Abstract
Although the roots of Ras sprouted from the rich history of retrovirus research, it was the discovery of mutationally activated RAS genes in human cancer in 1982 that stimulated an intensive research effort to understand Ras protein structure, biochemistry and biology. While the ultimate goal has been developing anti-Ras drugs for cancer treatment, discoveries from Ras have laid the foundation for three broad areas of science. First, they focused studies on the origins of cancer to the molecular level, with the subsequent discovery of genes mutated in cancer that now number in the thousands. Second, elucidation of the biochemical mechanisms by which Ras facilitates signal transduction established many of our fundamental concepts of how a normal cell orchestrates responses to extracellular cues. Third, Ras proteins are also founding members of a large superfamily of small GTPases that regulate all key cellular processes and established the versatile role of small GTP-binding proteins in biology. We highlight some of the key findings of the last 28 years.
Introduction
RAS genes were identified initially as viral genes transduced from the rodent genome and responsible for the highly oncogenic properties of RNA tumor viruses.Citation1 The study of these viral genes, their cellular counterparts, and the 21 kDa (p21) proteins encoded by these genes established key and fundamental properties of cellular Ras proteins. The identification in 1982 of mutationally activated and potently transforming human RAS genes in human cancer cell lines began an intensive focus on studying Ras structure, biochemistry and biology that continues to this day.Citation2 Fueled by the high frequency of RAS mutations in a wide spectrum of human cancers, including three of the four most deadly cancers in the U.S. (lung, colon and pancreatic cancer),Citation3 there has been an intensive search for the still elusive “anti-Ras” therapy for cancer treatment.Citation4 Dissection of the function of Ras proteins as simple on-off binary switches for diverse extracellular signaling cascades has established many of our current fundamental paradigms of signal transduction. The recent identification of germline RAS mutations in a class of developmental syndromes (RASopathies) expands the contribution of aberrant Ras signaling into other human disorders.Citation5 Finally, that Ras proteins represent simply the tip of the iceberg, and are the founding members of a superfamily of Ras-related small GTPases that now number more than 150,Citation6 has established that small GTPases are versatile and key regulators of virtually all fundamental cellular processes. In this review, we take a retrospective view of the rich history of Ras research, provide a snapshot of the current state-of-the art, and speculate on what is in store for the future. We provide a chronology ( and Suppl. Table 1) of representative key discoveries regarding the biochemistry and structure of Ras proteins, the mechanisms of Ras signal transduction, the Ras superfamily, and the implication of aberrant Ras activation in cancer and developmental syndromes.
The Retrovirus Years
The identification of Ras emerged during the extensive study of acutely transforming retroviruses isolated from mice, rats, cats, monkeys, chickens and turkeys. These oncogenic viruses cause rapid formation of sarcomas in infected animals and potently transform cells in culture. Discoveries of the potently oncogenic Harvey murine sarcoma virus in 1964Citation7 and the Kirsten murine sarcoma virus in 1967Citation8 provided our first glimpses of oncogenic genetic elements that only many years later were established as comprising the human HRAS and KRAS oncogenes, respectively. That the transforming properties of these sarcoma viruses resulted from transduction of normal cellular rat sequences into their own genomes was predicted by Scolnick and colleagues in 1973,Citation11 a time when the tools to prove this were lacking. Initially the genes that we now know as RAS were named as variants of src. Later their ability to cause rat sarcomas became the basis for their current gene names, and their discoverers' names became the basis for distinguishing each of them from the other: Harvey and Kirsten viral ras genes, or H-ras and K-ras, with their protein names cited as Ha-Ras or H-Ras and Ki-Ras or K-Ras. In a seminal discovery published in 1976, Varmus, Bishop, Vogt and colleagues determined that the potent viral src oncogene is a normal chicken gene transduced by the virus into its own genome, thereby converting a normal gene into a potent oncogenic agent.Citation9 Viral oncogenes of several other acutely transforming retroviruses were found to encode proteins that only much later were determined to be key components of Ras signaling networks, including the Raf and Akt serine/threonine kinases, the epidermal growth factor receptor (EGFR) tyrosine kinase, the catalytic subunit of class I phosphatidylinositol 3-kinase (PI3-K) (p110α) and the Ets transcription factor.
Scolnick and colleagues in the late 1970s to the early 1980s performed an extensive series of pioneering studies that determined the cellular origin of the viral H-ras and K-ras genes,Citation10,Citation11 and that these genes encode 21 kDa proteinsCitation12 that bind GDP and GTPCitation13 and are associated with the plasma membrane.Citation14 They also identified the cellular counterparts of the viral genes;Citation15,Citation16 that Ras proteins expressed in vertebrate cellsCitation17 also bind GDP and GTP and are membrane-associated;Citation18 that, when overexpressed, these proteins can also transform cells,Citation19 and that preferential binding to GTP is key for transformation.Citation13,Citation20 These studies established many of the fundamental biochemical and cellular properties of Ras as membrane-associated GTP-binding proteins and served as an important basis for the many subsequent studies that would expand on their findings ().
The Human Cancer Years
Despite recognition of the highly oncogenic potency of acutely transforming retroviruses, the realization that human cancers are not initiated by such infectious agents dampened enthusiasm for studying them as the basis of human cancer development. Instead, a new direction arose from the finding that biologically active eukaryotic DNA could be transferred into mammalian cells by transfection, following precipitation of the DNA using calcium phosphate.Citation21,Citation22 A critical key to the success of these experiments in demonstrating whether the transfected DNA had transforming properties was the use of NIH/3T3 mouse fibroblasts as the recipient cells.Citation23 Although immortalized, NIH/3T3 cells nevertheless retained some normal growth properties (density-dependent growth inhibition, high dependence on serum growth factors), and failed to propagate when deprived of substratum (e.g., did not form colonies in soft agar) or to form tumors when inoculated into immunocompromised mice.Citation23 However, these cells did display high sensitivity to retrovirus-induced “focus formation”, such that morphologically altered cells that were no longer contact-inhibited grew as easily visualized foci of transformed cells over the background monolayer of untransfected “normal” cells (). The ability of NIH/3T3 cells to become morphologically and growth-transformed by a single viral oncogene provided a sensitive one-hit biological assay for the activated oncogenes that were speculated to be present in DNA obtained from tumor but not from normal cells. NIH/3T3 cells (also referred to as NIH 3T3 or NIH3T3) have therefore been the longtime workhorse cell culture model for these studies, and were instrumental in characterizing RAS and many other oncogenes.
In 1979 Weinberg and colleagues showed that DNA isolated from chemically transformed rodent fibroblasts caused morphologic transformation of NIH/3T3 mouse fibroblasts.Citation24 In 1981, using the same approach, Krontiris and Cooper identified transforming activity with DNA isolated from the human EJ bladder carcinoma cell lineCitation25 (also referred to as T24 cellsCitation26), and Weinberg and colleagues also detected transforming activity in carcinoma and leukemia cell lines.Citation27,Citation28 Wigler and colleagues then made similar observations using DNA from lung and colon carcinoma cell lines,Citation29 followed by similar discoveries in human sarcoma cell lines.Citation30 The hunt was then on to identify and isolate the human oncogenes that transformed NIH/3T3 cells.
In 1982, this seemingly distinct direction of study unexpectedly collided with the retrovirus oncogene studies. Three groups made the discovery that the transforming genes identified in the NIH/3T3 DNA transfection assays were the same RAS genes identified earlier in Kirsten and Harvey sarcoma viruses.Citation31–Citation33 By the end of 1982, three groups had established the molecular basis of HRAS gene activation in the EJ/T24 bladder carcinoma cell line,Citation26 unexpectedly and remarkably by a single missense mutation in codon 12, which was also found in the viral H-ras and K-ras genes ().Citation34–Citation36 Mutation of codon 12 was also established as the mechanism of activation of KRAS from lung and colon tumor cells.Citation37 Additionally, a third transforming human RAS gene, not identified previously in any retroviruses, was discovered in neuroblastoma-derived DNA and was designated NRAS ().Citation38,Citation39 The identification of mutant RAS genes in patient tumors but not in normal tissue was an important validation that the RAS mutations identified in tumor cell lines were not simply artifacts of in vitro cell passage.Citation40,Citation41 This surprisingly simple mechanism of Ras protein activation then focused attention on the issue of what one amino acid substitution would do to the as yet to be determined biochemical and biological functions of Ras proteins.
The discovery of RAS oncogene activation by DNA and protein mutation also stimulated an intense focus on pursuing the origins of cancer at the molecular level. This led to the subsequent identification of oncogene activation, and of tumor suppressor gene inactivation, by alterations in protein structure and function. This bias and emphasis continues to this day, such that genome-wide DNA sequencing of the cancer genome is now a major mandate in the “war against cancer.”
Subsequent studies using the NIH/3T3 transfection assay, and later analyses using DNA sequencing methods, detected activated RAS genes in a wide spectrum of human tumor cell lines as well as in primary patient tumor material.Citation42 Most striking was the high frequency of RAS mutations found in colon,Citation43,Citation44 lungCitation45 and pancreaticCitation46,Citation47 cancers. In addition to mutations at codon 12, RAS mutations were later identified at codons 13 and 61. Although mutations affecting other regions of the proteins have been found at very low frequencies, these three codons are the sites of 97–99% of all RAS mutations in cancer () and thus comprise the three hot spots of Ras activation. Also strikingly, not all Ras isoforms are equally likely to be mutated. Among the RAS isoforms, missense mutations are found most commonly in KRAS (85%), less commonly in NRAS (12%), and rarely in HRAS (3%) (). Interestingly, the missense mutation frequency at each position varies widely between isoforms, with G12 mutations the most common in KRAS and HRAS, while Q61 mutations are the most common in NRAS ().
Analysis of the current data available at the COSMIC database (www.sanger.ac.uk/genetics/CGP/cosmic/) reveals that HRAS, the first activated RAS gene detected and characterized, is the least frequently mutated in human cancers (3%), whereas mutation of KRAS is the most prevalent (21%), followed by mutations in NRAS (8%) (). Activated RAS genes were also identified in tumors that arose from mutagenic treatment of rodents with chemical carcinogens.Citation48,Citation49
The potent ability of mutant RAS genes alone to convert immortalized NIH/3T3 cells to invasive and tumorigenic cells gave an initially simple and misleading perception of the genetic basis of cancer. This notion was revised subsequently by the finding that mutant H-Ras alone was unable to transform primary rodent fibroblasts, but instead, required concurrent activation of an oncogene (e.g., Myc) or inactivation of a tumor suppressor (e.g., p53) for Ras-mediated transformation of these cells.Citation50,Citation51 This requirement for a cooperating second hit was later extended by cell culture studies of primary human fibroblasts and epithelial cells, in which hTERT-mediated immortalization, inactivation of the p53 and Rb tumor suppressors, and protein phosphatase 2A inactivation were all found to make required contributions to Ras-dependent transformation.Citation52 ,Citation53 In 1987, studies in transgenic mouse models supported both the causal role of Ras activation in cancer development as well as the need for cooperating genetic events. Similarly, such studies showed the cooperation of viral H-ras and myc in mammary tumor formation.Citation54,Citation55
The requirement for complementing genetic events is consistent with the accumulation of genetic mutations in human colon and pancreatic cancer ( and B, respectively), and that cancer incidence increases with age. In pancreatic cancer, KRAS mutation is an early and initiating event, leading some to speculate wrongly that KRAS is not required for tumor maintenance and is not a suitable drug target in this disease. However, the importance of subsequent additional mutations can clearly be seen in mouse models, where endogenous KRAS activation alone induces tumor progression,Citation55 but where concurrent inactivation of INK4A/ARF, TP53 or SMAD4 causes greatly accelerated and more advanced tumor development.Citation56–Citation60
With advances in sequencing technology, it has become feasible to take an unbiased approach to begin to establish the full genetic complexity of the cancer cell genome. With the genomewide sequencing of breast and colon cancers,Citation61,Citation62 pancreatic cancerCitation63 and glioblastoma,Citation64,Citation65 a picture has emerged showing that cancers arise through a combination of the frequent mutation of a small common set of genes (mountains) and the infrequent mutation of many other genes (hills) specific to the individual cancers. Although the cancer cell genome harbors many gene mutations, the vast majority of these are passenger mutations that do not serve a causal role, whereas approximately 15–20 driver mutations contribute to cancer progression. A key finding that has arisen from these studies, in particular for colon and pancreatic cancer, is that mutations in KRAS are the biggest oncogene mountains in these two deadly cancers.
One issue that has been puzzling is the preferential mutational activation of specific RAS isoforms in different cancers. For example, there is near-exclusive mutational activation of KRAS in pancreatic, colon and lung cancers. Does this simply reflect distinct carcinogenic assaults that preferentially favor mutation of a certain isoform, or does tumor formation occur only through mutation of certain isoforms? While the evidence is still very limited, one provocative study that addressed this question using mouse models found that activation of KRAS but not NRAS promoted colonic tumorigenesis.Citation66 More such studies will be needed to determine if indeed mutation of a specific RAS isoform is critical to initiate oncogenesis in specific tissue types.
The Family Years
Evolutionary conservation of Ras.
The conservation of Ras in invertebrate species amenable to genetic analyses has contributed significantly to delineation of the roles and mechanisms of Ras in vertebrate signal transduction (). Two functionally redundant Ras proteins (Ras1 and Ras2) were identified as required for spore viability in the budding yeast S. cerevisiae.Citation67,Citation68 Interestingly, while Ras1 and Ras2 do show strong sequence identity with human Ras proteins, they possess additional divergent C-terminal sequences and, at ∼40 kDa, are much larger proteins. In the fission yeast S. pombe, one ∼21 kDa Ras protein (Ras1) has been identified as required for sporulation and mating.Citation69,Citation70 Two Drosophila RAS genes encoding ∼21 kDa proteins were identified initially (Dras1 and Dras2),Citation71 and DRas1 was shown later to be the authentic Ras ortholog and a regulator of eye development. The one RAS gene (encoding LET-60) in C. elegans controls vulval development.Citation72,Citation73 Finally, Ras proteins have also been studied in other model organisms, including the slime mold DictyosteliumCitation74 and zebrafish (N-Ras and K-Ras orthologs).Citation75,Citation76 However, no RAS genes are present in plant genomes.
The Ras superfamily: GTPases are versatile regulators.
In addition to their conservation in evolution, the RAS genes are the founding members and prototypes for a large family of RAS-related genes found in both invertebrates and vertebrates.Citation77,Citation6 Sequence identity and/or functional relationships subdivide the human family into at least five distinct branches: Ras, Rho, Rab, Arf and Ran ( and Suppl. Table 2). Much of the confusing nomenclature of small GTPases reflects the common practice of naming of each newly identified gene product in a manner that acknowledges its relationship to Ras, the founding member of the family, or to the tissue or cell line in which it was initially identified.
In 1983, Ypt1 became the first Ras-related protein to be added to the family, when it was found fortuitously in S. cerevisiae as the gene product of an open reading frame located between the genes encoding tubulin and actin, and was speculated to share nucleotide-binding properties with the “strikingly homologous” p21 Ras proteins.Citation78 Similarly, in 1985 came the fortuitous discovery of Ras homologous (Rho) GTPases in the snail Aplysia, in which Rho was identified inadvertently during a search for genes homologous to the alpha subunit of human chorionic gonadotropin.Citation79 Using Aplysia Rho as a DNA probe, this group then identified Rho orthologs in S. cerevisiae (Rho1 and Rho2),Citation80 Drosophila, rat and humans (RhoA, RhoB and RhoC).Citation81,Citation82
The fortuitous discovery of evolutionarily-conserved Ras and Ras-related genes prompted deliberate efforts to identify additional Ras-related genes. In particular, Tavitian and colleagues performed DNA oligonucleotide hybridization using probes corresponding to the GTP-binding motif 57-DTAGQEE/D-63. They identified sequences encoding a simian Ras-like protein (RalA) in 1986Citation83 and mammalian orthologs of Ypt1 from a rat brain cDNA library (Rab1–4) in 1987.Citation84 Utilizing Drosophila Dras3 as a probe, they identified two Ras proximate proteins (Rap1 and Rap2) in 1988.Citation85 In 1987, another group performing low-stringency hybridization to viral H-ras identified both human and mouse Ras-related (R-ras) genes.Citation86 Takai and colleagues utilized biochemical approaches to identify six membrane-bound small molecular weight GTP-binding proteins (designated smg),Citation87 including K-Ras and RhoB, and independently discovered Rab3ACitation88 and Rap1A.Citation89
First discovered in 1980 as an ADP ribosylation factor (Arf) in cholera toxin-catalyzed ADP-ribosylation of adenylate cyclase, Arf was determined in 1986 to be a GTP-binding proteinCitation90–Citation92 and four years later to function in Golgi transport.Citation93 Similarly, the related gene SAR1 (secretion-associated and Ras-related) was identified in a genomic library functional screen for suppressors of the ER-Golgi transport mutant of SEC12 in S. cerevisiaeCitation94 and determined later to be a GTP-binding protein.Citation95 Finally, Ran was initially designated TC4 upon its identification in a hybridization screen for RAS-related genes using teratocarcinoma cells.Citation96 The later determination that the gene encodes a Ras-related nuclear protein became the basis for the name Ran.Citation97–Citation99
More recently, with the sequencing of human and other genomes, in silico database searches have identified the complete repertoire of genes encoding Ras-related small GTPases.Citation77,Citation6 These searches revealed 56 members in C. elegansCitation100 and 90 in DrosophilaCitation101 (Suppl. Table 2). Interestingly, while the genomes of the flowering plant Arabidopsis thaliana and of the rice Oryza sativa were reported to contain Ras-related small GTPases of the Rab, Rho, Arf and Ran families, no Ras GTPases have been found in any plants.Citation102,Citation101
As the Ras-related gene families have expanded, an obvious speculation in the field has been whether other members are also involved in cancer and other human disorders. This has led to research showing that numerous Ras superfamily proteins, while not mutated in human cancer, nevertheless do serve critical roles in oncogenic growth. In particular, numerous members of the Rho family and their regulators have been implicated in cancer as well as in other human diseases. This role was foreshadowed by the discovery of activated and transforming RhoGEFs (e.g., Dbl, Vav, Ect2) in the same NIH/3T3 focus formation assays used to identify mutant Ras genes.Citation103
Several members of the Ras branch have been implicated in cancer (). In particular, Ras homologue enriched in brain (Rheb) was identified by differential cloning techniques applied to identify genes whose transcription is rapidly induced in rat brain neurons upon synaptic activity.Citation104 Rheb was determined to be an activator of mTOR and to be activated downstream of the PI3K signaling pathway.Citation105 Rheb is also activated by loss of function of the tuberous sclerosis complex proteins (Tsc1/Tsc2), which function as a Rheb-specific GAP. Tuberous sclerosis is a rare genetic disorder associated with tumor formation in many different organs, primarily in the skin, brain, eyes, heart, kidney and lungs.
Many of the findings of Ras structure and biochemistry have provided invaluable clues to dissecting the function and regulation of these Ras superfamily members. In turn, the other branches of the Ras superfamily () have provided key foundations for our understanding of how many normal cellular processes are regulated. A survey of several invertebrate genomes finds that the Rab family is the largest branch in all species characterized ( and Suppl. Table 2). All species also have significantly large Arf and Rho families. In contrast, as mentioned above, while C. elegans, Drosophila and S. cerevisiae all possess Ras families, the two plant genomes lack any Ras family proteins. At 64 human members, Rab family proteins comprise the largest branch. They control vesicular transport to regulate membrane and protein traffic in the secretory and endocytic pathways.Citation106 Members of the Arf family (Arf, Arl and Sar), comprised of 29 human members,Citation107 are also involved in regulation of vesicular trafficking and of endocytosis and exocytosis. The Ran branch, unique in being composed of only a single human member, is involved in regulation of nucleocytoplasmic transport in interphase cells and in organization of the spindle apparatus during mitosis. With some variations and exceptions, Ras superfamily proteins function as GDP/GTP-regulated binary switches. Regardless of regulatory mechanism, they all transduce information through signaling cascades.
The Signal Transduction Years
1993 was a milestone year and “signaling achievement” for the delineation of the “classic” Ras signaling pathway, in which the EGF receptor tyrosine kinase was shown to activate Ras, with activated Ras in turn then stimulating the ERK mitogen-activated protein kinase (MAPK) cascade. While it is now well-appreciated that this single linear pathway represents a gross oversimplification of the complex Ras signaling network, these early findings provided key foundations for understanding the biochemical regulation of small GTPases and heterotrimeric G protein alpha subunits, as well as the mechanistic basis for maintenance of cellular signaling integrity. Here we highlight the key findings that established the mode of regulation of the Ras GDP/GTP cycle, how Ras is activated by extracellular stimuli and how activated Ras stimulates cytoplasmic signaling to relay the signal to the nucleus to cause changes in gene expression.
Regulation of the Ras GDP/GTP cycle: GAPs and GEFs.
The ability of viral Ras proteins to bind both GDP and GTP provided the first clues to the biochemical function of cellular Ras proteins.Citation13,Citation108 This finding prompted speculation that, like G alpha subunits, normal Ras proteins may be GDP/GTP-regulated binary switches involved in signal transduction.Citation109 The later finding that mutant Ras proteins are impaired ∼10-fold in their intrinsic GTP hydrolysis (GTPase) activityCitation110,Citation20,Citation111,Citation112 provided the first clue that their aberrant function in cancer cells involves preferential binding to GTP. However, impairment of intrinsic GTPase activity alone was not sufficient to explain Ras transforming activity in vivo.Citation113 A major milestone in understanding Ras function was the discovery by Trahey and McCormick in 1987 that a cytosolic GTPase activating protein (GAP) activity was responsible for a 300-fold acceleration of the hydrolysis of GTP bound to normal Ras but not to tumor-associated mutant Ras proteins.Citation114 The first RasGAP protein, p120 RasGAP, was identified and characterized the next year.Citation115–Citation117 In 1990, pursuit of the tumor suppressor protein(s) involved in neurofibromatosis type I (NF1) unexpectedly revealed the existence of a second RasGAP, neurofibromin (Nf1).Citation118–Citation120 Subsequently, additional RasGAPs have been identified.Citation121 These observations, together with the findings that the GTP-bound form of Ras is the activated state, established that the key biochemical defect of mutant Ras proteins is GAP insensitivity, resulting in impaired GAP-stimulated GTP hydrolysis that in turn leads to persistent accumulation of the active, GTP-bound protein.
Determination of the crystal structure of H-Ras also contributed significant insight into how Ras functions as a GDP/GTP-regulated switch. The first structure of the “G domain” (lacking the C-terminal hypervariable sequence, which is disordered and interferes with crystallization) was reported in 1988,Citation122 although a second report by Wittinghofer and colleagues was more accurateCitation123 and the original structure was later revised accordingly.Citation124 Structural studies identified two key regions of conformational differences between GDP- and GTP-bound H-Ras.Citation125,Citation126 Referred to as switch I and II, these regions coincide with sequences critical for Ras interaction with its regulators and effectors. Subsequent structural determinations of Ras in complex with GAP, GEF and Ras-interacting domains of effectors revealed the critical role of switch I and II in these interactions. The structure of H-Ras in complex with the RasGAP catalytic domain of p120RasGAP then identified a molecular basis for the biochemical consequences of the activating substitutions at G12 and Q61: mimicry of the GTP-bound state and GAP insensitivity.Citation127
Concurrent with the discovery of GAPs, the race was also on to determine how normal Ras proteins are activated. The search for regulators of the other side of the GDP/GTP cycle began with clues from studies in yeast and flies. In 1987, genetic studies in S. cerevisiae were instrumental in identifying CDC25 as an upstream activator of Ras proteins,Citation128,Citation129 although it was not yet known that the biochemical basis for this activation was the ability of CDC25 to act as a guanine nucleotide exchange factor (GEF) that enhanced removal of GDP from Ras, thereby preferentially allowing the more abundant GTP to bind in its place. Ironically, the first evidence that CDC25 may function as a RasGEF came from biochemical analysis of a related gene product, SDC25,Citation130 yet SDC25 is a dispensable Ras regulator in S. cerevisiae.Citation131 Subsequently, CDC25 itself was shown to function biochemically as a RasGEFCitation132 ( and ). Similarly, genetic studies in Drosophila identified Son of Sevenless (SOS) function downstream of the Sevenless and EGFR tyrosine kinasesCitation133 and later analyses determined that SOS shares homology with yeast CDC25.Citation134 In 1992, utilizing the information from the yeast and fly CDC25 genes, several groups identified mammalian CDC25-homologous RasGRF and Sos proteins.Citation135–Citation138 In 1996, structural elucidation of the Sos1-Ras interaction showed that Sos1 disrupts nucleotide binding by displacing Switch 1 and distorting Switch 2,Citation139 providing a mechanistic basis for GEF-enhanced GDP release and rebinding of GTP. Members of a third class of RasGEFs, Ras guanine nucleotide releasing proteins (RasGRPs), were identified initially in functional screens for activated oncogenesCitation140–Citation142 ().
Ras activation by diverse extracellular stimuli: mechanisms of signaling convergence.
Another important step in dissecting the role of Ras signal transduction was the positioning of Ras downstream of the EGFR and other cell surface receptor tyrosine kinases. A first suggestion came from the observation that EGFR stimulation increased Ras-GTP binding.Citation143 Further evidence came from seminal studies by Stacey and colleagues using the Y13-259 Ras neutralizing antibody.Citation144,Citation145 Microinjection of Y13-259 blocked serum stimulation of cell cycle progression in untransformed NIH/3T3 cells and the ability of membrane-associated viral tyrosine kinases (Fms/CSF-1 receptor, Fes and Src) but not cytoplasmic serine/threonine kinases (Raf, Mos), to transform these cells. The resulting model, in which Ras functions downstream of membrane-associated kinases and upstream of cytoplasmic kinases, would take another eight years to be validated experimentally.
Biochemical and genetic studies also positioned Ras downstream of receptor tyrosine kinases. For example, C. elegans Ras (LET-60) could overcome a defect in EGFR (LET-23) function in vulval development,Citation72,Citation146,Citation73 Drosophila Ras1 was found to function downstream of the Sevenless receptor tyrosine kinase to regulate eye development,Citation147 and ligand stimulation of EGFR or of the platelet-derived growth factor receptor (PDGFR) caused transient increases in GTP-bound Ras in mammalian cells.Citation148–Citation150
Led by further genetic studies in C. elegans and Drosophila, the linkage between the EGFR and Ras was completed with the discovery of the Grb2 adaptor protein (). Grb2, comprised of a central Src homology 2 (SH2) domain that associates with tyrosine phosphorylated peptide sequences, and that is flanked by two SH3 domains that recognize proline-rich sequences, lacks any catalytic function and instead functions solely as an adaptor protein that facilitates the formation of protein complexes. Identified initially as a gene (Sem-5) involved in C. elegans vulval development, a mammalian counterpart was identified independently as a protein that bound to activated EGFR (Grb2).Citation151 These observations prompted a flurry of eight studies showing that Grb2, through its SH3 domains, associates with Sos1, thus completing the connection between EGFR and Ras.Citation152–Citation159 That Grb2:Sos1 association with the activated EGFR promotes Ras activation in part by relocalization of the normally cytosolic Sos1 to the plasma membrane where Ras is situated was supported by studies showing that membrane-targeted Sos1 could activate Ras.Citation160,Citation161 This observation established an important concept in signal transduction: that regulation of subcellular location is an important mechanism to regulate the function of signaling proteins. Subsequent studies revealed further variations on the Grb2-Sos-Ras signaling mechanism, whereby Grb2 recognizes other adaptor proteins (e.g., Shc, IRS-1, Gab2), creating links with yet other receptor tyrosine kinases.Citation162
Other classes of RasGEFs are regulated by distinct signaling mechanisms as a consequence of the divergent sequences flanking their conserved CDC25 homology RasGEF catalytic domainsCitation163 (). In particular, the RasGRPs possess a C1 domain that binds diacylglycerol (DAG). Therefore, phospholipase C (PLC)-catalyzed production of the membrane-associated DAG lipid recruits RasGRP to the plasma membrane to activate Ras. Differential activation of PLC isoforms downstream of distinct classes of signaling modulators such as receptor tyrosine kinases (PLCγ), G protein-coupled receptors (PLCβ) or Ras/Rho small GTPases (PLCε)Citation164 thus provides another mechanistic basis for an amazing diversity of extracellular stimuli to converge on Ras activation.
Ras downstream effector signaling: mechanisms of signaling divergence.
Identification of the first downstream effector of Ras also came from studies in S. cerevisiae, in which adenylyl cyclase (Cyr1) was identified as an effector of RAS1 and RAS2.Citation165 Adenylyl cyclase stimulates the production of cAMP, which then initiates protein phosphorylation cascades and yeast cell growth. Given the ability of mammalian Ras to substitute for S. cerevisiae RAS proteins to activate yeast adenylate cyclase and conversely of activated yeast RAS to transform mouse NIH/3T3 cells,Citation166–Citation168 it was assumed that effector utilization would also be interchangeable between S. cerevisiae and mammals. Disappointingly, and perhaps surprisingly in light of the conservation of regulatory RasGEFs and RasGAPs identified subsequently, adenylate cyclase was firmly excluded as the effector of mammalian Ras.Citation169,Citation165 Another eight years would pass before the final link in the canonical Ras pathway was discovered.
In 1993, a remarkable convergence of information from the genetic dissection of Ras signaling in DrosophilaCitation170 and in C. elegansCitation171 with information on the biochemistry of Ras proteins determined in mammalian cells identified the Raf serine/threonine kinase as a protein that bound preferentially to activated Ras-GTP (), suggesting that it might play a role in selective transmission of signals from the active form of Ras.Citation172–Citation175,Citation171 Since Raf had been identified previously as a viral oncogene,Citation176–Citation178 much was already known about its function. This association, together with other observations of Ras- and Raf-dependent activation of the ERK1 and ERK2 MAPKs and the ability of Raf to activate MEK1 and MEK2,Citation179–Citation183 defined the Raf-MEK-ERK protein kinase cascade downstream of Ras. Interestingly, whereas Ras activates adenylyl cyclase in budding yeast (S. cerevisiae), Ras binding to Byr2 in fission yeast (S. pombe) activates a protein kinase cascade analogous to that seen in mammals.Citation184
The striking conservation of the Raf-MEK-ERK cascade downstream of Ras in both Drosophila and C. elegans (), where activation of this pathway alone could phenocopy the roles of Ras to regulate eye development and vulval cell fate, respectively, suggested that perhaps a full delineation of Ras effector signaling had been achieved. However, even with the discovery of Raf, there were already indications that other Ras effectors would be found.Citation185 By 1994, the p110 catalytic subunits of class I PI3-K were recognized as the second class of validated Ras effectorsCitation186 (). That p110α and a key downstream target of PI3K (the AKT serine/threonine kinase) were also identified independently as retrovirus oncogenes and that mutationally activated alleles of the gene that encodes p110. (PIK3CA) have been found in human cancers further support an important role for this effector in Ras-mediated oncogenesis.
A structural feature common to the majority of Ras effectors is the presence of a Ras-binding domain (RBD) or Ras association (RA) domain that promotes association of the effector with GTP-bound Ras. Because most researchers screening for Ras binding partners utilized 3′-extended cDNA libraries, their screens favored the isolation of potential effectors such as RalGEFs that contain a C-terminal RA domain.Citation187–Citation189 In contrast, the Raf RBD is positioned at the N-terminus of the protein, and hence, was better represented in the random-primed library used by Vojtek and Cooper.Citation185 Initial studies of the role of RalGEFs () and of their substrates, the Ras family RalA and RalB small GTPases (), suggested only a minor role for RalGEF-Ral cascades in Ras transformation of NIH3T3 cells.Citation190,Citation191 Only when subsequent studies were done in human cells did a significant role emerge for RalGEF-Ral signaling in oncogenesis.Citation192,Citation193 The importance of Ral GTPases in pancreatic, prostate, bladder and other human cancers has now been establishedCitation194. That RalGDS, a RalGEF, is required for development of skin carcinomas induced by mutant HRAS also supports the importance of this effector pathway in Ras-mediated oncogenesis.Citation195 Thus, although no mutations in RalGEF-Ral signaling components have been found in human cancer, the RalGEF-Ral pathway has emerged as perhaps the third best validated effector in Ras oncogenicity.Citation194
Two other classes of Ras effectors have been implicated as positive mediators of Ras oncogenesis (). First, in silico searches for novel proteins with homology to the Raf-RBD identified Tiam1, a RacGEF (), as an effector of Ras. Binding to Ras promotes Tiam1 RacGEF activity, and Tiam1 deficiency impairs development of mutant HRAS-driven skin carcinomas.Citation196,Citation197 Second, a novel RA domain-containing isoform of phospholipase C (PLC210) was identified initially in a yeast two-hybrid screen for LET-60-interacting proteins.Citation198 Subsequently, three groups searching BLAST databases independently identified a human ortholog of PLC210 (PLCε) and showed that it is an effector for human H-Ras stimulation of DAG and calcium release.Citation199–Citation201 Using the same chemical carcinogen approach as was done with RalGDS and Tiam1, it was later shown that mice deficient in PLCε were viable but impaired in HRAS-induced skin carcinoma development.Citation202
Finally, members of the RA domain-containing RASSF family have been implicated as effectors and mediators of Ras-induced apoptosis ( and ), with the strongest evidence for Nore1/RASFF5.Citation203 Although RASSF1 to RASSF6 harbor a C-terminal RA domain and RASSF7 to RASSF10 are characterized by an N-terminal RA domain, only a subset of these proteins has been validated to serve as Ras effectors and to initiate apoptosis. Promoter methylation commonly extinguishes the expression of RASSF family members in cancer. The first linkage with Ras came from the discovery of Nore1 (now also called RASSF5) in a yeast two-hybrid library screen for H-Ras effectors.Citation204 Association of activated Ras with Nore1, in complex with the proapoptotic protein kinase MST1. was shown to induce apoptosis.Citation205 Subsequently, evaluation of other RASSF proteins for their ability to associate with Ras revealed limited evidence that RASSF1, RASSF2, RASSF4 and RASSF6 can also serve as Ras effectors in apoptosis.Citation206–Citation209 A full determination of their physiological role as effectors of Ras, whether in apoptosis or oncogenesis, awaits analyses of endogenous RASSF protein function in cell culture and mouse models driven by endogenous mutant Ras.
The first structure of a Ras effector complex was determined using the isolated RBD of Raf-1 in complex with Rap1A, a Ras family protein highly related to Ras (),Citation210 which ironically does not itself activate Raf-1. The Raf-RBD exhibits a 1000-fold preferential affinity for Ras-GTP over Ras-GDP.Citation211 This property was exploited later for the development of the now-classic pulldown assay to measure Ras activation in cell lysates;Citation212 analogous pulldown assays were developed later for other Ras and Rho family small GTPases. Subsequent studies determined the structure of the isolated RA domains of RalGDS,Citation213,Citation214 PLC epsilon,Citation213 and RASSF1Citation215 as well as the RBD of p110γ.Citation216 Overall, these studies revealed the similar ubiquitin-fold structure of the different Ras-interacting domains and both similar and distinct features of their interactions with the switch I and II domains of H-Ras.
Posttranslational mechanisms of regulation: location, location, location in space and time.
Nearly all Ras superfamily proteins have very well-conserved and highly related isoforms that differ from each other almost exclusively in their “variable” membrane targeting domains rather than in their “constant” G domains (). Thus, Ras activity is not controlled simply by its GDP/GTP cycle, a concept that has been clear since the 1984 studiesCitation217,Citation218 showing that the C-terminus of Ras is required for proper membrane association, lipid binding and biological activity.
The general concept that Ras proteins must be localized correctly in order to become activated and to be biologically active is well accepted, but a complete picture of what “localized correctly” looks like has yet to be filled in. The simplified localization paradigm that has evolved over the past two decades is that Ras proteins are synthesized as soluble precursors which then undergo several steps of post-translational modification () that facilitate their required transit to and tenure at the plasma membrane, where they are activated by GEFs and inactivated by GAPs, and interact with effectors to transmit their signals downstream (). The full story, both for the family as a whole and for individual family members, is more complex and interesting, especially in terms of dynamic spatiotemporal regulation rather than static positioning, and is still emerging as we write.
It was known as early as 1979Citation12 that viral Ras proteins were cytosolic when newly synthesized, but did not remain so. In 1980, an important study used electron microscopy to illustrate the presence of H-Ras at the inner leaflet of the plasma membrane, and that characterization remained the default for many years.Citation14 To determine the nature of the alteration to newly synthesized Ras that allowed it to become membrane-bound, labeling studies were done by analogy to Src proteins, which were known to be modified by the saturated fatty acid myristate. Therefore Ras was evaluated for modification by fatty acid acylation, and both myristic acid and its slightly longer cousin, the C16 palmitic acid, were tested. In 1982, it was clear that v-H-Ras could incorporate 3H-palmitic acidCitation219 but not 3H-myristic acid, thereby firmly establishing that these two classes of proteins were modified in a distinct manner. Two years later, Willumsen, Lowy and colleagues used palmitic acid labeling as well as deletion and structural mutants to characterize the processing that led to Ras becoming membrane-associated. They observed correctly that the C-terminus of v-H-Ras is necessary for its plasma membrane localization,Citation217 and specifically that residue Cys186Citation218 is required for Ras to bind lipid as well as to associate with membranes and exert transforming activity. They also correctly speculated that “no more than 3 [C-terminal] residues are cleaved during processing” (), and validated that v-H-Ras can incorporate 3H-palmitate. The latter finding led them and others to speculate or declare that Cys186 is the site of palmitoylation, as that lipid was the only one known at the time to be relevant to Ras.Citation220 In 1987, pulse-chase studies showed that the acylation of N-Ras is highly dynamic, with a half-life turnover of about 20 min.Citation221 This result suggested another possible mechanism for control of Ras activity.
However, also in 1987, Tamanoi and colleagues identified a mutant in S. cerevisiae, dpr1, that was unable to modify Ras despite retention of fatty acid acylation.Citation222 This finding suggested that acylation cannot be the first processing step of Ras, but other modifications were unknown at the time. Over the next couple of years, several studies emerged indicating that Ras is carboxyl-methylated in species from yeast to man,Citation223–Citation225 and that this modification does not proceed until after C-terminal proteolysis. Indeed, sequencing demonstrated that the 1984 predictionCitation218 of removal of three amino acids was correct.Citation226 The significance of this number would become apparent when the Ras converting enzyme Rce1 was identified. Meanwhile, each of these steps was shown to positively influence Ras membrane binding, in studies using techniques as varied as aqueous-detergent fractionation, differential centrifugation and immunofluorescence with anti-Ras antibodies. However, the puzzle was still missing an important piece.
Only in 1989, when it was recognized after much comparison of sequence alignments that Ras proteins share a C-terminal CAAX motif (C = cysteine, A = aliphatic amino acid, X = terminal amino acid) with yeast a-factor, a protein known to be modified by a farnesyl isoprenoid lipid, was it determined that Ras proteins are also modified by farnesylationCitation227–Citation229 and that farnesylation is an obligate first step to allow palmitoylation to then occur on additional cysteine(s) upstream of Cys186. Without farnesylation at Cys186 (for example, by replacement with a Cys>Ser “SAAX” mutation), Ras proteins become cytosolic. In contrast, Ras proteins that are farnesylated but do not undergo subsequent processing steps are localized to endomembranes, as has been clarified over many years of experimentation.
Beginning in 1989, Hancock, Marshall and colleagues demonstrated that H-Ras and N-Ras, but not K-Ras, are modified both by farnesylation at Cys186 and by palmitoylation at one (N-Ras) or two (H-Ras) cysteine residues almost immediately upstream of the farnesylated cysteine, and that palmitoylation enhances membrane association and biological activity.Citation228 The same group then established or contributed to several guiding principles that are relevant for many signaling proteins whose membrane targeting is modulated by lipidation: (1) a “second signal” found in the hypervariable domain is required in addition to farnesylation,Citation228 (2) the second signal consists of either a polybasic domain (K-Ras4B) or palmitoylation (H-Ras, N-Ras and K-Ras4A),Citation230 thereby immediately highlighting the distinctive regulation of K-Ras4B, and (3) the minimal plasma membrane targeting signal of Ras is the CAAX or CAAL (CAAL specifies geranylgeranylation rather than farnesylation) plus a hypervariable domain-derived second signal. Together, these two motifs are sufficient to target either Ras or a cytosolic heterologous protein, such as GFP, to membranes.Citation231 Once these principles were established, it was demonstrated that a fusion protein of the serine/threonine kinase and Ras effector, Raf, with the C-terminal ∼20 amino acids of Ras [designated “Raf-CAAX”], was activated simply by virtue of being tethered to the plasma membrane without being brought there specifically by full length Ras.Citation232,Citation233 Such a result suggested that a key role of Ras is to bring Raf to the plasma membrane for activation.
Exactly where Ras proteins are localized when they are not yet fully processed and at the plasma membrane has been clarified gradually. In 1999 Philips and colleagues showed that H-Ras and N-Ras, which can both be palmitoylated, trafficked through the Golgi on their way to the plasma membrane, whereas K-Ras, a nonpalmitoylated protein, did not.Citation234 This study and othersCitation235 also confirmed, consistent with the principles laid down nearly a decade before, that the CAAX-containing motif was necessary and sufficient to direct the proteins to endomembranes for carboxymethylation.
Whether Ras could signal from internal membranes was answered more definitively with the development of new tools such as the use of GFP-Raf-RBD for “bystander FRET” to detect Ras-GTP, FRET-based Raichu probesCitation236,Citation237 and others. A 2002 study showed not only that Ras proteins can signal when associated with the endoplasmic reticulum and Golgi, but that they preferentially use different effectors depending on the endomembrane from which they are signaling.Citation238 The next year, monitoring localization of the Raf-RBD probe for activated Ras revealed highly selective activation of Ras in response to growth factor signaling via PLCgamma and the RasGEF RasGRP1, rapidly at the plasma membrane and then later at the Golgi.Citation239 Biochemical studies could also be performed using Ras proteins physically tethered to sites of interest to show site-specific activation and deactivation, depending on the available regulatory proteins at those sites.Citation240 Regardless of the specific details, Ras activation and deactivation has been shown repeatedly to be tightly regulated, both spatially and temporally, and this tight regulation is due to modulating the localization of both Ras and its interacting partners.
The question of how Ras proteins “get around” was also beginning to be addressed. In 2005, time-lapse microscopy and photobleaching experiments showed that palmitoylation traps Ras proteins on membranes where they can undergo vesicular transport, and that depalmitoylation allows their release for recycling from the cytosol back to endomembranes.Citation241 A similar study also emphasized more clearly the importance of deacylation for cytosolic transit.Citation242 Collectively, these results supported the importance of the dynamic nature of palmitoylation, as shown nearly 10 years earlier,Citation221 to the turnover and transit of H-Ras and N-Ras between the cytosol, endomembranes including especially the Golgi complex, and the plasma membrane.
Other posttranslational modifications have been shown to influence Ras subcellular localization and differential engagement of effectors. For example, Bar-Sagi and colleagues showed that H-Ras modification by mono- and di-ubiquitination of H-Ras, but not of K-Ras, stabilizes its association with endosomes and modulates its ability to activate ERK MAPK signaling.Citation243
C-terminal phosphorylation has also emerged as an important additional regulator of Ras localization and function. Although the viral Ras proteins were identified as threonine phosphoproteins,Citation12 surprisingly, the cellular proteins were poorly serine phosphorylated.Citation18 This biochemical difference was found later to be due to a missense mutation in residue 59 in the viral Ras proteins, producing an autophosphorylation mechanism whereby the substituted threonine served as a phosphate acceptor from the bound GTP.Citation244 However, in 1987 Rosen and colleaguesCitation245 showed that K-Ras4B became phosphorylated upon PKC activation by phorbol ester. The site was not identified, and the modification was apparently not investigated further until 2006, when Philips, Cox and colleagues showed that PKC. phosphorylated K-Ras4B on Ser181 in the hypervariable membrane targeting domain, relocalizing it from the plasma membrane to internal membranes including Golgi, ER and mitochondria, and converting it at the latter site to an apoptosis-inducing protein.Citation246 Related mechanisms of Ras family GTPase regulation of subcellular location and function upon phosphorylation of the C-terminal membrane targeting domain have also been described for the Rnd3 and RalA small GTPases,Citation247–Citation251 as well as for Rap1Citation252 and RhoA.Citation253,Citation254
Originally, because of the strong sequence identity and shared effector utilization, there was a general acceptance that the different Ras isoforms were essentially identical in function. Thus, while KRAS and NRAS mutations are seen more frequently in human cancers, the study of H-Ras has dominated much of the early history of Ras research. This bias towards H-Ras was encouraged by the ready availability of H-Ras expression vectors and mutant proteins, recombinant protein and antibodies. However, in recent years, a shift has begun towards a focus on K-Ras. The essential requirement for mouse development of KRASCitation255,Citation256 but not NRASCitation257 or HRAS,Citation258 or both,Citation259 or KRAS4A,Citation260 has certainly helped to fuel this shift. While HRAS was able to substitute for KRAS to support mouse development, the occurrence of cardiovascular defects in adult mice nevertheless supported functional differences.Citation261
There has been a steady accumulation of evidence that the Ras isoforms are functionally distinct. First, as discussed above, trafficking is distinct for the different isoforms, exposing them to different pools of regulators and effectors.Citation262,Citation263 Second, palmitoylation also has been shown to contribute greatly to the differential trafficking of H-Ras and N-Ras compared to K-Ras by directing them into specific nanoclusters, or “rasosomes.”Citation264–Citation267
In addition, each Ras isoform is chaperoned by its own preferred binding partner. In 2001, the proposal was made that galectins could act as chaperones for Ras by virtue of their extensive structural similarity with RhoGDI.Citation268 Galectin-1 has a farnesyl-binding pocket that is strikingly similar to the geranylgeranyl-binding pocket of the RhoGDI/Cdc42 interaction,Citation268 and is a preferred binding partner for and enhances the activities of H-RasCitation269,Citation267 whereas Galectin-3 is a preferred binding partner for K-Ras.Citation270,Citation271 Collectively these studies have shown that the interactions between Ras proteins and galectins, which themselves modulate transformation and are often overexpressed in cancers, are independent of galectin carbohydrate-binding interactions, and that galectins modulate both the spatial distribution of Ras proteins in nanoclusters and their effector utilization. Most recently, mathematical modeling and FRET/FLIM microscopy have been used to identify a role for the “constant” or G domain of Ras proteins for orientation with respect to the plasma membrane, suggesting synergy between this type of localization recognition and the specificity conferred by the hypervariable domain.Citation262
Ras-Targeted Therapies: The Search Continues
Early efforts to target Ras for cancer treatment focused on the fundamental defect of mutated Ras, which is the refractory response to downregulation by GAPs and consequently the persistent binding of GTP (). However, efforts to develop a small molecule “GAP” that is active on mutant Ras met with failure. Additionally, efforts to develop GTP-competitive antagonists of Ras proteins analogous to the ATP-competitive inhibitors of protein kinases have been precluded by the picomolar binding affinity of Ras for GTP. While exploration of possible approaches to direct targeting of mutant Ras continue, most current efforts to develop small molecule anti-Ras strategies are focused on indirect approaches.
Farnesyltransferase inhibitors—“What a long strange trip it's been.”
One of the most promising early directions for the development of anti-Ras inhibitors centered on ways to prevent Ras posttranslational lipid modification and plasma membrane association (). As mentioned above, the discovery in 1989 that Ras proteins are farnesylated prompted a huge effort to target this modification.Citation272 Because the farnesyl pyrophosphate that contributes this lipid group to proteins is an obligate intermediate component of the mevalonate-cholesterol biosynthetic pathway, whose synthesis can be blocked by cholesterol-lowering drugs already in clinical use (e.g., lovastatin), the determination that Ras is farnesylated stimulated considerable excitement that an approach for pharmacologic inhibition of Ras function might soon be at hand.
The HMG-CoA reductase inhibitor lovastatin was the first statin approved by the FDA in 1987 for lowering cholesterol to prevent cardiovascular disease in patients with hypercholesterolemia. However, because the clinically effective concentration for lowering cholesterol biosynthesis is much lower than that needed to block Ras farnesylation,Citation273 the use of statins as specific Ras inhibitors would likely be ineffective. This in turn prompted the search for the enzyme required for the addition of the farnesyl group to Ras, which culminated in the isolation, by Goldstein, Brown and colleagues, of farnesyltransferase (FTase) in 1990.Citation274 The finding that the Ras CAAX tetrapeptide sequence alone was effective in blocking FTase activity initiated intensive efforts by the pharmaceutical industry to develop cell-permeable CAAX peptidomimetics as possible anti-Ras inhibitors.Citation272 The development of CAAX mimetics, as well as the use of high throughput screens to identify small molecule inhibitors of FTase from libraries of previously synthesized as well as natural compounds resulted in numerous potent and selective cell-penetrant FTase inhibitors (FTIs).
The early dramatic success of FTIs in cell-basedCitation275,Citation276 and mouse model studies, when evaluated in H-Ras-driven models, fueled considerable excitement that an anti-Ras inhibitor would soon reach the clinic. One of the most dramatic observations was the ability of FTI treatment to cause regression of H-Ras-driven mouse mammary tumors.Citation277 However, this excitement was soon tempered by a key study showing that tumor cell line sensitivity to FTI growth inhibition in vitro did not correlate with Ras mutation status,Citation278 and dashed by the unexpected findings that K-Ras4B and N-Ras proteins not only are resistant to FTI treatment,Citation279–Citation281 but in the presence of FTI also undergo alternative prenylation (modification by a related geranylgeranyl isoprenoid lipid), whose addition is catalyzed by a related, FTI-insensitive enzyme, geranylgeranyltransferase-I (GGTase-I), under conditions of FTI treatmentCitation282,Citation283 (). That the two Ras proteins most commonly mutated in human cancers could escape the inhibitory activities of FTIs provided an explanation for the lack of clinical efficacy of FTIs in clinical trials. This was seen perhaps most dramatically and disappointingly in trials for pancreatic cancer, where essentially all cancers are KRAS mutant.Citation284
The disappointing demise of FTIs as anti-Ras inhibitors also stimulated an appreciation that the three Ras proteins are not identical in function. As mentioned, the early bias in studying H-Ras was due largely to the ready availability of antibodies, recombinant protein and molecular constructs for H-Ras, and to the general perception that the Ras proteins were essentially identical in function as oncoproteins. With additional observations, in particular the ability of activated KRAS but not NRAS to drive colon tumorigenesis, recent studies have shifted to the study of KRAS.
One very unexpected outcome of the development of FTIs is the fortuitous development of a drug that may be useful for the treatment of Hutchinson-Gilford Progeria Syndrome (also called Progeria).Citation285 Progeria is a very rare, fatal genetic condition characterized by an appearance of accelerated aging in children. Children with Progeria die of atherosclerosis at an average age of 13 years. With fewer than 50 patients worldwide, commercial development of drug specifically for this premature aging disorder would never occur otherwise. Progeria is caused by the incomplete posttranslational processing of lamin A, another FTase substrate. However, after farnesylation, the farnesylated C-terminus is removed by proteolytic cleavage to generate the normal final protein. In progeria, this normal modification does not occur, leaving lamin A farnesylated. Lamin A and other nuclear lamins are the structural proteins of the nuclear lamina, an intermediate filament network that provides scaffolding for the cell nucleus. Therefore, FTI treatment may be one approach to prevent accumulation of this mutant lamin A protein, and three clinical trials to evaluate this possibility have been initiated with the FTI lonafarnib that has already been evaluated extensively in clinical trials with cancer patients (www.progeriaresearch.org). While the high expectations that FTIs would be effective anti-Ras inhibitors has not come to pass, if FTIs do find clinical usefulness for this disease, or perhaps targeted to FTase enzymes important in parasitic diseases such as malaria or sleeping sickness, it would provide a positive outcome to what has been a very disappointing era of anti-Ras drug discovery.
Finally, the genetic validation in cell culture and mouse models of their importance to Ras-mediated growth transformation has stimulated interest in targeting both Rce1 and Icmt, the two CAAX-signaled processing steps after farnesylation ().Citation286–Citation289 The results of these studies may also reflect the fact that Ras transformation can be dependent on multiple other CAAX-terminating small GTPases (e.g., Ral, RhoA, Rac1) whose functions also depend on these processing steps ( and C). Efforts to develop inhibitors of these enzymes are ongoing. Citation290
Competitors of Ras-chaperone interactions.
An entirely different approach to blocking modifications has been taken by Kloog and colleagues, who have developed the small molecule farnesylthiosalicyclic acid (FTS) into a clinical candidate, salirasib, that has gone to Phase I/II trials in hematopoietic disease as well as cancers of the pancreas and lung. The basis for their enthusiasm for salirasib includes the proposed mechanism of action, which is competition for Ras membrane-binding sites and/or galectin binding,Citation291 as well as the apparent efficacy without toxicity in models of a wide range of pathological conditions including those dependent on oncogenic K-Ras.Citation290
Inhibitors of Ras effector signaling.
Currently, the greatest ongoing efforts in anti-Ras drug discovery have focused on blocking Ras effector signaling (). Driven by considerable experimental validation for their critical role in Ras-driven oncogenesis, and by the finding of mutated BRAF and PIK3CA in human cancers, the Raf-MEK-ERK and PI3K-AKT-mTOR signaling pathways have been the most intensely targeted. A survey of www.clinicaltrials.gov finds at least 38 inhibitors of these pathways in phase I/II clinical evaluation, which have been reviewed recently.Citation4
Driven by the conservation of the Raf-MEK-ERK cascade in evolution, by the finding that BRAF and RAS mutations occur in nonoverlapping frequency in the same cancers (e.g., melanomas, colon cancer),Citation292 and finally, by the success of protein kinase inhibitors for cancer treatment, inhibitors of MEK and Raf have been the most intensively pursued. CI-1040 (PD 184352) was the first small-molecule MEK1/2 inhibitor that proceeded to clinical testing. Although it was well-tolerated, CI-1040 exerted insufficient anti-tumor activity in phase II evaluation and was discontinued.Citation293 Subsequently, other MEK inhibitors with improved potency and pharmacologic properties have entered clinical evaluation. Although developed originally as a Raf inhibitor,Citation294 sorafenib was subsequently found to have broad activity,Citation295 in particular, for tyrosine kinase receptors involved in tumor angiogenesis such as those for VEGF and PDGF. FDA approval of sorafenib in 2005 for advanced renal cell cancer, where neither RAS nor RAF mutations are seen, is most likely based on its anti-angiogenic rather than anti-Raf activity. More recently, a mutant BRAF-selective inhibitor (PLX4032) has shown promising activity in melanomas expressing mutant BRAF.Citation296 However, three recent studies reported that this and other Raf inhibitors displayed the paradoxical ability to activate, rather than block, ERK activation in RAS mutant tumor cells,Citation297–Citation299 a finding foreshadowed by two 1999 studies reporting that multiple Raf kinase inhibitors unexpectedly activated Raf, possibly due to depleting a negative feedback mechanism.Citation300,Citation301 In another approach, small molecules have been identified that inhibit the Ras-Raf interaction,Citation302, Citation303 although it is unclear whether such molecules can be developed into effective drugs.
Synthetic lethal partners of mutant KRAS—New targets for anti-Ras inhibitor development?
Recently, large-scale interfering RNA screencs have been applied to take a functional and unbiased approach to identify therapeutic targets for anti-Ras inhibition.Citation304–Citation306 These screens are based on the concept of synthetic lethality, in which two genes are defined as synthetically lethal if mutation of either gene alone is compatible with viability but the simultaneous mutation of both genes leads to death.Citation307 Mutationally-activated RAS genes thus represent one gene and RNA interference-mediated ablation in cancer cells of the expression of a second gene provides the second hit. Since normal cells lack mutant RAS, genes identified in this manner should in principle be selectively lethal for tumors but not normal cells.
One study of a limited RNAi library targeting 1011 genes with a focus on protein kinases identified the STK33 serine/threonine kinase as a synthetic lethal partner of mutated KRAS.Citation305 A diverse spectrum of KRAS mutant tumor cell lines showed STK33 dependency. The identification of STK33 illustrates an important strength of this type of functional screen, since no alteration in its expression, no mutations, and no transforming activity of STK33 was detected. Hence, STK33 would not have been identified by the more classical criteria for cancer-causing genes. In a second study, a broader genome-wide screen was done targeting 32,293 unique human transcripts.Citation304 The genes identified encode functionally diverse proteins that regulate several biological processes, especially mitosis. For example, one such gene encodes Polo-like kinase 1 (Plk1), a serine-threonine kinase with a key role in mitosis. Inhibitors of Plk1 have been in development for cancer treatment, and RAS mutant cells showed increased sensitivity to a Plk1-selective inhibitor. A third limited screen identified TANK-binding kinase 1 (TBK1) as synthetic lethal partner of mutant KRAS.Citation305 The TBK1 serine/threonine kinase can activate the NF-kappaB transcription factor and support cell survival. This role is likely to be associated with TBK1 function downstream of RalB, a critical effector of Ras-mediated survival signaling in human cells.Citation308 Finally, in a different approach, a fourth study identified a gene signature for KRAS dependency, which included genes encoding the Syk and Ron tyrosine kinases.Citation309 To validate this screen, they showed that KRAS mutant tumor cell lines were more sensitive to induction of apoptosis by treatment with a small molecule inhibitor of Syk.
These studies identified several protein kinases as synthetic lethal partners of mutant KRAS, and as such suggest highly tractable targets that may accelerate the development of these new leads for “anti-Ras” drugs. Overall, the diversity of genes identified in these screens will surely provide many additional intriguing candidates for anti-Ras therapeutic development.
Ras and Developmental Syndromes
An unexpected recent discovery has been that germline mutations both in RAS and in components upstream and downstream of Ras signaling are associated with a class of developmental syndromes now referred to as RASopathies ( and Suppl. Table 3).Citation310 Germline HRAS mutations were first identified in Costello syndrome,Citation311 followed by KRAS mutations in cardio-facio-cutaneous (CFC)Citation312 and Noonan syndromes.Citation313,Citation314 These syndromes each exhibit unique features, but additionally, because of a common genetic basis in Ras signaling, also share overlapping characteristics. These include craniofacial dysmorphology, cardiac malformations, cutaneous, musculokeletal and ocular abnormalities. Finally, whereas persons with Costello syndrome are at increased risk for malignant tumors,Citation315 persons with Noonan or CFC syndrome have no or only a slightly increased risk of cancer, as discussed below.
In addition to mutational activation of Ras, loss-of-function mutations in the p120 or neurofibromin RasGAPs or gain-of-function mutations in Sos1 also cause Ras hyperactivation in RASopathies.Citation5 Mutations in the Shp2 protein tyrosine phosphatase can also lead to downstream activation of Ras. The occurrence of mutations in RAS and RAF1, BRAF, MEK1 and MEK2 suggests that hyperactivation of the ERK MAPK cascade is the key effector mechanism by which Ras activation promotes these disorders. Thus, inhibitors of Raf, MEK and other inhibitors of this pathway developed for cancer treatment may fortuitously be useful for the treatment of these disorders.Citation316
An interesting feature of these diseases that distinguishes them from Ras- and Ras pathway-driven cancers is that the mutations seen in KRAS and BRAF in RASopathies are largely atypical of those seen in human cancers, and that mutations in Ras signaling components seen in RASopathies are not typically found in cancers at all. The KRAS mutations occur not at the three hot spots of codons 12, 13 or 61, but instead tend to be those that result in less potently activated Ras proteins (). The V600E mutation that represents ∼80% of the BRAF mutations seen in cancer is not seen in the ∼75% of individuals with CFC syndrome who have mutant BRAF. Mutational activation of Sos1,Citation310 or of MEK1 or MEK2 is also uncommon.Citation317,Citation61–Citation63 One possible explanation is that germline mutations that potently activate K-Ras signaling, which has been proposed to preferentially activate the Raf kinase cascade, compared to H-Ras activation of the PI3K cascade, are deleterious for human development and therefore individuals with those mutations would not survive to birth. While some analyses in mouse models have supported this premise,Citation318 others found that potently activated RAS alleles are well-tolerated.Citation319 However, the situation may be different for the frequent (∼85%) germline HRAS mutations in patients with Costello syndrome, where ∼90% of the mutations (G12S and G12A) also occur in human tumors (). Mice with a germline HRAS(G12V) mutation exhibited abnormalities observed in Costello syndrome patients and displayed mammary hyperplasia but developed tumors rarely,Citation320 despite expressing a strongly activating and tumor-associated mutant form of H-Ras. One possible explanation for this surprising result comes from a study in which zebrafish with germline expression of HRAS(G12V) displayed Costello-like symptoms and oncogene-induced senescence (OIS), whereas overexpression of the same protein driven in larvae off a heat-shock promoter resulted in hyperproliferation that required p53 to drive OIS.Citation321 Perhaps HRAS-driven tumorigenicity in Costello patients requires loss of p53. A few NRAS mutations have recently been described to occur in Noonan syndromeCitation322 and are also distinct from the hotspot mutations seen most commonly in cancer (). It will be of great interest to determine the basis for isoform- and mutation-selective dependence on aberrant Ras signaling in cancers versus RASopathies.
The Future
When describing the rich history of Ras, we have often found it appropriate to cite the wise sayings of Yogi Berra. The finding that Ras utilizes a multitude of effectors beyond Raf, with more likely to be found, emphasized that “it ain't over 'til it's over”. The disappointing failure of efforts to develop FTIs as Ras inhibitors reflected the mindset that “when you come to a fork in the road, take it”. That we have reached numerous stages where we felt our knowledge of Ras was complete, only to find yet new wrinkles, has repeatedly prompted the sentiment that “the future is too hard to predict.” The finding that Raf inhibitors paradoxically activate Raf is an example of unanticipated signaling whose mechanistic basis is only now being worked out (“we made too many wrong mistakes”). What is in store for the future? Certainly Ras will teach us new ways in which the cell signaling circuitry is regulated. We anticipate yet more striking differences in the regulation and roles of the three Ras isoforms in normal biology and disease, at the subcellular to organismal levels. Recently, the structure of human K-Ras4B has been determined, and although not published it is available online (www.thesgc.org/structures/structure_description/3GFT/). Unexpectedly, despite the complete sequence identity of H-Ras and K-Ras4B switch II regions, the side chain orientation in this loop is described to be strikingly different in the two proteins. As essentially all previous structural studies have focused on H-Ras, future structural studies of K-Ras complexes will likely identify molecular interactions, in addition to subcellular localization, that reflect biological differences. Functional genetic screens in cell culture and in vivo model systems will continue to identify genes that unexpectedly influence Ras-dependent biology. Although Ras is presently considered an “intractable” drug target, it is still possible that new thinking and technology may produce effective approaches to directly target Ras for cancer treatment. We anticipate that development of better mouse models of Ras-driven oncogenesis will provide more accurate preclinical identification of therapeutic approaches that will prove effective in the cancer patient, compared to current mouse models in which genetic ablation of candidate targets for Ras inhibition evaluates prevention rather than “treatment” of a pre-existing cancer. Additionally, genetic ablation of a given target is clearly not equivalent to pharmacologic ablation of that target. As we head into the future, one Yogi-ism to avoid is, “You've got to be very careful if you don't know where you are going, because you might not get there”. Anticipating that unexpected findings may ultimately prove at least as informative as expected ones will give us the courage to take both logical and perhaps seemingly illogical approaches and directions, as we try to solve the remaining riddles of Ras.
Figures and Tables
Figure 1 Timeline of representative key discoveries in Ras research. See Suppl. Table 1 for references.
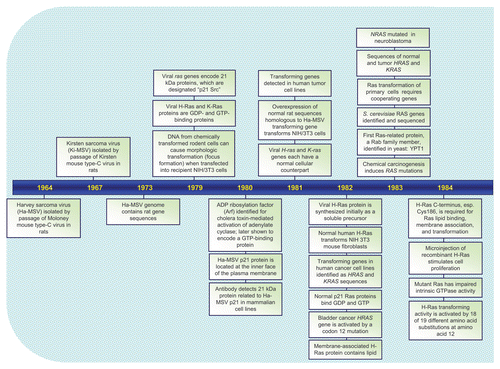
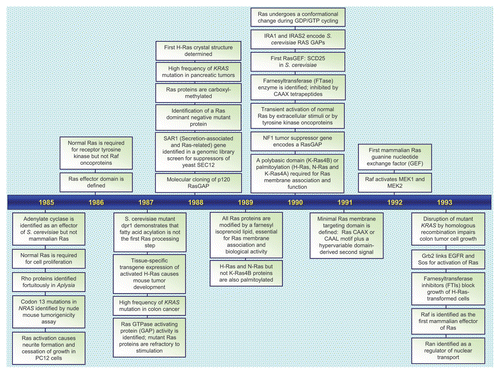
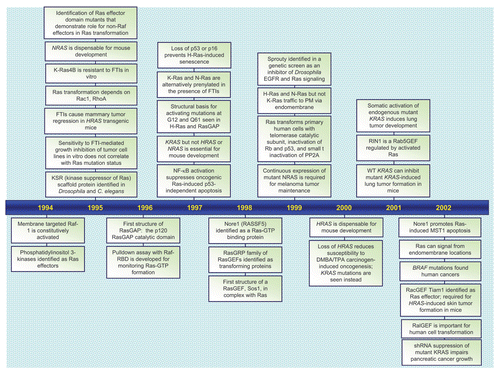
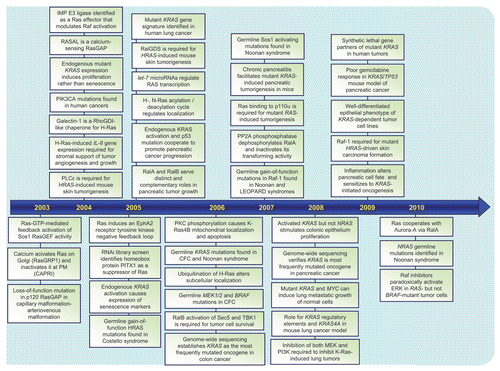
Figure 2 Ras is a GDP/GTP-regulated binary switch. (A) The three RAS genes encode four 188–189 amino acid proteins that share 82–90% overall sequence identity; KRAS encodes two splice variants due to alternative exon 4 utilization, leading to divergent C-terminal sequences. Exons 4A and 4B encode 39 and 38 amino acids, respectively, with 19 identical and 4 conserved substitutions. K-Ras4A is most similar to viral K-ras while K-Ras4B is the predominant isoform expressed in human cells. Residues 1–164 comprise the G domain that contains six conserved sequence motifs shared with other Ras superfamily and GTP-binding proteins. These motifs are involved in binding either phosphate/Mg2+ (PM) or the guanine base (G) of GDP and GTP. Residues in Switch I (aa 30–38) and II (aa 60–76) change in conformation during GDP/GTP cycling. The core effector binding domain (E; residues 32–40) and flanking sequences are involved in effector binding specificity. (B) Regulators of the Ras GDP/GTP cycle. RasGEFs stimulate GDP/GTP exchange. With the 10-fold higher cellular concentrations of GTP over GDP, the net result of RasGEF stimulation is formation of active Ras-GTP. Ras-GTP binds preferentially to downstream effectors. RasGAPs accelerate the intrinsic GTP hydrolysis activity of Ras to promote formation of inactive Ras-GDP. Shown are “classic” missense mutants of Ras proteins that have been useful for dissection of Ras function. The Ras(S17N) dominant negative sequesters and blocks RasGEF activity, preventing Ras activation. The G12V and Q61L mutations, found in human cancers, impair GAP-stimulated GTP hydrolysis. The T35S, E37G and Y40C effector domain mutants (EDMs) differentially impair effector binding. The T35S mutant retains efficient binding to Raf but not PI3K or RalGEF, whereas the E37G mutant retains efficient binding to RalGEF but not Raf or PI3K, and the Y40C mutant retains efficient binding to PI3K but not Raf or RalGEF.
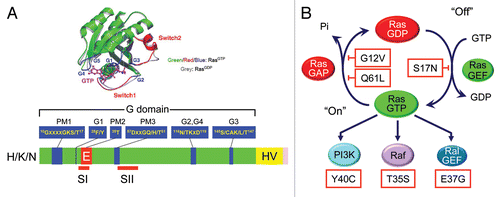
Figure 3 Detection of activated and mutated HRAS in human EJ/T24 bladder carcinoma cells. The NIH/3T3 focus formation assay was used to detect activated oncogenes present in human tumor but not normal genomic DNA. High molecular weight DNA was isolated from the EJ/T24 human bladder carcinoma cell line, converted to a calcium phosphate precipitant, and added to the growth medium of a monolayer of NIH/3T3 cells. After 14 days, foci of morphologically and growth transformed cells can be detected in cultures treated with DNA from tumor cells but not in parallel cultures treated with DNA from normal human cells. The active HRAS fragment from EJ/T24 bladder cells lies within a 4.6 kDa XhoI-SphI fragment. Human H-Ras protein is encoded by sequences spanning four exons. Exon 1 encodes amino acids 1–37. Sequence comparison of the bladder carcinoma-derived HRAS DNA identified a single base substitution at codon 12, resulting in a single missense mutation (G12V).
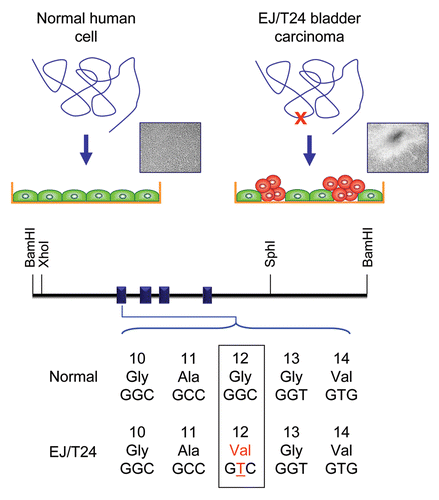
Figure 4 Ras mutations in cancer and developmental disorders. Missense mutations in (A) H-Ras, (B) K-Ras and (C) N-Ras in human cancers were compiled from COSMIC (www.sanger.ac.uk/genetics/CGP/cosmic/). Each specific substitution seen at residue 12, 13 or 61 is indicated separately (pink boxes placed above the Ras protein ribbon), followed in parentheses by the number of cancers identified to have that mutation. The numbers and types of missense mutations in each Ras isoform found in developmental syndromes (RASopathies) were compiled from The Ras/MAPK Syndrome Homepage (www.medgen.med.tohoku.ac.jp/RasMapk%20syndromes.html). Specific substitutions are indicated (green boxes) below the Ras protein ribbon and numbers are given in parentheses after each mutation. (D) Distribution of Ras missense mutations in cancer. The distribution of mutations in H-Ras, K-Ras and N-Ras was calculated from data in COSMIC depicted in (A–C). The percentages of missense mutations at 12, 13, 61 and all other positions were determined for H-Ras (629 total mutations), K-Ras (15,594 total) and N-Ras (2,189 total).
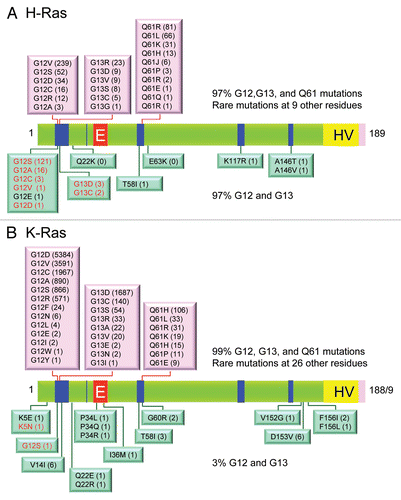
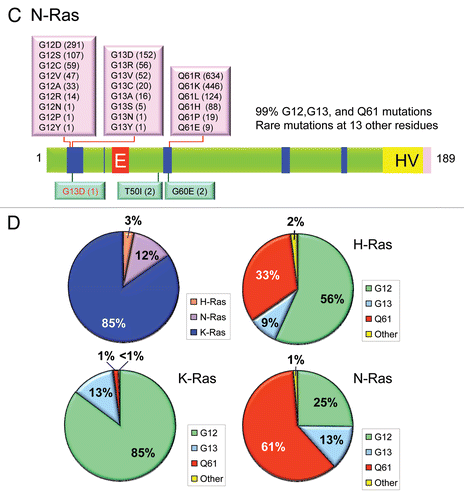
Figure 5 Colorectal and pancreatic cancer progression. (A) Colorectal cancer progression and gene mutations. Colonic epithelial cells undergo a histologic transition from normal to malignant state that is driven by specific genetic events including inactivation of tumor suppressors (APC, SMAD4 and TP53) and activation of the KRAS oncogene.Citation323 The three stages of adenomas represent tumors of increasing size, dysplasia, and villous content. (B) Pancreatic cancer progression and gene mutations. Multiple tumor types arise from the exocrine pancreas, of which greater than 95% are pancreatic ductal adenocarcinoma (PDAC). Normal ductal epithelium progression to infiltrating cancer (left to right) is illustrated through a series of histologically defined precursor lesions (PanINs) that show increasing degrees of disruption of cellular morphology, nuclear atypia and dysplastic growth.Citation324 High grade PanIN-3 progresses to invasive PDAC. Activating point mutations in the KRAS gene occur early, inactivation of the p16/INK4A gene occurs at an intermediate stage, and inactivation of the TP53, SMAD4/DPC4 and BRCA2 genes occurs relatively late. Oncogenes are indicated in green text and tumor suppressors in red text.
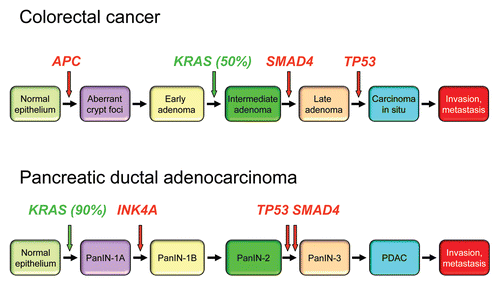
Figure 6 Conservation of Ras proteins in evolution. (A) Domain architecture of Ras proteins and the boundaries of the constant G domains were determined using SMART (http://smart.embl-heidelberg.de/). (B) Clustal/W was then used to align the G domain sequences, and the percent of amino acid identity was determined and used to generate the dendrogram.
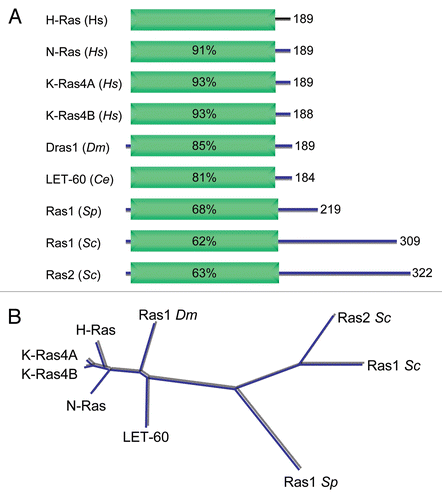
Figure 7 Human and invertebrate Ras superfamily proteins. (A) The numbers of members of each species and of each Ras superfamily branch were obtained from the references cited in Suppl. Table 2, and then used to generate the graphs. Numbers indicate total members per family. The graph of human family proteins was generated using the numbers in reference Citation6. (B) Human Ras family proteins. Dendrogram generated by Clustal/W and adapted from reference Citation325. (C) Human Rho family proteins. Adapted from reference Citation326.
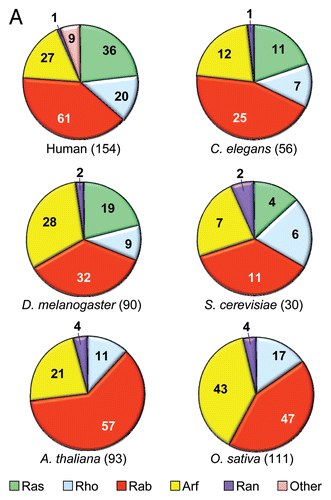
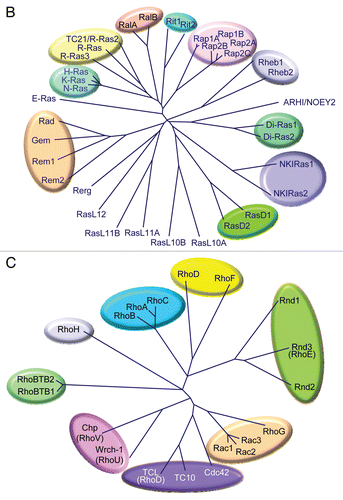
Figure 8 Ras interactome. Proteins that regulate Ras GDP/GTP cycling, catalyze posttranslational modification, or serve as immediate downstream effectors are indicated. Compiled in part from Table 1 in reference Citation121.
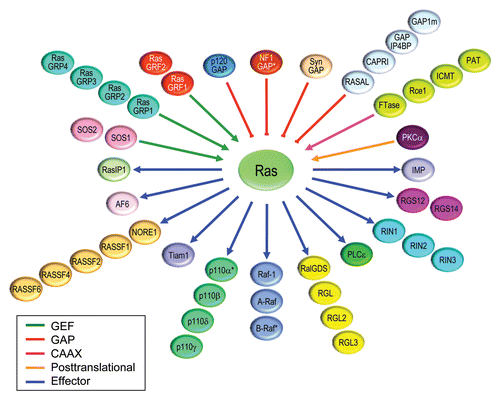
Figure 9 Ras signaling. (A) Conservation of Raf-MEK-ERK cascade downstream of Ras. (B) Ras effector pathways implicated in Ras-mediated oncogenesis. Compiled in part from literature cited in reference Citation327.
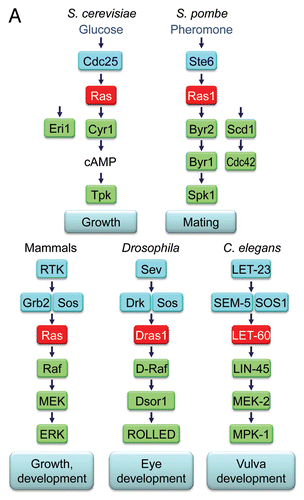

Figure 10 Ras posttranslational processing and membrane association. (A) The C-terminal 24–25 residues comprise the membrane targeting sequence which is comprised of the C-terminal CAAX box, required for posttranslational lipid modification, and the hypervariable (HV) domain that includes a second membrane targeting sequence element (palmitoylated cysteine(s) or polybasic stretches). (B) CAAX motif-signaled posttranslational processing. The cysteine residue of the CAAX box (red) is modified posttranslationally by cytosolic farnesyltransferase (FTase)-catalyzed covalent addition of a C15 farnesyl isoprenoid. Processing is completed on the cytosolic leaflet of the endoplasmic reticulum (ER) by Rce1 (Ras and a-factor converting enzyme-1)-catalyzed proteolytic removal of the AAX residues and then by Icmt (isoprenylcysteine carboxyl methyltransferase)-catalyzed carboxylmethylation of the now terminal farnesylated cysteine residue. FTase inhibitor (FTI) treatment blocks farnesylation and all subsequent CAAX modifications, rendering Ras cytosolic and inactive. Similarly, a serine substitution of the cysteine residue of the CAAX motif (“SAAX”) also prevents farnesylation and all CAAX-signaled modifications. The X residue of the CAAX motif dictates prenyltransferase specificity. Substitution of the X residue with leucine generates mutants that undergo modification by GGTase-I and addition of a C20 geranylgeranyl lipid instead of a C15 farnesyl isoprenoid. (C) Ras plasma membrane association is dependent on a second targeting element. The CAAX-signaled modifications alone are not sufficient to promote plasma membrane association. Additional sequence elements in the HV provide a second membrane targeting signal. In H-Ras, N-Ras and K-Ras4A, cysteine residues in the HV region are modified posttranslationally by ER-associated protein acyltransferases (PATs) that promote covalent addition of a C16 palmitate fatty acid. In K-Ras4B, lysine-rich polybasic amino acids comprise the second signal, which facilitate association with the negatively charged head groups of phosphatidylserine and phosphatidylinositol in the cytosolic face of the plasma membrane. Whereas H-Ras and N-Ras (and presumably K-Ras4A) traffic through the classical secretory pathway through the Golgi to the plasma membrane, K-Ras4B bypasses the Golgi and transits to the plasma membrane by a poorly characterized mechanism. Additionally, the differing second signals of K-Ras4B and H-Ras dictate localization to distinct plasma membrane subdomains, that may lead to distinct effector signaling.
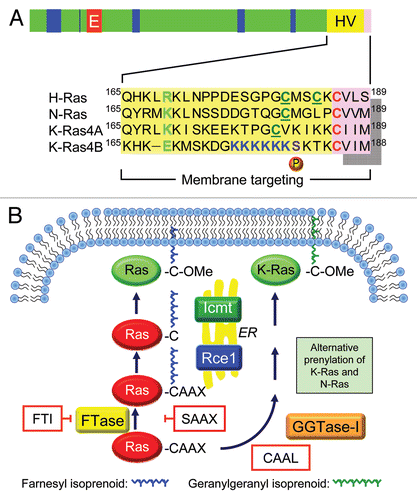
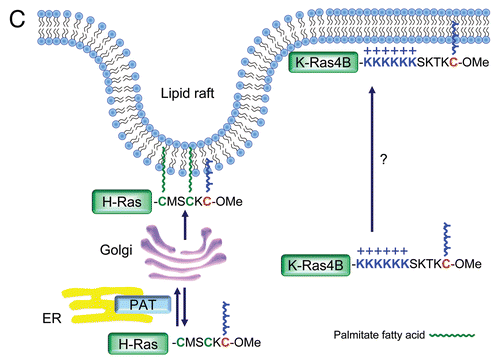
Figure 11 Ras signaling components mutated in RASopathies. Generated based on information summarized in Suppl. Table 3 and from references cited in reference Citation5.
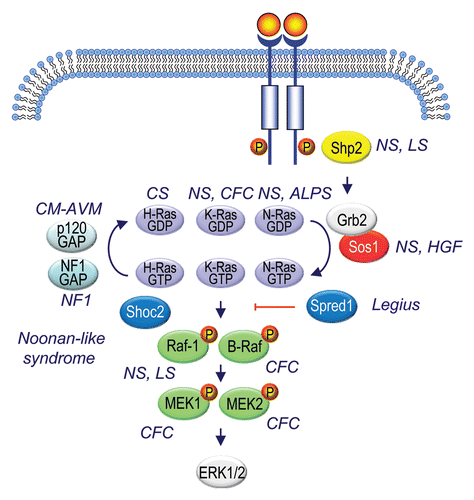
Additional material
Download Zip (307.3 KB)Acknowledgements
We are grateful to Ed Scolnick and Jay Gibbs for their insight into the early years of Ras history. We apologize to those colleagues whose work we could not cite due to length restrictions. We thank Lanika DeGraffenreid for assistance in manuscript preparation. Our research is supported by grants from the National Cancer Institute (CA042978, CA067771, CA106991, CA109550 and CA127152), the Emerald Foundation and the Susan G. Komen Foundation.
References
- Malumbres M, Barbacid M. RAS oncogenes: the first 30 years. Nat Rev Cancer 2003; 3:459 - 465
- Karnoub AE, Weinberg RA. Ras oncogenes: split personalities. Nat Rev Mol Cell Biol 2008; 9:517 - 531
- Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ. Cancer statistics, 2009. CA Cancer J Clin 2009; 59:225 - 249
- Yeh JJ, Madigan JP, Campbell PM, DeGraffenreid L, Der CJ. Targeting Ras for anti-cancer drug discovery. The Handbook of Cell Signaling 2009; Academic Press 2837 - 2857
- Tidyman WE, Rauen KA. The RASopathies: developmental syndromes of Ras/MAPK pathway dysregulation. Curr Opin Genet Dev 2009; 19:230 - 236
- Wennerberg K, Rossman KL, Der CJ. The Ras superfamily at a glance. J Cell Sci 2005; 118:843 - 846
- Harvey JJ. An Unidentified Virus Which Causes the Rapid Production of Tumours in Mice. Nature 1964; 204:1104 - 1105
- Kirsten WH, Mayer LA. Morphologic responses to a murine erythroblastosis virus. J Natl Cancer Inst 1967; 39:311 - 335
- Stehelin D, Varmus HE, Bishop JM, Vogt PK. DNA related to the transforming gene(s) of avian sarcoma viruses is present in normal avian DNA. Nature 1976; 260:170 - 173
- Scolnick EM, Rands E, Williams D, Parks WP. Studies on the nucleic acid sequences of Kirsten sarcoma virus: a model for formation of a mammalian RNA-containing sarcoma virus. J Virol 1973; 12:458 - 463
- Scolnick EM, Parks WP. Harvey sarcome virus: a second murine type C sarcoma virus with rat genetic information. J Virol 1974; 13:1211 - 1219
- Shih TY, Weeks MO, Young HA, Scholnick EM. Identification of a sarcoma virus-coded phosphoprotein in nonproducer cells transformed by Kirsten or Harvey murine sarcoma virus. Virology 1979; 96:64 - 79
- Scolnick EM, Papageorge AG, Shih TY. Guanine nucleotide-binding activity as an assay for src protein of rat-derived murine sarcoma viruses. Proc Natl Acad Sci USA 1979; 76:5355 - 5359
- Willingham MC, Pastan I, Shih TY, Scolnick EM. Localization of the src gene product of the Harvey strain of MSV to plasma membrane of transformed cells by electron microscopic immunocytochemistry. Cell 1980; 19:1005 - 1014
- Ellis RW, Defeo D, Shih TY, Gonda MA, Young HA, Tsuchida N, et al. The p21 src genes of Harvey and Kirsten sarcoma viruses originate from divergent members of a family of normal vertebrate genes. Nature 1981; 292:506 - 511
- Chang EH, Gonda MA, Ellis RW, Scolnick EM, Lowy DR. Human genome contains four genes homologous to transforming genes of Harvey and Kirsten murine sarcoma viruses. Proc Natl Acad Sci USA 1982; 79:4848 - 4852
- Langbeheim H, Shih TY, Scolnick EM. Identification of a normal vertebrate cell protein related to the p21 src of Harvey murine sarcoma virus. Virology 1980; 106:292 - 300
- Papageorge A, Lowy D, Scolnick EM. Comparative biochemical properties of p21 ras molecules coded for by viral and cellular ras genes. J Virol 1982; 44:509 - 519
- Chang EH, Furth ME, Scolnick EM, Lowy DR. Tumorigenic transformation of mammalian cells induced by a normal human gene homologous to the oncogene of Harvey murine sarcoma virus. Nature 1982; 297:479 - 483
- Gibbs JB, Sigal IS, Poe M, Scolnick EM. Intrinsic GTPase activity distinguishes normal and oncogenic ras p21 molecules. Proc Natl Acad Sci USA 1984; 81:5704 - 5708
- Hill M, Hillova J. Recombinational events between exogenous mouse DNA and newly synthesized DNA strands of chicken cells in culture. Nat New Biol 1971; 231:261 - 265
- Wigler M, Pellicer A, Silverstein S, Axel R. Biochemical transfer of single-copy eucaryotic genes using total cellular DNA as donor. Cell 1978; 14:725 - 731
- Jainchill JL, Aaronson SA, Todaro GJ. Murine sarcoma and leukemia viruses: assay using clonal lines of contact-inhibited mouse cells. J Virol 1969; 4:549 - 553
- Shih C, Shilo BZ, Goldfarb MP, Dannenberg A, Weinberg RA. Passage of phenotypes of chemically transformed cells via transfection of DNA and chromatin. Proc Natl Acad Sci USA 1979; 76:5714 - 5718
- Krontiris TG, Cooper GM. Transforming activity of human tumor DNAs. Proc Natl Acad Sci USA 1981; 78:1181 - 1184
- O'Toole CM, Povey S, Hepburn P, Franks LM. Identity of some human bladder cancer cell lines. Nature 1983; 301:429 - 430
- Murray MJ, Shilo BZ, Shih C, Cowing D, Hsu HW, Weinberg RA. Three different human tumor cell lines contain different oncogenes. Cell 1981; 25:355 - 361
- Shih C, Padhy LC, Murray M, Weinberg RA. Transforming genes of carcinomas and neuroblastomas introduced into mouse fibroblasts. Nature 1981; 290:261 - 264
- Perucho M, Goldfarb M, Shimizu K, Lama C, Fogh J, Wigler M. Human-tumor-derived cell lines contain common and different transforming genes. Cell 1981; 27:467 - 476
- Marshall CJ, Hall A, Weiss RA. A transforming gene present in human sarcoma cell lines. Nature 1982; 299:171 - 173
- Santos E, Tronick SR, Aaronson SA, Pulciani S, Barbacid M. T24 human bladder carcinoma oncogene is an activated form of the normal human homologue of BALB- and Harvey-MSV transforming genes. Nature 1982; 298:343 - 347
- Der CJ, Krontiris TG, Cooper GM. Transforming genes of human bladder and lung carcinoma cell lines are homologous to the ras genes of Harvey and Kirsten sarcoma viruses. Proc Natl Acad Sci USA 1982; 79:3637 - 3640
- Parada LF, Tabin CJ, Shih C, Weinberg RA. Human EJ bladder carcinoma oncogene is homologue of Harvey sarcoma virus ras gene. Nature 1982; 297:474 - 478
- Taparowsky E, Suard Y, Fasano O, Shimizu K, Goldfarb M, Wigler M. Activation of the T24 bladder carcinoma transforming gene is linked to a single amino acid change. Nature 1982; 300:762 - 765
- Reddy EP, Reynolds RK, Santos E, Barbacid M. A point mutation is responsible for the acquisition of transforming properties by the T24 human bladder carcinoma oncogene. Nature 1982; 300:149 - 152
- Tabin CJ, Bradley SM, Bargmann CI, Weinberg RA, Papageorge AG, Scolnick EM, et al. Mechanism of activation of a human oncogene. Nature 1982; 300:143 - 149
- Capon DJ, Seeburg PH, McGrath JP, Hayflick JS, Edman U, Levinson AD, et al. Activation of Ki-ras2 gene in human colon and lung carcinomas by two different point mutations. Nature 1983; 304:507 - 513
- Shimizu K, Goldfarb M, Suard Y, Perucho M, Li Y, Kamata T, et al. Three human transforming genes are related to the viral ras oncogenes. Proc Natl Acad Sci USA 1983; 80:2112 - 2116
- Hall A, Marshall CJ, Spurr NK, Weiss RA. Identification of transforming gene in two human sarcoma cell lines as a new member of the ras gene family located on chromosome 1. Nature 1983; 303:396 - 400
- Feig LA, Bast RC Jr, Knapp RC, Cooper GM. Somatic activation of rasK gene in a human ovarian carcinoma. Science 1984; 223:698 - 701
- Santos E, Martin-Zanca D, Reddy EP, Pierotti MA, Della Porta G, Barbacid M. Malignant activation of a K-ras oncogene in lung carcinoma but not in normal tissue of the same patient. Science 1984; 223:661 - 664
- Pulciani S, Santos E, Lauver AV, Long LK, Aaronson SA, Barbacid M. Oncogenes in solid human tumours. Nature 1982; 300:539 - 542
- Bos JL, Fearon ER, Hamilton SR, Verlaan-de Vries M, van Boom JH, van der Eb AJ, et al. Prevalence of ras gene mutations in human colorectal cancers. Nature 1987; 327:293 - 297
- Forrester K, Almoguera C, Han K, Grizzle WE, Perucho M. Detection of high incidence of K-ras oncogenes during human colon tumorigenesis. Nature 1987; 327:298 - 303
- Rodenhuis S, van de Wetering ML, Mooi WJ, Evers SG, van Zandwijk N, Bos JL. Mutational activation of the K-ras oncogene. A possible pathogenetic factor in adenocarcinoma of the lung. N Engl J Med 1987; 317:929 - 935
- Smit VT, Boot AJ, Smits AM, Fleuren GJ, Cornelisse CJ, Bos JL. KRAS codon 12 mutations occur very frequently in pancreatic adenocarcinomas. Nucleic Acids Res 1988; 16:7773 - 7782
- Almoguera C, Shibata D, Forrester K, Martin J, Arnheim N, Perucho M. Most human carcinomas of the exocrine pancreas contain mutant c-K-ras genes. Cell 1988; 53:549 - 554
- Sukumar S, Notario V, Martin-Zanca D, Barbacid M. Induction of mammary carcinomas in rats by nitroso-methylurea involves malignant activation of H-ras-1 locus by single point mutations. Nature 1983; 306:658 - 661
- Balmain A, Pragnell IB. Mouse skin carcinomas induced in vivo by chemical carcinogens have a transforming Harvey-ras oncogene. Nature 1983; 303:72 - 74
- Land H, Parada LF, Weinberg RA. Tumorigenic conversion of primary embryo fibroblasts requires at least two cooperating oncogenes. Nature 1983; 304:596 - 602
- Ruley HE. Adenovirus early region 1A enables viral and cellular transforming genes to transform primary cells in culture. Nature 1983; 304:602 - 606
- Hahn WC, Counter CM, Lundberg AS, Beijersbergen RL, Brooks MW, Weinberg RA. Creation of human tumour cells with defined genetic elements. Nature 1999; 400:464 - 468
- Hahn WC, Dessain SK, Brooks MW, King JE, Elenbaas B, Sabatini DM, et al. Enumeration of the simian virus 40 early region elements necessary for human cell transformation. Mol Cell Biol 2002; 22:2111 - 2123
- Quaife CJ, Pinkert CA, Ornitz DM, Palmiter RD, Brinster RL. Pancreatic neoplasia induced by ras expression in acinar cells of transgenic mice. Cell 1987; 48:1023 - 1034
- Sinn E, Muller W, Pattengale P, Tepler I, Wallace R, Leder P. Coexpression of MMTV/v-Ha-ras and MMTV/c-myc genes in transgenic mice: synergistic action of oncogenes in vivo. Cell 1987; 49:465 - 475
- Aguirre AJ, Bardeesy N, Sinha M, Lopez L, Tuveson DA, Horner J, et al. Activated Kras and Ink4a/Arf deficiency cooperate to produce metastatic pancreatic ductal adenocarcinoma. Genes Dev 2003; 17:3112 - 3126
- Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, et al. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell 2005; 7:469 - 483
- Bardeesy N, Cheng KH, Berger JH, Chu GC, Pahler J, Olson P, et al. Smad4 is dispensable for normal pancreas development yet critical in progression and tumor biology of pancreas cancer. Genes Dev 2006; 20:3130 - 3146
- Izeradjene K, Combs C, Best M, Gopinathan A, Wagner A, Grady WM, et al. Kras(G12D) and Smad4/Dpc4 haploinsufficiency cooperate to induce mucinous cystic neoplasms and invasive adenocarcinoma of the pancreas. Cancer Cell 2007; 11:229 - 243
- Kojima K, Vickers SM, Adsay NV, Jhala NC, Kim HG, Schoeb TR, et al. Inactivation of Smad4 accelerates Kras(G12D)-mediated pancreatic neoplasia. Cancer Res 2007; 67:8121 - 8130
- Sjoblom T, Jones S, Wood LD, Parsons DW, Lin J, Barber TD, et al. The consensus coding sequences of human breast and colorectal cancers. Science 2006; 314:268 - 274
- Wood LD, Parsons DW, Jones S, Lin J, Sjoblom T, Leary RJ, et al. The genomic landscapes of human breast and colorectal cancers. Science 2007; 318:1108 - 1113
- Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, Angenendt P, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science 2008; 321:1801 - 1806
- Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P, et al. An integrated genomic analysis of human glioblastoma multiforme. Science 2008; 321:1807 - 1812
- Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W, et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med 2009; 360:765 - 773
- Haigis KM, Kendall KR, Wang Y, Cheung A, Haigis MC, Glickman JN, et al. Differential effects of oncogenic K-Ras and N-Ras on proliferation, differentiation and tumor progression in the colon. Nat Genet 2008; 40:600 - 608
- Dhar R, Nieto A, Koller R, DeFeo-Jones D, Scolnick EM. Nucleotide sequence of two rasH related-genes isolated from the yeast Saccharomyces cerevisiae. Nucleic Acids Res 1984; 12:3611 - 3618
- Powers S, Kataoka T, Fasano O, Goldfarb M, Strathern J, Broach J, et al. Genes in S. cerevisiae encoding proteins with domains homologous to the mammalian ras proteins. Cell 1984; 36:607 - 612
- Fukui Y, Kaziro Y. Molecular cloning and sequence analysis of a ras gene from Schizosaccharomyces pombe. EMBO J 1985; 4:687 - 691
- Fukui Y, Kozasa T, Kaziro Y, Takeda T, Yamamoto M. Role of a ras homolog in the life cycle of Schizosaccharomyces pombe. Cell 1986; 44:329 - 336
- Neuman-Silberberg FS, Schejter E, Hoffmann FM, Shilo BZ. The Drosophila ras oncogenes: structure and nucleotide sequence. Cell 1984; 37:1027 - 1033
- Beitel GJ, Clark SG, Horvitz HR. Caenorhabditis elegans ras gene let-60 acts as a switch in the pathway of vulval induction. Nature 1990; 348:503 - 509
- Han M, Sternberg PW. let-60, a gene that specifies cell fates during C. elegans vulval induction, encodes a ras protein. Cell 1990; 63:921 - 931
- Reymond CD, Gomer RH, Mehdy MC, Firtel RA. Developmental regulation of a Dictyostelium gene encoding a protein homologous to mammalian ras protein. Cell 1984; 39:141 - 148
- Cheng R, Bradford S, Barnes D, Williams D, Hendricks J, Bailey G. Cloning, sequencing, and embryonic expression of an N-ras proto-oncogene isolated from an enriched zebrafish (Danio rerio) cDNA library. Mol Mar Biol Biotechnol 1997; 6:40 - 47
- Liu L, Zhu S, Gong Z, Low BC. K-ras/PI3K-Akt signaling is essential for zebrafish hematopoiesis and angiogenesis. PLoS One 2008; 3:e2850
- Colicelli J. Human RAS superfamily proteins and related GTPases. Sci STKE 2004; 2004:RE13
- Gallwitz D, Donath C, Sander C. A yeast gene encoding a protein homologous to the human c-has/bas proto-oncogene product. Nature 1983; 306:704 - 707
- Madaule P, Axel R. A novel ras-related gene family. Cell 1985; 41:31 - 40
- Madaule P, Axel R, Myers AM. Characterization of two members of the rho gene family from the yeast Saccharomyces cerevisiae. Proc Natl Acad Sci USA 1987; 84:779 - 783
- Yeramian P, Chardin P, Madaule P, Tavitian A. Nucleotide sequence of human rho cDNA clone 12. Nucleic Acids Res 1987; 15:1869
- Chardin P, Madaule P, Tavitian A. Coding sequence of human rho cDNAs clone 6 and clone 9. Nucleic Acids Res 1988; 16:2717
- Chardin P, Tavitian A. The ral gene: a new ras related gene isolated by the use of a synthetic probe. EMBO J 1986; 5:2203 - 2208
- Touchot N, Chardin P, Tavitian A. Four additional members of the ras gene superfamily isolated by an oligonucleotide strategy: molecular cloning of YPT-related cDNAs from a rat brain library. Proc Natl Acad Sci USA 1987; 84:8210 - 8214
- Pizon V, Chardin P, Lerosey I, Olofsson B, Tavitian A. Human cDNAs rap1 and rap2 homologous to the Drosophila gene Dras3 encode proteins closely related to ras in the ‘effector’ region. Oncogene 1988; 3:201 - 204
- Lowe DG, Capon DJ, Delwart E, Sakaguchi AY, Naylor SL, Goeddel DV. Structure of the human and murine R-ras genes, novel genes closely related to ras proto-oncogenes. Cell 1987; 48:137 - 146
- Kikuchi A, Yamashita T, Kawata M, Yamamoto K, Ikeda K, Tanimoto T, et al. Purification and characterization of a novel GTP-binding protein with a molecular weight of 24,000 from bovine brain membranes. J Biol Chem 1988; 263:2897 - 2904
- Matsui Y, Kikuchi A, Kondo J, Hishida T, Teranishi Y, Takai Y. Nucleotide and deduced amino acid sequences of a GTP-binding protein family with molecular weights of 25,000 from bovine brain. J Biol Chem 1988; 263:11071 - 11074
- Kawata M, Matsui Y, Kondo J, Hishida T, Teranishi Y, Takai Y. A novel small molecular weight GTP-binding protein with the same putative effector domain as the ras proteins in bovine brain membranes. Purification, determination of primary structure, and characterization. J Biol Chem 1988; 263:18965 - 18971
- Enomoto K, Gill DM. Cholera toxin activation of adenylate cyclase. Roles of nucleoside triphosphates and a macromolecular factor in the ADP ribosylation of the GTP-dependent regulatory component. J Biol Chem 1980; 255:1252 - 1258
- Kahn RA, Gilman AG. Purification of a protein cofactor required for ADP-ribosylation of the stimulatory regulatory component of adenylate cyclase by cholera toxin. J Biol Chem 1984; 259:6228 - 6234
- Kahn RA, Gilman AG. The protein cofactor necessary for ADP-ribosylation of Gs by cholera toxin is itself a GTP binding protein. J Biol Chem 1986; 261:7906 - 7911
- Stearns T, Willingham MC, Botstein D, Kahn RA. ADP-ribosylation factor is functionally and physically associated with the Golgi complex. Proc Natl Acad Sci USA 1990; 87:1238 - 1242
- Nakano A, Brada D, Schekman R. A membrane glycoprotein, Sec12p, required for protein transport from the endoplasmic reticulum to the Golgi apparatus in yeast. J Cell Biol 1988; 107:851 - 863
- Nakano A, Muramatsu M. A novel GTP-binding protein, Sar1p, is involved in transport from the endoplasmic reticulum to the Golgi apparatus. J Cell Biol 1989; 109:2677 - 2691
- Drivas GT, Shih A, Coutavas E, Rush MG, D'Eustachio P. Characterization of four novel ras-like genes expressed in a human teratocarcinoma cell line. Mol Cell Biol 1990; 10:1793 - 1798
- Becker Y. Antiviral drugs. Mode of action and chemotherapy of viral infections of man. Monogr Virol 1976; 11:1 - 130
- Melchior F, Paschal B, Evans J, Gerace L. Inhibition of nuclear protein import by nonhydrolyzable analogues of GTP and identification of the small GTPase Ran/TC4 as an essential transport factor. J Cell Biol 1993; 123:1649 - 1659
- Moore MS, Blobel G. The GTP-binding protein Ran/TC4 is required for protein import into the nucleus. Nature 1993; 365:661 - 663
- Lundquist EA. Small GTPases. WormBook 2006; 1 - 18
- Jiang SY, Ramachandran S. Comparative and evolutionary analysis of genes encoding small GTPases and their activating proteins in eukaryotic genomes. Physiol Genomics 2006; 24:235 - 251
- Vernoud V, Horton AC, Yang Z, Nielsen E. Analysis of the small GTPase gene superfamily of Arabidopsis. Plant Physiol 2003; 131:1191 - 1208
- Rossman KL, Der CJ, Sondek J. GEF means go: turning on RHO GTPases with guanine nucleotide-exchange factors. Nat Rev Mol Cell Biol 2005; 6:167 - 180
- Yamagata K, Sanders LK, Kaufmann WE, Yee W, Barnes CA, Nathans D, et al. rheb, a growth factor- and synaptic activity-regulated gene, encodes a novel Ras-related protein. J Biol Chem 1994; 269:16333 - 16339
- Aspuria PJ, Tamanoi F. The Rheb family of GTP-binding proteins. Cell Signal 2004; 16:1105 - 1112
- Stenmark H. Rab GTPases as coordinators of vesicle traffic. Nat Rev Mol Cell Biol 2009; 10:513 - 525
- Kahn RA, Cherfils J, Elias M, Lovering RC, Munro S, Schurmann A. Nomenclature for the human Arf family of GTP-binding proteins: ARF, ARL, and SAR proteins. J Cell Biol 2006; 172:645 - 650
- Shih TY, Papageorge AG, Stokes PE, Weeks MO, Scolnick EM. Guanine nucleotide-binding and autophosphorylating activities associated with the p21src protein of Harvey murine sarcoma virus. Nature 1980; 287:686 - 691
- Hurley JB, Simon MI, Teplow DB, Robishaw JD, Gilman AG. Homologies between signal transducing G proteins and ras gene products. Science 1984; 226:860 - 862
- McGrath JP, Capon DJ, Goeddel DV, Levinson AD. Comparative biochemical properties of normal and activated human ras p21 protein. Nature 1984; 310:644 - 649
- Sweet RW, Yokoyama S, Kamata T, Feramisco JR, Rosenberg M, Gross M. The product of ras is a GTPase and the T24 oncogenic mutant is deficient in this activity. Nature 1984; 311:273 - 275
- Manne V, Bekesi E, Kung HF. Ha-ras proteins exhibit GTPase activity: point mutations that activate Ha-ras gene products result in decreased GTPase activity. Proc Natl Acad Sci USA 1985; 82:376 - 380
- Der CJ, Finkel T, Cooper GM. Biological and biochemical properties of human rasH genes mutated at codon 61. Cell 1986; 44:167 - 176
- Trahey M, McCormick F. A cytoplasmic protein stimulates normal N-ras p21 GTPase, but does not affect oncogenic mutants. Science 1987; 238:542 - 545
- Trahey M, Wong G, Halenbeck R, Rubinfeld B, Martin GA, Ladner M, et al. Molecular cloning of two types of GAP complementary DNA from human placenta. Science 1988; 242:1697 - 1700
- Gibbs JB, Schaber MD, Allard WJ, Sigal IS, Scolnick EM. Purification of ras GTPase activating protein from bovine brain. Proc Natl Acad Sci USA 1988; 85:5026 - 5030
- Vogel US, Dixon RA, Schaber MD, Diehl RE, Marshall MS, Scolnick EM, et al. Cloning of bovine GAP and its interaction with oncogenic ras p21. Nature 1988; 335:90 - 93
- Ballester R, Marchuk D, Boguski M, Saulino A, Letcher R, Wigler M, et al. The NF1 locus encodes a protein functionally related to mammalian GAP and yeast IRA proteins. Cell 1990; 63:851 - 859
- Martin GA, Viskochil D, Bollag G, McCabe PC, Crosier WJ, Haubruck H, et al. The GAP-related domain of the neurofibromatosis type 1 gene product interacts with ras p21. Cell 1990; 63:843 - 849
- Xu GF, Lin B, Tanaka K, Dunn D, Wood D, Gesteland R, et al. The catalytic domain of the neurofibromatosis type 1 gene product stimulates ras GTPase and complements ira mutants of S. cerevisiae. Cell 1990; 63:835 - 841
- Mitin N, Rossman KL, Der CJ. Signaling interplay in Ras superfamily function. Curr Biol 2005; 15:R563 - R574
- de Vos AM, Tong L, Milburn MV, Matias PM, Jancarik J, Noguchi S, et al. Three-dimensional structure of an oncogene protein: catalytic domain of human c-H-ras p21. Science 1988; 239:888 - 893
- Pai EF, Kabsch W, Krengel U, Holmes KC, John J, Wittinghofer A. Structure of the guanine-nucleotide-binding domain of the Ha-ras oncogene product p21 in the triphosphate conformation. Nature 1989; 341:209 - 214
- Tong L, Milburn MV, de Vos AM, Kim SH. Structure of ras proteins. Science 1989; 245:244
- Milburn MV, Tong L, deVos AM, Brunger A, Yamaizumi Z, Nishimura S, et al. Molecular switch for signal transduction: structural differences between active and inactive forms of protooncogenic ras proteins. Science 1990; 247:939 - 945
- Schlichting I, Almo SC, Rapp G, Wilson K, Petratos K, Lentfer A, et al. Time-resolved X-ray crystallographic study of the conformational change in Ha-Ras p21 protein on GTP hydrolysis. Nature 1990; 345:309 - 315
- Scheffzek K, Lautwein A, Kabsch W, Ahmadian MR, Wittinghofer A. Crystal structure of the GTPase-activating domain of human p120GAP and implications for the interaction with Ras. Nature 1996; 384:591 - 596
- Robinson LC, Gibbs JB, Marshall MS, Sigal IS, Tatchell K. CDC25: a component of the RAS-adenylate cyclase pathway in Saccharomyces cerevisiae. Science 1987; 235:1218 - 1221
- Broek D, Toda T, Michaeli T, Levin L, Birchmeier C, Zoller M, et al. The S. cerevisiae CDC25 gene product regulates the RAS/adenylate cyclase pathway. Cell 1987; 48:789 - 799
- Crechet JB, Poullet P, Mistou MY, Parmeggiani A, Camonis J, Boy-Marcotte E, et al. Enhancement of the GDP-GTP exchange of RAS proteins by the carboxyl-terminal domain of SCD25. Science 1990; 248:866 - 868
- Boy-Marcotte E, Ikonomi P, Jacquet M. SDC25, a dispensable Ras guanine nucleotide exchange factor of Saccharomyces cerevisiae differs from CDC25 by its regulation. Mol Biol Cell 1996; 7:529 - 539
- Jones S, Vignais ML, Broach JR. The CDC25 protein of Saccharomyces cerevisiae promotes exchange of guanine nucleotides bound to ras. Mol Cell Biol 1991; 11:2641 - 2646
- Rogge RD, Karlovich CA, Banerjee U. Genetic dissection of a neurodevelopmental pathway: Son of sevenless functions downstream of the sevenless and EGF receptor tyrosine kinases. Cell 1991; 64:39 - 48
- Bonfini L, Karlovich CA, Dasgupta C, Banerjee U. The Son of sevenless gene product: a putative activator of Ras. Science 1992; 255:603 - 606
- Wei W, Mosteller RD, Sanyal P, Gonzales E, McKinney D, Dasgupta C, et al. Identification of a mammalian gene structurally and functionally related to the CDC25 gene of Saccharomyces cerevisiae. Proc Natl Acad Sci USA 1992; 89:7100 - 7104
- Bowtell D, Fu P, Simon M, Senior P. Identification of murine homologues of the Drosophila son of sevenless gene: potential activators of ras. Proc Natl Acad Sci USA 1992; 89:6511 - 6515
- Shou C, Farnsworth CL, Neel BG, Feig LA. Molecular cloning of cDNAs encoding a guanine-nucleotide-releasing factor for Ras p21. Nature 1992; 358:351 - 354
- Martegani E, Vanoni M, Zippel R, Coccetti P, Brambilla R, Ferrari C, et al. Cloning by functional complementation of a mouse cDNA encoding a homologue of CDC25, a Saccharomyces cerevisiae RAS activator. EMBO J 1992; 11:2151 - 2157
- Boriack-Sjodin PA, Margarit SM, Bar-Sagi D, Kuriyan J. The structural basis of the activation of Ras by Sos. Nature 1998; 394:337 - 343
- Tognon CE, Kirk HE, Passmore LA, Whitehead IP, Der CJ, Kay RJ. Regulation of RasGRP via a phorbol ester-responsive C1 domain. Mol Cell Biol 1998; 18:6995 - 7008
- Ebinu JO, Bottorff DA, Chan EY, Stang SL, Dunn RJ, Stone JC. RasGRP, a Ras guanyl nucleotide-releasing protein with calcium- and diacylglycerol-binding motifs. Science 1998; 280:1082 - 1086
- Reuther GW, Lambert QT, Rebhun JF, Caligiuri MA, Quilliam LA, Der CJ. RasGRP4 is a novel Ras activator isolated from acute myeloid leukemia. J Biol Chem 2002; 277:30508 - 30514
- Kamata T, Feramisco JR. Epidermal growth factor stimulates guanine nucleotide binding activity and phosphorylation of ras oncogene proteins. Nature 1984; 310:147 - 150
- Mulcahy LS, Smith MR, Stacey DW. Requirement for ras proto-oncogene function during serum-stimulated growth of NIH 3T3 cells. Nature 1985; 313:241 - 243
- Smith MR, DeGudicibus SJ, Stacey DW. Requirement for c-ras proteins during viral oncogene transformation. Nature 1986; 320:540 - 543
- Aroian RV, Koga M, Mendel JE, Ohshima Y, Sternberg PW. The let-23 gene necessary for Caenorhabditis elegans vulval induction encodes a tyrosine kinase of the EGF receptor subfamily. Nature 1990; 348:693 - 699
- Fortini ME, Simon MA, Rubin GM. Signalling by the sevenless protein tyrosine kinase is mimicked by Ras1 activation. Nature 1992; 355:559 - 561
- Satoh T, Endo M, Nakafuku M, Nakamura S, Kaziro Y. Platelet-derived growth factor stimulates formation of active p21ras.GTP complex in Swiss mouse 3T3 cells. Proc Natl Acad Sci USA 1990; 87:5993 - 5997
- Gibbs JB, Marshall MS, Scolnick EM, Dixon RA, Vogel US. Modulation of guanine nucleotides bound to Ras in NIH3T3 cells by oncogenes, growth factors, and the GTPase activating protein (GAP). J Biol Chem 1990; 265:20437 - 20442
- Satoh T, Endo M, Nakafuku M, Akiyama T, Yamamoto T, Kaziro Y. Accumulation of p21ras.GTP in response to stimulation with epidermal growth factor and oncogene products with tyrosine kinase activity. Proc Natl Acad Sci USA 1990; 87:7926 - 7929
- Lowenstein EJ, Daly RJ, Batzer AG, Li W, Margolis B, Lammers R, et al. The SH2 and SH3 domain-containing protein GRB2 links receptor tyrosine kinases to ras signaling. Cell 1992; 70:431 - 442
- Olivier JP, Raabe T, Henkemeyer M, Dickson B, Mbamalu G, Margolis B, et al. A Drosophila SH2-SH3 adaptor protein implicated in coupling the sevenless tyrosine kinase to an activator of Ras guanine nucleotide exchange, Sos. Cell 1993; 73:179 - 191
- Simon MA, Dodson GS, Rubin GM. An SH3-SH2-SH3 protein is required for p21Ras1 activation and binds to sevenless and Sos proteins in vitro. Cell 1993; 73:169 - 177
- Egan SE, Giddings BW, Brooks MW, Buday L, Sizeland AM, Weinberg RA. Association of Sos Ras exchange protein with Grb2 is implicated in tyrosine kinase signal transduction and transformation. Nature 1993; 363:45 - 51
- Gale NW, Kaplan S, Lowenstein EJ, Schlessinger J, Bar-Sagi D. Grb2 mediates the EGF-dependent activation of guanine nucleotide exchange on Ras. Nature 1993; 363:88 - 92
- Li N, Batzer A, Daly R, Yajnik V, Skolnik E, Chardin P, et al. Guanine-nucleotide-releasing factor hSos1 binds to Grb2 and links receptor tyrosine kinases to Ras signalling. Nature 1993; 363:85 - 88
- Rozakis-Adcock M, Fernley R, Wade J, Pawson T, Bowtell D. The SH2 and SH3 domains of mammalian Grb2 couple the EGF receptor to the Ras activator mSos1. Nature 1993; 363:83 - 85
- Buday L, Downward J. Epidermal growth factor regulates p21ras through the formation of a complex of receptor, Grb2 adapter protein, and Sos nucleotide exchange factor. Cell 1993; 73:611 - 620
- Chardin P, Camonis JH, Gale NW, van Aelst L, Schlessinger J, Wigler MH, et al. Human Sos1: a guanine nucleotide exchange factor for Ras that binds to GRB2. Science 1993; 260:1338 - 1343
- Quilliam LA, Huff SY, Rabun KM, Wei W, Park W, Broek D, et al. Membrane-targeting potentiates guanine nucleotide exchange factor CDC25 and SOS1 activation of Ras transforming activity. Proc Natl Acad Sci USA 1994; 91:8512 - 8516
- Aronheim A, Engelberg D, Li N, al-Alawi N, Schlessinger J, Karin M. Membrane targeting of the nucleotide exchange factor Sos is sufficient for activating the Ras signaling pathway. Cell 1994; 78:949 - 961
- Giubellino A, Burke TR Jr, Bottaro DP. Grb2 signaling in cell motility and cancer. Expert Opin Ther Targets 2008; 12:1021 - 1033
- Buday L, Downward J. Many faces of Ras activation. Biochim Biophys Acta 2008; 1786:178 - 187
- Harden TK, Sondek J. Regulation of phospholipase C isozymes by ras superfamily GTPases. Annu Rev Pharmacol Toxicol 2006; 46:355 - 379
- Toda T, Uno I, Ishikawa T, Powers S, Kataoka T, Broek D, et al. In yeast, RAS proteins are controlling elements of adenylate cyclase. Cell 1985; 40:27 - 36
- DeFeo-Jones D, Tatchell K, Robinson LC, Sigal IS, Vass WC, Lowy DR, et al. Mammalian and yeast ras gene products: biological function in their heterologous systems. Science 1985; 228:179 - 184
- Kataoka T, Powers S, Cameron S, Fasano O, Goldfarb M, Broach J, et al. Functional homology of mammalian and yeast RAS genes. Cell 1985; 40:19 - 26
- Broek D, Samiy N, Fasano O, Fujiyama A, Tamanoi F, Northup J, et al. Differential activation of yeast adenylate cyclase by wild-type and mutant RAS proteins. Cell 1985; 41:763 - 769
- Birchmeier C, Broek D, Wigler M. ras proteins can induce meiosis in Xenopus oocytes. Cell 1985; 43:615 - 621
- Dickson B, Sprenger F, Morrison D, Hafen E. Raf functions downstream of Ras1 in the Sevenless signal transduction pathway. Nature 1992; 360:600 - 603
- Han M, Golden A, Han Y, Sternberg PW. C. elegans lin-45 raf gene participates in let-60 ras-stimulated vulval differentiation. Nature 1993; 363:133 - 140
- Van Aelst L, Barr M, Marcus S, Polverino A, Wigler M. Complex formation between RAS and RAF and other protein kinases. Proc Natl Acad Sci USA 1993; 90:6213 - 6217
- Warne PH, Viciana PR, Downward J. Direct interaction of Ras and the amino-terminal region of Raf-1 in vitro. Nature 1993; 364:352 - 355
- Zhang XF, Settleman J, Kyriakis JM, Takeuchi-Suzuki E, Elledge SJ, Marshall MS, et al. Normal and oncogenic p21ras proteins bind to the amino-terminal regulatory domain of c-Raf-1. Nature 1993; 364:308 - 313
- Moodie SA, Willumsen BM, Weber MJ, Wolfman A. Complexes of Ras.GTP with Raf-1 and mitogen-activated protein kinase kinase. Science 1993; 260:1658 - 1661
- Rapp UR, Goldsborough MD, Mark GE, Bonner TI, Groffen J, Reynolds FH Jr, et al. Structure and biological activity of v-raf, a unique oncogene transduced by a retrovirus. Proc Natl Acad Sci USA 1983; 80:4218 - 4222
- Kan NC, Flordellis CS, Mark GE, Duesberg PH, Papas TS. A common onc gene sequence transduced by avian carcinoma virus MH2 and by murine sarcoma virus 3611. Science 1984; 223:813 - 816
- Jansen HW, Lurz R, Bister K, Bonner TI, Mark GE, Rapp UR. Homologous cell-derived oncogenes in avian carcinoma virus MH2 and murine sarcoma virus 3611. Nature 1984; 307:281 - 284
- Gallego C, Gupta SK, Heasley LE, Qian NX, Johnson GL. Mitogen-activated protein kinase activation resulting from selective oncogene expression in NIH 3T3 and rat 1a cells. Proc Natl Acad Sci USA 1992; 89:7355 - 7359
- Kyriakis JM, App H, Zhang XF, Banerjee P, Brautigan DL, Rapp UR, et al. Raf-1 activates MAP kinase-kinase. Nature 1992; 358:417 - 421
- Wood KW, Sarnecki C, Roberts TM, Blenis J. ras mediates nerve growth factor receptor modulation of three signal-transducing protein kinases: MAP kinase, Raf-1, and RSK. Cell 1992; 68:1041 - 1050
- Howe LR, Leevers SJ, Gomez N, Nakielny S, Cohen P, Marshall CJ. Activation of the MAP kinase pathway by the protein kinase raf. Cell 1992; 71:335 - 342
- Dent P, Haser W, Haystead TA, Vincent LA, Roberts TM, Sturgill TW. Activation of mitogen-activated protein kinase kinase by v-Raf in NIH 3T3 cells and in vitro. Science 1992; 257:1404 - 1407
- Masuda T, Kariya K, Shinkai M, Okada T, Kataoka T. Protein kinase Byr2 is a target of Ras1 in the fission yeast Schizosaccharomyces pombe. J Biol Chem 1995; 270:1979 - 1982
- Vojtek AB, Hollenberg SM, Cooper JA. Mammalian Ras interacts directly with the serine/threonine kinase Raf. Cell 1993; 74:205 - 214
- Rodriguez-Viciana P, Warne PH, Dhand R, Vanhaesebroeck B, Gout I, Fry MJ, et al. Phosphatidylinositol-3-OH kinase as a direct target of Ras. Nature 1994; 370:527 - 532
- Spaargaren M, Bischoff JR. Identification of the guanine nucleotide dissociation stimulator for Ral as a putative effector molecule of R-ras, H-ras, K-ras, and Rap. Proc Natl Acad Sci USA 1994; 91:12609 - 12613
- Kikuchi A, Demo SD, Ye ZH, Chen YW, Williams LT. ralGDS family members interact with the effector loop of ras p21. Mol Cell Biol 1994; 14:7483 - 7491
- Hofer F, Fields S, Schneider C, Martin GS. Activated Ras interacts with the Ral guanine nucleotide dissociation stimulator. Proc Natl Acad Sci USA 1994; 91:11089 - 11093
- Urano T, Emkey R, Feig LA. Ral-GTPases mediate a distinct downstream signaling pathway from Ras that facilitates cellular transformation. EMBO J 1996; 15:810 - 816
- White MA, Vale T, Camonis JH, Schaefer E, Wigler MH. A role for the Ral guanine nucleotide dissociation stimulator in mediating Ras-induced transformation. J Biol Chem 1996; 271:16439 - 16442
- Hamad NM, Elconin JH, Karnoub AE, Bai W, Rich JN, Abraham RT, et al. Distinct requirements for Ras oncogenesis in human versus mouse cells. Genes Dev 2002; 16:2045 - 2057
- Chien Y, White MA. RAL GTPases are linchpin modulators of human tumour-cell proliferation and survival. EMBO Rep 2003; 4:800 - 806
- Bodemann BO, White MA. Ral GTPases and cancer: linchpin support of the tumorigenic platform. Nat Rev Cancer 2008; 8:133 - 140
- Gonzalez-Garcia A, Pritchard CA, Paterson HF, Mavria G, Stamp G, Marshall CJ. RalGDS is required for tumor formation in a model of skin carcinogenesis. Cancer Cell 2005; 7:219 - 226
- Lambert JM, Lambert QT, Reuther GW, Malliri A, Siderovski DP, Sondek J, et al. Tiam1 mediates Ras activation of Rac by a PI(3)K-independent mechanism. Nat Cell Biol 2002; 4:621 - 625
- Malliri A, van der Kammen RA, Clark K, van der Valk M, Michiels F, Collard JG. Mice deficient in the Rac activator Tiam1 are resistant to Ras-induced skin tumours. Nature 2002; 417:867 - 871
- Shibatohge M, Kariya K, Liao Y, Hu CD, Watari Y, Goshima M, et al. Identification of PLC210, a Caenorhabditis elegans phospholipase C, as a putative effector of Ras. J Biol Chem 1998; 273:6218 - 6222
- Kelley GG, Reks SE, Ondrako JM, Smrcka AV. Phospholipase C(epsilon): a novel Ras effector. EMBO J 2001; 20:743 - 754
- Lopez I, Mak EC, Ding J, Hamm HE, Lomasney JW. A novel bifunctional phospholipase c that is regulated by Galpha 12 and stimulates the Ras/mitogen-activated protein kinase pathway. J Biol Chem 2001; 276:2758 - 2765
- Song C, Hu CD, Masago M, Kariyai K, Yamawaki-Kataoka Y, Shibatohge M, et al. Regulation of a novel human phospholipase C, PLCepsilon, through membrane targeting by Ras. J Biol Chem 2001; 276:2752 - 2757
- Bai Y, Edamatsu H, Maeda S, Saito H, Suzuki N, Satoh T, et al. Crucial role of phospholipase Cepsilon in chemical carcinogen-induced skin tumor development. Cancer Res 2004; 64:8808 - 8810
- Avruch J, Xavier R, Bardeesy N, Zhang XF, Praskova M, Zhou D, et al. Rassf family of tumor suppressor polypeptides. J Biol Chem 2009; 284:11001 - 11005
- Vavvas D, Li X, Avruch J, Zhang XF. Identification of Nore1 as a potential Ras effector. J Biol Chem 1998; 273:5439 - 5442
- Khokhlatchev A, Rabizadeh S, Xavier R, Nedwidek M, Chen T, Zhang XF, et al. Identification of a novel Ras-regulated proapoptotic pathway. Curr Biol 2002; 12:253 - 265
- Vos MD, Ellis CA, Bell A, Birrer MJ, Clark GJ. Ras uses the novel tumor suppressor RASSF1 as an effector to mediate apoptosis. J Biol Chem 2000; 275:35669 - 35672
- Vos MD, Ellis CA, Elam C, Ulku AS, Taylor BJ, Clark GJ. RASSF2 is a novel K-Ras-specific effector and potential tumor suppressor. J Biol Chem 2003; 278:28045 - 28051
- Eckfeld K, Hesson L, Vos MD, Bieche I, Latif F, Clark GJ. RASSF4/AD037 is a potential ras effector/tumor suppressor of the RASSF family. Cancer Res 2004; 64:8688 - 8693
- Allen NP, Donninger H, Vos MD, Eckfeld K, Hesson L, Gordon L, et al. RASSF6 is a novel member of the RASSF family of tumor suppressors. Oncogene 2007; 26:6203 - 6211
- Nassar N, Horn G, Herrmann C, Scherer A, McCormick F, Wittinghofer A. The 2.2 A crystal structure of the Ras-binding domain of the serine/threonine kinase c-Raf1 in complex with Rap1A and a GTP analogue. Nature 1995; 375:554 - 560
- Herrmann C, Martin GA, Wittinghofer A. Quantitative analysis of the complex between p21ras and the Ras-binding domain of the human Raf-1 protein kinase. J Biol Chem 1995; 270:2901 - 2905
- Taylor SJ, Shalloway D. Cell cycle-dependent activation of Ras. Curr Biol 1996; 6:1621 - 1627
- Huang L, Hofer F, Martin GS, Kim SH. Structural basis for the interaction of Ras with RalGDS. Nat Struct Biol 1998; 5:422 - 426
- Vetter IR, Linnemann T, Wohlgemuth S, Geyer M, Kalbitzer HR, Herrmann C, et al. Structural and biochemical analysis of Ras-effector signaling via RalGDS. FEBS Lett 1999; 451:175 - 180
- Stieglitz B, Bee C, Schwarz D, Yildiz O, Moshnikova A, Khokhlatchev A, et al. Novel type of Ras effector interaction established between tumour suppressor NORE1A and Ras switch II. EMBO J 2008; 27:1995 - 2005
- Pacold ME, Suire S, Perisic O, Lara-Gonzalez S, Davis CT, Walker EH, et al. Crystal structure and functional analysis of Ras binding to its effector phosphoinositide 3-kinase gamma. Cell 2000; 103:931 - 943
- Willumsen BM, Christensen A, Hubbert NL, Papageorge AG, Lowy DR. The p21 ras C-terminus is required for transformation and membrane association. Nature 1984; 310:583 - 586
- Willumsen BM, Norris K, Papageorge AG, Hubbert NL, Lowy DR. Harvey murine sarcoma virus p21 ras protein: biological and biochemical significance of the cysteine nearest the carboxy terminus. EMBO J 1984; 3:2581 - 2585
- Sefton BM, Trowbridge IS, Cooper JA, Scolnick EM. The transforming proteins of Rous sarcoma virus, Harvey sarcoma virus and Abelson virus contain tightly bound lipid. Cell 1982; 31:465 - 474
- Buss JE, Sefton BM. Direct identification of palmitic acid as the lipid attached to p21ras. Mol Cell Biol 1986; 6:116 - 122
- Magee AI, Gutierrez L, McKay IA, Marshall CJ, Hall A. Dynamic fatty acylation of p21N-ras. EMBO J 1987; 6:3353 - 3357
- Fujiyama A, Matsumoto K, Tamanoi F. A novel yeast mutant defective in the processing of ras proteins: assessment of the effect of the mutation on processing steps. EMBO J 1987; 6:223 - 228
- Clarke S, Vogel JP, Deschenes RJ, Stock J. Posttranslational modification of the Ha-ras oncogene protein: evidence for a third class of protein carboxyl methyltransferases. Proc Natl Acad Sci USA 1988; 85:4643 - 4647
- Gutierrez L, Magee AI, Marshall CJ, Hancock JF. Post-translational processing of p21ras is two-step and involves carboxyl-methylation and carboxy-terminal proteolysis. EMBO J 1989; 8:1093 - 1098
- Deschenes RJ, Stimmel JB, Clarke S, Stock J, Broach JR. RAS2 protein of Saccharomyces cerevisiae is methylesterified at its carboxyl terminus. J Biol Chem 1989; 264:11865 - 11873
- Fujiyama A, Tamanoi F. RAS2 protein of Saccharomyces cerevisiae undergoes removal of methionine at N terminus and removal of three amino acids at C terminus. J Biol Chem 1990; 265:3362 - 3368
- Schafer WR, Kim R, Sterne R, Thorner J, Kim SH, Rine J. Genetic and pharmacological suppression of oncogenic mutations in ras genes of yeast and humans. Science 1989; 245:379 - 385
- Hancock JF, Magee AI, Childs JE, Marshall CJ. All ras proteins are polyisoprenylated but only some are palmitoylated. Cell 1989; 57:1167 - 1177
- Casey PJ, Solski PA, Der CJ, Buss JE. p21ras is modified by a farnesyl isoprenoid. Proc Natl Acad Sci USA 1989; 86:8323 - 8327
- Hancock JF, Paterson H, Marshall CJ. A polybasic domain or palmitoylation is required in addition to the CAAX motif to localize p21ras to the plasma membrane. Cell 1990; 63:133 - 139
- Hancock JF, Cadwallader K, Paterson H, Marshall CJ. A CAAX or a CAAL motif and a second signal are sufficient for plasma membrane targeting of ras proteins. EMBO J 1991; 10:4033 - 4039
- Leevers SJ, Paterson HF, Marshall CJ. Requirement for Ras in Raf activation is overcome by targeting Raf to the plasma membrane. Nature 1994; 369:411 - 414
- Stokoe D, Macdonald SG, Cadwallader K, Symons M, Hancock JF. Activation of Raf as a result of recruitment to the plasma membrane. Science 1994; 264:1463 - 1467
- Choy E, Chiu VK, Silletti J, Feoktistov M, Morimoto T, Michaelson D, et al. Endomembrane trafficking of ras: the CAAX motif targets proteins to the ER and Golgi. Cell 1999; 98:69 - 80
- Apolloni A, Prior IA, Lindsay M, Parton RG, Hancock JF. H-ras but not K-ras traffics to the plasma membrane through the exocytic pathway. Mol Cell Biol 2000; 20:2475 - 2487
- Mochizuki N, Yamashita S, Kurokawa K, Ohba Y, Nagai T, Miyawaki A, et al. Spatio-temporal images of growth-factor-induced activation of Ras and Rap1. Nature 2001; 411:1065 - 1068
- Lu A, Tebar F, Alvarez-Moya B, Lopez-Alcala C, Calvo M, Enrich C, et al. A clathrin-dependent pathway leads to KRas signaling on late endosomes en route to lysosomes. J Cell Biol 2009; 184:863 - 879
- Chiu VK, Bivona T, Hach A, Sajous JB, Silletti J, Wiener H, et al. Ras signalling on the endoplasmic reticulum and the Golgi. Nat Cell Biol 2002; 4:343 - 350
- Bivona TG, Perez De Castro I, Ahearn IM, Grana TM, Chiu VK, Lockyer PJ, et al. Phospholipase Cgamma activates Ras on the Golgi apparatus by means of RasGRP1. Nature 2003; 424:694 - 698
- Arozarena I, Matallanas D, Berciano MT, Sanz-Moreno V, Calvo F, Munoz MT, et al. Activation of H-Ras in the endoplasmic reticulum by the RasGRF family guanine nucleotide exchange factors. Mol Cell Biol 2004; 24:1516 - 1530
- Goodwin JS, Drake KR, Rogers C, Wright L, Lippincott-Schwartz J, Philips MR, et al. Depalmitoylated Ras traffics to and from the Golgi complex via a nonvesicular pathway. J Cell Biol 2005; 170:261 - 272
- Rocks O, Peyker A, Kahms M, Verveer PJ, Koerner C, Lumbierres M, et al. An acylation cycle regulates localization and activity of palmitoylated Ras isoforms. Science 2005; 307:1746 - 1752
- Jura N, Scotto-Lavino E, Sobczyk A, Bar-Sagi D. Differential modification of Ras proteins by ubiquitination. Mol Cell 2006; 21:679 - 687
- John J, Frech M, Wittinghofer A. Biochemical properties of Ha-ras encoded p21 mutants and mechanism of the autophosphorylation reaction. J Biol Chem 1988; 263:11792 - 11799
- Ballester R, Furth ME, Rosen OM. Phorbol ester- and protein kinase C-mediated phosphorylation of the cellular Kirsten ras gene product. J Biol Chem 1987; 262:2688 - 2695
- Bivona TG, Quatela SE, Bodemann BO, Ahearn IM, Soskis MJ, Mor A, et al. PKC regulates a farnesyl-electrostatic switch on K-Ras that promotes its association with Bcl-XL on mitochondria and induces apoptosis. Mol Cell 2006; 21:481 - 493
- Wu JC, Chen TY, Yu CT, Tsai SJ, Hsu JM, Tang MJ, et al. Identification of V23RalA-Ser194 as a critical mediator for Aurora-A-induced cellular motility and transformation by small pool expression screening. J Biol Chem 2005; 280:9013 - 9022
- Riento K, Totty N, Villalonga P, Garg R, Guasch R, Ridley AJ. RhoE function is regulated by ROCK I-mediated phosphorylation. EMBO J 2005; 24:1170 - 1180
- Sablina AA, Chen W, Arroyo JD, Corral L, Hector M, Bulmer SE, et al. The tumor suppressor PP2A Abeta regulates the RalA GTPase. Cell 2007; 129:969 - 982
- Madigan JP, Bodemann BO, Brady DC, Dewar BJ, Keller PJ, Leitges M, et al. Regulation of Rnd3 localization and function by protein kinase C alpha-mediated phosphorylation. Biochem J 2009; 424:153 - 161
- Lim KH, Brady DC, Kashatus DF, Ancrile BB, Der CJ, Cox AD, et al. Aurora-A phosphorylates, activates, and relocalizes the small GTPase RalA. Mol Cell Biol 2010; 30:508 - 523
- Lerosey I, Pizon V, Tavitian A, de Gunzburg J. The cAMP-dependent protein kinase phosphorylates the rap1 protein in vitro as well as in intact fibroblasts, but not the closely related rap2 protein. Biochem Biophys Res Commun 1991; 175:430 - 436
- Lang P, Gesbert F, Delespine-Carmagnat M, Stancou R, Pouchelet M, Bertoglio J. Protein kinase A phosphorylation of RhoA mediates the morphological and functional effects of cyclic AMP in cytotoxic lymphocytes. EMBO J 1996; 15:510 - 519
- Forget MA, Desrosiers RR, Gingras D, Beliveau R. Phosphorylation states of Cdc42 and RhoA regulate their interactions with Rho GDP dissociation inhibitor and their extraction from biological membranes. Biochem J 2002; 361:243 - 254
- Johnson L, Greenbaum D, Cichowski K, Mercer K, Murphy E, Schmitt E, et al. K-ras is an essential gene in the mouse with partial functional overlap with N-ras. Genes Dev 1997; 11:2468 - 2481
- Koera K, Nakamura K, Nakao K, Miyoshi J, Toyoshima K, Hatta T, et al. K-ras is essential for the development of the mouse embryo. Oncogene 1997; 15:1151 - 1159
- Umanoff H, Edelmann W, Pellicer A, Kucherlapati R. The murine N-ras gene is not essential for growth and development. Proc Natl Acad Sci USA 1995; 92:1709 - 1713
- Ise K, Nakamura K, Nakao K, Shimizu S, Harada H, Ichise T, et al. Targeted deletion of the H-ras gene decreases tumor formation in mouse skin carcinogenesis. Oncogene 2000; 19:2951 - 2956
- Esteban LM, Vicario-Abejon C, Fernandez-Salguero P, Fernandez-Medarde A, Swaminathan N, Yienger K, et al. Targeted genomic disruption of H-ras and N-ras, individually or in combination, reveals the dispensability of both loci for mouse growth and development. Mol Cell Biol 2001; 21:1444 - 1452
- Plowman SJ, Arends MJ, Brownstein DG, Luo F, Devenney PS, Rose L, et al. The K-Ras 4A isoform promotes apoptosis but does not affect either lifespan or spontaneous tumor incidence in aging mice. Exp Cell Res 2006; 312:16 - 26
- Potenza N, Vecchione C, Notte A, De Rienzo A, Rosica A, Bauer L, et al. Replacement of K-Ras with H-Ras supports normal embryonic development despite inducing cardiovascular pathology in adult mice. EMBO Rep 2005; 6:432 - 437
- Abankwa D, Gorfe AA, Inder K, Hancock JF. Ras membrane orientation and nanodomain localization generate isoform diversity. Proc Natl Acad Sci USA 107:1130 - 1135
- Casar B, Arozarena I, Sanz-Moreno V, Pinto A, Agudo-Ibanez L, Marais R, et al. Ras subcellular localization defines extracellular signal-regulated kinase 1 and 2 substrate specificity through distinct utilization of scaffold proteins. Mol Cell Biol 2009; 29:1338 - 1353
- Plowman SJ, Muncke C, Parton RG, Hancock JF. H-ras, K-ras, and inner plasma membrane raft proteins operate in nanoclusters with differential dependence on the actin cytoskeleton. Proc Natl Acad Sci USA 2005; 102:15500 - 15505
- Rotblat B, Yizhar O, Haklai R, Ashery U, Kloog Y. Ras and its signals diffuse through the cell on randomly moving nanoparticles. Cancer Res 2006; 66:1974 - 1981
- Tian T, Harding A, Inder K, Plowman S, Parton RG, Hancock JF. Plasma membrane nanoswitches generate high-fidelity Ras signal transduction. Nat Cell Biol 2007; 9:905 - 914
- Belanis L, Plowman SJ, Rotblat B, Hancock JF, Kloog Y. Galectin-1 is a novel structural component and a major regulator of h-ras nanoclusters. Mol Biol Cell 2008; 19:1404 - 1414
- Rotblat B, Niv H, Andre S, Kaltner H, Gabius HJ, Kloog Y. Galectin-1(L11A) predicted from a computed galectin-1 farnesyl-binding pocket selectively inhibits Ras-GTP. Cancer Res 2004; 64:3112 - 3118
- Paz A, Haklai R, Elad-Sfadia G, Ballan E, Kloog Y. Galectin-1 binds oncogenic H-Ras to mediate Ras membrane anchorage and cell transformation. Oncogene 2001; 20:7486 - 7493
- Elad-Sfadia G, Haklai R, Balan E, Kloog Y. Galectin-3 augments K-Ras activation and triggers a Ras signal that attenuates ERK but not phosphoinositide 3-kinase activity. J Biol Chem 2004; 279:34922 - 34930
- Ashery U, Yizhar O, Rotblat B, Elad-Sfadia G, Barkan B, Haklai R, et al. Spatiotemporal organization of Ras signaling: rasosomes and the galectin switch. Cell Mol Neurobiol 2006; 26:471 - 495
- Cox AD, Der CJ. Farnesyltransferase inhibitors: promises and realities. Curr Opin Pharmacol 2002; 2:388 - 393
- Sinensky M, Beck LA, Leonard S, Evans R. Differential inhibitory effects of lovastatin on protein isoprenylation and sterol synthesis. J Biol Chem 1990; 265:19937 - 19941
- Reiss Y, Goldstein JL, Seabra MC, Casey PJ, Brown MS. Inhibition of purified p21ras farnesyl:protein transferase by Cys-AAX tetrapeptides. Cell 1990; 62:81 - 88
- James GL, Goldstein JL, Brown MS, Rawson TE, Somers TC, McDowell RS, et al. Benzodiazepine peptidomimetics: potent inhibitors of Ras farnesylation in animal cells. Science 1993; 260:1937 - 1942
- Kohl NE, Mosser SD, deSolms SJ, Giuliani EA, Pompliano DL, Graham SL, et al. Selective inhibition of ras-dependent transformation by a farnesyltransferase inhibitor. Science 1993; 260:1934 - 1937
- Kohl NE, Omer CA, Conner MW, Anthony NJ, Davide JP, deSolms SJ, et al. Inhibition of farnesyltransferase induces regression of mammary and salivary carcinomas in ras transgenic mice. Nat Med 1995; 1:792 - 797
- Sepp-Lorenzino L, Ma Z, Rands E, Kohl NE, Gibbs JB, Oliff A, et al. A peptidomimetic inhibitor of farnesyl:protein transferase blocks the anchorage-dependent and -independent growth of human tumor cell lines. Cancer Res 1995; 55:5302 - 5309
- James GL, Brown MS, Cobb MH, Goldstein JL. Benzodiazepine peptidomimetic BZA-5B interrupts the MAP kinase activation pathway in H-Ras-transformed Rat-1 cells, but not in untransformed cells. J Biol Chem 1994; 269:27705 - 27714
- James GL, Goldstein JL, Brown MS. Polylysine and CVIM sequences of K-RasB dictate specificity of prenylation and confer resistance to benzodiazepine peptidomimetic in vitro. J Biol Chem 1995; 270:6221 - 6226
- Fiordalisi JJ, Johnson RL 2nd, Weinbaum CA, Sakabe K, Chen Z, Casey PJ, et al. High affinity for farnesyltransferase and alternative prenylation contribute individually to K-Ras4B resistance to farnesyltransferase inhibitors. J Biol Chem 2003; 278:41718 - 41727
- Rowell CA, Kowalczyk JJ, Lewis MD, Garcia AM. Direct demonstration of geranylgeranylation and farnesylation of Ki-Ras in vivo. J Biol Chem 1997; 272:14093 - 14097
- Whyte DB, Kirschmeier P, Hockenberry TN, Nunez-Oliva I, James L, Catino JJ, et al. K- and N-Ras are geranylgeranylated in cells treated with farnesyl protein transferase inhibitors. J Biol Chem 1997; 272:14459 - 14464
- Macdonald JS, McCoy S, Whitehead RP, Iqbal S, Wade JL 3rd, Giguere JK, et al. A phase II study of farnesyl transferase inhibitor R115777 in pancreatic cancer: a Southwest oncology group (SWOG 9924) study. Invest New Drugs 2005; 23:485 - 487
- Worman HJ, Fong LG, Muchir A, Young SG. Laminopathies and the long strange trip from basic cell biology to therapy. J Clin Invest 2009; 119:1825 - 1836
- Bergo MO, Ambroziak P, Gregory C, George A, Otto JC, Kim E, et al. Absence of the CAAX endoprotease Rce1: effects on cell growth and transformation. Mol Cell Biol 2002; 22:171 - 181
- Bergo MO, Gavino BJ, Hong C, Beigneux AP, McMahon M, Casey PJ, et al. Inactivation of Icmt inhibits transformation by oncogenic K-Ras and B-Raf. J Clin Invest 2004; 113:539 - 550
- Wahlstrom AM, Cutts BA, Karlsson C, Andersson KM, Liu M, Sjogren AK, et al. Rce1 deficiency accelerates the development of K-RAS-induced myeloproliferative disease. Blood 2007; 109:763 - 768
- Wahlstrom AM, Cutts BA, Liu M, Lindskog A, Karlsson C, Sjogren AK, et al. Inactivating Icmt ameliorates K-RAS-induced myeloproliferative disease. Blood 2008; 112:1357 - 1365
- Blum R, Cox AD, Kloog Y. Inhibitors of chronically active ras: potential for treatment of human malignancies. Recent Pat Anticancer Drug Discov 2008; 3:31 - 47
- Rotblat B, Ehrlich M, Haklai R, Kloog Y. The Ras inhibitor farnesylthiosalicylic acid (Salirasib) disrupts the spatiotemporal localization of active Ras: a potential treatment for cancer. Methods Enzymol 2008; 439:467 - 489
- Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, et al. Mutations of the BRAF gene in human cancer. Nature 2002; 417:949 - 954
- Rinehart J, Adjei AA, Lorusso PM, Waterhouse D, Hecht JR, Natale RB, et al. Multicenter phase II study of the oral MEK inhibitor, CI-1040, in patients with advanced non-small-cell lung, breast, colon, and pancreatic cancer. J Clin Oncol 2004; 22:4456 - 4462
- Lyons JF, Wilhelm S, Hibner B, Bollag G. Discovery of a novel Raf kinase inhibitor. Endocr Relat Cancer 2001; 8:219 - 225
- Wilhelm SM, Carter C, Tang L, Wilkie D, McNabola A, Rong H, et al. BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res 2004; 64:7099 - 7109
- Garber K. Cancer research. Melanoma drug vindicates targeted approach. Science 2009; 326:1619
- Hatzivassiliou G, Song K, Yen I, Brandhuber BJ, Anderson DJ, Alvarado R, et al. RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature
- Poulikakos PI, Zhang C, Bollag G, Shokat KM, Rosen N. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature
- Heidorn SJ, Milagre C, Whittaker S, Nourry A, Niculescu-Duvas I, Dhomen N, et al. Kinase-dead BRAF and oncogenic RAS cooperate to drive tumor progression through CRAF. Cell 140:209 - 221
- Hall-Jackson CA, Eyers PA, Cohen P, Goedert M, Boyle FT, Hewitt N, et al. Paradoxical activation of Raf by a novel Raf inhibitor. Chem Biol 1999; 6:559 - 568
- Hall-Jackson CA, Goedert M, Hedge P, Cohen P. Effect of SB 203580 on the activity of c-Raf in vitro and in vivo. Oncogene 1999; 18:2047 - 2054
- Gonzalez-Perez V, Reiner DJ, Alan JK, Mitchell C, Edwards LJ, Khazak V, et al. Genetic and functional characterization of putative Ras/Raf interaction inhibitors in C. elegans and mammalian cells. J Mol Signal 5:2
- Kato-Stankiewicz J, Hakimi I, Zhi G, Zhang J, Serebriiskii I, Guo L, et al. Inhibitors of Ras/Raf-1 interaction identified by two-hybrid screening revert Ras-dependent transformation phenotypes in human cancer cells. Proc Natl Acad Sci USA 2002; 99:14398 - 14403
- Luo J, Emanuele MJ, Li D, Creighton CJ, Schlabach MR, Westbrook TF, et al. A genome-wide RNAi screen identifies multiple synthetic lethal interactions with the Ras oncogene. Cell 2009; 137:835 - 848
- Scholl C, Frohling S, Dunn IF, Schinzel AC, Barbie DA, Kim SY, et al. Synthetic lethal interaction between oncogenic KRAS dependency and STK33 suppression in human cancer cells. Cell 2009; 137:821 - 834
- Barbie DA, Tamayo P, Boehm JS, Kim SY, Moody SE, Dunn IF, et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature 2009; 462:108 - 112
- Kaelin WG Jr. Synthetic lethality: a framework for the development of wiser cancer therapeutics. Genome Med 2009; 1:99
- Chien Y, Kim S, Bumeister R, Loo YM, Kwon SW, Johnson CL, et al. RalB GTPase-mediated activation of the IkappaB family kinase TBK1 couples innate immune signaling to tumor cell survival. Cell 2006; 127:157 - 170
- Singh A, Greninger P, Rhodes D, Koopman L, Violette S, Bardeesy N, et al. A gene expression signature associated with “K-Ras addiction” reveals regulators of EMT and tumor cell survival. Cancer Cell 2009; 15:489 - 500
- Swanson KD, Winter JM, Reis M, Bentires-Alj M, Greulich H, Grewal R, et al. SOS1 mutations are rare in human malignancies: implications for Noonan Syndrome patients. Genes Chromosomes Cancer 2008; 47:253 - 259
- Aoki Y, Niihori T, Kawame H, Kurosawa K, Ohashi H, Tanaka Y, et al. Germline mutations in HRAS protooncogene cause Costello syndrome. Nat Genet 2005; 37:1038 - 1040
- Niihori T, Aoki Y, Narumi Y, Neri G, Cave H, Verloes A, et al. Germline KRAS and BRAF mutations in cardio-facio-cutaneous syndrome. Nat Genet 2006; 38:294 - 296
- Carta C, Pantaleoni F, Bocchinfuso G, Stella L, Vasta I, Sarkozy A, et al. Germline missense mutations affecting KRAS Isoform B are associated with a severe Noonan syndrome phenotype. Am J Hum Genet 2006; 79:129 - 135
- Schubbert S, Zenker M, Rowe SL, Boll S, Klein C, Bollag G, et al. Germline KRAS mutations cause Noonan syndrome. Nat Genet 2006; 38:331 - 336
- Gripp KW. Tumor predisposition in Costello syndrome. Am J Med Genet C Semin Med Genet 2005; 137C:72 - 77
- Rauen KA, Schoyer L, McCormick F, Lin AE, Allanson JE, Stevenson DA, et al. Proceedings from the 2009 genetic syndromes of the Ras/MAPK pathway: From bedside to bench and back. Am J Med Genet A 152A:4 - 24
- Bansal A, Ramirez RD, Minna JD. Mutation analysis of the coding sequences of MEK-1 and MEK-2 genes in human lung cancer cell lines. Oncogene 1997; 14:1231 - 1234
- Tuveson DA, Shaw AT, Willis NA, Silver DP, Jackson EL, Chang S, et al. Endogenous oncogenic K-ras(G12D) stimulates proliferation and widespread neoplastic and developmental defects. Cancer Cell 2004; 5:375 - 387
- Guerra C, Mijimolle N, Dhawahir A, Dubus P, Barradas M, Serrano M, et al. Tumor induction by an endogenous K-ras oncogene is highly dependent on cellular context. Cancer Cell 2003; 4:111 - 120
- Schuhmacher AJ, Guerra C, Sauzeau V, Canamero M, Bustelo XR, Barbacid M. A mouse model for Costello syndrome reveals an Ang II-mediated hypertensive condition. J Clin Invest 2008; 118:2169 - 2179
- Santoriello C, Deflorian G, Pezzimenti F, Kawakami K, Lanfrancone L, d'Adda di Fagagna F, et al. Expression of H-RASV12 in a zebrafish model of Costello syndrome causes cellular senescence in adult proliferating cells. Dis Model Mech 2009; 2:56 - 67
- Cirstea IC, Kutsche K, Dvorsky R, Gremer L, Carta C, Horn D, et al. A restricted spectrum of NRAS mutations causes Noonan syndrome. Nat Genet 2009; 42:27 - 29
- Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell 1990; 61:759 - 767
- Hruban RH, Goggins M, Parsons J, Kern SE. Progression model for pancreatic cancer. Clin Cancer Res 2000; 6:2969 - 2972
- Hanker AB, Der CJ. The Roles of Ras Family Small GTPases in Breast Cancer. Handbook of Cell Signaling 2009; Academic Press 2763 - 2772
- Stevens EV, Der CJ. Van Golen K. Rho History. The Rho GTPases in Cancer 2010; Springer
- Repasky GA, Chenette EJ, Der CJ. Renewing the conspiracy theory debate: does Raf function alone to mediate Ras oncogenesis?. Trends Cell Biol 2004; 14:639 - 647