). Uniform incorporation was confirmed by LC/MS analysis of whole cell lysates (Fig. S1). For DENV-2 infection, cells were infected with DENV-2 NGC (a kind gift from Mary Ng, NUS) in serum-free DMEM for 1 hour using a MOI of 0.5. The inoculum was replaced with fresh media and incubated for 24 hours prior to analysis.
Plasmid constructs, T7 templates and in vitro transcription.
The 5′ and 3′ UTRs from DENV-2 NGC were amplified by PCR using oligonucleotides specific to nts 1–172 and 10,242–10,725. The oligo used to amplify the 5′ UTR also contained the T7 promoter sequence before the start of the 5′ UTR. The 5′ UTR was fused to an oligo containing the streptavidin and tobramycin aptamers and spacer sequences using splice overlap extension (SOE) PCR.Citation34,Citation57 PCR templates were used for MEGAscript T7 RNA polymerase in vitro transcription reactions (Ambion/Applied Biosystems) to generate RNA according to the manufacturer's instructions. Details of the oligonucleotides and templates are available upon request.
Tobramycin RNA affinity chromatography.
Tobramycin RNA affinity chromatography was adapted from Hartmuth et al. 2004. NHS-activated sepharose beads (GE Healthcare) were washed four times with 1 mM HCl and resuspended in coupling buffer (0.2 M NaHCO3, 0.5 M NaCl, pH 8.3) containing 5 mM tobramycin. Following incubation overnight at 4°C with head-to-tail rotation, beads were spun down and resuspended in blocking buffer. After 1 hour incubation at 4°C, beads were washed 3 times and resuspended in PBS. RNA in RNA binding buffer (20 mM Tris, pH 7.0, 1 mM CaCl2, 1 mM MgCl2, 75 mM NaCl, 145 mM KCl, 0.1 mg/mL yeast tRNA, 0.2 mM DTT) was heated to 95°C for 5 min, cooled to room temperature, combined with tobramycin beads and incubated at 4°C with end over end rotation, 1 hour. Beads were centrifuged and washed two times with RNA washing buffer (20 mM Tris, pH 7.0, 1 mM CaCl2, 1 mM MgCl2, 75 mM NaCl, 145 mM KCl, 0.1% NP-40, 0.2 mM DTT). Cell lysates were pre-cleared using tobramycin beads before the RNA beads were combined with lysate in cell lysis buffer (50 mM Tris, pH 7.5, 150 mM NaCl, 1% NP-40, 1x protease and phosphatase inhibitor (Roche)). Beads and lysate were incubated 1–1.5 hr at 4°C with end over end rotation. Beads were centrifuged and resuspended in 145 mM protein washing buffer (20 mM Tris, pH 7.0, 1 mM CaCl2, 1 mM MgCl2, 75 mM NaCl, 145 mM KCl, 0.5% NP-40), and transferred to a Pierce Micro-Spin column (Thermo). Beads were washed four times with 145 mM protein washing buffer and incubated for 5 min at room temp with elution buffer (20 mM Tris, pH 7.0, 1 mM CaCl2, 3 mM MgCl2, 145 mM KCl, 5 mM tobramycin, 0.2 mM DTT). Eluate was collected by centrifugation.
Mass spectrometry and data analysis.
Eluted protein complexes from RNA affinity chromatography were separated by 1D-SDS-PAGE and digested with trypsin using established procedures.Citation58 Samples were analyzed on an Orbitrap or Orbitrap XL mass spectrometer (Thermo Fisher) coupled to a Proxeon Easy-nLC. Survey full scan MS spectra (m/z 300–1,400) were acquired with a resolution of R = 60,000 at m/z 400, an AGC target of 1e6 ions, and a maximum injection time of 500 ms. The ten most intense peptide ions in each survey scan with an ion intensity above 2,000 counts and a charge state ≥2 were sequentially isolated to a target value of 1e4 and fragmented in the linear ion trap by collisionally induced dissociation (CID) using a normalized collision energy of 35%. A dynamic exclusion was applied using a maximum exclusion list of 500 with one repeat count, repeat and exclusion duration of 30 seconds. Proteins were searched using Mascot version 2.2 (Matrix Science, London, UK) against a concatenated target/decoy database, prepared by sequence reversing the human International Protein Index (IPI) (version 3.52, 73,928 sequences, www.ebi. ac.uk) and adding common contaminants such as human keratins, porcine trypsin and proteases to give a total of 148,380 sequences. Cysteine carbamidomethylation was searched as a fixed modification, N-acetylation and oxidized methionine were searched as variable modifications. Labeled arginine and lysine were specified as fixed or variable modifications, depending on the prior knowledge about the parent ion. SILAC peptide and protein quantification was performed automatically with MaxQuant version 1.0.13.13 using default parameter settings.Citation37 Maximum false discovery rates (FDR) were set to 0.01 for both protein and peptide.
String 8.3 analysis.
The proteins identified by quantitative mass spectrometry were analyzed using the String 8.3 database.Citation40 The String database is composed of known and predicted interactions based on genomic context, high-throughput and coexpression analysis, and prior knowledge from the scientific literature. Proteins identified with a p value < 1e-3 were analyzed using default settings and mapped according to confidence of an existing interaction.
Antibodies.
Rabbit antibodies against DDX6 and G3BP2 were obtained from Bethyl Laboratories. Mouse monoclonal antibodies against G3BP1, TIAL1 and non-specific IgG were obtained from BD Biosciences. Rabbit antibody against USP10 and mouse monoclonal antibody against SLBP were obtained from Abcam. The J2 anti-dsRNA antibody was obtained from English and Scientific Consulting. Rabbit antibodies against GAPDH were obtained from Cell Signalling. The pan-flavivirus mouse monoclonal antibody to E protein was produced from the 4G2 hybridoma. The 3H5 mouse monoclonal antibody to DENV E protein was produced from the 3H5 hybridoma. Rabbit antibodies against Caprin1 were a kind gift from Bernard Moss and George Katsafanas (NIH, USA).Citation59 Goat anti-rabbit DyLight 800 and Goat anti-mouse DyLight 680 antibodies (Thermo) were used for detection of primary antibodies in western or viral focus assays. Secondary antibodies used for immunofluorescence microscopy are described below.
Immunofluorescence microscopy.
HuH-7 cells were plated on coverslips in 24-well plates. 24 hours later, cells were infected with DENV-2 NGC at an MOI of 1.0. 24 hours later, uninfected and infected cells were fixed with 4% formaldehyde in PBS, permeabilized with 0.1% Triton X-100 in PBS and blocked in 0.05% Tween-20 and 4% normal goat serum in PBS. Coverslips were incubated with antibody in blocking buffer and washed using 0.05% Tween-20 in PBS. For DDX6, Caprin1, G3BP2 and USP10, primary antibody was detected using a FITC-conjugated anti-rabbit secondary (Jackson Immunoresearch). Mouse monoclonal antibody to SLBP or G3BP1 was detected using an Alexa 488-conjugated anti-mouse secondary (Invitrogen). The dsRNA antibody was directly conjugated with Alexa 594 using the Zenon antibody conjugation kit (Invitrogen). Cells were fixed for 10 min in 4% paraformaldehyde in PBS after incubation with the conjugated dsRNA antibody. DAPI (Molecular Probes) was included in the final wash to visualize cell nuclei. Coverslips were sealed and visualized using an Olympus IX71 epifluorescent microscope and DP71 digital camera. Images were processed using the ImageJ software package.
siRNAs.
The siRNAs used for knockdown were obtained from Invitrogen. 25 nM siRNA was transfected using RNAiMAX (Invitrogen) according to manufacturer's instructions 48 hours prior to infection. The control siRNA, siGFP, targets the coding sequence of GFP RNA (5′-CGG CAA GCT GAC CCT GAA GTT CAT-3′). The siRNAs for DDX6 knockdown target three different sequences in DDX6 RNA (siDDX6_1; 5′-GGA ACT ATG AAG ACT TAA ATT-3′, siDDX6_2; 5′-AGG ACT CAT TTA TTG AGC AAG ACT T-3′, siDDX6_3; 5′-CAA CTG GGT TAT TCT TGC TTC TAT A-3′). The target sequence for siDDX6_1 is based on Chu and Rana, 2006.Citation25 The siRNA targeting PTB was the P1 siRNA used in Anwar et al. 2009.
Western blots.
Samples were separated by SDS-PAGE, transferred to Immobilon FL (Millipore) and probed with indicated antibodies in PBS containing 5% milk and 0.5% Tween 20. Blots were washed with 0.5% Tween 20 in PBS and primary antibodies were detected by incubating with anti-rabbit DyLight800 and anti-mouse DyLight680 antibodies. Blots were washed in 0.5% Tween 20 in PBS and a final wash was performed in PBS prior to imaging using the Odyssey infrared imaging system (LiCor). Where appropriate, bands were quantified using the Odyssey software package and P values were calculated in GraphPad Prism 4 using a one-way analysis of variance (ANOVA) and Dunnett's Multiple Comparison Test.
Focus forming assays.
Tissue culture media from infected HuH-7 cells was diluted 10-fold in serum-free RPMI 6400 (Gibco) and used to infect BHK-21 cell monolayers in 24 well plates for 1 hour. After 1 hour, infectious media was replaced with RPMI 6400 containing 1% Aquacide II (Calbiochem). Cells were incubated for four days and fixed in 4% formaldehyde in PBS, permeabilized using 0.5% Triton X-100 in PBS and blocked using 0.1% Tween 20 and 1% normal goat serum in PBS. Viral foci were detected using the 3H5 monoclonal antibody to DENV E protein and anti-mouse DyLight 680 secondary antibody. Viral foci were visualized using the Odyssey infrared imaging system. p values were calculated in GraphPad Prism 4 using a one-way analysis of variance (ANOVA) and Dunnett's Multiple Comparison Test.
Quantitative real-time PCR.
Quantitative real-time PCR was performed as described in Anwar et al. (2009). 25 ng of an in vitro transcript derived form the D. melanogaster Boule mRNA was added to each sample prior to Trizol extraction and used as a normalization control. p values were calculated in GraphPad Prism 4 using a one-way analysis of variance (ANOVA) and Dunnett's Multiple Comparison Test.
RNA immunoprecipitation.
RNA immunoprecipitation was performed as described in Anwar et al. (2009). Oligonucleotides used to amplify DENV-2 and β-actin RNAs are the same as in Anwar et al.. Real-time PCR was performed as described above.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Figures and Tables
Figure 1 Identification of cellular proteins that interact with the DENV-2 5′ and 3′ UTRs. (A) Schematic of DNA template used to generate RNA for chromatography. The control construct contains the T7 promoter fused to sequences from DENV-2 NGC NS2A coding sequence (nts 3,504–3,674 and nts 3,739–4,221), with the tobramycin aptamer inserted between the NS2A sequences. The 5′ and 3′ UTR construct contains the T7 promoter fused to the DENV-2 NGC 5′ UTR (nts 1–172), tobramycin aptamer sequence and the 3′ UTR from DENV-2 NGC (nts 10,242–10,724). Following in vitro transcription using T7 RNA polymerase, RNA was purified, heated to 95°c, gradually cooled to room temperature and bound to tobramycin-sepharose beads. (B) Scatter plot comparing the results from two independent quantitative mass spectrometric analyses of DENV-2 5′ and 3′ UTR-interacting proteins. In the experiment plotted on the y-axis (forward experiment), the control RNA was incubated with lysate from cells labeled with light arginine and lysine (K0R0) and the DENV-2 5′ and 3′ UTR RNA was incubated with lysate from cells labeled with heavy arginine and lysine (K8R10). In the experiment plotted on the x-axis (reverse experiment), the control RNA was incubated with heavy lysate and the DENV-2 5′ and 3′ UTR RNA was incubated with light lysate. The product of ratio significance in the forward and reverse experiment (pforward x preverse (p) < 1e–10), and minimum log2 SILAC ratio of 2 for forward experiment and maximum ratio of -2 for reverse experiment were used to select the most confident interacting partners (red dots in upper left quadrant).
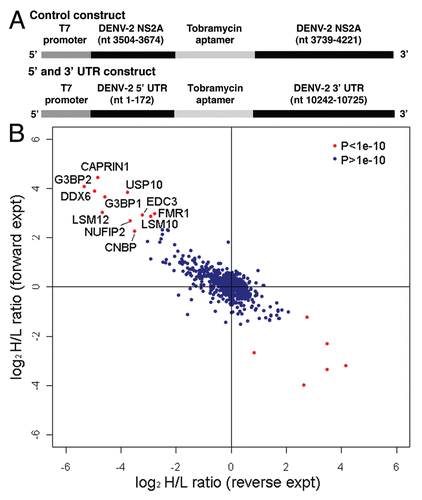
Figure 2 DDX6, Caprin1, G3BP1, G3BP2 and USP10 colocalize with sites of DENV replication. HuH-7 cells on coverslips were infected with DENV-2 NGC at an MOI of 1.0 for 24 hours prior to fixation and probing with antibody specific for either SLBP (A and B), DDX6 (C and D), Caprin1 (E and F), G3BP1 (G and H), G3BP2 (I and J) or USP10 (K and L) and dsRNA as a marker for sites of DENV replication as previously shown by Welsch et al. The coverslips probed for SLBP and dsRNA were prepared at a different time, but under similar conditions, than the coverslips used in the rest of the localization studies described here. However, the same preparation of coverslips was also probed for DDX6 and dsRNA with the same colocalization pattern as observed with the other coverslips probed for DDX6 and dsRNA. In the final wash, DAPI was added to visualize cell nuclei and coverslips were sealed prior to visualization using an Olympus IX71 epifluorescent microscope and DP71 digital camera. Images were processed using the ImageJ software package.
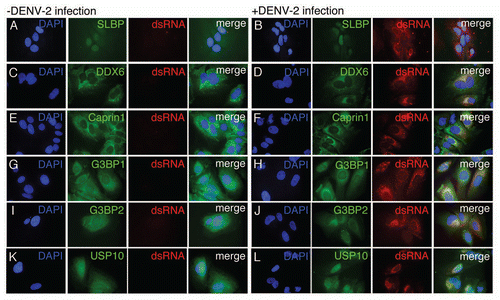
Figure 3 DDX6 is required for efficient assembly or release of infectious Dengue virus. (A) Analysis of protein expression of cellular DDX6 and GAPDH proteins at 24 hours post-infection. HuH-7 cells were transfected with 25 nM siRNAs targeting GFP (siGFP) or DDX6 (siDDX6_1, siDDX6_2 and siDDX6_3) and incubated for 48 hours. cells were infected with DENV-2 NGC at an MOI of 0.5 and harvested at 24 hours post-infection. Western blots were probed using rabbit polyclonal antibodies specific for DDX6 and GAPDH. Following incubation with secondary antibody, blots were visualized using the Odyssey Infrared Imaging system. (B) Quantification of western blots for DDX6 protein normalized to GAPDH levels. Quantification was performed using the Odyssey Infrared imaging software package and the level of DDX6 protein in each sample was normalized to GAPDH. DDX6 levels are plotted as a percent relative to cells treated with siGFP. The raw data from quantification was used for one-way ANOVA (p < 0.0001) and Dunnett's Multiple Comparison Test, where p < 0.01 is indicated by an asterisk. All p values were calculated with respect to the siGFP control. (C) Focus forming units (ffu) of tissue culture supernatants 24 hours post-infection. Tissue culture media was diluted and used to infect monolayers of BHK21 cells. Four days post-infection, cells were fixed and analyzed for DENV E protein expression, as described in the Materials and Methods. Virus foci were counted and plotted as ffu/mL. Data was analyzed using a one-way ANOVA (p = 0.0022) and Dunnett's Multiple Comparison Test, where p < 0.01 is indicated by an asterisk. All p values were calculated with respect to the siGFP control. (D) DENV-2 RNA level in the tissue culture media 24 hours post-infection. Total RNA was extracted from tissue culture media of infected cells, reverse transcribed using random primers, and used in Real-time PCR reactions. 25 ng of an in vitro transcript derived form the D. melanogaster Boule mRNA was added to each sample prior to Trizol extraction and used as a normalization control. A standard curve was generated by reverse transcribing 1 ng of in vitro transcribed RNA corresponding to the DENV-2 amplicon. Number of DENV-2 RNA molecules was calculated from the standard curve and plotted. Data was analyzed using a one-way ANOVA (p = 0.0024) and Dunnett's Multiple comparison Test, where p < 0.01 is indicated by an asterisk. All p values were calculated with respect to the siGFP control.
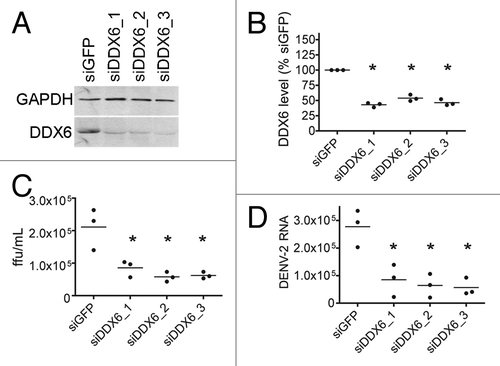
Figure 4 DDX6 interacts with DENV-2 RNA in vivo during infection. (A) RNA immunoprecipitation was performed using either a non-specific IgG or DDX6-specific antibody on uninfected and infected HuH-7 cell lysates. Cells were infected with DENV-2 NGC at an MOI of 0.5 and harvested 48 hours post-infection. Antibodies bound to protein A beads were incubated with uninfected or infected cell lysates and washed 5x with RIPA buffer containing 1 M NaCl. RNA was isolated using Trizol and analyzed by RT-PCR using primers specific for β-actin, c-myc or DENV sequences. (B) RNA immunoprecipitation was repeated in triplicate and processed as in (A), except analysis was performed using Real-time PCR for DENV. To facilitate normalization, an in vitro transcript was added prior to Trizol extraction as a control for recovery. Data was analyzed using a one-way ANOVA (p = 0.0045) and Dunnett's Multiple Comparison Test, where p < 0.01 is indicated by an asterisk.
Figure 5 DDX6 interacts with the DENV-2 3′ UTR via DB2 and DB1 structures. (A–D) Interaction sites for DDX6 were determined using the RNA affinity chromatography method. Deletion mutants were generated from the original 5′ and 3′ UTR construct by PCR and used as templates for T7 in vitro transcription. The left part illustrates the secondary structures present in the DENV-2 3′ UTR based on structural studies and Mfold secondary structure prediction.Citation11,Citation60,Citation61 The deletion mutations used to map 3′ UTR interaction sites are illustrated by black lines under the predicted secondary structures. T7 transcripts were purified, heated to 95°C, cooled and bound to tobramycin-sepharose beads. RNA-bound beads were incubated with cell lysate followed by washing and elution prior to analysis by western blotting for DDX6.
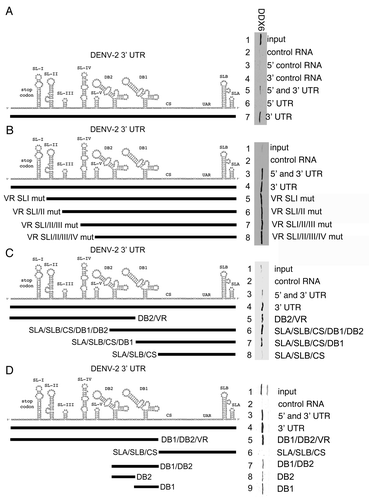
Figure 6 DDX6 interacts with the DENV-2 DB2 and DB1 via pseudoknot structures. (A) Schematic of the DB2 and DB1 structures in the 3′ UTR and sequences predicted to mediate formation of pseudoknot structures (pk2′, pk2, pk1′ and pk1).Citation11,Citation44 (B) RNA affinity chromatography was performed similarly as in except point mutations in the predicted pseudoknot-forming sequences were made in the full-length 3′ UTR. The templates were in vitro transcribed, purified, heated to 95°C, cooled and bound to tobramycin-sepharose beads. RNA-bound beads were incubated with cell lysate followed by washing and elution prior to analysis by western blotting for DDX6.
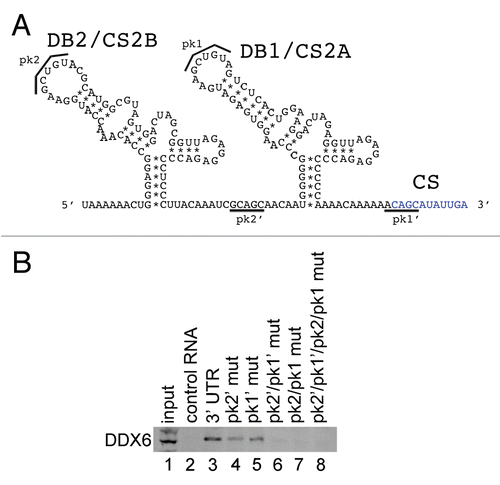
Figure 7 DDX6, Caprin1, G3BP1, G3BP2 and USP10 bind to different sequences in the 3′ UTR of DENV-2. Interaction sites for DDX6, Caprin1, G3BP1, G3BP2 and USP10 were determined using the RNA affinity chromatography method. Deletion mutants were generated similarly to those described in . The left part illustrates the secondary structures present in the DENV-2 3′ UTR based on structural studies and Mfold secondary structure prediction.Citation11,Citation60,Citation61 The deletion mutations used to map 3′ UTR interaction sites are illustrated by black lines under the predicted secondary structures. T7 transcripts were purified, heated to 95°C, cooled and bound to tobramycin-sepharose beads. RNA-bound beads were incubated with cell lysate followed by washing and elution prior to analysis by western blotting for TIAL1 (negative control), DDX6 (positive control), Caprin1, G3BP1, G3BP2 and USP10.
Table 1 List of DENV-2 UTR-interacting proteins identified by mass spectrometry and their respective SILAC ratios with p values from independent forward and reverse experiments
Additional material
Download Zip (7.1 MB)Acknowledgements
The authors also would like to thank Shelton Bradrick (Duke University, USA) for the pTNT-FLuc firefly luciferase construct, Matt Marengo (Duke University, USA) for advice on statistical methods, Azlinda Bte Anwar (Duke-NUS Graduate Medical School, Singapore) for critical discussion and proofreading, and Subhash Vasudevan (Duke-NUS Graduate Medical School, Singapore) for technical advice. This research was supported by the Agency for Science, Technology and Research of Singapore and Singapore Ministry of Health Awards R-913-200-002-304 (to M.G.B.), and grants from the Agency for Science, Technology and Research of Singapore (to W.B.).
References
- Lindenbach B, Thiel HJ, Rice CM. Flaviviridae: The Viruses and Their Replication 2007; Philadelphia Lippincott, Williams and Wilkins
- Brinton MA, Dispoto JH. Sequence and secondary structure analysis of the 5′-terminal region of flavivirus genome RNA. Virology 1988; 162:290 - 299
- Brinton MA, Fernandez AV, Dispoto JH. The 3′-nucleotides of flavivirus genomic RNA form a conserved secondary structure. Virology 1986; 153:113 - 121
- Grange T, Bouloy M, Girard M. Stable secondary structures at the 3′-end of the genome of yellow fever virus (17 D vaccine strain). FEBS Lett 1985; 188:159 - 163
- Hahn CS, Hahn YS, Rice CM, Lee E, Dalgarno L, Strauss EG, Strauss JH. Conserved elements in the 3′ untranslated region of flavivirus RNAs and potential cyclization sequences. J Mol Biol 1987; 198:33 - 41
- Takegami T, Washizu M, Yasui K. Nucleotide sequence at the 3′ end of Japanese encephalitis virus genomic RNA. Virology 1986; 152:483 - 486
- Filomatori CV, Lodeiro MF, Alvarez DE, Samsa MM, Pietrasanta L, Gamarnik AV. A 5′ RNA element promotes dengue virus RNA synthesis on a circular genome. Genes Dev 2006; 20:2238 - 2249
- Clyde K, Barrera J, Harris E. The capsid-coding region hairpin element (cHP) is a critical determinant of dengue virus and West Nile virus RNA synthesis. Virology 2008; 379:314 - 323
- Clyde K, Harris E. RNA secondary structure in the coding region of dengue virus type 2 directs translation start codon selection and is required for viral replication. J Virol 2006; 80:2170 - 2182
- Holden KL, Harris E. Enhancement of dengue virus translation: role of the 3′ untranslated region and the terminal 3′ stem-loop domain. Virology 2004; 329:119 - 133
- Alvarez DE, De Lella Ezcurra AL, Fucito S, Gamarnik AV. Role of RNA structures present at the 3′UTR of dengue virus on translation, RNA synthesis and viral replication. Virology 2005; 339:200 - 212
- Blaney JE Jr, Durbin AP, Murphy BR, Whitehead SS. Development of a live attenuated dengue virus vaccine using reverse genetics. Viral Immunol 2006; 19:10 - 32
- Chiu WW, Kinney RM, Dreher TW. Control of translation by the 5′- and 3′-terminal regions of the dengue virus genome. J Virol 2005; 79:8303 - 8315
- Alvarez DE, Filomatori CV, Gamarnik AV. Functional analysis of dengue virus cyclization sequences located at the 5′ and 3′ UTRs. Virology 2008; 375:223 - 235
- Friebe P, Harris E. Interplay of RNA elements in the dengue virus 5′ and 3′ ends required for viral RNA replication. J Virol 2010; 84:6103 - 6118
- Holden KL, Stein DA, Pierson TC, Ahmed AA, Clyde K, Iversen PL, Harris E. Inhibition of dengue virus translation and RNA synthesis by a morpholino oligomer targeted to the top of the terminal 3′ stem-loop structure. Virology 2006; 344:439 - 452
- Garcia-Montalvo BM, Medina F, del Angel RM. La protein binds to NS5 and NS3 and to the 5′ and 3′ ends of Dengue 4 virus RNA. Virus Res 2004; 102:141 - 150
- Yocupicio-Monroy M, Padmanabhan R, Medina F, del Angel RM. Mosquito La protein binds to the 3′ untranslated region of the positive and negative polarity dengue virus RNAs and relocates to the cytoplasm of infected cells. Virology 2007; 57:29 - 40
- Paranjape SM, Harris E. Y box-binding protein-1 binds to the dengue virus 3′-untranslated region and mediates antiviral effects. J Biol Chem 2007; 282:30497 - 30508
- Emara MM, Brinton MA. Interaction of TIA-1/TIAR with West Nile and dengue virus products in infected cells interferes with stress granule formation and processing body assembly. Proc Natl Acad Sci USA 2007; 104:9041 - 9046
- Emara MM, Liu H, Davis WG, Brinton MA. Mutation of mapped TIA-1/TIAR binding sites in the 3′ terminal stem-loop of West Nile virus minus-strand RNA in an infectious clone negatively affects genomic RNA amplification. J Virol 2008; 82:10657 - 10670
- Anwar A, Leong KM, Ng ML, Chu JJ, Garcia-Blanco MA. The polypyrimidine tract-binding protein is required for efficient dengue virus propagation and associates with the viral replication machinery. J Biol Chem 2009; 284:17021 - 17029
- Krishnan MN, Ng A, Sukumaran B, Gilfoy FD, Uchil PD, Sultana H, et al. RNA interference screen for human genes associated with West Nile virus infection. Nature 2008; 455:242 - 245
- Sessions OM, Barrows NJ, Souza-Neto JA, Robinson TJ, Hershey CL, Rodgers MA, et al. Discovery of insect and human dengue virus host factors. Nature 2009; 458:1047 - 1050
- Chu CY, Rana TM. Translation repression in human cells by microRNA-induced gene silencing requires RCK/p54. PLoS Biol 2006; 4:210
- Minshall N, Kress M, Weil D, Standart N. Role of p54 RNA helicase activity and its C-terminal domain in translational repression, P-body localization and assembly. Mol Biol Cell 2009; 20:2464 - 2472
- Weston A, Sommerville J. Xp54 and related (DDX6-like) RNA helicases: roles in messenger RNP assembly, translation regulation and RNA degradation. Nucleic Acids Res 2006; 34:3082 - 3094
- Ladomery M, Wade E, Sommerville J. Xp54, the Xenopus homologue of human RNA helicase p54, is an integral component of stored mRNP particles in oocytes. Nucleic Acids Res 1997; 25:965 - 973
- Andrei MA, Ingelfinger D, Heintzmann R, Achsel T, Rivera-Pomar R, Luhrmann R. A role for eIF4E and eIF4E-transporter in targeting mRNPs to mammalian processing bodies. RNA 2005; 11:717 - 727
- Beckham CJ, Parker R. P bodies, stress granules and viral life cycles. Cell Host Microbe 2008; 3:206 - 212
- Mas A, Alves-Rodrigues I, Noueiry A, Ahlquist P, Diez J. Host deadenylation-dependent mRNA decapping factors are required for a key step in brome mosaic virus RNA replication. J Virol 2006; 80:246 - 251
- Jangra RK, Yi M, Lemon SM. DDX6 (Rck/p54) is required for efficient hepatitis C virus replication but not for internal ribosome entry site-directed translation. J Virol 2010; 84:6810 - 6824
- Scheller N, Mina LB, Galao RP, Chari A, Gimenez-Barcons M, Noueiry A, et al. Translation and replication of hepatitis C virus genomic RNA depends on ancient cellular proteins that control mRNA fates. Proc Natl Acad Sci USA 2009; 106:13517 - 13522
- Hartmuth K, Vornlocher HP, Luhrmann R. Tobramycin affinity tag purification of spliceosomes. Methods Mol Biol 2004; 257:47 - 64
- Butter F, Scheibe M, Morl M, Mann M. Unbiased RNA-protein interaction screen by quantitative proteomics. Proc Natl Acad Sci USA 2009; 106:10626 - 10631
- Ong SE, Blagoev B, Kratchmarova I, Kristensen DB, Steen H, Pandey A, Mann M. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol Cell Proteomics 2002; 1:376 - 386
- Cox J, Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol 2008; 26:1367 - 1372
- Anderson P, Kedersha N. RNA granules: post-transcriptional and epigenetic modulators of gene expression. Nat Rev Mol Cell Biol 2009; 10:430 - 436
- Black DL. Mechanisms of alternative pre-messenger RNA splicing. Annu Rev Biochem 2003; 72:291 - 336
- Jensen LJ, Kuhn M, Stark M, Chaffron S, Creevey C, Muller J, et al. STRING 8—a global view on proteins and their functional interactions in 630 organisms. Nucleic Acids Res 2009; 37:412 - 416
- Welsch S, Miller S, Romero-Brey I, Merz A, Bleck CK, Walther P, et al. Composition and three-dimensional architecture of the dengue virus replication and assembly sites. Cell Host Microbe 2009; 5:365 - 375
- Pandey NB, Sun JH, Marzluff WF. Different complexes are formed on the 3′ end of histone mRNA with nuclear and polyribosomal proteins. Nucleic Acids Res 1991; 19:5653 - 5659
- Akao Y, Yoshida H, Matsumoto K, Matsui T, Hogetu K, Tanaka N, Usukura J. A tumour-associated DEAD-box protein, rck/p54 exhibits RNA unwinding activity toward c-myc RNAs in vitro. Genes Cells 2003; 8:6716
- Olsthoorn RC, Bol JF. Sequence comparison and secondary structure analysis of the 3′ noncoding region of flavivirus genomes reveals multiple pseudoknots. RNA 2001; 7:1370 - 1377
- Manzano M, Reichert ED, Polo S, Falgout B, Kasprzak W, Shapiro BA, Padmanabhan R. Identification of cis-acting elements in the 3′-untranslated region of the dengue virus type 2 RNA that modulate translation and replication. J Biol Chem 2011; 286:22521 - 22534
- Li W, Li Y, Kedersha N, Anderson P, Emara M, Swiderek KM, et al. Cell proteins TIA-1 and TIAR interact with the 3′ stem-loop of the West Nile virus complementary minus-strand RNA and facilitate virus replication. J Virol 2002; 76:11989 - 2000
- Wang PG, Kudelko M, Lo J, Siu LY, Kwok KT, Sachse M, et al. Efficient assembly and secretion of recombinant subviral particles of the four dengue serotypes using native prM and E proteins. PLoS One 2009; 4:8325
- Ariumi Y, Kuroki M, Kushima Y, Osugi K, Hijikata M, Maki M, et al. Hepatitis C Virus Hijacks P-body and Stress Granule Components Around Lipid Droplets. J Virol 2011;
- Miyanari Y, Atsuzawa K, Usuda N, Watashi K, Hishiki T, Zayas M, et al. The lipid droplet is an important organelle for hepatitis C virus production. Nat Cell Biol 2007; 9:1089 - 1097
- Samsa MM, Mondotte JA, Iglesias NG, Assuncao-Miranda I, Barbosa-Lima G, Da Poian AT, et al. Dengue virus capsid protein usurps lipid droplets for viral particle formation. PLoS Pathog 2009; 5:1000632
- Gibbings DJ, Ciaudo C, Erhardt M, Voinnet O. Multivesicular bodies associate with components of miRNA effector complexes and modulate miRNA activity. Nat Cell Biol 2009; 11:1143 - 1149
- Tahbaz N, Kolb FA, Zhang H, Jaronczyk K, Filipowicz W, Hobman TC. Characterization of the interactions between mammalian PAZ PIWI domain proteins and Dicer. EMBO Rep 2004; 5:189 - 194
- Gibbings D, Voinnet O. Control of RNA silencing and localization by endolysosomes. Trends Cell Biol 2010; 20:491 - 501
- Thomas MG, Loschi M, Desbats MA, Boccaccio GL. RNA granules: the good, the bad and the ugly. Cell Signal 23:324 - 334
- Anderson P, Kedersha N. RNA granules. J Cell Biol 2006; 172:803 - 808
- Alves-Rodrigues I, Mas A, Diez J. Xenopus Xp54 and human RCK/p54 helicases functionally replace yeast Dhh1p in brome mosaic virus RNA replication. J Virol 2007; 81:4378 - 4380
- Srisawat C, Engelke DR. Streptavidin aptamers: affinity tags for the study of RNAs and ribonucleoproteins. RNA 2001; 7:632 - 641
- Shevchenko A, Tomas H, Havlis J, Olsen JV, Mann M. In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat Protoc 2006; 1:2856 - 2860
- Katsafanas GC, Moss B. Colocalization of transcription and translation within cytoplasmic poxvirus factories coordinates viral expression and subjugates host functions. Cell Host Microbe 2007; 2:221 - 228
- Polacek C, Foley JE, Harris E. Conformational changes in the solution structure of the dengue virus 5′ end in the presence and absence of the 3′ untranslated region. J Virol 2009; 83:1161 - 1166
- Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res 2003; 31:3406 - 3415